
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: miR-199a-3p knockdown inhibits dedifferentiated liposarcoma (DDLPS) cell viability and enhances apoptosis through targeting casein kinase-1 alpha (CK1α)
Ye Cao†
a,
Jiajia Zheng†b and
Chentao Lv *a
*a
aDepartment of General Surgery, Shanghai Public Health Clinical Center, No. 921 Rd. Tongxin, Hongkou, 200083, Shanghai, China. E-mail: y9919002tang@163.com; Tel: +86-13651613217
bDepartment of General Surgery, Zhongshan Hospital & Red Cross Hospital, Xuhui, 200030, Shanghai, China
First published on 23rd July 2019
Abstract
Dedifferentiated liposarcoma (DDLPS) is an aggressive tumor with high mortality. More insight into the biology of DDLPS tumorigenesis is needed to devise novel therapeutic approaches. Previous data showed that miRNA-199a-3p (miR-199a-3p) was strongly upregulated in DDLPS tissues. However, the biological role of miR-199a-3p in DDLPS remains unknown. In this study, we detected miR-199a-3p expression using RT-qPCR and observed that miR-199a-3p was more highly expressed in DDLPS tissues and cell lines (SW872 and LPS141). Functionally, MTT assay, flow cytometry and western blot results demonstrated that knockdown of miR-199a-3p inhibited DDLPS cell viability, enhanced apoptosis rate, and decreased expression of apoptosis-related genes Bax and cleaved caspase 3, as well as increased Bcl-2 expression in vitro. Moreover, xenograft tumors were generated and miR-199a-3p knockdown could suppress DDLPS xenograft tumor growth accompanying decreased proliferating cell nuclear antigen (PCNA) level and increased cleaved caspase 3 level in vivo. Mechanically, luciferase reporter assay and RNA immunoprecipitation (RIP) identified that CK1α was targeted and downregulated by miR-199a-3p. Expression of CK1α was lower in DDLPS tissues. Besides, there was a negative linear correlation between expressions of miR-199a-3p and CK1α in DDLPS tissues. Rescue experiments indicated that CK1α silencing could abolish the effect of miR-199a-3p knockdown on cell viability and apoptosis in DDLPS cells in vitro. In conclusion, knockdown of miR-199a-3p inhibits DDLPS cell viability and enhances apoptosis through targeting CK1α in vitro and in vivo. Our results suggest miR-199a-3p/CK1α axis may be a novel pathogen of DDLPS.
Introduction
Sarcomas are malignant heterogeneous tumors of mesenchymal derivation. Liposarcoma (LPS) is the most common soft tissue sarcoma and accounts for approximately 25% of all adult sarcomas.1 In addition, LPS is classified into three subtypes according to histopathological examination: well-differentiated/dedifferentiated (WD/DDLPS), myxoid/round cell and pleomorphic. WD/DDLPS are characterized by chromosome 12q amplification in about 90% of cases, whereas DDLPS exhibits more aggressive biological behavior with frequent multiple recurrences and subsequent metastasis in spite of multimodal treatment approaches.2 DDLPS has been thought to arise from WDLPS because the dedifferentiated component of these tumors always co-exists with an adjacent region of WDLPS and because WDLPS often progresses and recurs as DDLPS.3,4 Current treatment modalities (surgery, chemotherapy, and radiotherapy) all have limitations and surgical resection is the primary treatment for WD/DDLPS;5,6 but DDLPS exhibits 80% local recurrence and 20% metastasis in patients despite an aggressive treatment with surgery and systemic chemotherapy.5 Therefore, in order to successfully target specific tumorigenic drivers, molecularly driven studies are needed to improve the identification and increase the understanding of genetic and epigenetic deregulations in LPS, especially in DDLPS.microRNAs (miRNAs) are short (approximately 22 nucleotides) non-protein coding RNA molecules that regulate posttranscriptional gene expression by targeting the 3′ untranslated region of messenger RNAs, which contributes to various biological processes such as development, differentiation, apoptosis and proliferation. In soft tissue sarcoma, various miRNAs are differentially expressed, which are related to development, progression and invasion.7 Advanced evidences have verified that dyregulation of miRNAs is involved in the initiation and progression of LPS.8 The discovery of the regulatory functions of miRNAs in LPS progression promotes several therapeutically actionable molecular markers to be identified in WD/DDLPS.9 Moreover, miRNAs as potential diagnostic biomarkers attract much focus. Circulating miRNAs may function as fast and reliable differential diagnosis in LPS. For example, miR-25-3p and miR-92a-3p are observed to be secreted by LPS cells through extracellular vesicles (EVs) and to stimulate LPS progression, providing a novel insight that exosome-derived miRNAs may be useful as potential biomarkers of LPS outcome and as a possible approach to monitor treatment efficiency.10 miR-155 is a LPS oncogene and plasma levels of miRNA-155 serves as a powerful diagnostic marker for DDLPS.11,12
miR-199a-3p is encoded by two genes, miR-199a-1 and miR-199a-2.13 miR-199a-3p takes part in coagulative cascade, swelling and pain in diabetic peripheral neuropathy,14 myometrial contractility during pregnancy and labour,15 cardiomyocyte proliferation and cardiac regeneration during the embryonic life,16 as well as cancers.17–19 miR-199a-3p, together with miR-199a-5p, has been reported to be either upregulated or downregulated in a variety of human cancers.20 miR-199a-3p regulates various cellular processes and represents an important tumor suppressor in hepatocellular carcinoma (HCC).17 Multivariate analysis showed that miR-199a-3p associated with overall and progression-free survival, residual tumor volume and platinum-free interval in advanced epithelial ovarian cancer.18 Pencheva et al. identified convergent multi-miRNAs, including miR-1908, miR-199a-5p, and miR-199a-3p, targeting ApoE as endogenous promoters of metastatic invasion, angiogenesis, and colonization in melanoma.19 In adult soft tissue sarcomas leiomyosarcoma (LMS) and undifferentiated pleomorphic sarcoma (UPS), there were 5 miRNAs miR-199b-5p, miR-320a, miR-126, miR-22 and miR-199a-3p differentially expressed according to a miRNA analysis.21 Compared with normal fat tissue, miR-199a-3p was strongly upregulated in WD/DDLPS, and level of this miRNA was higher in DDLPS with 6.0-fold (WDLPS: 3.7-fold).13 However, there is lack of research on the role of miR-199a-3p in LPS.
To address the present lack of fundamental study, we investigated the expression of miR-199a-3p in DDLPS tissues and cell lines, as well as its regulatory mechanism through target gene. Furthermore, effect of miR-199a-3p on tumor growth was also explored in xenograft experiments. In this current study, we examined the role of miR-199a-3p as an oncogene in DDLPS.
Materials and methods
Clinical specimens
Human liposarcoma (sample subtype: DDLPS) tissues and paired normal fat tissues were obtained from thirty patients (age: 43–73 years; male: 46.7%) during surgical resection at Shanghai Public Health Clinical Center after informed consent given by each individual. The tumor tissues were both local recurrence and primary. All tissues were snap-frozen in liquid nitrogen for future use. All protocol involving human subjects were approved by the Ethics Committee of Shanghai Public Health Clinical Center. All experiments were performed in accordance with the Guidelines of “National Institutes of Health for the care and use of laboratory Animals” and Experiments were approved by the ethics committee of “Shanghai Public Health Clinical Center”. Informed consents were obtained from human participants of this study.Cell and cell culture
Human DDLPS cell lines SW872 (HTB-92) was purchased from American Tissue Type Culture Collection (ATCC; Rockville, MD, USA), and LPS141 was from Dr Jonathan Fletcher (Brigham and Women's Hospital, Boston, MA, USA). Human white pre-adipocytes primary cultures were provided from PromoCell (Heidelberg, Germany). All cells were cultured and passaged in high glucose dulbecco's modified eagle medium (DMEM; Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Gibco) and 1% antibiotics (100 U per ml penicillin and 100 μg ml−1 streptomycin; Invitrogen, Carlsbad, CA, USA). Pre-adipocytes were differentiated into adipocytes per manufacturer's instructions using a commercial pre-adipocyte differentiation media (serum free media containing: insulin, dexamethasone, IBMX, L-thyroxine, ciglitazone, and heparin) and adipocyte nutrition media (3% FBS) (supplemented media containing: insulin, dexamethasone, and 3-isobutyl-1-methylxanthine). Adipogenic differentiation was confirmed by markers of adipocyte, and FABP4 and adiponectin expressions were detected via RT-qPCR. All cells were cultured at 37 °C in a humidified environment containing 5% CO2 for indicated times.Cell transfection
SW872 and LPS141 cells were seeded into 6-well plate (Corning, NY, USA) and incubated overnight. Lipofectamine™ 2000 (Invitrogen) transfection reagent was used for transfection. miR-199a-3p mimic, miR-199a-3p inhibitor, special siRNA against CK1α, and the indicated negative controls were acquired from GenePharma (Shanghai, China). After transfection for 24 h, the cells were cultured for further functional experiments.MTT assay
The cell viability of SW872 and LPS141 cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma, Louis, MO, USA) staining after transfected for 1 d, 2 d and 3 d. 5 mg l−1 MTT was added to each well and the cultures were incubated for another 4 h at 37 °C. The supernatant was aspirated, and formazan crystals were dissolved in 150 μl dimethyl sulfoxide (DMSO; Sigma). The absorbance at 490 nm was measured with SpectraMax M4 (Molecular devices, Shanghai, China). All experiments were performed in triplicate.Flow cytometry
Apoptosis rate was analyzed by Annexin V-FITC/PI kit (Beyotime, Shanghai, China) on flow cytometry. Apoptotic cells (SW872 and LPS141 cells) were labelled with FITC-Annexin V and PI for 30 min in the dark and fluorescence was analyzed on cytoFLEX LX flow cytometer (Beckman-Counter Electronics, Jiangsu, China) using CytExpert software. Quadrants were positioned on Annexin V/PI plots to distinguish apoptotic cells (Annexin V+/PI−, Annexin V+/PI+).Real time quantitative PCR (RT-qPCR)
For examination of CK1α mRNA expression and miR-199a-3p, total RNAs from tissues and cultured cells were extracted with TRIzol reagent (Invitrogen) and the first-strand cDNA was synthesized using PrimeScript RT reagent kit (Takara, Shiga, Japan). The qPCR was performed with SYBR green qPCR master mix (Promega, Madison, WI, USA) on the ABI PRISM 7500 Real-time PCR System (Applied Biosystems, Foster city, CA, USA). Relative levels of the mRNAs were normalized to that of GAPDH mRNA, and the U6 small nuclear RNA (U6) was used as an internal control to mature miR-199a-3p. The reactions were performed in triplicate for each sample at least three independent runs and the primers involved are as follows: CK1α isoform 2: 5′-AGTGGCAGTGAAGCTAGAATCT-3′ and 5′-CGCCCAATACCCATTAGGAAGTT-3′; GAPDH: 5′-CGAGCCACATCGCTCAGACA-3′ (sense) and 5′-GTGGTGAAGACGCCAGTGGA-3′ (antisense). The detection of levels of miR-199a-3p depended on hsa-miR-199a-3p qPCR Primer Mix (Takara, Dalian, China). The ΔΔCt method was applied for all real-time PCR analysis.Western blot
Total protein from tumors and cultured cells was extracted in RIPA lysis buffer (Beyotime), and the protein concentrations were determined by Bradford protein assay reagent (Bio-Rad, Shanghai, China). 20 μg samples were loaded for the standard procedures of western blot assay. GAPDH on the same membrane was an internal standard to normalize protein levels. The primary antibodies were purchased from Cell Signaling Technology (CST; Danvers, Massachusetts, USA) and as follows: CK1α (#2655, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000), Bcl-2 (#2872, 1:1000), Bax (#2772, 1:1000), cleaved caspase 3 (#9661, 1:1000), PCNA (#2586, 1:2000), and GAPDH (#97166, 1:1000).
:1000), Bcl-2 (#2872, 1:1000), Bax (#2772, 1:1000), cleaved caspase 3 (#9661, 1:1000), PCNA (#2586, 1:2000), and GAPDH (#97166, 1:1000).
Luciferase reporter assay and RNA immunoprecipitation (RIP)
Human CK1α 3′ UTR fragment containing the potential binding sites of hsa-miR-199a-3p was cloned by PCR methods into pGL4 vector (Invitrogen), as well as the mutated CK1α 3′ UTR sequence. The pGL3-mutant type plasmid was constructed by replacing the ACUACUGU of the complementary sequence in the CK1α sequence. SW872 and LPS141 cells were transfected according to the following groups: CK1α wt+miR-NC mimic (miR-NC), CK1α wt+miR-199a-3p mimic (miR-199a-3p), CK1α wut+miR-NC, CK1α wut+miR-199a-3p. After 48 h incubation, cells were collected to measure Firefly and Renilla luciferase activity using the dual-luciferase reporter assay system (Promega). All the data were the average of at least three independent transfections.RIP was performed with SW872 and LPS141 cells extract after transfection of miR-199a-3p/NC mimic. Magna RIP™ RNA-binding protein immunoprecipitation kit (Millipore, Bradford, MA, USA) was chosen to detect expression of CK1α mRNA from the samples bound to the Ago2 or IgG antibody. All operation obeyed the standard instructions.
Xenograft experiment
microOFF™ hsa-miR-199a-3p antagomir (miR-199a-3p antagomir) and its negative control (miR-NC antagomir) were acquired from RiBoBio (Guangzhou, China). SW872 and LPS141 cells were stably transfected with miR-NC antagomir or miR-199a-3p antagomir. BALB/c nude mice (4 wk-old, female) were obtained from Model Animal Research Center of Nanjing University. The animal experiments were approved by the Institutional Review Board of Shanghai Public Health Clinical Center and were carried out in accordance with National Institutions of Health Guide for Care and Use of Laboratory Animals. Equal numbers (106) of stably transfected cells in 0.2 mL of saline were subcutaneously injected into left forelimb (6 mice per group) for 6 weeks. The tumors was measured per week with caliper. The tumor volume was calculated using the formula: V (mm3) = 1/2ab2 (a is the longest tumor axis and b is the shortest tumor axis). The mice were practiced with euthanasia on week 6 and the weight of tumors were evaluated with electronic balance. Immediately, the xenograft tumors were stored in −80 °C for further isolation of total protein and total RNA.Statistical analyses
Data were presented as mean ± standard error of mean (SEM). Statistical analyses were performed using Graphpad 6.0. The P-values were evaluated using a one-way analysis of variance (ANOVA). P < 0.05 was considered to indicate a statistically significant difference.Results
miR-199a-3p is higher expressed in DDLPS tissues and cell lines
Previous data showed that mir-199a-3p was strongly dyregulated over 3-fold in both WDLPS and DDLPS compared with normal fat tissue samples.13 Consistent with this finding, we observed a higher expression of miR-199a-3p (4.5-fold) in DDLPS tissues (Fig. 1A). Moreover, levels of miR-199a-3p in DDLPS cell lines were also detected, and miR-199a-3p was significantly upregulated in SW872 (4.7-fold) and LPS141 cells (5.6-fold) than that in normal adipocytes (Fig. 1B). The data of markers of adipocyte differentiated from pre-adipocyte was not exhibited. These data suggest that miR-199a-3p may play potential role in human DDLPS.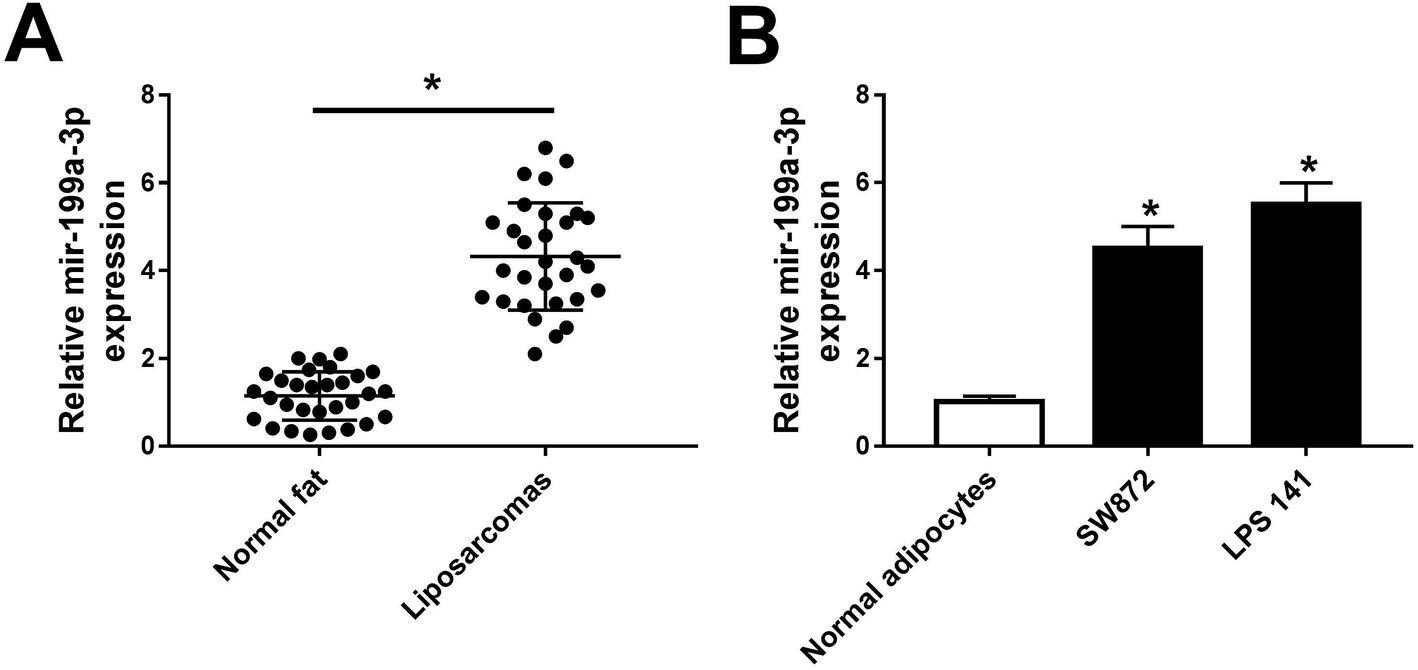 | ||
| Fig. 1 Expression of miR-199a-3p in dedifferentiated liposarcoma (DDLPS) tissues and cell lines. (A) miR-199a-3p levels were detected by RT-qPCR in normal fat tissues and DDLPS tissues. (B) miR-199a-3p levels were detected by RT-qPCR in normal adipocytes and DDLPS cell lines SW872 and LPS141. Data are plotted as mean ± SEM for miRNA performed in triplicate. *, n = 3, p < 0.05 compared to with normal samples. | ||
miR-199a-3p knockdown inhibits DDLPS cell viability and enhances apoptosis in vitro
miR-199a-3p was previously found to act as a tumor suppressor in majority of cancers and as an oncogene in gastric cancer.17–19,22 Here, its contribution to DDLPS biology had been elucidated. miR-199a-3p expression was knocked down utilizing an anti-sense construct to determine the contribution of this miRNA to DDLPS cell progression in vitro. As shown in Fig. 2A, miR-199a-3p was lower expressed in SW872 and LPS141 cells after transient transfection of miR-199a-3p inhibitor (anti-miR-199a-3p) when compared to that in negative control group (anti-NC). Transient miR-199a-3p knockdown was found to significantly inhibit cell viability, promote apoptotic cells in SW872 and LPS141 cells (Fig. 2B and C). Furthermore, expression of apoptosis-related genes was altered either; decreased Bcl-2 and increased Bax and cleaved caspase 3 occurred in miR-199a-3p-knocked down cells (Fig. 2D and E). These results indicate miR-199a-3p knockdown inhibits DDLPS cell viability and enhances apoptosis in vitro.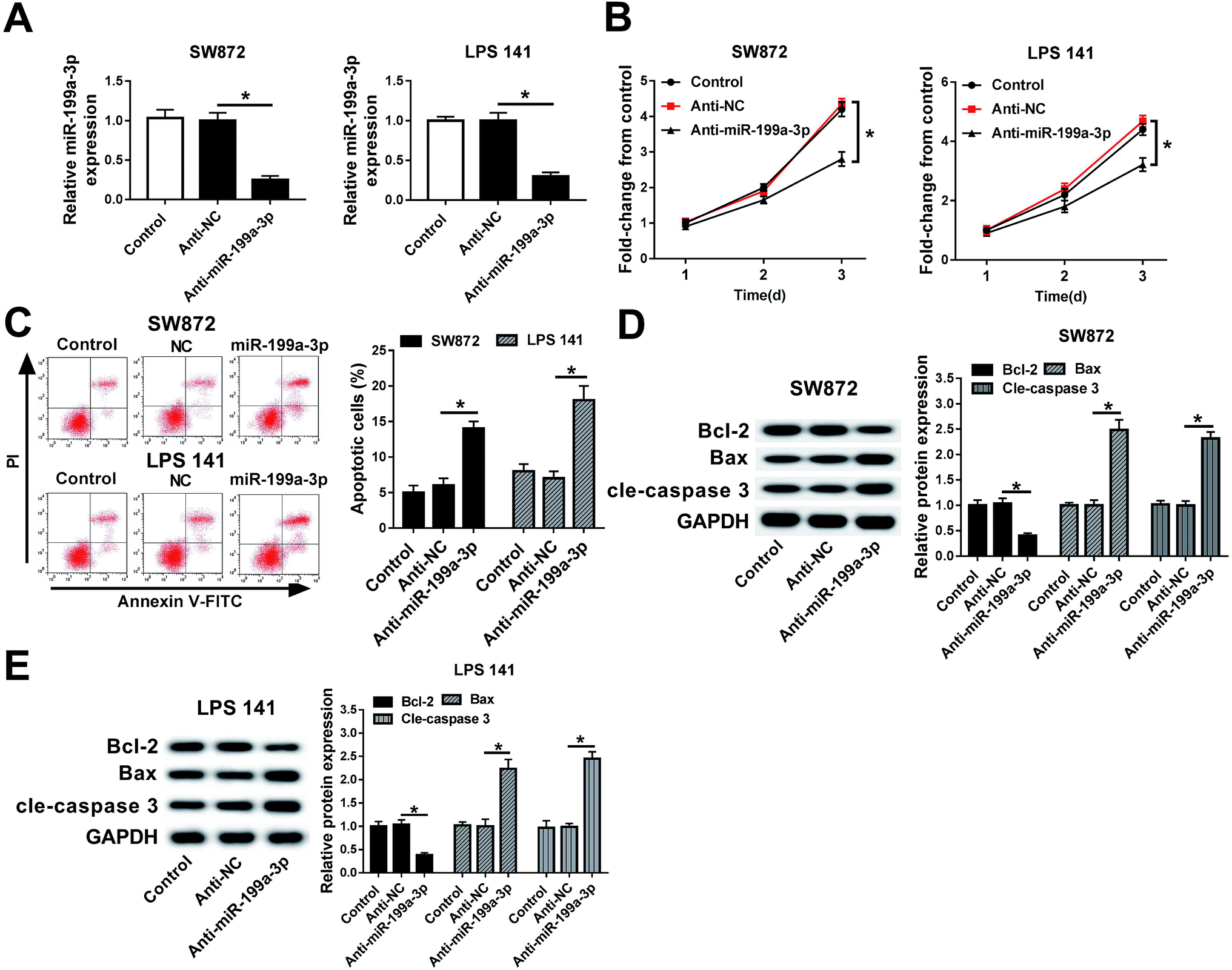 | ||
| Fig. 2 Effects of miR-199a-3p knockdown on DDLPS cell viability and apoptosis. SW872 and LPS141 cells were lower expressed of miR-199a-3p by transiently transfected with miR-199a-3p inhibitor (anti-miR-199a-3p). (A) RT-qPCR analyses identified the decrease of miR-199a-3p level. (B) Cell viability was measured with cell viability assay. OD490 values were recorded and normalized to untreated cells (control). (C) Apoptotic cells were detected and calculated by flow cytometry. (D and E) Western blot assay validated the expression of apoptosis-related genes. The quantitation of Bcl-2, Bax and cleaved caspase 3 (cle-caspase 3) was determined with an image analyzer and normalized to control group. Data are plotted as mean ± SEM. *, n = 3, p < 0.05 comparing to cells transfected with anti-miR-NC (anti-NC). | ||
miR-199a-3p knockdown inhibits DDLPS tumor growth in vivo
To examine the relevance of miR-199a-3p knockdown in vivo, lower expression of miR-199a-3p in DDLPS cells were obtained by stable transfection of miR-199a-3p antagomir (anti-miR-199a-3p) into SW872 and LPS141 cells; then stably transfected cells were subcutaneously injected into the left forelimb of nude mice (n = 6), separately. Tumor growth was monitored on xenograft volume, and the area of tumors was distinctively reduced after injection for 5–6 weeks, compared with miR-NC antagomir (anti-NC) group; tumor weight was less in miR-199a-3p knockdown group (Fig. 3A and B). Next, expression of miR-199a-3p and markers of cell proliferation and apoptosis was examined in xenografts. Together with the inhibition of miR-199a-3p, proliferating cell nuclear antigen (PCNA) expression was impaired and cleaved caspase 3 was elevated in tumors induced by miR-199a-knocked down DDLPS cells (Fig. 3C and D). Such findings demonstrate that miR-199a-3p knockdown inhibits DDLPS tumor growth in vivo, implying the tumor suppressive role of miR-199a-3p in DDLPS.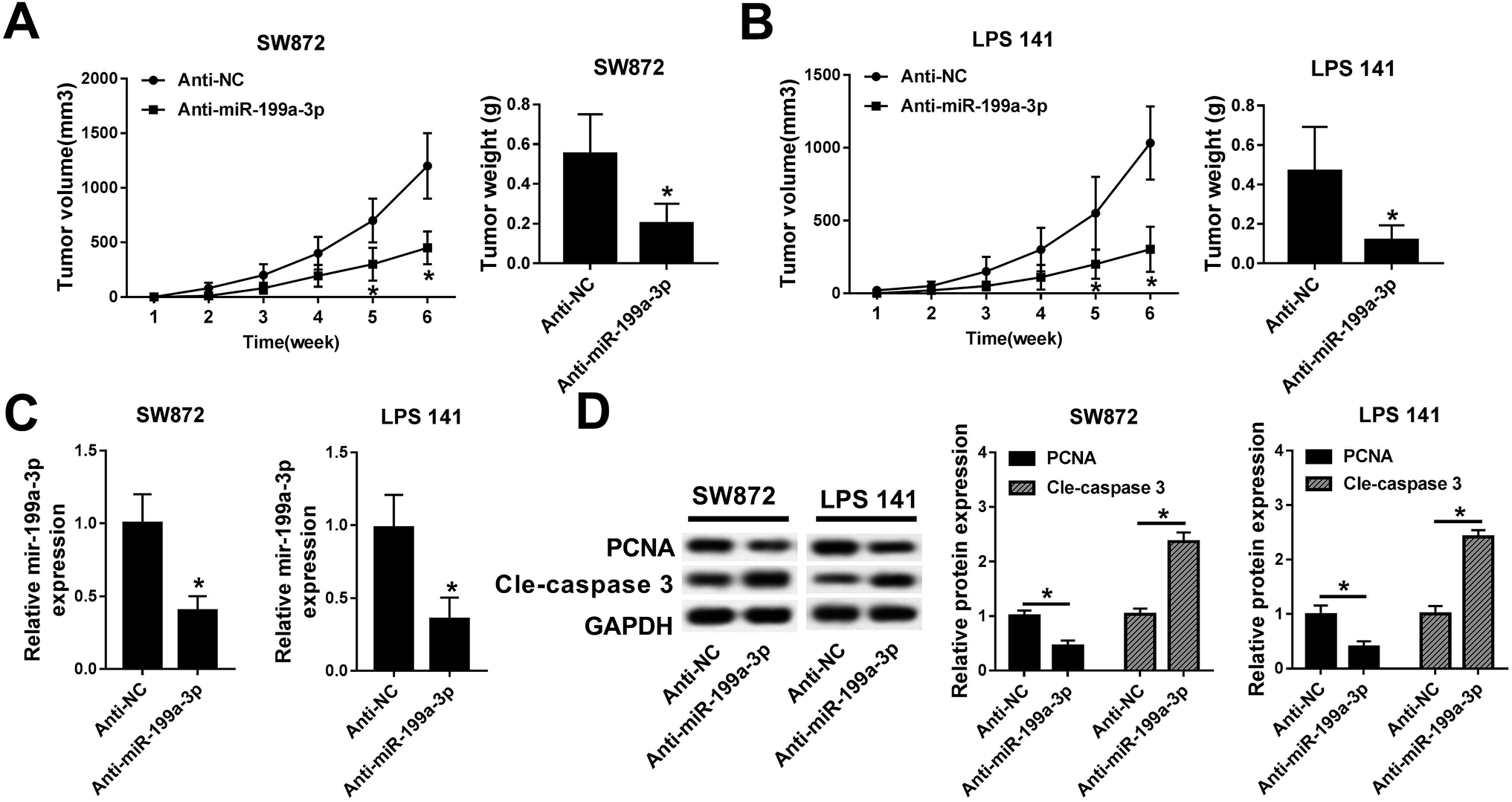 | ||
| Fig. 3 Effects of miR-199a-3p knockdown on DDLPS xenograft growth. SW872 and LPS141 cells were stably transfected miR-199a-3p/NC antagomir (anti-miR-199a-3p/NC), followed by subcutaneous injection into left forelimb of BALB/c nude mice (n = 6). (A) Tumor volumes were measured every week and tumor curve was drawn. (B–D) After injection for 6 weeks, the mice were performed euthanasia and tumors were separated. (B) Tumor weight was recorded. (C) RT-qPCR analyses identified miR-199a-3p expression in tumors. (D) Western blot assay validated the expression of proliferating cell nuclear antigen (PCNA) and cle-caspase 3 in tumors and the quantitation was determined with an image analyzer. Data are plotted as mean ± SEM. *, n = 3, p < 0.05 comparing to cells stably transfected with miR-NC antagomir (anti-NC). | ||
CK1α is a target gene of miR-199a-3p and downregulated in DDLPS tissues
Next, the regulatory effect of miR-199a-3p in DDLPS remains to be explored. Among multiple potential targets, this search on Targetscan Human identified CK1α, a key regulator of the Wnt/β-catenin pathway, to be a predicted miR-199a-3p target, containing a highly conserved motif (2452–2458 bp, NM_001892.5) matching with the miR-199a-3p “seed sequence” (6 nucleotide: CAGUAGU) (Fig. 4A). Then, the target sequence on CK1α wild type (CK1α-wt) was cloned into pMIR vector, as well as the mutant type of CK1α (CK1α-mut). Luciferase reporter assay was used to validate the binding of miR-199a-3p on CK1α, and SW872 and LPS141 cells were co-transfected with CK1α-wt/mut and miR-199a-3p/NC mimic (miR-199a-3p/NC). The luciferase activity was remarkably reduced when overexpressed miR-199a-3p and CK1α-WT (Fig. 4B); however, there was no differences in CK1α-mut groups. RNA immunoprecipitation (RIP) assay further identified the target binding of miR-199-3p and CK1α (Fig. 4C). Western blot showed CK1α expression was inhibited by miR-199-3p mimic, promoted by anti-miR-199a-3p in both SW872 and LPS141 cells (Fig. 4D). More importantly, CK1α mRNA expression was downregulated and negatively linear correlative with miR-199a-3p in DDLPS tissues (Fig. 4E and F).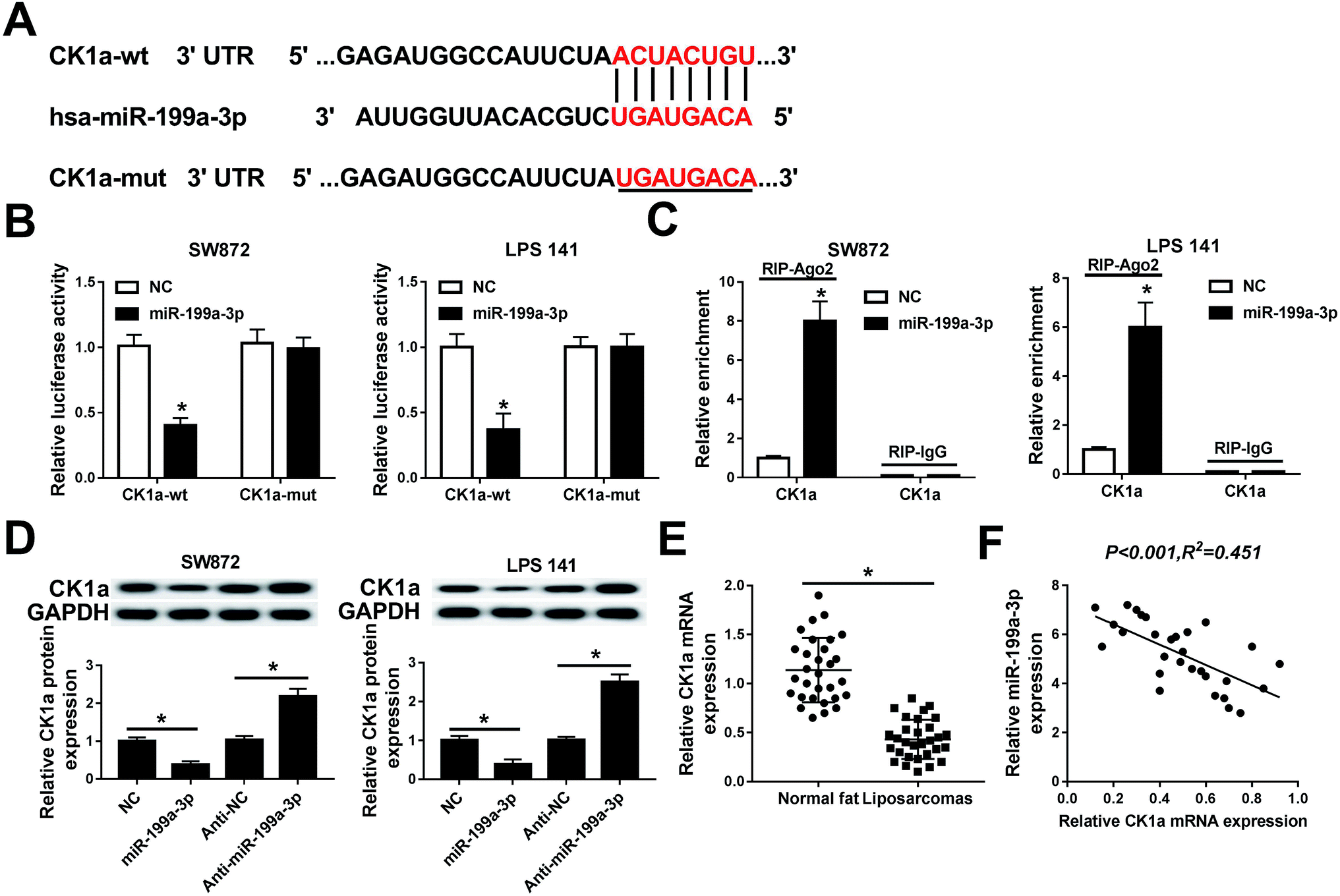 | ||
| Fig. 4 Experimental validation of casein kinase-1α (CK1α) as a target gene of miR-199a-3p. (A) The putative target site (red) of CK1α mRNA determined by computational predictions on Targetscan Human. (B) Luciferase reporter assay was used to validate the binding of miR-199a-3p on CK1α wild type 3′ UTR (CK1α-wt) in SW872 and LPS141 cells. (C) RNA immunoprecipitation (RIP) assay was performed to further identify the target binding. (D) Western blot analyzed the effect of miR-199a-3p on CK1α expression in SW872 and LPS141 cells. (E) Expression of CK1α mRNA in normal fat tissues and DDLPS tissues was determined by RT-qPCR. (F) Spearman rank correlation test analyzed the relationship between miR-199a-3p and CK1α mRNA expression. Data are plotted as mean ± SEM. *, n = 3, p < 0.05 comparing to cells transfected with miR-NC mimic (NC) or anti-NC. | ||
CK1α mediates the role of miR-199a-3p knockdown in DDLPS cells
Rescue experiments were launched to clarify the role of CK1α in mediating the biological action of miR-199a-3p in DDLPS cells. SW872 and LPS141 cells were divided into four transfection groups: anti-NC, anti-miR-199a-3p, anti-miR-199a-3p+scramble, anti-miR-199a-3p+siCK1α. As shown in Fig. 5A, the upregulation of anti-miR-199a-3p on CK1α expression was impaired by siCK1α. In the condition of miR-199a-3p knockdown, cell viability was inhibited, which was blocked by silencing of CK1α (Fig. 5B). Downregulation of CK1α could abolish the promotion effect of anti-miR-199a-3p on apoptosis rate as indicated in Fig. 5C. Furthermore, expression of Bax and cleaved caspase 3 was increased by miR-199a-3p knockdown, and subsequently decreased by CK1α silencing (Fig. 5D and E). These results declare that miR-199a-3p knockdown inhibits DDLPS cell viability and enhances apoptosis through upregulating CK1α in SW872 and LPS141 cells.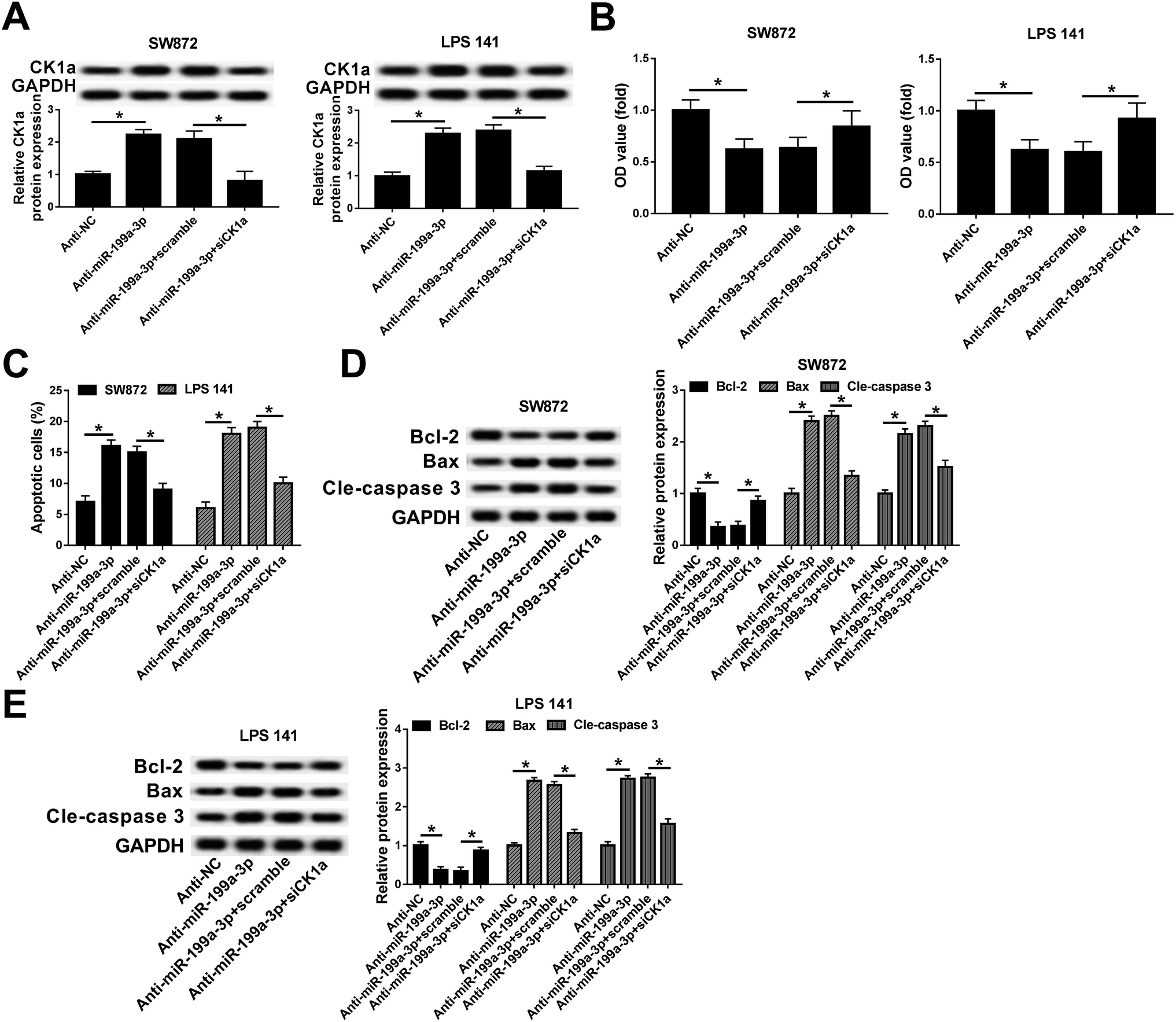 | ||
| Fig. 5 Role of CK1α silencing on DDLPS cell viability and apoptosis induced by miR-199a-3p. SW872 and LPS141 cells were transfected with siRNA against human CK1α (siCK1α) and effect of CK1α downregulation in miR-199a-3p-knocked down cells were evaluated. (A) Expression of CK1α was detected with RT-qPCR. (B) Cell viability was examined by MTT assay. (C) Apoptotic cells were measured with flow cytometry. (D and E) Western blot assay validated the expression of apoptosis-related genes. The quantitation of Bcl-2, Bax and cle-caspase 3 was determined with an image analyzer. Data are plotted as mean ± SEM. *, n = 3, p < 0.05 comparing to cells transfected with anti-NC or cells co-transfected with anti-miR-199a-3p and scrambled siRNA (scramble). | ||
Discussion
WD/DDLPS, the most prevalent histological subtype of LPS is largely resistant to conventional chemotherapy and radiotherapy, so there is a pressing need to develop new targeted therapies for LPS patients with recurrent disease.13 Instead of mRNA expression profiling, miRNA profiles have been reported to hold many advantages in human cancers, including LPS.23 Moreover, miRNA expression profiling is correlated with clinical and biological characteristics of tumors, such as tissue type, differentiation status, aggressiveness and response to therapy.24 Functional characterization of miRNAs provides more insights into the pathogenesis of LPS and promotes the identification of novel drug targets or prognostic biomarkers.11,25 However, many of the key driver genes critical to liposarcomagenesis remain to be discovered. In this study, we showed miR-199a-3p was significantly overexpressed in DDLPS tumor tissues and cell lines and its knockdown inhibited viability and facilitated apoptosis in vitro through targeting CK1α; the growth of xenograft tumors induced by SW872 and LPS141 cells were impaired by miR-199a-3p inhibition, as well.miR-199a gene takes important part in human WD/DDLPS. miR-199a-3p and miR-199a-5p are both encoded by two genes, miR-199a-1 and miR-199a-2. miR-199a-2 is in a cistron with miR-214, which was not upregulated in WDLPS (or in DDLPS) according to Agilent microarrays.13 Therefore, Ugras et al. believed that most of the upregulation likely originated from miR-199a-1, which is monocistronic. In that research, miR-199a-3p/5p were highly expressed with more than 3.7-fold in WD/DDLPS and possessed even higher levels in DDLPS with 6.0-fold and 7.7-fold, compared with normal fat tissue samples. On the contrary, miR-199a-2 and miR-214, both located in intron 14 of the DNM3 gene at 1q24.3, was also reported to be amplified in 33% WDLPS (7/21) and 35% DDLPS (6/17).26 However, there is lack of research on the role of miR-199a-3p in LPS, as well as the precise mechanism. Here, we were inspired by the upregulation of miR-199a-3p in DDLPS tissues and cells, and we investigated the biological effect of miR-199a-3p knockdown in DDLPS in vitro and in vivo. Cell viability and tumor growth were decreased and apoptosis was elevated by miR-199a-3p knockdown. Furthermore, it was figured out for the first time that miR-199a-3p regulated the occurrence and development of DDLPS through targeting CK1α. Interestingly, Guled et al. believed differentially expressed miRNAs miR-199a-3p, miR-199b-5p, miR-320a, miR-126, miR-22 in LMS had all been implicated in mesenchymal stem cells (MSCs) differentiation in several studies.21 In particular, miR-199a-3p was identified to be upregulated post-chondrogenic induction by over a two-fold change in adipose-derived MSCs.27 These results suggest miR-199a-3p may take vital influence in differentiation in WD/DDLPS.
miR-199a-3p displays dual role in different tumors. Prior work has implicated miR-199a-3p plays tumor suppressive role in majority of cancers. For example, miR-199a-3p inhibited hepatocellular carcinoma (HCC) cell proliferation, migration, invasion and angiogenesis, and induced apoptosis by targeting YAP1, VEGFA, VEGFR1, VEGFR2, HGF, MMP2 and CD151.17,28 Besides, miR-199a-3p displayed tumor suppressor functions in papillary thyroid carcinoma, bladder cancer and breast cancer.29 At the same time, miR-199a-3p serves as oncogene in several tumors, either. miR-199a-3p was investigated to positively correlate with the radioresistance of esophageal cancer cells. Proliferation and survival explained to be promoted with miR-199a-3p in both endothelial cells and breast cancer cells.30 As a novel tumor promoter in gastric cancer (GC), miR-199a-3p was involved in increasing proliferation and inhibiting apoptosis by targeting ZHX1 in GC cells and xenograft tumors.22 In adult soft tissue sarcomas, higher expression of miR-199a-3p existed in LMS than UPS, and in DDLPS than WDLPS.13,21 In this study, we observed the upregulation of miR-199a-3p in DDLPS tumor tissues and cell lines, which suggesting the potential oncogenic role of miR-199a-3p in DDLPS. Moreover, loss-of functional experiments showed that miR-199a-3p knockdown functioned tumor suppressive role in DDLPS in vitro and in vivo. These findings supported the oncogenic role of miR-199a-3p in DDLPS and provide novel knowledge of miR-199a-3p in tumors, especially in DDLPS.
CK1α is a member of CK1 family, which expressed in many eukaryotes.31 The sequence of CK1α is highly conserved and participates in diverse cellular processes, such as cell cycle progression, apoptosis and differentiation. Additionally, CK1α was needed in cell signaling pathways, including Wnt/β-catenin,32 Hedgehog,33 NF-κB pathway.34 Although the mechanism underlying remaining to be studied, CK1α phosphorylated cytoplasmic β-catenin at Ser45 in the absence of Wnt signal, promoting the degradation of β-catenin.32 Research identified CK1a was a key regulator of the Wnt/β-catenin pathway.35,36 Furthermore, CK1α had been a novel target in Wnt/β-catenin pathway to develop drugs against tumors.35 Suppression of CK1α in melanoma cells induced a switch in Wnt/β-catenin signaling to promote metastasis.36 miR-155 targeting CK1α was a LPS oncogene and downregulation of miR-155 retarded tumor cell growth, decreased colony formation, and induced G1/S cell cycle arrest in vitro; moreover, tumor growth in murine xenografts was blocked by miR-155 inhibition in vivo through upregulating CK1α and downregulating β-Catenin and cyclin D1.12 Protein expression of CK1α exhibited markedly lower in a cohort of snap-frozen human DDLPS tissues. In the present study, we observed significantly decreased CK1α mRNA levels among DDLPS tissue samples compared to normal fat tissues. CK1α expression was downregulated by miR-199a-3p. The expression of CK1α mRNA was negatively linear correlative with miR-199a-3p. Functionally, CK1α silencing, at least partially, abolished the effect of miR-199a-3p on viability and apoptosis in SW872 and LPS141 cells. In consideration that mountains of reports identified upregulated canonical Wnt signaling in a large cohort of human sarcomas including LPS, the possibility of miR-199a-3p dyregulation in affecting Wnt/β-catenin pathway would be illuminated.
It is declared that 24% of the DDLPS and no WDLPS had 1p32.2 (JUN) amplifications and that amplifications in GLI1 (12q13.3), JUN, and MAP3K5 (6q23.3) were mutually exclusive and occurred predominately in the DDLPS.26 These data seems to remind us that these molecules (JUN, GLI1 and MAPK3K5) could be promising targets for identification of precise mechanism underlying miR-199a-3p in DDLPS. Therefore, more molecular mechanisms of miR-199a-3p activity in DDLPS remains to be fully announced.
In conclusion, we find that miR-199a-3p functions as an oncogene in DDLPS by targeting CK1α. Knockdown of miR-199a-3p suppresses cell viability and tumor growth, and facilitates apoptosis via CK1α upregulation. These data help to make fundamental knowledge of miR-199a-3p in DDLPS and provide its regulatory mechanism, for the first time, through miR-199a-3p/CK1α axis. miR-199a-3p could be a potential biomarkers of development, progression and prognosis for DDLPS.
Conflicts of interest
There are no conflicts of interest to declare.References
- R. Conyers, S. Young and D. M. Thomas, Sarcoma, 2011, 2011, 483154, DOI:10.1155/2011/483154.
- N. Mandahl, L. Magnusson, J. Nilsson, B. Viklund, E. Arbajian, F. V. von Steyern, A. Isaksson and F. Mertens, Mol. Cytogenet., 2017, 10, 25, DOI:10.1186/s13039-017-0325-5.
- S. Singer, C. R. Antonescu, E. Riedel and M. F. Brennan, Ann. Surg., 2003, 238, 358–370 Search PubMed.
- H. L. Evans, E. H. Soule and R. K. Winkelmann, Cancer, 1979, 43, 574–584 CrossRef CAS PubMed.
- E. Fabre-Guillevin, J. M. Coindre, S. Somerhausen Nde, F. Bonichon, E. Stoeckle and N. B. Bui, Cancer, 2006, 106, 2725–2733 CrossRef PubMed.
- F. C. Eilber, F. R. Eilber, J. Eckardt, G. Rosen, E. Riedel, R. G. Maki, M. F. Brennan and S. Singer, Ann. Surg., 2004, 240, 686–695 Search PubMed.
- M. A. Smolle, A. Leithner, F. Posch, J. Szkandera, B. Liegl-Atzwanger and M. Pichler, Int. J. Mol. Sci., 2017, 18, 9 Search PubMed.
- R. Sun, J. K. Shen, E. Choy, Z. Yu, F. J. Hornicek and Z. Duan, Discov. Med., 2015, 20, 311–324 Search PubMed.
- K. L. Bill, L. Casadei, B. C. Prudner, H. Iwenofu, A. M. Strohecker and R. E. Pollock, Cell. Mol. Life Sci., 2016, 73, 3711–3718 CrossRef CAS PubMed.
- L. Casadei, F. Calore, C. J. Creighton, M. Guescini, K. Batte, O. H. Iwenofu, A. Zewdu, D. A. Braggio, K. L. Bill, P. Fadda, F. Lovat, G. Lopez, P. Gasparini, J. L. Chen, R. D. Kladney, G. Leone, D. Lev, C. M. Croce and R. E. Pollock, Cancer Res., 2017, 77, 3846–3856 CrossRef CAS PubMed.
- A. Boro, D. Bauer, W. Born and B. Fuchs, Am. J. Cancer Res., 2016, 6, 544–552 CAS.
- P. Zhang, K. Bill, J. Liu, E. Young, T. Peng, S. Bolshakov, A. Hoffman, Y. Song, E. G. Demicco, D. L. Terrada, C. J. Creighton, M. L. Anderson, A. J. Lazar, G. G. Calin, R. E. Pollock and D. Lev, Cancer Res., 2012, 72, 1751–1762 CrossRef CAS PubMed.
- S. Ugras, E. Brill, A. Jacobsen, M. Hafner, N. D. Socci, P. L. Decarolis, R. Khanin, R. O'Connor, A. Mihailovic, B. S. Taylor, R. Sheridan, J. M. Gimble, A. Viale, A. Crago, C. R. Antonescu, C. Sander, T. Tuschl and S. Singer, Cancer Res., 2011, 71, 5659–5669 CrossRef CAS PubMed.
- L. Sommese, G. Benincasa, M. Lanza, A. Sorriento, C. Schiano, R. Lucchese, R. Alfano, G. F. Nicoletti and C. Napoli, J. Diabetes Complicat., 2018, 32, 1076–1084 CrossRef PubMed.
- N. E. Renthal, K. C. Williams and C. R. Mendelson, Nat. Rev. Endocrinol., 2013, 9, 391–401 CrossRef CAS PubMed.
- G. Chen, H. Li, X. Li, B. Li, L. Zhong, S. Huang, H. Zheng, M. Li, G. Jin, W. Liao, Y. Liao, Y. Chen and J. Bin, J. Mol. Cell. Cardiol., 2018, 122, 152–164 CrossRef CAS PubMed.
- E. Callegari, L. D'Abundo, P. Guerriero, C. Simioni, B. K. Elamin, M. Russo, A. Cani, C. Bassi, B. Zagatti, L. Giacomelli, S. Blandamura, F. Moshiri, S. Ultimo, A. Frassoldati, G. Altavilla, L. Gramantieri, L. M. Neri, S. Sabbioni and M. Negrini, Mol. Ther.--Nucleic Acids, 2018, 11, 485–493 CrossRef CAS PubMed.
- M. Petrillo, G. F. Zannoni, L. Beltrame, E. Martinelli, A. DiFeo, L. Paracchini, I. Craparotta, L. Mannarino, G. Vizzielli, G. Scambia, M. D'Incalci, C. Romualdi and S. Marchini, Ann. Oncol., 2016, 27, 625–634 CrossRef CAS PubMed.
- N. Pencheva, H. Tran, C. Buss, D. Huh, M. Drobnjak, K. Busam and S. F. Tavazoie, Cell, 2012, 151, 1068–1082 CrossRef CAS PubMed.
- S. Fiorino, M. L. Bacchi-Reggiani, M. Visani, G. Acquaviva, A. Fornelli, M. Masetti, A. Tura, F. Grizzi, M. Zanello, L. Mastrangelo, R. Lombardi, L. Di Tommaso, A. Bondi, S. Sabbatani, A. Domanico, C. Fabbri, P. Leandri, A. Pession, E. Jovine and D. de Biase, World J. Gastroenterol., 2016, 22, 3907–3936 CrossRef CAS PubMed.
- M. Guled, L. Pazzaglia, I. Borze, N. Mosakhani, C. Novello, M. S. Benassi and S. Knuutila, Genes, Chromosomes Cancer, 2014, 53, 693–702 CrossRef CAS PubMed.
- Z. Wang, X. Ma, Q. Cai, X. Wang, B. Yu, Q. Cai, B. Liu, Z. Zhu and C. Li, FEBS Lett., 2014, 588, 4504–4512 CrossRef CAS PubMed.
- T. Shimoji, H. Kanda, T. Kitagawa, K. Kadota, R. Asai, K. Takahashi, N. Kawaguchi, S. Matsumoto, Y. Hayashizaki, Y. Okazaki and K. Shinomiya, Biochem. Biophys. Res. Commun., 2004, 314, 1133–1140 CrossRef CAS PubMed.
- U. Gyvyte, S. Juzenas, V. Salteniene, J. Kupcinskas, L. Poskiene, L. Kucinskas, S. Jarmalaite, K. Stuopelyte, R. Steponaitiene, G. Hemmrich-Stanisak, M. Hubenthal, A. Link, S. Franke, A. Franke, D. Pangonyte, V. Lesauskaite, L. Kupcinskas and J. Skieceviciene, OncoTargets Ther., 2017, 8, 37225–37238 Search PubMed.
- A. Fricke, A. F. V. Cimniak, P. V. Ullrich, C. Becherer, C. Bickert, D. Pfeifer, J. Heinz, G. B. Stark, H. Bannasch, D. Braig and S. U. Eisenhardt, Cancer Biomarkers, 2018, 22, 199–207 CAS.
- W. D. Tap, F. C. Eilber, C. Ginther, S. M. Dry, N. Reese, K. Barzan-Smith, H. W. Chen, H. Wu, F. R. Eilber, D. J. Slamon and L. Anderson, Genes, Chromosomes Cancer, 2011, 50, 95–112 CrossRef CAS PubMed.
- Z. Zhang, Y. Kang, Z. Zhang, H. Zhang, X. Duan, J. Liu, X. Li and W. Liao, Osteoarthritis and Cartilage, 2012, 20, 1638–1646 CrossRef CAS PubMed.
- A. Ghosh, D. Dasgupta, A. Ghosh, S. Roychoudhury, D. Kumar, M. Gorain, R. Butti, S. Datta, S. Agarwal, S. Gupta, G. Krishna Dhali, A. Chowdhury, T. D. Schmittgen, G. C. Kundu and S. Banerjee, Cell Death Dis., 2017, 8, e2706, DOI:10.1038/cddis.2017.123.
- X. Fan, S. Zhou, M. Zheng, X. Deng, Y. Yi and T. Huang, Biomed. Pharmacother., 2017, 88, 507–514 CrossRef CAS PubMed.
- C. Zang, F. Zhao, L. Hua and Y. Pu, Cancer Cell Int., 2018, 18, 186 CrossRef CAS PubMed.
- U. Knippschild, A. Gocht, S. Wolff, N. Huber, J. Lohler and M. Stoter, Cell. Signal., 2005, 17, 675–689 CrossRef CAS PubMed.
- S. Amit, A. Hatzubai, Y. Birman, J. S. Andersen, E. Ben-Shushan, M. Mann, Y. Ben-Neriah and I. Alkalay, Genes Dev., 2002, 16, 1066–1076 CrossRef CAS PubMed.
- Q. Shi, S. Li, S. Li, A. Jiang, Y. Chen and J. Jiang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5651–E5660 CrossRef CAS PubMed.
- N. Bidere, V. N. Ngo, J. Lee, C. Collins, L. Zheng, F. Wan, R. E. Davis, G. Lenz, D. E. Anderson, D. Arnoult, A. Vazquez, K. Sakai, J. Zhang, Z. Meng, T. D. Veenstra, L. M. Staudt and M. J. Lenardo, Nature, 2009, 458, 92–96 CrossRef CAS PubMed.
- C. A. Thorne, A. J. Hanson, J. Schneider, E. Tahinci, D. Orton, C. S. Cselenyi, K. K. Jernigan, K. C. Meyers, B. I. Hang, A. G. Waterson, K. Kim, B. Melancon, V. P. Ghidu, G. A. Sulikowski, B. LaFleur, A. Salic, L. A. Lee, D. M. Miller 3rd and E. Lee, Nat. Chem. Biol., 2010, 6, 829–836 CrossRef CAS PubMed.
- T. Sinnberg, M. Menzel, S. Kaesler, T. Biedermann, B. Sauer, S. Nahnsen, M. Schwarz, C. Garbe and B. Schittek, Cancer Res., 2010, 70, 6999–7009 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |