DOI:
10.1039/C9RA01329F
(Paper)
RSC Adv., 2019,
9, 14391-14399
Enhanced photocatalytic reduction of CO2 to CO over BiOBr assisted by phenolic resin-based activated carbon spheres†
Received
21st February 2019
, Accepted 1st April 2019
First published on 8th May 2019
Abstract
Photocatalytic reduction of CO2 using solar energy to decrease CO2 emission is a promising clean renewable fuel production technology. Recently, Bi-based semiconductors with excellent photocatalytic activity and carbon-based carriers with large specific surface areas and strong CO2 adsorption capacity have attracted extensive attention. In this study, activated carbon spheres (ACSs) were obtained via carbonization and steam activation of phenolic resin-based carbon spheres at 850 °C synthesized by suspension polymerization. Then, the BiOBr/ACSs sample was successfully prepared via a simple impregnation method. The as-prepared samples were characterized by XRD, SEM, EDX, DRS, PL, EIS, XPS, BET, CO2 adsorption isotherm and CO2-TPD. The BiOBr and BiOBr/ACSs samples exhibited high CO selectivity for photocatalytic CO2 reduction, and BiOBr/ACSs achieved a rather higher photocatalytic activity (23.74 μmol g−1 h−1) than BiOBr (2.39 μmol g−1 h−1) under simulated sunlight irradiation. Moreover, the analysis of the obtained results indicates that in this photocatalyst system, due to their higher micropore surface area and larger micropore volume, ACSs provide enough physical adsorption sites for CO2 adsorption, and the intrinsic structure of ACSs can offer effective electron transfer ability for a fast and efficient separation of photo-induced electron–hole pairs. Finally, a possible enhanced photocatalytic mechanism of BiOBr/ACSs was investigated and proposed. Our findings should provide new and important research ideas for the construction of highly efficient photocatalyst systems for the reduction of CO2 to solar fuels and chemicals.
1. Introduction
With the rapid development of industrial economy, the global environmental pollution and energy crisis have become extremely significant challenges that humans are facing in the short-term.1 More importantly, the heavy reliance of economic development on the fossil fuel energy will bring about more than 30.4 Gt a−1 of CO2 emissions into the atmosphere,2 and the CO2 emissions will probably increase up to 43 Gt a−1 by 2035 without factitious CO2 conversion utilization. Moreover, CO2 has been considered as a dominating factor for the anthropogenic climate change to cause the “Greenhouse Effect”; therefore, it is necessary to find out suitable ways for the maximum utilization of CO2.3,4 At present, solar photocatalytic technology, as a great potential way to solve the abovementioned problem, has received significant attentions for the reduction of CO2 to useful high-energy fuels (e.g. CO, CH3OH, CH4, HCOOH, etc.).5–10 Among them, CO, as a crucial feedstock in the d-metal-catalyzed Fischer–Tropsch synthetic processes, possesses a significant fuel value (ΔcH = −283.0 kJ mol−1) and is also easily converted to CH3OH; therefore, it is attracting significant interest from researchers.11 Most importantly, it is imperative to find a suitable and promising photocatalyst system with high selectivity and activity for CO2 reduction under the action of solar light.
BiOBr, as one of the Bi-based semiconductors with several advantages, including earth abundance, stability, economy and non-toxicity, exhibits a unique layered structure, excellent electrical and optical properties as well as a suitable indirect band gap (∼2.7 eV) to endow effective photocatalytic activity and stability.12–17 It was believed that the Bi 6s and O 2p states could form a large number of dispersed hybridized valence bands, which facilitated the migration and oxidation of photo-generated holes; this induced the efficient separation of photo-generated electron–hole (e−–h+) pairs and then improved the photocatalytic efficiency.18 In 2016, Ye et al. found that the as-prepared BiOBr sample with ultrathin thickness and a Bi-rich structure could convert CO2 to CO (2.67 μmol g−1 h−1) and CH4 (0.16 μmol g−1 h−1).19 After this, in 2017, they reported that {001}-dominated BiOBr nanosheets showed 100% selectivity for the conversion of CO2 to CO and the highest CO yield of 4.45 μmol g−1 h−1 under simulated sunlight irradiation.20 Most importantly, in 2018, Xie et al. synthesized defect-engineered BiOBr atomic layers with a rather high CO yield (87.4 μmol g−1 h−1) for the photocatalytic reduction of CO2 under visible light-driven irradiation.21 These remarkable findings well confirmed that BiOBr, as a promising new-type of CO2 reduction material, could be responsible for the excellent selectivity and activity of the photocatalytic reduction of CO2 to CO. As is known, the separation efficiency of photogenerated e−–h+ pairs and the adsorption performance of CO2 on the catalyst surface have been two significant and crucial factors for the photocatalytic reduction of CO2.4,22–24 However, for a single BiOBr photocatalyst, the low separation efficiency of the electron–hole pairs and the weak CO2 adsorption capacity limit its development and applications in the photocatalytic reduction of CO2. Therefore, it is urgent to construct an ideal photocatalyst system with excellent light absorption, efficient separation of photogenerated e−–h+ pairs and high CO2 adsorption capacity.25
According to abovementioned problems, the selection of materials with higher adsorption capacity, larger specific surface area and stronger charge transfer ability to be loaded onto high-efficiency photocatalysts should be a very significant and interesting issue in the research and development of photocatalytic CO2 reduction; in most of the studies, it has been found that carbon-based carriers (such as activated carbon,26 carbon nanotubes,27 graphene,28 and carbon nanodots29) meet these requirements due to their unique electronic structures, stable chemical structures, large specific surface areas, strong adsorption capacities, high thermal conductivity and electron mobilities.30 Especially, activated carbon spheres (ACSs) with excellent electron transfer capability, suitable sphere-type structure and size, high loading density and mechanical strength, have attracted widespread attention as a kind of promising and valuable adsorbent for hazardous materials in the liquid and gas phases as well as catalyst supports. For example, Liu et al. found that micrometer-sized carbon spheres, rich in O-containing functional groups, exhibited a remarkably enhanced adsorption capacity for Cr(VI), 0.4834 mmol g−1, about 4 times that of unmodified AC.31 Wickramaratne et al. prepared a series of activated carbon spheres (ACSs) through the carbonization of phenolic resin spheres using the one-pot modified Stöber and CO2 activation method, which exhibited high surface areas (from 730 to 2930 m2 g−1) and CO2 adsorption capacities at 1 bar (4.55 and 8.05 mmol g−1, at 25 °C and 0 °C, respectively).32 Moreover, Rivera-Utrilla et al. and Ao et al. synthesized TiO2-AC33 and BiOBr-AC26 photocatalysts, respectively, with greatly improved photocatalytic performances owing to the higher BET surface area of the AC support with strong adsorption capacity for pollutants and significant influence on the optical absorption capacity and crystal size of the catalysts. Therefore, due to their high specific surface areas and strong CO2 adsorption capacity, ACSs can become rather ideal carriers to support high-efficiency photocatalysts to effectively enhance the performance of photocatalytic CO2 reduction.
Among them, phenolic resins have been considered as promising polymer precursor materials to produce ACSs with high surface areas because of their low inorganic impurity and negligible ash contents. In our study, a new method is reported for the preparation of ACSs. At first, phenolic resin-based spheres were synthesized by suspension polymerization using m-cresol and formaldehyde as precursor materials and adding ethylene glycol or poly(ethylene glycol) as an additive; then, the millimeter-sized ACSs with high specific surface area were prepared by the steam activation method. Subsequently, a layer of BiOBr was uniformly loaded on the ACSs surface using a simple impregnation method. The phase structure, morphology, elemental analysis, optical absorption characteristics, specific surface areas and CO2 adsorption ability of the as-prepared BiOBr/ACSs were analyzed by XRD, SEM, EDX, XPS, DRS, PL, EIS, BET, CO2 adsorption isotherm and CO2-TPD. In addition, the correlative performances of photocatalytic CO2 reduction were evaluated under simulated sunlight irradiation. Finally, the photocatalytic mechanism of action of the BiOBr/ACSs sample has been investigated and proposed; the results obtained herein should provide new and important research ideas and great guiding significance for the construction of highly-efficient photocatalyst systems for photocatalytic CO2 reduction.
2. Experimental
2.1 Chemicals
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O) and potassium bromide (KBr) were purchased form Tianjin Sinopharm Chemical Reagent (Co. Ltd.) and Tianjin Zhiyuan Reagent (Co. Ltd.), respectively. m-Cresol was obtained from Aladdin. Formaldehyde, ethylene glycol (EG), and hexamethylenetetramine were provided by Tianjin Kemiou Chemical Reagent (Co. Ltd.). Triethylamine and polyvinyl alcohol were provided by Tianjin Fangde Technology (Co. Ltd.) and Shanxi Sanwei Group (Co. Ltd.). All reagents were of analytical grade. Deionized water was used throughout this study.
2.2 Preparation
BiOBr. In a typical synthesis, 0.01 mmol of Bi(NO3)3·5H2O was dissolved in 40 mL of ethylene glycol (EG) at 50 °C, and 0.01 mmol of KBr was dissolved in 20 mL of deionized water under continuous stirring until the solution became transparent. Subsequently, KBr was poured into the abovementioned solution. After stirring for 5 h, the suspension was filtered, repeatedly washed with deionized water and absolute ethanol, and finally dried at 60 °C overnight.
ACSs. A three-necked flask was set in an oil bath, and 200 mL of deionized water was added to it. Then, 30 mL of m-cresol, 33 mL of formaldehyde, 15 mL of ethylene glycol and 1.5 mL of trimethylamine were successively added to the three-necked flask. After this, 1.5 g of polyvinyl alcohol and 1.8 g of hexamethylenetetramine were added under continuous stirring when the temperature increased to 110 °C. After stirring for 3 h, the phenolic resin sphere samples were poured out, obtained, heated at 850 °C under a N2 atmosphere, and then activated by steam for 1 h to obtain ACSs.
BiOBr/ACSs. The BiOBr/ACSs samples were prepared by an impregnation method. Typically, 2 g of ACSs was added to a 0.05% polyvinyl alcohol solution and stirred for 1.5 h. The product was obtained by filtration. After this, 1 g of BiOBr was added to 20 mL of deionized water, the mixture was stirred for 10 min and ultrasonicated for 10 min, such that BiOBr was evenly dispersed in deionized water. The ACSs were added to the BiOBr solution and stirred for 20 min. After the reaction was completed, the product was filtered and dried at 60 °C for 12 h. The samples obtained were named BiOBr/ACSs, as shown in Scheme 1.
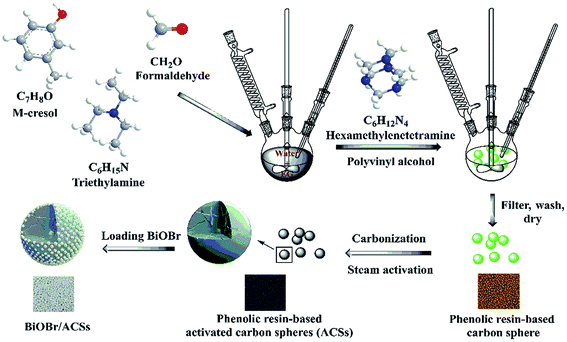 |
| | Scheme 1 Preparation process of BiOBr/ACSs. | |
2.3 Characterization
The crystal phase of the as-prepared samples was investigated by X-ray diffraction (XRD) (DX-2700 diffractometer, Japan) with Cu Kα radiation in the 2θ range of 10–80°. The morphologies and element mapping of the prepared samples were investigated via the Nanosem 430 (FEI, America) field emission scanning electron microscope at the operating voltage of 10 kV with an energy dispersive X-ray (EDX) microanalysis system. XPS spectra of these materials were obtained via Thermo Fisher ESCALAB 250 xi, England, using Al Kα radiation (1486.6 eV). Binding energies were calculated with respect to C(1s) at 284.8 eV. Binding energies were measured with a precision of ±0.05 eV. The UV-vis diffuse reflection spectra (UV-vis DRS) were obtained using a UV-vis spectrophotometer (UV-3600, Shimadzu, Japan). Photoluminescence (PL) spectra were obtained using the Hitachi F-7000 Fluorescence spectrophotometer. The specific surface area and porous structure of all the samples were determined by N2 adsorption/desorption using the V-Sorb 4800P instrument. The CO2 adsorption isotherm was obtained using an automated gas sorption analyzer (Quantachrome Autosorb-1, USA). Temperature-programmed desorption of CO2 (CO2-TPD) was measured as follows: 0.1 g of sample was pretreated at 400 °C for 1 h under a He atmosphere and then naturally cooled down to 30 °C. Next, the samples were placed under a CO2 atmosphere for 0.5 h to ensure sufficient CO2 adsorption. Before desorption, the sample was purged with He for 3 h and then desorbed at the rate of 10 °C min−1 to 350 °C under a He atmosphere.
2.4 Photocatalytic CO2 reduction
Photocatalytic CO2 reduction experiments were carried out in a gas-closed circulation system (CEL-SPH2N-D9, Beijing China Education Au-Light Co., Ltd.) irradiated with a 300 W xenon lamp. Herein, 50 mL of deionized water was added to the quartz glass reactor, and a certain amount of catalyst was uniformly dispersed in the quartz glass reactor. Before turning on the light, the photoreactor system needed a thorough vacuum treatment, and CO2 gas was introduced into the circulation system. During the light irradiation of 9 h, the yield of CO produced was analyzed every hour by on-line gas chromatography (GC-7920, Beijing China Education Au-Light Co., Ltd.) with a flame ionization detector. Gas products were analyzed by an external standard method.
3. Results and discussion
3.1 Phase structure analysis
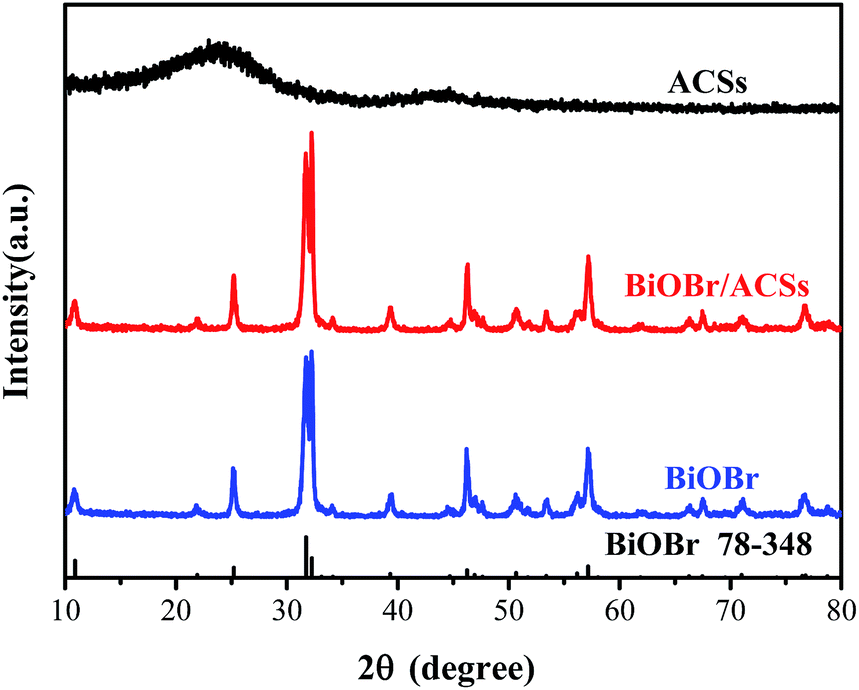
Fig. 1 shows the XRD patterns of the as-prepared BiOBr, ACSs, and BiOBr/ACSs samples. The XRD peaks of the BiOBr sample are well indexed to the tetragonal phase BiOBr (JCPDF no. 73-348),13 and no impurity peak is detected; this indicates high purity of as-prepared BiOBr sample. It has been found that the XRD patterns of BiOBr obtained after loading BiOBr onto ACSs do not shift significantly; this implies that the effect of ACSs on the phase structure of the BiOBr sample is negligible.
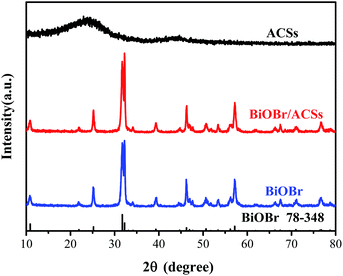 |
| | Fig. 1 XRD patterns of ACSs, BiOBr/ACSs and BiOBr samples. | |
3.2 Morphological structure and energy dispersive X-ray (EDX) analysis
Fig. 2 shows the SEM images of the prepared samples. In Fig. 2(a and b), it is found that the BiOBr sample exhibits a self-assembled sphere-structure with the single sphere diameter of approximately 0.8–1.2 μm. Moreover, the surface of the ACSs activated by steam is smooth and free from cracks with good sphericity (Fig. 2c), and an excellent pore-structure distribution is obtained via the carbonization process (inset in upper-right corner of Fig. 2e); the abovementioned conditions are favorable for the activation agent to enter the interior of the sample during the activation process, and finally, the formation of ACSs with different, new and expanding pore-distributions is achieved. For the BiOBr/ACSs sample (Fig. 2d), a large amount of white BiOBr powder obviously adheres to the ACSs surface (Fig. S1†), and BiOBr loading has almost no influence on the internal pore-structure distribution of ACSs, as shown in the partially magnified half-section of BiOBr/ACSs (Fig. 2f). In the high-magnification SEM image of BiOBr/ACSs (Fig. S1†), we can observe that BiOBr supported on the surface of ACSs still maintains its self-assembled spherical nanosheet structure; this indicates that there is almost no change in the morphology of BiOBr and ACSs before and after loading.
 |
| | Fig. 2 Low (a) and high (b) magnification SEM images of BiOBr, low-magnification SEM images of ACSs (c) and BiOBr/ACSs (d) (inset c: an image of ACSs; inset d: an image of BiOBr/ACSs), a high-magnification SEM image of ACSs (e) and BiOBr/ACSs (f) cross profiles (inset: low-magnification SEM images of ACSs (e) and BiOBr/ACSs (f) cross profile); (g and h) EDX mappings of the BiOBr/ACSs samples, C (i), O (j), Bi (k), and Br (l) elements. | |
To further clarify the chemical compositions of the samples, the corresponding energy dispersive X-ray spectroscopy (EDX) elemental mappings (Fig. 2g–h) and EDX spectra (Fig. S2†) of the C, O, Bi and Br (Fig. 2i–l) elements were obtained. The atomic ratio of the Bi, O and Br elements is very close to the reasonable value of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, and the observed Bi, O and Br elemental mappings well indicate that BiOBr can be evenly distributed and wrapped on the ACSs surface; thus, the excellent BiOBr/ACSs catalyst system can be formed.
:1:1, and the observed Bi, O and Br elemental mappings well indicate that BiOBr can be evenly distributed and wrapped on the ACSs surface; thus, the excellent BiOBr/ACSs catalyst system can be formed.
3.3 XPS spectra
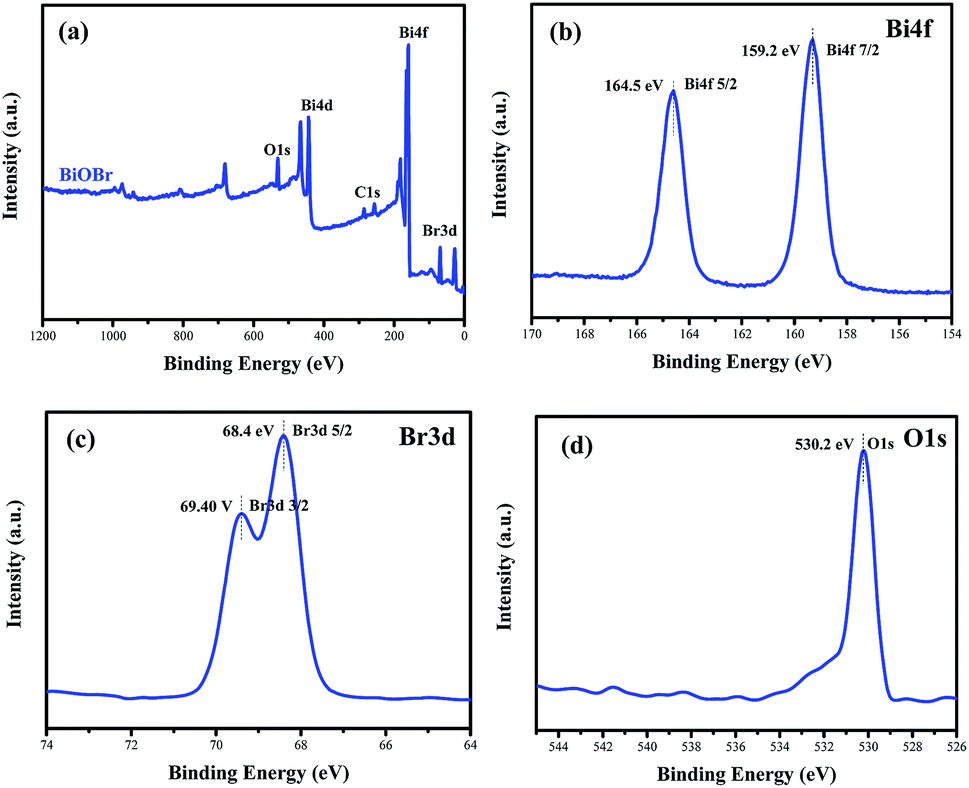
To confirm the elemental compositions and valence states of the as-prepared samples, XPS survey spectra of the BiOBr sample were obtained, as shown in Fig. 3. It can be clearly seen from the wide survey scan data shown in Fig. 3a that Bi, O and Br are present without other prominent impurities; this rules out the possibility of the existence of adventitious carbon-based contaminants, and this is in good agreement with the abovementioned EDX scan results. The high-resolution XPS Bi 4f spectrum of BiOBr is presented in Fig. 3b. The two peaks at 164.5 and 159.2 eV are associated with Bi 4f5/2 and Bi 4f7/2, respectively, indicating that the Bi3+ valence state exists in the BiOBr nanosheets. As shown in Fig. 3d, the O 1s core level spectrum can be well fitted with the peak at 530.2 eV that belongs to the lattice oxygen O2− originating from the Bi–O bond.34 The XPS spectrum of Br 3d (Fig. 3c) exhibits two major peaks with the binding energies at 68.4 and 69.4 eV, corresponding to Br 3d5/2 and Br 3d3/2 of Br−.
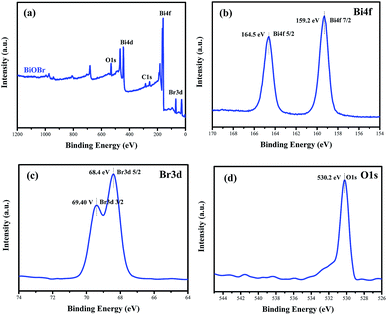 |
| | Fig. 3 Survey XPS spectra (a) and high-resolution XPS spectra of BiOBr: Bi 4f (b), Br 3d (c) and O 1s (d). | |
3.4 Optical absorption properties
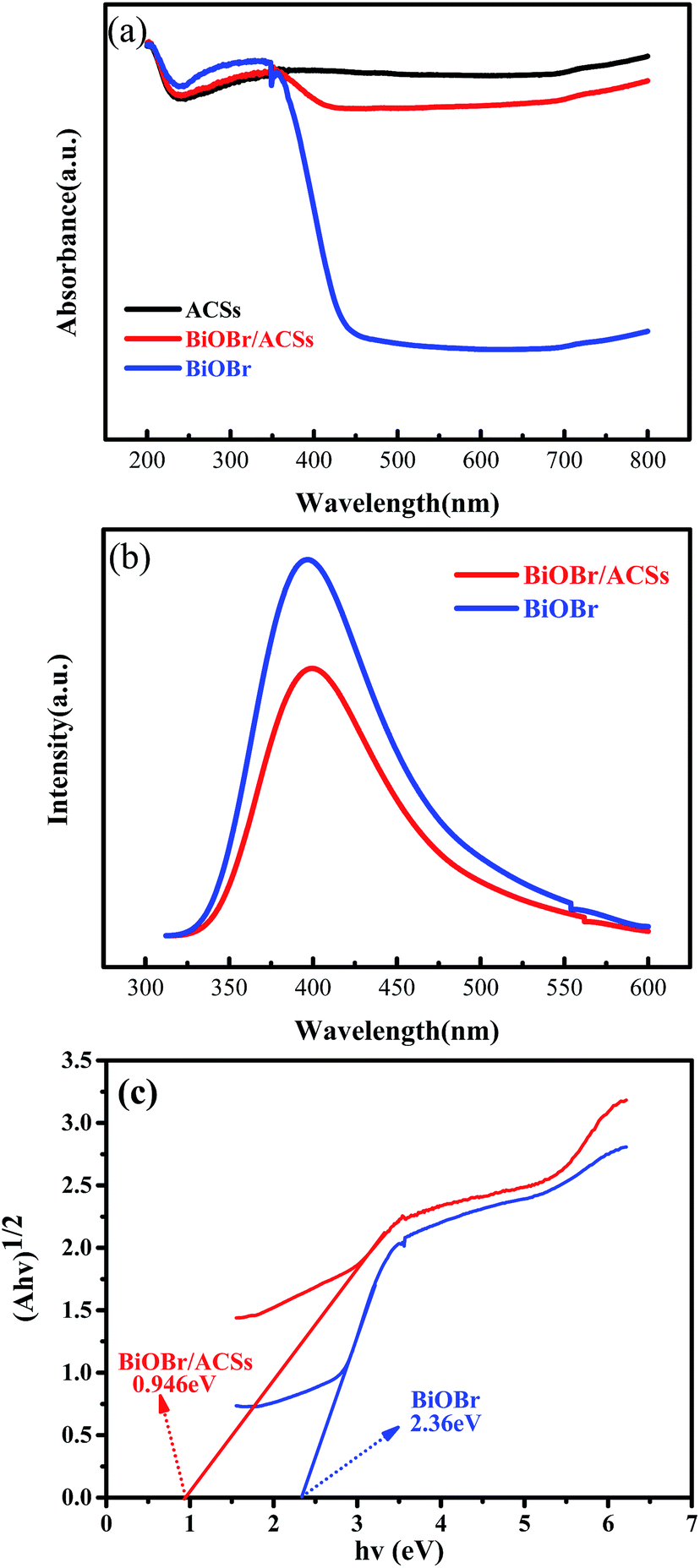
The optical absorption properties of the samples were analyzed via the DRS technology and PL spectroscopy, as illustrated in Fig. 4. The band gap (Eg) of BiOBr and BiOBr/ACSs can be calculated by the equation (αhν)n = A(hν − Eg).35,36 Fig. 4c displays the curve of (αhν)1/2 vs. hν according to the obtained BiOBr and BiOBr/ACSs DRS spectra, and the extrapolation intercept provides the Eg values for BiOBr and BiOBr/ACSs of 2.36 and 0.946 eV, respectively. After the introduction of ACSs, it can be clearly found that the BiOBr/ACSs sample has stronger optical absorption in the visible region than pure BiOBr. Thus, the addition of ACSs could improve the visible light-driven efficiency of BiOBr, enhancing the photocatalytic activity of BiOBr. Moreover, for the photocatalytic CO2 reduction process, the separation efficiency of photo-generated carriers should be an important factor. Herein, the PL spectra were obtained to evaluate the separation efficiency of the photo-generated e−–h+ pairs for the BiOBr and BiOBr/ACSs samples, as shown in Fig. 4b. As is known, the higher the PL intensity, the higher the probability of recombination of photo-generated e−–h+ pairs. It can be found that both pure BiOBr and BiOBr/ACSs samples have a diffraction peak at 400 nm; however, the latter reveals decreased intensity when compared with the former; this confirms an improved separation efficiency of the e−–h+ pairs after the introduction of ACSs.37 The EIS measurement results (Fig. S3†) reveal that the Nyquist circle of BiOBr/ACSs is smaller than that of BiOBr; this indicates that BiOBr/ACSs have lower resistance than pure BiOBr; this can accelerate the interfacial charge-transfer process.38
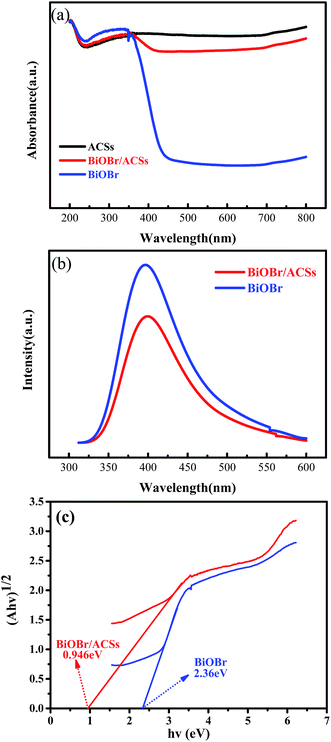 |
| | Fig. 4 (a) DRS of the as-prepared samples; (b) PL spectra of BiOBr and BiOBr/ACSs samples; and (c) plots of (αhν)1/2 versus photon energy (hν). | |
3.5 CO2 adsorption performance and porous structure parameters
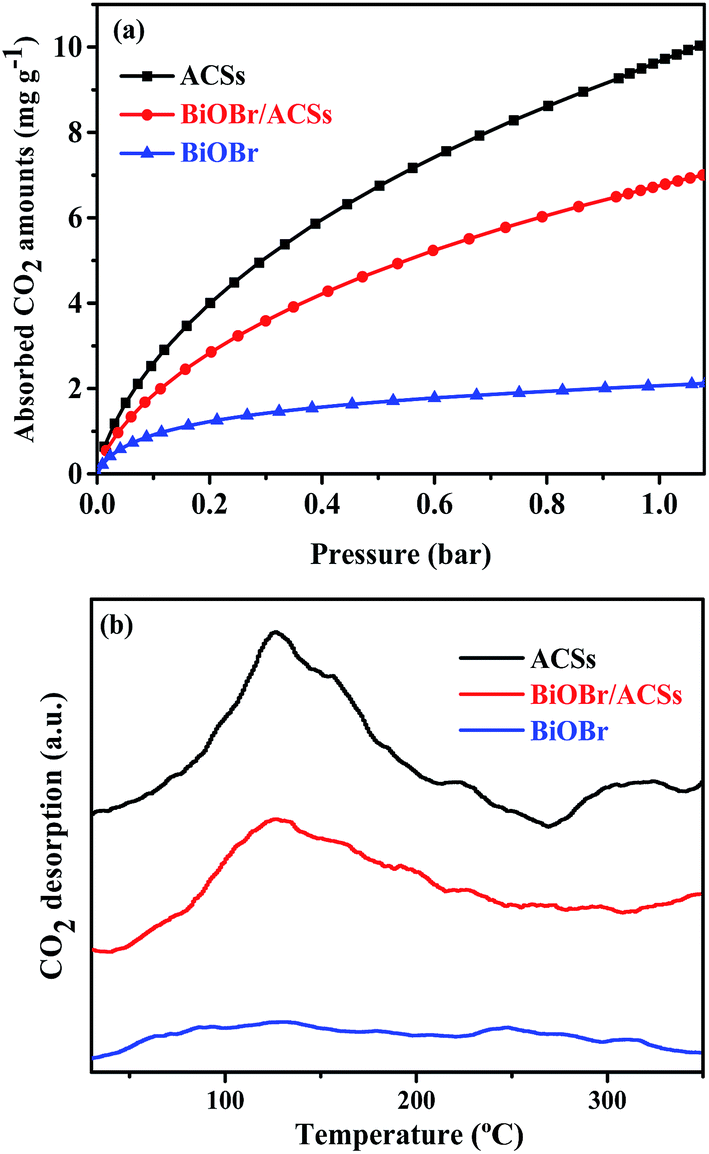
The CO2 adsorption performance of a catalyst has been considered as one of the most key factors influencing the catalytic activity in the photocatalytic CO2 reduction process.39 The CO2 adsorption isotherms and CO2 temperature-programmed desorption (CO2-TPD) spectra for the BiOBr, ACSs and BiOBr/ACSs samples are provided in Fig. 5. The CO2 adsorption isotherms of the as-prepared sample at 298 K are shown in Fig. 5a. It is clearly seen that the CO2 adsorption capacity at 298 K over ACSs can reach 10.03 mg g−1, much higher than that of BiOBr/ACSs (7.0 mg g−1) and BiOBr (2.23 mg g−1); this should be ascribed to higher specific surface area and larger micropore volume of the ACSs and BiOBr/ACSs. In Fig. 5b, it can be found that pure ACSs possess strongest CO2 adsorption capacity, and the desorption peaks of CO2 occur at 50–150 °C and 260–330 °C, which should be mainly attributed to strong physical adsorption and weak chemisorption, respectively.40,41 In addition, pure BiOBr exhibits a notably weak CO2 desorption curve; however, after loading BiOBr on the ACSs surface, the intensity of the CO2 adsorption and desorption curves of the BiOBr/ACSs sample is obviously enhanced; this will be conducive to make an increasing number of CO2 molecules participate in the surface-activation and catalytic reaction process; this will improve the catalytic reaction efficiency and rate.42
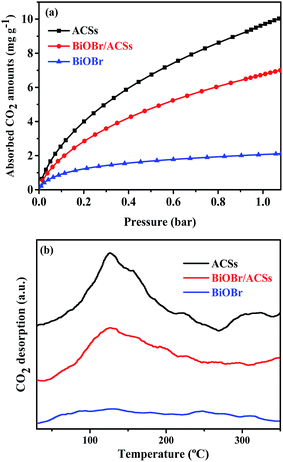 |
| | Fig. 5 (a) CO2 adsorption isotherms and (b) CO2 temperature-programmed desorption (CO2-TPD) of the as-prepared samples. | |
Usually, a material with high surface area, large micropore volume and more basic surface functions should have higher CO2 capture performance.43,44 Table 1 summarizes the corresponding structural parameters, including BET surface areas, average pore sizes, micropore and total pore volumes, as calculated from the N2 adsorption isotherms, of all the as-prepared samples. As can be observed from Table 1, the ACSs and BiOBr/ACSs samples have rather the high specific BET surface areas of 936.42 and 792.56 m2 g−1, total pore volumes of 0.37 and 0.31 cm3 g−1, and a high micropore ratio (>83%), respectively. These higher micropore surface area and larger micropore volume can provide more physical activation sites for CO2 adsorption, and the kinetic diameter of CO2 is 0.33 nm such that the CO2 adsorption capacity should ultimately be controlled by the micropore volume.43,45 These are the main reasons why ACSs exhibit higher CO2 adsorption ability than the BiOBr/ACSs and BiOBr samples.
Table 1 Porous structure parameters of the as-prepared BiOBr, ACSs and BiOBr/ACSs samples
| Samples |
SBET (m2 g−1) |
Vtotal (cm3 g−1) |
Vmicro (cm3 g−1) |
Vsemi − Vmicro (cm3 g−1) |
Dp (nm) |
| ACSs |
936.42 |
0.37 |
0.34 |
0.32 |
1.58 |
| BiOBr/ACSs |
792.56 |
0.31 |
0.26 |
0.25 |
1.56 |
| BiOBr |
21.13 |
0.11 |
0.008 |
— |
20.82 |
3.6 Photocatalytic CO2 reduction
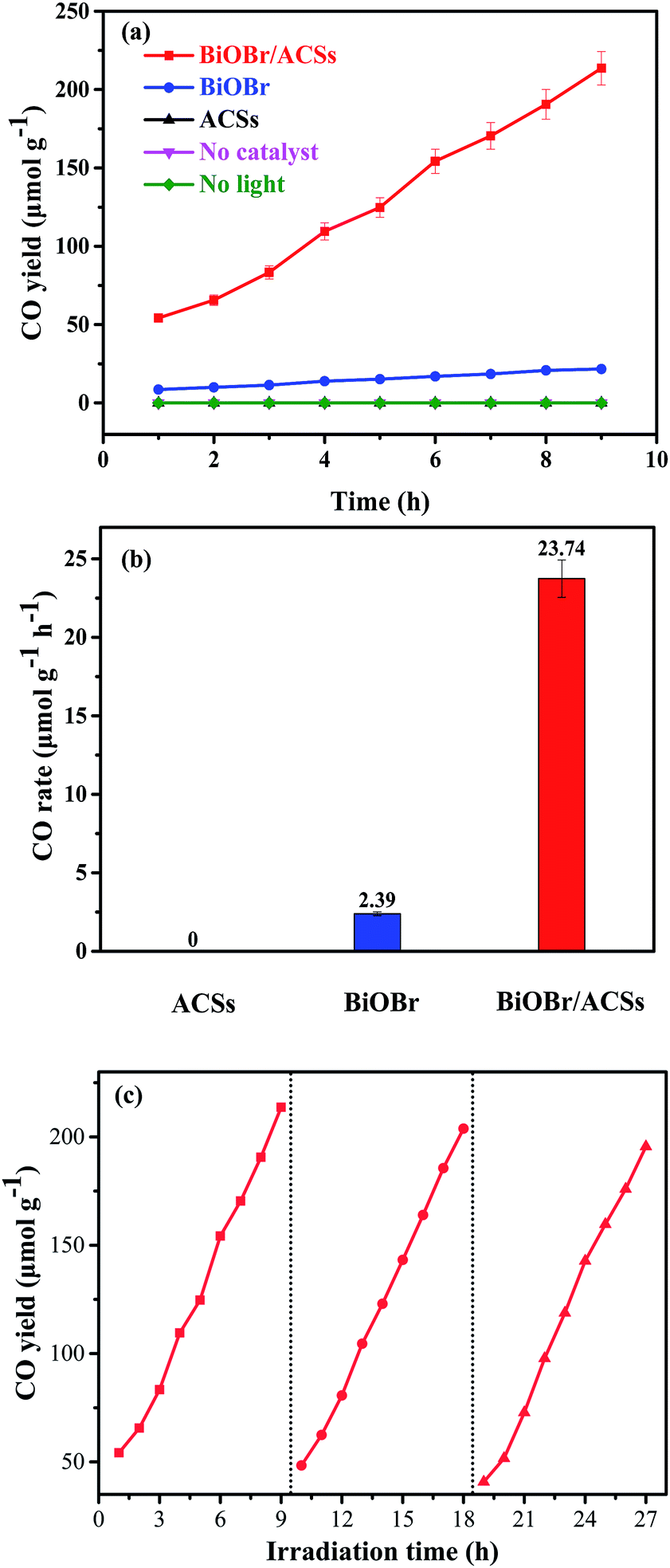
Fig. 6 shows the plots of a series of experiments for the photocatalytic CO2 reduction over ACSs, BiOBr/ACSs and BiOBr samples under simulated sunlight irradiation. It can be found that CO2 cannot be reduced to CO in the absence of the BiOBr photocatalyst or in the dark during these controlled experiments. Only a small amount of CO gas was detected in the reaction system when BiOBr was used as a single photocatalyst under simulated sunlight irradiation (Fig. 6a). Surprisingly, the BiOBr/ACSs sample exhibits significant photocatalytic CO2 reduction activity and has fast-raised CO gas production efficiency with an increase in the irradiation time, with the CO yield of 213.67 μmol g−1 after 9 h, about 9.9 times that of pure BiOBr (21.51 μmol g−1). For the BiOBr/ACSs photocatalyst, the enhanced performance of photocatalytic CO2 reduction should mainly be related to the lower electron–hole charge recombination rate (Fig. 4b), higher micropore surface area (792.56 m2 g−1) and larger micropore volume that can provide more physical activation sites for CO2 adsorption (Fig. 5).
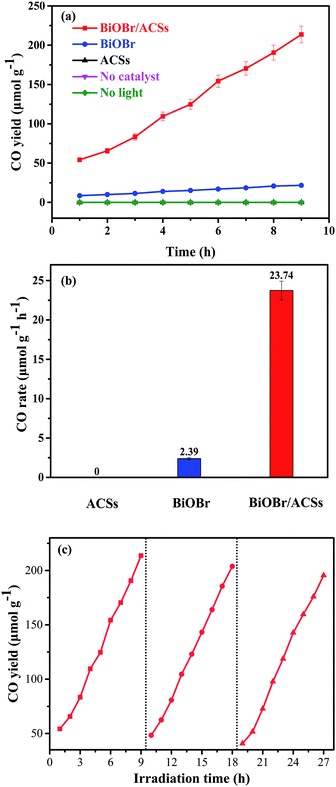 |
| | Fig. 6 Photocatalytic CO2 reduction over BiOBr and BiOBr/ACSs samples under UV-vis irradiation: (a) CO yield, (b) CO rate and (c) cyclic experiments. | |
Furthermore, the relevant cyclic experiments (Fig. 6c) further reveal that the photocatalytic activity of the BiOBr/ACSs sample only decreases about 8.28% after three cycles (9 h per cycle). Thus, our as-prepared BiOBr/ACSs sample should exhibit high activity, stability and reusability for photocatalytic CO2 reduction.
3.7 Photocatalytic mechanism
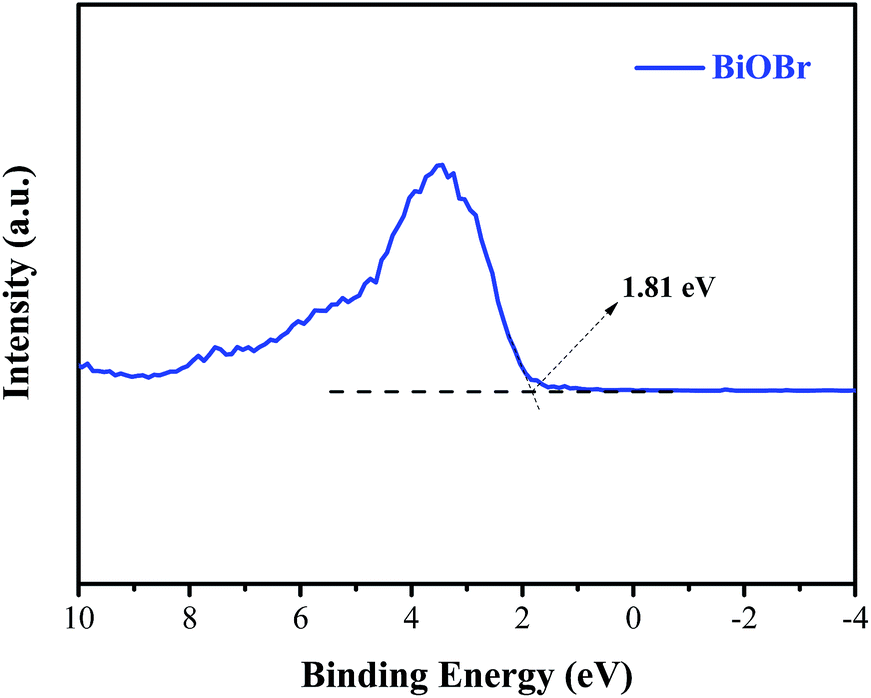
Generally, the conduction band minimum (CBM) of a semiconductor decided its photocatalytic reduction ability, and a higher CBM will endow the semiconductor with better photocatalytic reduction ability.20 Fig. 7 shows the valence band (VB) XPS spectra of the as-prepared BiOBr sample. It is found that the VB maximum of BiOBr is 1.81 eV. Combined with the DRS data of Eg (about 2.36 eV) for BiOBr, the calculated CBM should be −0.55 eV (obtained using the equation ECB = EVB − Eg),1 higher than the E0(CO2/CO) value of −0.53 eV. This is the main reason why a certain photocatalytic CO2 reduction activity (2.39 μmol g−1 h−1) of pure BiOBr exists.
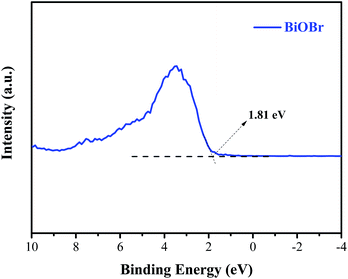 |
| | Fig. 7 Valence-band XPS spectra of BiOBr. | |
Based on the abovementioned experimental results and theoretical analyses, a possible mechanism for the enhanced photocatalytic activity of BiOBr/ACSs has been proposed in Scheme 2. As is known, the photocatalytic conversion of CO2 to CO should belong to a two-electron reduction reaction process at the gas–solid interface,4,46 where CO2 adsorption on the catalyst surface and the efficient separation of photoinduced e−–h+ pairs are two rather important factors affecting the CO2 reduction reaction. At first, BiOBr/ACSs can adsorb large amounts of CO2 when the system is filled with CO2 gas; this is mainly attributed to the contribution of a sufficient reaction zone from ACSs with larger specific surface area and higher pore volume than that of pure BiOBr (Table 1); consequently, a higher number of CO2 physical adsorption sites are achieved on the catalyst surface via the assistance of ACSs. Then, under simulated sunlight irradiation, BiOBr/ACSs have a more efficient e−–h+ pair separation than BiOBr (as seen in Fig. 4b); this should be related to the intrinsic electron transport capacity of ACSs,47,48 bringing about more efficient electron transfer to avoid the fast recombination of photoinduced electron–hole pairs. Finally, the valence band (VB) electrons (e−) of BiOBr are excited to its CBM, and CO2 activation is achieved; moreover, the holes (h+) left in VBM react with H2O to provide H+ (h+ + H2O → H+ + 1/2O2) for the reduction of CO2 to CO (2H+ + CO2 + 2e− → 2CO + H2O). Therefore, our findings on the enhanced photocatalytic CO2 reduction activity of BiOBr assisted by ACSs should provide new ideas and an interesting guidance for the construction of high-efficiency photocatalyst systems in the field of CO2 reduction to useful high-energy fuels.
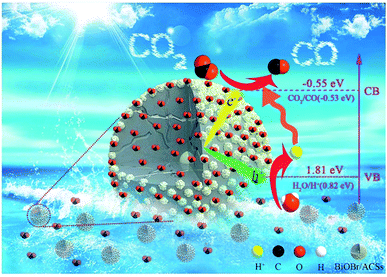 |
| | Scheme 2 Possible mechanism of the photocatalytic CO2 reduction to CO over BiOBr/ACSs. | |
4. Conclusions
In summary, phenolic resin spheres were prepared by suspension polymerization, and the ACSs were obtained via carbonization and steam activation; moreover, the BiOBr/ACSs photocatalyst was built using ACSs as a carrier. The photocatalytic results showed that BiOBr/ACSs had highest photocatalytic activity, with a yield of 213.67 μmol g−1 after 9 hours, about 9.9 times that of BiOBr (21.51 μmol g−1), and a corresponding CO production rate of approximately 23.74 μmol g−1 h−1 and 2.39 μmol g−1 h−1. Through the characterization of the prepared samples, we found that the following excellent characteristics of ACSs improved the ability of BiOBr to reduce CO2: (1) ACSs had no effect on the phase structure of BiOBr; (2) the rich pore structure (>83% micropore ratio) and high specific surface area (936.42 m2 g−1) of ACSs could provide more CO2 adsorption sites on the catalyst surface and adsorb more CO2 molecules (10.03 mg g−1); this allowed BiOBr to be under a CO2-enriched atmosphere; (3) the addition of ACSs could reduce the recombination rate of photo-generated electron–hole pairs, improving the effective electron mobility of BiOBr and thus enhancing the photocatalytic activity. Our findings should provide a new insight and method for the design and construction of highly active Bi-based photocatalytic materials assisted by ACSs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21506144, 21676178, 21706179), the Natural Science Foundation of Shanxi Province (201701D221037, 201801D211008), and the Foundation of State Key Laboratory of Coal Conversion (J18-19-605). In addition, the author is particularly grateful to Dr Jinbo Xue for his guidance on the article.
Notes and references
- C. h. Ding, L. Q. Ye, Q. Zhao, Z. G. Zhong, K. C. Liu, H. Q. Xie, K. Y. Bao, X. G. Zhang and Z. X. Huang, J. CO2 Util., 2016, 14, 135–142 CrossRef CAS.
- G. Centi and S. Perathoner, ChemSusChem, 2010, 3, 195–208 CrossRef CAS.
- E. J. Maginn, J. Phys. Chem. Lett., 2010, 1, 3478–3479 CrossRef.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- S. Kamimura, Y. Sasaki, M. Kanaya, T. Tsubota and T. Ohno, RSC Adv., 2016, 6, 112594–112601 RSC.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- M. Rakowski DuBois and D. L. DuBois, Acc. Chem. Res., 2009, 42, 1974–1982 CrossRef CAS.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2132–2133 CrossRef CAS.
- Y. Yang, C. Zhang, C. Lai, G. M. Zeng, D. L. Huang, M. Cheng, J. J. Wang, F. Chen, C. Y. Zhou and W. P. Xiong, Adv. Colloid Interface Sci., 2018, 254, 76–93 CrossRef CAS.
- M. Hu, R. Li, X. C. Zhang, C. M. Zhang, H. Zhang, C. M. Fan and J. Zhu, RSC Adv., 2017, 7, 50079–50086 RSC.
- R. Li, X. Y. Gao, C. M. Fan, X. C. Zhang, Y. W. Wang and Y. F. Wang, Appl. Surf. Sci., 2015, 355, 1075–1082 CrossRef CAS.
- H. Tian, Y. Fan, Y. Zhao and L. Liu, RSC Adv., 2014, 4, 13061 RSC.
- H. F. Cheng, B. B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- H. P. Lin, W. W. Lee, S. T. Huang, L. W. Chen, T. W. Yeh, J. Y. Fu and C. C. Chen, J. Mol. Catal. A: Chem., 2016, 417, 168–183 CrossRef CAS.
- R. He, D. Xu, B. Cheng, J. Yu and W. Ho, Nanoscale Horiz., 2018, 3, 464–504 RSC.
- L. Q. Ye, X. L. Jin, C. Liu, C. H. Ding, H. Q. Xie, K. H. Chu and P. K. Wong, Appl. Catal., B, 2016, 187, 281–290 CrossRef CAS.
- D. Wu, L. Q. Ye, H. Y. Yip and P. K. Wong, Catal. Sci. Technol., 2017, 7, 265–271 RSC.
- J. Wu, X. D. Li, W. Shi, P. Q. Ling, Y. F. Sun, X. C. Jiao, S. Gao, L. Liang, J. Q. Xu, W. S. Yan, C. M. Wang and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 8719–8723 CrossRef CAS PubMed.
- J. R. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- M. Q. Yang and Y. J. Xu, Nanoscale Horiz., 2016, 1, 185–200 RSC.
- X. X. Chang, T. Wang and J. L. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- Y. Bai, L. Q. Ye, L. Wang, X. Shi, P. Q. Wang, W. Bai and P. K. Wong, Appl. Catal., B, 2016, 194, 98–104 CrossRef CAS.
- Y. H. Ao, H. Tang, P. F. Wang, C. Wang, J. Hou and J. Qian, Composites, Part B, 2014, 59, 96–100 CrossRef CAS.
- X. H. Xia, Z. J. Jia, Y. Yu, Y. Liang, Z. Wang and L. L. Ma, Carbon, 2007, 45, 717–721 CrossRef CAS.
- Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865–2870 CrossRef CAS PubMed.
- B. Song, T. T. Wang, H. G. Sun, Q. Shao, J. K. Zhao, K. K. Song, L. H. Hao, L. Wang and Z. H. Guo, Dalton Trans., 2017, 46, 15769–15777 RSC.
- Z. Yang, X. Hao, S. Chen, Z. Ma, W. Wang, C. Wang, L. Yue, H. Sun, Q. Shao, V. Murugadoss and Z. Guo, J. Colloid Interface Sci., 2019, 533, 13–23 CrossRef CAS PubMed.
- S. X. Liu, J. Sun and Z. H. Huang, J. Hazard. Mater., 2010, 173, 377–383 CrossRef CAS PubMed.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849–1855 CrossRef CAS PubMed.
- R. Ocampo-Pérez, M. Sánchez-Polo, J. Rivera-Utrilla and R. Leyva-Ramos, Appl. Catal., B, 2011, 104, 177–184 CrossRef.
- X. C. Zhang, B. Q. Lu, R. Li, X. L. Li, X. Y. Gao and C. M. Fan, Sep. Purif. Technol., 2015, 154, 68–75 CrossRef CAS.
- E. A. Davis and N. F. Mott, Philos. Mag., 1970, 22, 0903–0922 CrossRef CAS.
- J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi B, 1966, 15(2), 627–637 CrossRef CAS.
- C. Gao, S. M. Chen, Y. Wang, J. W. Wang, X. S. Zheng, J. F. Zhu, L. Song, W. K. Zhang and Y. J. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- J. Xia, J. Di, S. Yin, H. Xu, J. Zhang, Y. Xu, L. Xu, H. Li and M. Ji, RSC Adv., 2014, 4, 82–90 RSC.
- J. Di, C. Zhu, M. Ji, M. Duan, R. Long, C. Yan, K. Gu, J. Xiong, Y. She, J. Xia, H. Li and Z. Liu, Angew. Chem., Int. Ed., 2018, 57, 14847–14851 CrossRef CAS PubMed.
- Y. L. Zhao, Y. C. Wei, X. X. Wu, H. L. Zheng, Z. Zhao, J. Liu and J. M. Li, Appl. Catal., B, 2018, 226, 360–372 CrossRef CAS.
- Y. Bai, P. Yang, P. Q. Wang, H. Q. Xie, H. F. Dang and L. Q. Ye, J. CO2 Util., 2018, 23, 51–60 CrossRef CAS.
- H. Cheng, B. Huang, Y. Liu, Z. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2012, 48, 9729–9731 RSC.
- C. M. Zhang, W. Song, Q. L. Ma, L. J. Xie, X. C. Zhang and H. Guo, Energy Fuels, 2016, 30, 4181–4190 CrossRef CAS.
- C. M. Zhang, W. Song, G. H. Sun, L. Xie, J. L. Wang, K. X. Li, C. G. Sun, H. Liu, C. E. Snape and T. Drage, Energy Fuels, 2013, 27, 4818–4823 CrossRef CAS.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112–116 RSC.
- E. Karamian and S. Sharifnia, J. CO2 Util., 2016, 16, 194–203 CrossRef CAS.
- Y. P. Zhai, Y. Q. Dou, D. Y. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS.
- M. Li, S. S. Xiang, X. Q. Chang and C. S. Chang, J. Solid State Electrochem., 2016, 21, 485–494 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01329f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Changming Zhanga,
Guangmin Rena,
Zhanfeng Zheng
*a,
Changming Zhanga,
Guangmin Rena,
Zhanfeng Zheng