
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRotational dynamics of the organic bridging linkers in metal–organic frameworks and their substituent effects on the rotational energy barrier†
Srimanta Pakhira *
*
Discipline of Physics, Indian Institute of Technology Indore (IITI), Simrol, Khandwa Road, Indore, 453552, MP, India. E-mail: spakhira@iiti.ac.in; srimantacu@gmail.com; Tel: +91-732-430-6830
First published on 21st November 2019
Abstract
Organic bridging linkers or ligands play an important role in gas and fuel storage, CO2 capture, and controlling the radical polymerization reactions in metal–organic frameworks (MOFs) nanochannels. The rotation of the linkers causes the expansion of the pore size and pore volume in MOFs. To understand the rotational behavior of organic linkers in MOFs and the substituent effects of the linkers, we investigated the equilibrium structure, stability, potential energy curves (PECs), and rotational energy barriers of the organic bridging linkers of a series of MOF model systems imposing three constrained imaginary planes. Both the dispersion-uncorrected and dispersion-corrected density functional theory (DFT and DFT-D i.e. B3LYP and B3LYP-D3) methods with the correlation consistent double-ζ quality basis sets have been applied to study the model MOF systems [Cu4(X)(Y)6(NH3)4] (where X = organic bridging linker, and Y = HCO2). The present study found that the structural parameters and rotational energy barrier of the model MOF containing 1,4-benzendicarboxylate (BDC) linker are in accord with previous experiments. This study reveals that rotational barriers significantly differ depending on the substituents of organic linkers, and the linker dynamical rotation provides information about the framework flexibility with various potential applications in porous materials science. Changing the linkers in the MOFs could be helpful for designing various new kinds of flexible MOFs which will have many important applications in gas storage and separation, catalysis, polymerization, sensing, etc.
1 Introduction
The past few decades have seen explosive and tremendous growth in the preparation, characterization, experimental synthesis, computational investigation, and study of porous structure materials known as metal−organic frameworks (MOFs) due to the possibility of obtaining a large variety of aesthetically interesting structures that could also be of great interest for applications in a number of fields related to porous materials. MOFs are a family of compounds consisting of metal ions or clusters or metal-containing units coordinated to organic linkers, which form extended network structures, using strong bonds (reticular synthesis) to create open crystalline frameworks with permanent porosity. They are crystalline hybrid porous materials where porosity and functionality can be controlled on the nanoscale by choice of the metal secondary building units (SBUs) and the organic bridging linker, also known as the bulkier rotational group. The structure of MOFs is characterized by an open framework that can be porous with transition-metal cations and multidentate organic linkers.MOFs have the potential to exhibit properties inherent to the building blocks, such as geometric rigidity, chemical functionality, or chirality. Their simple preparations are generally high yielding and scalable, and by careful use of the building blocks a certain degree of design can be wielded to produce targeted products from the vast number of MOFs that are potentially accessible. These MOFs are materials of wide interest in recent years because of their interesting chemistry and potential applications in various fields of science and technology as intriguing porous materials because of their high structural regularity and diversity, easy modification, high porosity, and structural flexibility.1–3 MOF structures often have sizable pores that can be filled with guest molecules, many of which are adsorbed to the internal surfaces. Thus, MOFs have emerged as a class of porous materials with enormous potential applications in various fields of science, especially nanoscience and nanotechnology, with possible industrial applications within catalysis, gas adsorption, separation and storage, selective separation, sensing, molecular recognition, chemical reaction, radical polymerization, etc.4–12 In the last decade a large number of MOFs containing various organic linkers were synthesized experimentally and studied theoretically as well as computationally. On account of these bright prospects, MOF materials have aroused more and more attention from chemistry and materials science. Additionally, scientists are always still researching and finding ways to design MOFs to target specific properties with significant achievements.
Several kinds of organic bridging ligands/linkers with multiple carboxylate groups have been used in MOFs to bridge between inorganic building units i.e. metal cation clusters.13,14 In particular, one of the simplest dicarboxylate organic linker, 1,4-benzendicarboxylate (BDC), is used to construct various MOFs such as MOF-5,15 Al-MOFs,16 [Cu2(BDC)2DABCO]17 and [Zn2(BDC)2DABCO]18 (where BDC = 1,4 benzene-dicarboxylate and DABCO = 1,4-diazabicyclo[2,2,2]octane) porous materials further promoted the development of this porous material science field, mainly due to their robust porosity. The crystallographic structures of all these MOF materials revealed a planar conformation of the BDC linker,17,18 and the planar rigidity of the BDC linker in the MOF-5 (also known as IRMOF-1) was further investigated by analysing the rotational energy barrier of the BDC linker. Yaghi and co-workers experimentally estimated the rotational energy barrier of the BDC linker in MOF-5 to be about 47.3 kJ ± 8.4 mol−1 by solid-state NMR measurements,19 which was later confirmed by density functional theory (DFT) calculations to be within the range 51.0–62.8 kJ mol−1 considering a model system.20–22 In contrast to the rigid BDC conformation, the flexible property of the framework in [Zn2(BDC)2DABCO] was also reported and thoroughly investigated.18,21 The breathing behaviour, or the guest molecule dependent framework deformation, of the [Zn2(BDC)2DABCO] MOF was first reported between the guest-free orthorhombic framework and the benzene-inclusion rhombic-grid one,18 and it has shown that the breathing behaviour of the MOFs is due to the rotation of the organic linkers.11
In recent years, a new kind of MOF material (such as [M2(X)2DABCO], where M = Cu or Zn, X = dicarboxylate ligand) has been synthesized by introducing substituent groups onto the organic bridging linkers. Their properties and possible applications in modern porous materials science have also been investigated. The crystallographic structures of the MOFs revealed that the ligand-dependent planar or nonplanar equilibrium structures of the ligand X is due to the O–C–C–C dihedral angle rotation of the MOFs.23,24 The rotational barrier of the 2-bromo-1,4-benzendicarboxylate (BrBDC, a Br substitution in the BDC) in the IRMOF-2 was observed to be 30.5 kJ mol−1 by dielectric spectroscopy and it was computed to be 19.7–31.4 kJ mol−1 by various ab initio methods considering a model system,22 indicating that the ligand or linker is no longer rigid but can frequently rotate during sub-microsecond MD simulations. Similarly, the 1,4-naphthalenedicarboxylate (NDC) linker is also used to construct MOFs. The rotational dynamic behaviour and the rotational barrier of the NDC in the MOF, [Zn2(NDC)2(DABCO)] were experimentally studied.25 The rotational barrier of the NDC linker was measured to be 53.0 kJ mol−1 by NMR spectroscopy.25 Recently, Pakhira et al. computationally studied the rotational barrier of the [Zn2(NDC)2(DABCO)] MOF, and found results comparable to experimental data.21 Therefore, Zn-based SBU containing MOFs (i.e. [Zn2(X)2DABCO]) were studied well, however Cu-based MOFs (i.e. [Cu2(X)2DABCO]) have not been studied yet in detail especially computationally or theoretically.
To the best of our knowledge, there are no computational or theoretical results to support the rotational barrier of the NDC in the [Cu2(NDC)2(DABCO)] MOF, which could provide important information for a precise design of the micropore with an imperative role in polymerization reactions and gas storage in this MOF. The breathing behaviour of these MOF materials can also be modulated by the substituent groups onto the organic bridging linkers.26 These kind of MOFs, [M2(X)2DABCO] (where M is metal and X is organic linkers), were used for radical polymerization reactions in the one-dimensional MOF nanochannels.11,27 Although Kitagawa and co-workers27 succeeded in the tacticity control of polymerization reactions using these linkers, it is still elusive how the different ligands X contribute to the tacticity control. Their experimental observation indicates that the linkers X, along with their substituents and rotation play the most important role in controlling the tacticity and the size of the micropore nanochannels of the MOFs.24–27 However, the effect of metal cations on the linker rotation, rotational energy barrier, and tacticity control are not well studied either experimentally or theoretically.
A recent experimental observation found the substituent effects of the organic bridging linkers play an important role in determining the rotational barrier of the organic linker in the model MOFs, and the flexibility of the organic linker in MOFs. Therefore, the rotational and dynamical behaviours of these organic linkers and substituent of the organic linkers in the MOFs should play an important role in controlling not only the tacticity of PMMA produced by the radical polymerization inside the MOF channels, but also gas storage, CO2 capture, gas separation, etc. However, to the best of our knowledge, no theoretical or computational comprehensive studies have been carried out in detail to study the electronic structure calculations, rotational and dynamical behaviour of the organic linkers, and the effect of the substituents of the linkers on the rotational barrier energy of various kinds of MOFs considering a model system with Cu2+ metal cations and different kinds of organic ligands such as BDC, DFBDC, TFBDC, DMBDC, DBrBDC and NDC (where DFBDC = 2,5-difluoro-1,4-benzenedicarboxylate, and TFBDC = 2,3,5,6-tetrafluoro-1,4-benzenedicarboxylate), as shown in Fig. 1. However, MOFs containing Zn2+ in a different coordination environment, such as MOF-69 and [Zn2(X)2DABCO]21 MOFs, have been shown to be more stable than those with purely tetrahedral Zn2+ ions. MOF-69 contains both tetrahedral and octahedral Zn centres which share oxygen to form infinite columns and exhibit chemical stability to exchange with a variety of solvents. In order to increase the chemical stability of MOFs, early efforts involved the employment of SBUs containing Cu2+ ions with d9 configuration. Compared to a d10 transition metal like Zn, metal ions with d9 configuration possess ligand field stabilization energy, regardless of what coordination environment those ions adopt. This increased ligand field stabilization energy should make the ligand substitution processes more favourable. Thus, we used Cu2+ ions instead of Zn2+ ions in the present study followed by the previous experimental works of Kitagawa and his co-workers.24
 | ||
| Fig. 1 Different kinds of dicarboxylate ligands or organic bridging linkers (X) used to construct the metal–organic frameworks (MOFs); where BDC = 1,4-benzendicarboxylate; DFBDC = 2,5-difluoro-1,4-benzenedicarboxylate; TFBDC = 2,3,5,6-tetrafluoro-1,4-benzenedicarboxylate; DMBDC = 2,3-dimethoxy-1,4-benzenedicarboxylate and NDC = 1,4-naphthalenedicarboxylate. | ||
To explore the rotational dynamics, substituent effects of the linkers on rotational energy barrier, framework flexibility, the design principle of MOFs using different kinds of linkers and tacticity control mechanism by the [Cu2(X)2DABCO] MOFs, we have computationally investigated the rotational behaviour of dicarboxylate ligands X to prepare accurate force field parameters for future MD simulation studies. For this purpose, we investigated and studied the electronic structure, stability, flexibility, potential energy curves (PECs), and rotational energy barrier of various organic linkers X (See Fig. 1) in the [Cu2(X)2DABCO] type of MOFs by analysing the following model MOF systems noted as [Cu4(X)(Y)6(NH3)4], where X = dicarboxylate ligand, and Y = HCO2. Due to our computational limitation (QM calculations can be done on ∼100 atoms) and the large size of the frameworks of MOFs, we have considered only one fragment of each MOF including one of the ligands X between the two metal units [Cu2(HCO2)3] in the MOFs for our model computational system. To mimic the actual environment in those kinds of model MOFs, each Cu atom was capped with one –NH3 and –HCO2 groups following the work of Pakhira et al.21 and Grosch and Paesani.28 Pires et al.29 studied a series of zirconium dicarboxylate-based MOFs and their application in the separation of ethane/ethylene mixtures with the effect of linker rotation. Towards understanding the rotational dynamics of the linkers in presence of the Cu2+-ions and the rotational barrier of the linkers, we have considered a cluster model [Cu2(X)2DABCO] MOF system to investigate the structure and rotation of the linkers from the molecular point of view, which will contribute to the creation of novel MOF model system computationally. To provide a fundamental insight, we have computationally considered five dicarboxylate ligands, and they are BDC, DFBDC (2,5-difluoro-1,4-benzenedicarboxylate), TFBDC (2,3,5,6-tetrafluoro-1,4-benzenedicarboxylate), DMBDC, and NDC by introducing various substituents onto the organic linkers, as shown in Fig. 1.
2 Computational details and methodology
First-principle based hybrid density functional theory (DFT) was used to perform all the computations.29–33 It is essential to include the long-range dispersion correction in the hybrid DFT30–42 to describe the systems properly, as there are many weak van der Waals interactions in MOF model systems. The improved version of semi-empirical Grimme-D3 (ref. 37–40) dispersion corrections were added in the present calculations in order to incorporate van der Waals (vdW) dispersion effects on the system and to estimate the vdW forces. Both the DFT (B3LYP)30 and DFT-D31 (B3LYP-D3) methods (which have been shown to give correct electronic properties of the MOFs) with the correlation consistent double-ζ (cc-pVDZ) quality basis set have been used to obtain the optimized geometries of the five model MOF systems, [Cu4(X)(Y)6(NH3)4] (where X = BDC, DFBDC, TFBDC, DMBDC and NDC, and Y = HCO2), (Fig. 2 and 3 and S1–S3 in the ESI†). The cc-pVDZ basis set has been used for C, H, N, O and F atoms,43 and the cc-pVDZ-pp basis set has been used for only Cu atoms.44 The harmonic vibrational frequencies at the optimized geometries were analyzed at the respective levels of theory to reveal the nature of the stable minima. The DFT and DFT-D methods were used for geometry optimization because densities and energies obtained with the methods are less affected by spin contamination than other approaches.31–36,45–48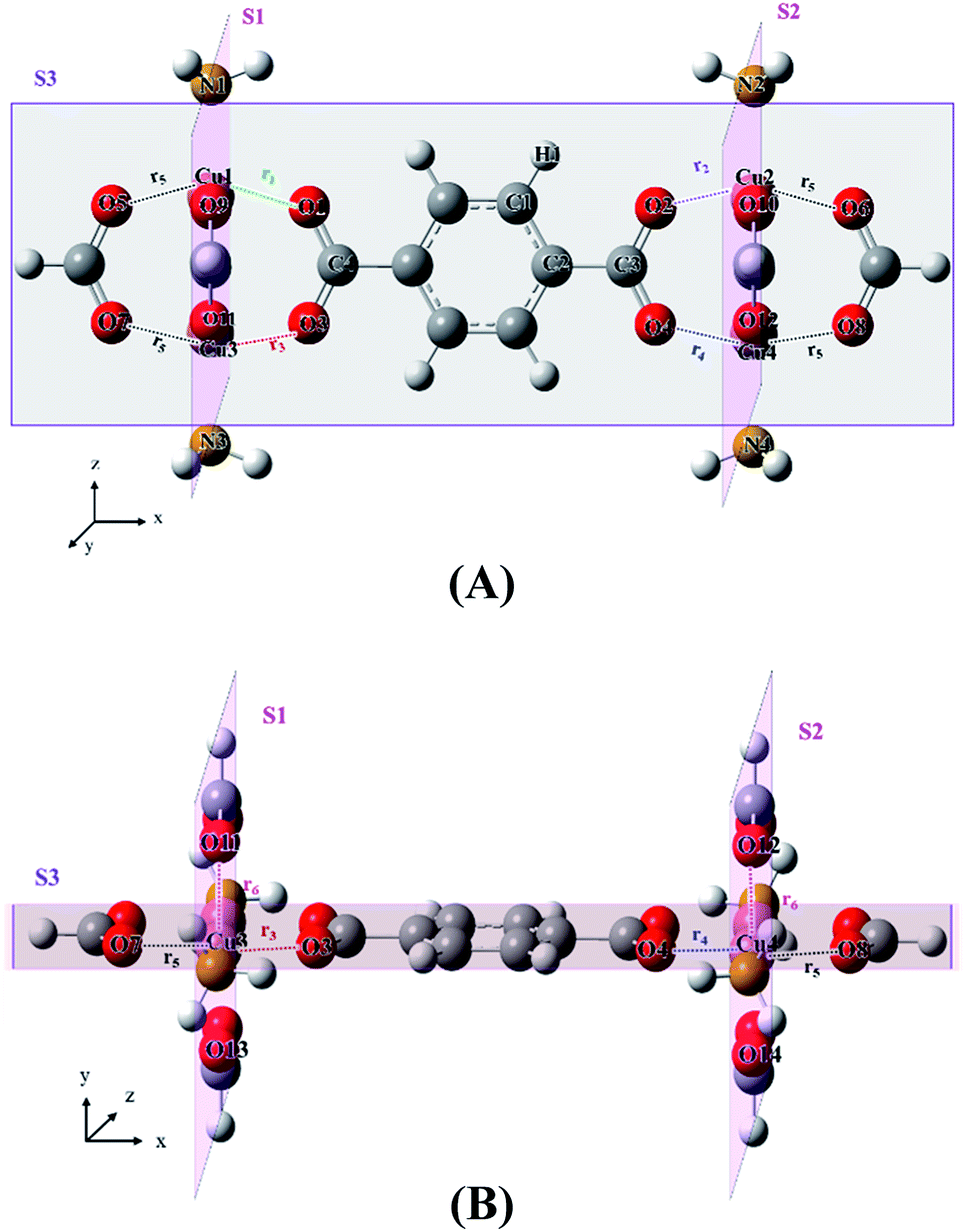 | ||
| Fig. 2 Optimized structure of the model [Cu4(BDC)(HCO2)6(NH3)4] MOF containing BDC organic linker (BDC = 1,4-benzenedicarboxylate); (A) top view, (B) side view. The dihedral angle χ of the BDC linker is represented by C1–C2–C3–O2. The atoms H, C, N, O and Cu were represented by the white, grey, dark yellow, red, and blackish red colours respectively, and S1, S2, and S3 are the imaginary planes. | ||
 | ||
| Fig. 3 Optimized structure of the model [Cu4(TFBDC)(HCO2)6(NH3)4] MOF containing TFBDC (2,3,5,6-tetrafluoro-1,4-benzenedicarboxylate); (A) top view, (B) side view. The dihedral angle χ of the BDC linker is represented by C1–C2–C3–O2. The atoms H, C, N, O, F, and Cu were represented by the white, grey, dark yellow, red, sky blue, and blackish red colours respectively, and S1, S2, and S3 are the imaginary planes. | ||
As stated earlier, Grimme and co-workers37–39 introduced an improved version of the semi-empirical hybrid DFT method, DFT-D3,38,40 which incorporates an additional term in the dispersion series and accounts all the vdW weak dispersion effects as well as interactions. Thereby some effects of the chemical environment are incorporated into an otherwise largely heuristic correction. The detailed description of the DFT-D methods can be found in ref. 37, 38, 48, and 49. In this work, “-D3” corrections have been incorporated into the double hybrid B3LYP functional. Grimme37–39 demonstrated that using the DFT-D method with the ζ quality basis sets much of the basis set superposition error (BSSE) calculation is avoided,37–39,48–50 which is otherwise essential for highly correlated methods like MP2 and coupled-cluster single double (triple) (CCSD(T)) methods. In fact, the BSSE is absorbed into the empirical potential showed by Grimme and co-workers.37 The threshold used for evaluating the convergence of the energy, forces, and electron density was 10−7 a.u. for each parameter. A very accurate geometry and thermochemistry can be obtained by this DFT-D functional when the systems are covalently bonded and weakly bound vdW dominated by long-range dispersion forces.35,36,48–50
During the optimization, we have considered planar rigidity constraint condition with three imaginary planes, S1 (y–z plane), S2 (y–z plane) and S3 (x–z plane), as shown in Fig. 2 and 3, to incorporate imaginary crystal effects as well as to keep the planar rigid MOFs structure without any distortion. In addition, this condition reduces the number of degrees of freedom and enables us to optimize and scan many MOF model systems with limited computational resources. The intersection angles between the planes S1 and S3, and S2 and S3 were kept at 90°. Two metal atoms (Cu1, Cu3), two HCO2 groups and two N atoms (N1 and N3) were situated on the plane S1. Other two metal atoms (Cu2, Cu4), two HCO2 groups and two N atoms (N2 and N4) were situated on the plane S2. The two carboxylate groups of the linker, the four Cu and four N atoms, and two HCO2 groups were situated on the plane S3. Atoms with the planar constraint can move on the constraint planes. The planes S1 and S2 are parallel and distances Cu1–Cu2, Cu3–Cu4, N1–N2, and N3–N4 (i.e. the distance between the planes S1 and S2) correspond to the lattice constant along the MOF channel direction and the distance between S1 and S2 planes was relaxed during the optimization. This means that the equilibrium distance between the S1 and S2 planes, which corresponds to the lattice constant, was optimized at both the levels of theory, and it was relaxed during the potential energy scan of the linkers in MOF. It should be noted that there is no planar constraint on the atoms of the organic bridging ligands/linkers i.e. rotational groups, or the benzene ring and its substituent groups. All the DFT and DFT-D calculations were performed with the general-purpose electronic structure quantum chemistry program Gaussian09 (ref. 51) suite with the default convergence criteria.
The potential energy curves (PECs) and rotational barrier energies of the organic ligands have been computed for all the MOF model systems at both the B3LYP, and B3LYP-D3 methods with the correlation consistent double-ζ quality basis sets imposing the abovementioned planar constraint conditions. As stated earlier, the BSSE is absorbed into the empirical potential so we have ignored the BSSE corrections in the present computation for the PECs and rotational barrier calculations. The rotation of the organic linkers is defined as a continuous transition where the spatial alignment of a dicarboxylate ligand is changed by turning around a rotational axis. Taking the optimized structure, the bridging ligands were rotated around the Cu cluster–cluster axis (dihedral angle χ(C1–C2–C3–O2), see Fig. 2 and 3) to scan PECs in the range of 0° to 90° with 5° increments. During the scan, all the degrees of freedom were relaxed with the fixed χ value and the planar constraint. The rotational energy barrier has been computed as the energy difference between the most stable and unstable positions of the organic linkers on the PECs calculated by the partially relaxed scan.
3 Results and discussions
3.1. Equilibrium geometries and structures of the model MOFs containing different dicarboxylate ligands/linkers
To obtain the equilibrium geometry and structural properties of the [Cu4(X)(Y)6(NH3)4] model MOF systems, we applied both the B3LYP and B3LYP-D3 methods, and a comparison has been drawn to study the consistency of both the levels of theory. An investigation has been carried out to study the equilibrium structural properties/parameters i.e. several bond distances, bond angles, and dihedral angles of the model MOF systems. Optimized equilibrium bond distances of the BDC and TFBDC models are tabulated in Table 1. A comparison has been drawn between our computational results with the previous experimental results as well as theoretical values wherever available, and good agreement was obtained. For example, in the BDC case, the average Cu–O equilibrium bond distances (i.e. average value of r1–r6) showed less than 0.01 Å difference between the B3LYP and B3LYP-3D methods, and it agrees within 0.04 Å with the previous experimental value where the experiment was performed by using Zn2+ ion instead of Cu2+ ion; see Table 1. The discrepancy between the experiments and theory are well below the DFT accuracy, and the average Cu–O bond distances in the [Cu4(X)(Y)6(NH3)4] model MOF systems, is about 0.05 Å lower than the average Zn–O bond lengths in the [Zn4(X)(Y)6(NH3)4] model MOF systems, reported by Pakhira et al. computationally.21 This result indicates that the Cu2+ cations increase the interactions between the carboxylate groups and the metal cations in the [Cu2(X)2DABCO] MOFs by reducing the equilibrium bond lengths, which causes more rigid frameworks than Zn-based [Zn2(X)2DABCO] MOFs. From the comparison between the DFT and DFT-D methods, we can conclude that the structural properties of the model MOFs obtained by both the methods, are reasonably in accordance with previous experimental as well as theoretical reported values. Therefore, as a representative result, several structural properties (equilibrium bond distances and angle) obtained by the B3LYP-D3 are reported in Table 2 and optimized structures are shown in Fig. 2 and 3 (BDC and TFBDC organic linkers) and S1–S3 (DFBDC, DMBDC and NDC linkers) in the ESI† at the same level of theory.| Systems parametersa | BDC | TFBDC | Others | ||||
|---|---|---|---|---|---|---|---|
| Methods | Others | Methods | |||||
| B3LYP | B3LYP-D3 | Expt. | Theo. | B3LYP | B3LYP-D3 | Expt. | |
| a All units are in Å.b Ref. 24.c Ref. 21.d Ref. 28. | |||||||
| r1 | 1.996 | 1.989 | 2.027b | 2.054c,d | 2.011 | 2.002 | — |
| r2 | 1.996 | 1.989 | — | — | 2.011 | 2.002 | — |
| r3 | 1.996 | 1.989 | — | — | 2.011 | 2.002 | — |
| r4 | 1.996 | 1.989 | — | — | 2.011 | 2.002 | — |
| r5 | 2.000 | 1.997 | — | — | 2.008 | 2.004 | — |
| r6 | 2.002 | 1.999 | — | — | 2.011 | 2.008 | — |
| RCu−N | 2.205 | 2.184 | — | — | 2.215 | 2.193 | — |
| Cu1–Cu2 | 10.951 | 10.926 | 10.824b | — | 10.969 | 10.932 | 10.866b |
| Cu1–Cu3 | 2.654 | 2.658 | 2.891 | 2.669 | — | ||
| Structural parameters | Dicarboxylate ligand in the model MOFs | ||||
|---|---|---|---|---|---|
| BDC | DFBDC | TFBDC | DMBDC | NDC | |
| r1 (Å) | 1.989 | 1.975 | 2.002 | 2.006 | 1.967 |
| r2 (Å) | 1.989 | 2.008 | 2.002 | 2.009 | 1.967 |
| r3 (Å) | 1.989 | 2.008 | 2.002 | 1.965 | 2.005 |
| r4 (Å) | 1.989 | 1.976 | 2.002 | 1.958 | 2.005 |
| r5 (Å) | 1.997 | 1.999 | 2.004 | 1.998 | 1.997 |
| r6 (Å) | 1.999 | 1.999 | 2.008 | 2.002 | 2.001 |
| RCu−N (Å) | 2.184 | 2.182 | 2.193 | 2.184 | 2.184 |
| Cu1–Cu2 (Å) | 10.926 | 10.969 | 10.932 | 10.944 | 10.936 |
| Cu1–Cu3 (Å) | 2.658 | 2.662 | 2.656 | 2.842 | 2.659 |
| O5–Cu1–O1 (°) | 168.7 | 169.1 | 168.7 | 168.9 | 168.5 |
| O7–Cu3–O3 (°) | 168.7 | 168.0 | 168.7 | 168.5 | 168.8 |
| Cu1–O1–C4 (°) | 122.8 | 123.0 | 121.5 | 121.6 | 123.9 |
| Cu3–O3–C4 (°) | 122.8 | 122.5 | 121.5 | 124.2 | 121.7 |
| Cu2–O2–C3 (°) | 122.8 | 122.5 | 121.5 | 121.6 | 123.9 |
| Cu4–O4–C3 (°) | 122.8 | 123.1 | 121.5 | 124.2 | 121.8 |
| H1–C1–C2 (°) | 118.8 | 119.2 | — | 118.1 | 119.1 |
| F–C–C (°) | — | 122.6 | 120.5 | — | — |
| O–C–C (°) | — | — | — | 122.8 | — |
| C–C–C (°) | — | — | — | — | 122.5 |
All the structural parameters of the BDC containing organic linkers of the simplest MOF model system are well harmonized with the earlier experimental reported values as depicted in Tables 1 and 2. The lattice constant along the one dimensional channel i.e. the equilibrium bond distance of the nonadjacent Cu1–Cu2 agrees well with the previous experimental values of Matsuda et al.24 (measured by X-ray crystallography) within 0.10 Å. The Cu1–Cu2 bond distances of the other model MOF systems containing DFBDC, TFBDC, DMBDC and NDC linkers are well harmonized with the experimental values of Matsuda et al.24 In other words, we can say that the present DFT-D calculations of the equilibrium geometries with the parameters of all the MOF model systems containing various organic linkers are in excellent agreement with the previous experiments and in some cases our computational results reproduce the earlier experimental values. In all of other model MOFs (which contain DFBDC, TFBDC, DMBDC and NDC ligands), there are small differences in the average bond distances of Cu–O, Cu1–Cu2 and Cu1–Cu3 and the average angels θ(Cu–O–C) and β(O–Cu–O) by the substitution of the organic linkers as shown in Table 2. The average angles of θ(Cu–O–C) and β(O–Cu–O) agree with previous experimental (125.5° and 158.6°)52 and theoretical (125.9° and 162.9° degrees)28 values. The deviation is well below the DFT accuracy. Therefore, we can say that there is no significant effect on the bond distances and angles when the organic ligands have been changed from BDC linker to other linkers i.e. DFBDC, TFBDC, DMBDC and NDC as depicted in Tables 1 and 2.
Optimized equilibrium dihedral angle χ(C1–C2–C3–O2) of all the model MOFs are tabulated in Table 3 along with the previous experimental values of Matsuda et al.24 for comparison. The optimized dihedral angle of both the BDC and DFBDC ligands is 0°, i.e. the organic linkers are lying on the same plane (here plane S3; see Fig. 2, and S1 in the ESI†). This result reveals that both the linker structures are planar, as it was observed by Matsuda et al. in their X-ray crystallography of the BDC and DFBDC ligands. In the other model systems containing TFBDC, DMBDC, and NDC linkers, the organic bridging linkers are inclined about the dihedral angle χ 46.6°, 25.100°, and 45.015°, respectively from the plane S3 computed at the B3LYP-D3 method as shown in Fig. 2, S2–S3 in the ESI.† The computed dihedral angles χ of these model MOF are in reasonable accord with X-ray crystal structure.24 Therefore, the present calculations showed that the MOF model systems containing TFBDC, DMBDC, and NDC linkers are not planar as the linkers are inclined. In conclusion, our optimization of the model MOF systems well reproduced the experimentally determined dihedral angle χ within 3°–10°, or the planar or nonplanar structures of the dicarboxylate ligands.
3.2. Electrostatic and vdW interactions determine the position of the organic linkers in MOFs
This study found that the equilibrium bond distances (Cu–O, Cu1–Cu2, and Cu1–Cu3) and angels (θ(Cu–O–C) and β(O–Cu–O)) in the Cu clusters of the model MOFs are not significantly altered by the substitution of the organic ligands in the MOF nanochannels. However, the structure of the bridging linkers or dicarboxylate ligands significantly fluctuates between the planar and nonplanar structures i.e. the linkers can be inclined at a particular angle depending on the nature and structure of the linkers. This phenomenon indicates that two interactions are playing an important role in determining the planarity and non-planarity of the organic ligands; (a) electrostatic interactions (including hydrogen bonding) and (b) van der Waals (vdW) interactions. Therefore, we can say that the intramolecular electrostatic interactions between the atoms in the carboxylate groups and the organic birding ligands like BDC and DFBDC must be strong at the planar structure because of the short distance. We found that there are four hydrogen bonded (O⋯H) attractive interactions to stabilize the planar structure in the model MOF containing BDC linker, the average O⋯H distance between H and O atoms (H1–O2 in Fig. 2A) is about 2.43 Å. Natural charge calculations were performed by Natural Bond Orbital (NBO) analysis, and the average charges of the atoms in the linkers are reported in Table 4. This NBO study indicates that there is a repulsion between the F and O atoms (since they have same polarity of charges) in the TFBDC linker as shown in F–O2 in Fig. 3A, and there are electrostatic repulsive interactions between the F and O atoms because of their strong electronegativity. Due to this repulsion in the model MOF system, the TFBDC ligand is inclined at 46.6°. In the cases of DFBDC and DMBDC organic linkers, there are two attractive interactions between the H and O atoms and two repulsive interactions between the F and O atoms in the benzene and O atoms in the carboxylate groups. The planar equilibrium structures of these two ligands suggest that the hydrogen bonded attractive interactions are stronger than the repulsive interactions.| Dicarboxylate linker in the model MOFs | Charges of various atoms in the linkers | Charge of O atoms in the dicarboxylate of the linker | |||
|---|---|---|---|---|---|
| H | C | O | F | ||
| BDC | 0.245 | −0.128 | — | — | −0.703 |
| DFBDC | 0.262 | 0.458 | — | −0.333 | −0.680 |
| TFBDC | — | −0.226 | — | −0.324 | −0.647 |
| DMBDC | −0.193 | −0.196 | −0.575 | — | −0.699 |
| NDC | 0.226 | −0.222 | — | — | −0.701 |
Apart from the electrostatic interaction or repulsion, there are vdW interactions, which play an important role in understanding the nonplanar structure of the NDC linker. The distance between the carboxylate O and closest naphthalene C atoms (2.871 Å) and that between the O and closest H atoms (2.217 Å) at the partially relaxed scan structure at χ = 0° are shorter than the sum of the vdW radii of the C and O atoms (3.569 Å), as well as the H and O atoms (3.120 Å) respectively. Here we have used the vdW radii from the general AMBER force field as shown in Table 5.53 A strong electrostatic repulsive force between the closet C atoms in the NDC linker and O atoms in the carboxylate group was observed. Thus, both the strong vdW repulsive forces and electrostatic forces have appeared at the planar NDC structure. Due to this strong vdW dispersion with electrostatic repulsion, the nonplanar equilibrium structure of the NDC linker containing MOF model system appears at the equilibrium dihedral angle (χ) 46.0° (Fig. S3† in the ESI†). Both the B3LYP and B3LYP-D3 methods tend to underestimate the dihedral angle χ of the model MOF containing NDC linker about 3–4° degree with the experimental value.24
| Dicarboxylate linker in the model MOFs | Distances at 0.0° B3LYP-D3 (Å) | Sum of vdW radiia (Å) | ||
|---|---|---|---|---|
| a The vdW radii are from general AMBER force field (ref. 53).b Distance between the O atom of the carboxylate group and the H or C atom which is in the rotational group and closest to the O atom. | ||||
| NDC | O–Hb | O–Cb | O–H | O–C |
| 1.935 | 2.714 | 3.120 | 3.569 | |
3.3. Assortment of rotational energy barrier of various organic linkers in MOFs
The rotational energy barriers were computed at both the DFT and DFT-D methods for the BDC, DFBDC, TFBDC, DMBDC, and NDC linkers in the model MOF systems and the potential energy curves (PECs) of the organic ligands were plotted as shown in Fig. 4 and S4–S6.† Now, we will discuss the rotational barrier energies of these organic linkers in the model MOFs, which are reported in Table 6. The rotational energy barriers of all the aforementioned organic ligands of the model MOFs have been computed from the PECs obtained by the partially relaxed scan, finding the most unstable position of the organic ligands. In the model systems with the planar dicarboxylate ligands (BDC and DFBDC), the energy minimum and maximum were found at the dihedral angles χ(C1–C2–C3–O2) 0° and 90° respectively (Fig. 4A and S4†). Meanwhile, in the model systems with non-planar dicarboxylate ligands (TFBDC, DMBDC, and NDC; see Fig. 4B and S5 and S6 in the ESI†), the energy minimum was found around the dihedral angles (χ) 30–45° and the two energy maximum angles were at 0° and 90° (Fig. 4B and S5 and S6 in the ESI†). Therefore, there is one rotational barrier at 90° for the planar organic linkers and two barriers at 0° and 90° for the nonplanar linkers such as TFBDC, DMBDC, and NDC. | ||
| Fig. 4 (A) Potential energy curves (PECs) of the model MOF containing BDC organic linker; and (B) PECs of the model MOF containing TFBDC linker. | ||
| Dicarboxylate linker in the model MOFsa | Rotational barrier at 90.0° | Rotational barrier at 0.0° | Others | |||
|---|---|---|---|---|---|---|
| Methods | Methods | |||||
| B3LYP | B3LYP-D3 | B3LYP | B3LYP-D3 | Expt. | Theo. | |
| a All units are in kJ mol−1.b Ref. 19 (in IRMOF-1).c Ref. 22 (in IRMOF-1 model system).d Ref. 25. | ||||||
| BDC | 51.09 | 51.22 | — | — | 47.5 ± 8.4b | 51.04–60.67c |
| DFBDC | 26.91 | 28.51 | — | — | — | — |
| TFBDC | 04.94 | 05.96 | 12.47 | 13.00 | — | — |
| DMBDC | 09.74 | 14.60 | 02.34 | 02.10 | — | — |
| NDC | 16.83 | 17.88 | 17.70 | 18.01 | 53d | — |
The present computation found that the BDC organic linker in the MOF model system has the highest rotational barrier energy, which reflects the strong stabilization of the planar structure by the four hydrogen bonded interactions. The calculated rotational energy barrier of the BDC linker is well harmonized with both the previous experimental and theoretical results shown in Table 6 as well. The barrier of the BDC linker in the model MOFs has been decreased about 6–5 kJ mol−1 when Cu2+ cations are used in the MOFs instead of Zn2+ cations. The next highest energy barrier of the MOF model system at χ = 0° is found in the DFBDC ligand and the calculated energy value using the B3LYP-D3 method is about 22.71 kJ mol−1 lower than BDC organic ligand indicating that this linker is more flexible than BDC. Therefore, we can say that the rotational barrier is reduced by an amount of 22.71 kJ mol−1 due to the F atom substitution in the BDC ligand of the model MOF system. This calculation found that the rotational barrier of the DFBDC linker in the [Cu2(X)2DABCO] MOFs has been decreased about 11 kJ mol−1 when Cu2+ cations were used in the frameworks compared to the [Zn2(X)2DABCO] MOFs reported by Pakhira et al.21 Therefore, we can say that not only the linkers the metal cations have an important role in determining the rotational barrier. The rotational energy barrier is further decreased when four H atoms are substituted by four F atoms in the BDC linker to form the TFBDC ligand. As a result, the model MOF containing the TFBDC organic ligand has two unstable positions and two rotational energy barriers at the dihedral angle χ = 0.0° and 90.0° reported in Table 6. The barriers are 5.96 and 13.0 kJ mol−1 at 0.0° and 90.0° computed with B3LYP-D3 method, respectively, and the barrier at 90° is the lowest rotational barrier of this TFBDC ligand among all other ligands which have rotational energy barrier at the same angle 90.0°. Similarly, we found that DMBDC linker has two rotational barriers about 14.60 and 2.10 kJ mol−1 at 90.0° and 0.0°, respectively, suggesting stronger repulsive interactions between the O atoms in the benzene than the O atoms in the carboxylate groups. We can say that the second rotational barrier lies below the accuracy of DFT-D method, and we can say –OCH3 group in the BDC linker can reduce the rotational energy barrier resulting in the DMBDC linker being more flexible in MOFs. The NDC organic linker in MOFs has two energy barriers at 0° and 90° like TFBDC linker and the two energy barriers at χ = 0° and 90° are very similar in the model MOF.
The calculated energy barrier of the BDC organic linkers in MOF is in good agreement with the previous experimental value of Gould et al.19 and theoretical value of Winston et al.22 However, the computed values of rotational energy barriers of the NDC linker at both the position χ = 0° and 90° are about more than 32 kJ mol−1 smaller than the reported experimental value.25 To investigate this large discrepancy, we considered the conditions of our partially relaxed scan, which can affect the rotational barriers. The main reason behind this discrepancy is that Horike et al. used Cu2+ in their experiment in crystal form of the MOF and the Cu1–Cu2 distance, which corresponds to the lattice constant of the MOF along the channel direction, was considered in only one dimension and we did not calculate three dimensional effects of the crystal. This lattice constant of the [Cu2(NDC2)2DABCO] MOF was about 10.936 Å, which is almost 0.20 Å lower than the [Zn2(NDC2)2DABCO] MOF. These distances are about 0.2–0.3 Å shorter than the experimental lattice constant, 10.819 Å, in the X-ray structure of [Cu2(NDC2)2DABCO].24 Thus, we have calculated the partially relaxed scan with the additional constraint of the Cu1–Cu2 distance at the experimental value 10.819 Å. However, the energy barrier with the Cu1–Cu2 distance constraint was about 18 kJ mol−1 at both the rotational dihedral angle χ = 0° and 90°, and the difference is less than 0.4 kJ mol−1 compared to those without the Cu1–Cu2 distance constraint. Next, we removed the planar constraint condition and calculated the barriers by optimizing the NDC model without constraint and with the constraint of χ = 0° and χ = 90°, and the barrier energy values did not change significantly. We found that both the Cu1–Cu2 distance and the planar constraint condition are not the important factors for the rotational barrier discrepancy. However, we have found a difference between the distances Cu1–Cu2 (11.061 Å) and Cu3–Cu4 (10.895 Å) (as shown see atom names in Fig. S3 in the ESI†) and in the optimized structure without constraints, meaning that S1 and S2 planes are not parallel. We have also found that the optimized dihedral angle χ decreased by an amount 5° from the constrained optimized geometry. This indicates that, in the solid-state real MOFs, the structures of the Cu clusters depend on the conformation of the adjacent NDC ligand. Therefore, it is plausible that larger models of MOF framework with multiple linkers is necessary to correctly predict the rotational barriers of NDC. The present computations found that the Cu2+ cations in the [Cu2(X)2DABCO] MOFs reduce the rotational barrier energy about 4 kJ mol−1 of the NDC linker compared to the barrier of the same NDC linker in the Zn2+ cations-based [Zn2(X)2DABCO] MOFs. We can say that our study reveals that the rotational energy barriers significantly differ depending on the organic linkers/ligands or rotational groups, metal cations, ranging from the highest 51.22 kJ mol−1 of BDC linker to the lowest one 2.10 kJ mol−1 of DMBDC. The barrier can also provide information on the flexibility of the MOFs as the lower energy barrier of the linkers means that the linker is more flexible. The high barrier indicates the system is more rigid resulting in the linker being less flexible. In fact, by the Arrhenius equation with the obtained rotational barrier and the assumed pre-exponential factor of BDC A = 1.6 × 1012 s−1,19 we can estimate the rotational frequency of the organic linkers. The estimated frequency at 343 K widely ranges from 2.6 × 103 s−1 in BDC linker to 4.1 × 1010 s−1 in TFBDC linker. The detailed description of the Arrhenius equation can be found in ref. 19. Among all the model MOFs studied here, we can say the TFBDC and DMBDC linkers are the most flexible, as they have lower rotational barrier.
3.4. Effect of linker (i.e. dicarboxylate ligand) rotation on MOF applications in framework flexibility
Framework flexibility is one of the most important properties and characteristics of MOFs, which is not only interesting, but also useful for a variety of potential applications such as H2-storage, energy storage, control the polymerization reaction in MOFs nanochannels, etc. The linker rotation, or the rotation of the organic ligand groups in MOFs, is one of the significant properties of flexible MOFs and this linker rotation causes the flexibility of MOFs. Düren and co-workers showed that the rotation of the organic ligands causes the expansion of the pore windows i.e. pore size and pore volume in the MOFs, and the size of adsorbed molecules were larger than expected from the MOF crystal structure.54 The pore size of the MOFs and along with its properties can also be fine-tuned through the fabricated shrewd design of the organic bridging ligands. The rotation of the organic linkers or ligands should also play an also important role in forming a breathing MOF.55–57 Therefore, the flexible property due to the linker rotation is so-called breathing and swelling phenomena as a function of host–guest interactions. The great advantage of these kinds of rotational motion of the organic linkers is that the core framework remains stable and only the organic bridging linker can rotate. Since the framework is permanent, this MOF design (changing only the organic linkers in the MOF while keeping the same framework) can be applied to prepare several MOFs in a variety of pore sizes, and they are able to switch between open and closed pore structures when exposed to an external stimulus. Therefore, both the size of the pore and the breathing subunit can be designed independently to maximize selectivity. In this study we found the model MOFs containing TFBDC and DMBDC are the most flexible MOFs among all the MOFs studied here, as their rotational barrier is the lowest among the all other model MOFs systems. Thus, these two linkers can be used to construct flexible MOFs. This investigation suggests to the experimentalists to synthesize the flexible MOFs by using TFBDC and DMBDC organic linkers, which must have an impact on controllable micropore volume. Therefore, framework flexibility based on the rotation of the linkers can be used to engineer special functions. These two MOFs with TFBDC and DMBDC linkers should have large pore expansion which can contain large number of guest molecules. We will study the H2-storage capacity and polymerization reactions using these MOFs with these TFBDC and DMBDC linkers, which will have a potential application in nanoporous materials science.Compared with conventional porous materials, MOFs with various organic linkers have much larger framework flexibility, which can give rise to not only various types of interesting structural responses and dynamic behaviours toward external stimuli, but also significantly improved performances for storage, separation, sensing, and other applications. Therefore, controlling the flexibility of MOFs, or rational design and synthesis of MOFs with specified flexibility and dynamism, are of practical importance. However, framework flexibility is simultaneously controlled by many factors, and trivial difference of a structural parameter or other factor related with the sample or environment can drastically change the response. In other words, framework flexibility can be more difficult to design or control, compared with the static features such as framework and pore structures. Our study suggests that changing the linkers with lower rotational energy barrier of the ligand especially TFBDC can be used in MOFs for controlling the flexibility of MOF materials. We speculate that the flexibility of a MOF may constitute an intrinsic impediment on its experimental realization. This highlights the importance of systematic prediction of large amplitude flexibility regimes in MOFs.
4. Conclusions
In summary, we have computationally studied the equilibrium structure, geometry, stability, PECs, and rotational energy barrier of the five [Cu4(X)(Y)6(NH3)4] model MOFs containing X = BDC, DFBDC, TFBDC, DMBDC, and NDC organic bridging linkers by using first-principles based dispersion corrected and uncorrected DFT methods with cc-pVDZ basis sets considering planar constraints with the three imaginary planes. Due to the limitation of computational resources and the restriction of the DFT methods, we considered the model [Cu4(X)(Y)6(NH3)4] MOF systems with only one organic bridging linker by ignoring the effects of other linkers which can interfere in the course of rotation of the organic ligands. Most of the discussion was based on the results at the B3LYP-D3 level of theory. The present study reveals that the structural and electronic properties of these model MOFs can be changed by substituting various organic linkers in MOFs. We found that the computed structural properties (such as bond distance, angle, and dihedral angle) of the BDC model MOF system are well harmonized with the previous experimental and theoretical values. It has been found that the BDC and DFBDC linkers in MOFs are planar since their equilibrium dihedral angle (χ) is at 0.0°. On the other hand, the dihedral angle of the TFBDC, DMBDC and NDC linkers is about at 46.6°, 25.10°, and 46.0°, respectively, resulting in non-planar structures. The highest and lowest rotational barriers were observed in the BDC and TFBDC linkers in the model MOFs, respectively, by conducting a partially relaxed scan. Two interactions, electrostatic (including the hydrogen bonded) and vdW interactions, must play important roles in forming the planar or non-planar stable structures of the MOFs and PECs. The PECs of the linker were computed by relaxed scan and the rotational barriers have been computed considering the same MOF model systems. We have found that the rotational energy barrier of the linkers in the model MOF systems has been significantly changed due to substituents onto the organic linkers. The results indicate that the rotational energy barrier as well as flexibility of the organic bridging ligands can be controlled by substituting the linkers in the MOFs, and also altering the metal cations. Hence, this study represents the first step toward the characterization of the MOFs flexibility in the dicarboxylate ligands theoretically. We can say that changing the organic bridging linkers in MOFs could offer an interesting research for the creation of porous materials in a rigid framework with several important material properties and possible applications. This study will help us in our future research to perform radical polymerization reactions in the microporous MOF channels, H2-storage, CH4 gas adsorption, and CO2 capture in the MOFs. Framework flexibility leads to a rotation of the pillar, which eventually ends up in the opening of the pore space. In conclusion, this study has shown that flexibility of the organic linkers in the MOFs would significantly differ depending on the linkers. This study also provides compelling insights into how to construct a metal–organic framework as a model system for computational analysis. Our results will provide a new insight into the field of porous materials science.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to acknowledge Indian Institute of Technology Indore for supporting this work. Dr Srimanta Pakhira thanks the Science and Engineering Research Board, Department of Science and Technology (SERB-DST), Govt. of India for providing his highly prestigious Ramanujan Faculty Fellowship under the scheme number SB/S2/RJN-067/2017, and for his Early Career Research Award (ECRA) under the Grant No. ECR/2018/000255. The author would like to thank Ms. Stephanie Marxsen, Florida State University, Florida, USA for her helpful discussions. The author thanks “Bioinformatics Resources and Applications Facility (BRAF), C-DAC, Pune, Maharashtra, India” for providing the computing facility. Dr Pakhira is grateful to Professor Pradeep Mathur, Director, Indian Institute of Technology Indore (IITI), Simrol, Khandwa Road, Indore, 453552, MP, India for supporting the research facility, helpful discussions, allowing him to carry out this research, and developing a keen interest in him for this field. His thanks are also extended to Professor Pradeep Mathur (Director, IIT Indore) who encouraged his whim of contributing to this periodical fundamental research and encouraging him in typing the manuscript.References
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 239, 424–428 CrossRef.
- Q. Li, C. H. Sue, S. Basu, A. K. Shveyd, W. Zhang, G. Barin, L. Fang, A. A. Sarjeant, J. F. Stoddart and O. M. Yaghi, Angew. Chem., Int. Ed., 2010, 49, 6751–6755 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- W. Morris, B. Leung, H. Furukawa, O. K. Yaghi, N. He, H. Hayashi, Y. Houndonougbo, M. Asta, B. B. Laird and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 11006–11008 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- T. G. Glover, G. W. Peterson, B. J. Schindler, D. Britt and O. M. Yaghi, Chem. Eng. Sci., 2011, 66, 163–170 CrossRef.
- T. Uemura, R. Nakanishi, S. Mochizuki, Y. Muratac and S. Kitagawa, Chem. Commun., 2015, 51, 9892–9895 RSC.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed.
- T. Uemura, Y. Ono, K. Kitagawa and S. Kitagawa, Macromolecules, 2008, 41, 87–94 CrossRef CAS.
- T. Uemura, Y. Ono, Y. Hijikata and S. Kitagawa, J. Am. Chem. Soc., 2010, 132, 4917–4924 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS.
- H. Li, X. Feng, D. Ma, M. Zhang, Y. Zhang, Y. Liu, J. Zhang and B. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 3160–3163 CrossRef CAS PubMed.
- K. Seki, S. Takamizawa and W. Mori, Chem. Lett., 2001, 30, 332–333 CrossRef.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS PubMed.
- S. L. Gould, D. Tranchemontagne, O. M. Yaghi and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2008, 130, 3246–3247 CrossRef CAS PubMed.
- M. Tafipolsky, S. Amirjalayer and R. Schmid, J. Comput. Chem., 2007, 28, 1169–1176 CrossRef CAS PubMed.
- S. Pakhira, M. Takayanagi and M. Nagaoka, J. Phys. Chem. C, 2015, 119, 28789–28799 CrossRef CAS.
- E. B. Winston, P. J. Lowell, J. Vacek, J. Chocholousova, J. Michl and J. C. Price, Phys. Chem. Chem. Phys., 2008, 10, 5188–5191 RSC.
- H. Chun, D. N. Dybetsev, H. Kim and K. Kim, Chem. – Eur. J., 2005, 11, 3521–3529 CrossRef CAS PubMed.
- R. Matsuda, W. Kosaka, R. Kitaura, Y. Kubota, M. Takata and S. Kitagawa, Microporous Mesoporous Mater., 2014, 189, 83–90 CrossRef CAS.
- S. Horike, R. Matsuda, D. Tanaka, S. Matsubara, M. Mizuno, K. Endo and S. Kitagawa, Angew. Chem., Int. Ed., 2006, 45, 7226–7230 CrossRef CAS PubMed.
- K. Uemura, Y. Yamasaki, F. Onishi, H. Kita and M. Ebihara, Inorg. Chem., 2010, 49, 10133–10143 CrossRef CAS PubMed.
- T. Uemura, Y. Ono, Y. Hijikata and S. Kitagawa, J. Am. Chem. Soc., 2010, 132, 4917–4924 CrossRef CAS PubMed.
- J. S. Grosch and F. Paesani, J. Am. Chem. Soc., 2012, 134, 4207–4215 CrossRef CAS PubMed.
- J. Pires, J. Fernandes, K. Dedecker, J. R. B. Gomes, G. Pérez-Sánchez, F. Nouar, C. Serre and M. L. Pinto, ACS Appl. Mater. Interfaces, 2019, 11, 27410–27421 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. Niu, S. Pakhira, K. Marcus, Z. Li, J. L. Mendoza-Cortes and Y. Yang, Adv. Energy Mater., 2018, 1800480 CrossRef.
- S. Pakhira, K. P. Lucht and J. L. Mendoza-Cortes, J. Phys. Chem. C, 2017, 121, 21160–21170 CrossRef CAS.
- S. Pakhira, R. I. Singh, O. Olatunji-Ojo, M. Frenklach and W. A. Lester Jr, J. Phys. Chem. A, 2016, 120, 3602–3612 CrossRef CAS PubMed.
- S. Pakhira, C. Sahu, K. Sen and A. K. Das, Struct. Chem., 2012, 24, 549–558 CrossRef.
- Y. Lei, S. Pakhira, K. Fujisawa, X. Wang, O. O. Iyiola, N. P. López, A. L. Elías, L. P. Rajukumar, C. Zhou, B. Kabius, N. Alem, M. Endo, R. Lv, J. L. Mendoza-Cortes and M. Terrones, ACS Nano, 2017, 11, 5103–5112 CrossRef CAS PubMed.
- S. Pakhira and J. L. Mendoza-Cortes, J. Phys. Chem. C, 2018, 122, 4768–4782 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- M. Parac, M. Etinski, M. Peric and S. Grimme, J. Chem. Theory Comput., 2005, 1, 1110–1118 CrossRef CAS.
- M. Bursch, E. Caldeweyher, A. Hansen, H. Neugebauer, S. Ehlert and S. Grimme, Acc. Chem. Res., 2019, 52, 258–266 CrossRef CAS PubMed.
- J. P. P. Ramalho, J. R. B. Gomes and F. Illasc, RSC Adv., 2013, 3, 13085–13100 RSC.
- S. Pakhira and J. L. Mendoza-Cortes, Phys. Chem. Chem. Phys., 2019, 21, 8785–8796 RSC.
- J. Hui, N. B. Schorr, S. Pakhira, Z. Qu, J. L. Mendoza-Cortes and J. Rodríguez-López, J. Am. Chem. Soc., 2018, 140, 13599–13603 CrossRef CAS PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 Search PubMed.
- J. Baker, A. Scheiner and J. Andzelm, Chem. Phys. Lett., 1993, 216, 380–388 CrossRef CAS.
- A. Montoya, T. N. Truong and A. F. Sarofim, J. Phys. Chem. A, 2000, 104, 6108–6110 CrossRef CAS.
- S. Pakhira, B. S. Lengeling, O. Olatunji-Ojo, M. Caffarel, M. Frenklach and W. A. Lester Jr, J. Phys. Chem., 2015, 119, 4214–4223 CrossRef CAS PubMed.
- S. Pakhira, K. Sen, C. Sahu and A. K. Das, J. Chem. Phys., 2013, 138, 164319 CrossRef PubMed.
- See http://www.uni-muenster.de/Chemie.oc/grimme/ for a FORTRAN program implementing the DFT-D3 method and a file with available C6 coefficients. WestfalischeWilhelms-Universitat Munster, OrganischChemischesInstitut, TheoretischeOrganischeChemie, Grimme Research Group, Corrensstrasse 40 D-48149 Munster, email: E-mail: grimmes@unimuenster.de.
- J. Hui, S. Pakhira, R. Bhargava, Z. J. Barton, X. Zhou, A. J. Chinderle, J. L. Mendoza-Cortes and J. Rodríguez-López, ACS Nano, 2018, 12, 2980–2990 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, and G. A. Petersson, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- K. Uemura, Y. Yamasaki, Y. Komagawa, K. Tanaka and H. Kita, Angew. Chem., Int. Ed., 2007, 46, 6662–6665 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldweli, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- D. Fairen-Jimenez, R. Galvelis, A. Torrisi, A. D. Gellan, M. T. Wharmby, P. A. Wright, C. Mellot-Draznieks and T. Düren, Dalton Trans., 2012, 41, 10752 RSC.
- C. R. Murdock, B. C. Hughes, Z. Lu and D. M. Jenkins, Coord. Chem. Rev., 2014, 258–259, 119–136 CrossRef CAS.
- L. Chen, J. P. S. Mowat, D. Fairen-Jimenez, C. A. Morrison, S. P. Thompson, P. A. Wright and T. Duren, J. Am. Chem. Soc., 2013, 135, 15763–15773 CrossRef CAS PubMed.
- A. J. Fletchera, K. M. Thomasa and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491–2510 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The equilibrium structures of the DFBDC, DMBDC and NDC linker containing model MOFs, PECs, optimized Cartesian coordinates and harmonic vibrational frequencies of the systems are provided. See DOI: 10.1039/c9ra01288e |
| This journal is © The Royal Society of Chemistry 2019 |