
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
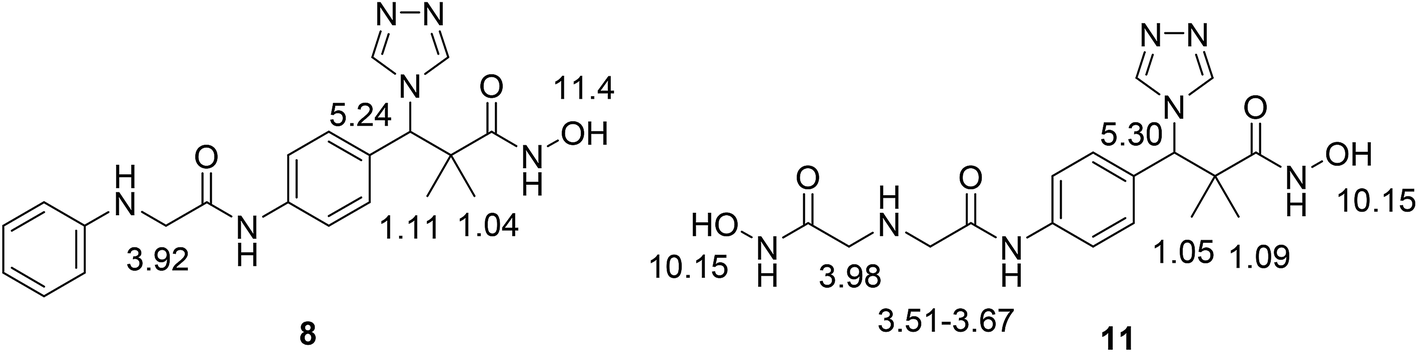
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and antiproliferative assay of triazolyl-2,2-dimethyl-3-phenylpropanoates as potential HDAC inhibitors
Samir El-Rayes *a,
Gomaa M. S.b,
Abouelmagd A.a,
Walid Fathallac and
Ibrahim. A. I. Alia
*a,
Gomaa M. S.b,
Abouelmagd A.a,
Walid Fathallac and
Ibrahim. A. I. Alia
aDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia, Egypt. E-mail: samir_elrayes@yahoo.com
bDepartment of Pharmaceutical Chemistry, College of Clinical Pharmacy, Imam Abdulrahman Bin Faisal University, P. O. Box 1982, Dammam 31441, Eastern Province, Kingdom of Saudi Arabia
cDepartment of Physics and Math., Faculty of Engineering, Port-Said University, Port-Said, Egypt
First published on 7th May 2019
Abstract
Recently, histone deacetylase (HDAC) inhibition has gained great importance in cancer treatment. We herein, describe the design, synthesis and biological testing of 16 compounds based on the structure modification of methyl 3-(4-(2-chloroacetamido)phenyl)-3-hydroxy-2,2-dimethylpropanoate (5) and methyl 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (14) as potent HDACIs. Two series were synthesized based on the structure of 3-(4-(2-chloroacetamido)phenyl)-3-hydroxy-2,2-dimethylpropanoate and 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate. The compounds were tested in vitro for their antiproliferative activity against HeLa cells. The results identified compounds 16b, 16c, 18 (IC50; 11.69, 0.69, 3.39 μM respectively) as potential good inhibitors compared to the standard drug doxorubicin (IC50; 2.29 μM). Those compounds also exhibited promising activity against other cancer cell lines namely; HCT-116, MCF-7, PC3, A549 and therefore were selected as hits for further optimization. The docking experiment results performed on the HDAC-2 crystal structure were in close agreement with the biological testing results which suggest that those compounds potentially work through HDAC inhibition.
Introduction
Cancer is one of the major health challenges worldwide. Cancer is an abnormal cell growth that shifts the controlled mechanisms of cell proliferation and differentiation associated with high mortality rate.1 Chemotherapy is one of the most effective approaches used to treat solid as well as hematological tumors.2,3 Quinazoline sulfonamide and quinazolindione4,5 are well established privileged structures in cancer chemotherapy that showed great activity against different types of cancer including breast cancer and pancreatic cancer. Acetylation of histones in a eukaryotic cell is involved in many cellular functions including proliferation, cell-cycle regulation and apoptosis.6 Histone acetylation is controlled by a balance between histone acetyl transferases (HATs) (associated with gene transcription) and histone deacetylase (HDAC) involved in deacetylation of ε-amino groups of lysine residue on histone tails (associated with gene silencing).7 Abnormal alterations in the histone acetylation process are associated with cancer development.8 Therefore, histone deacetylase (HDAC) inhibition is considered one of the promising strategies for the development of novel anticancer agents.9 HDACIs are classified into different classes depending on their structures namely aliphatic acids, hydroxamic acids, 2-aminoanilides, cyclic peptides, and electrophilic ketones.10 In general, most of the currently reported HDACIs consist of a zinc-binding group (ZBG) and a five- to six-carbon hydrophobic spacer attached to a hydrophobic group via a connection unit (Fig. 1a).11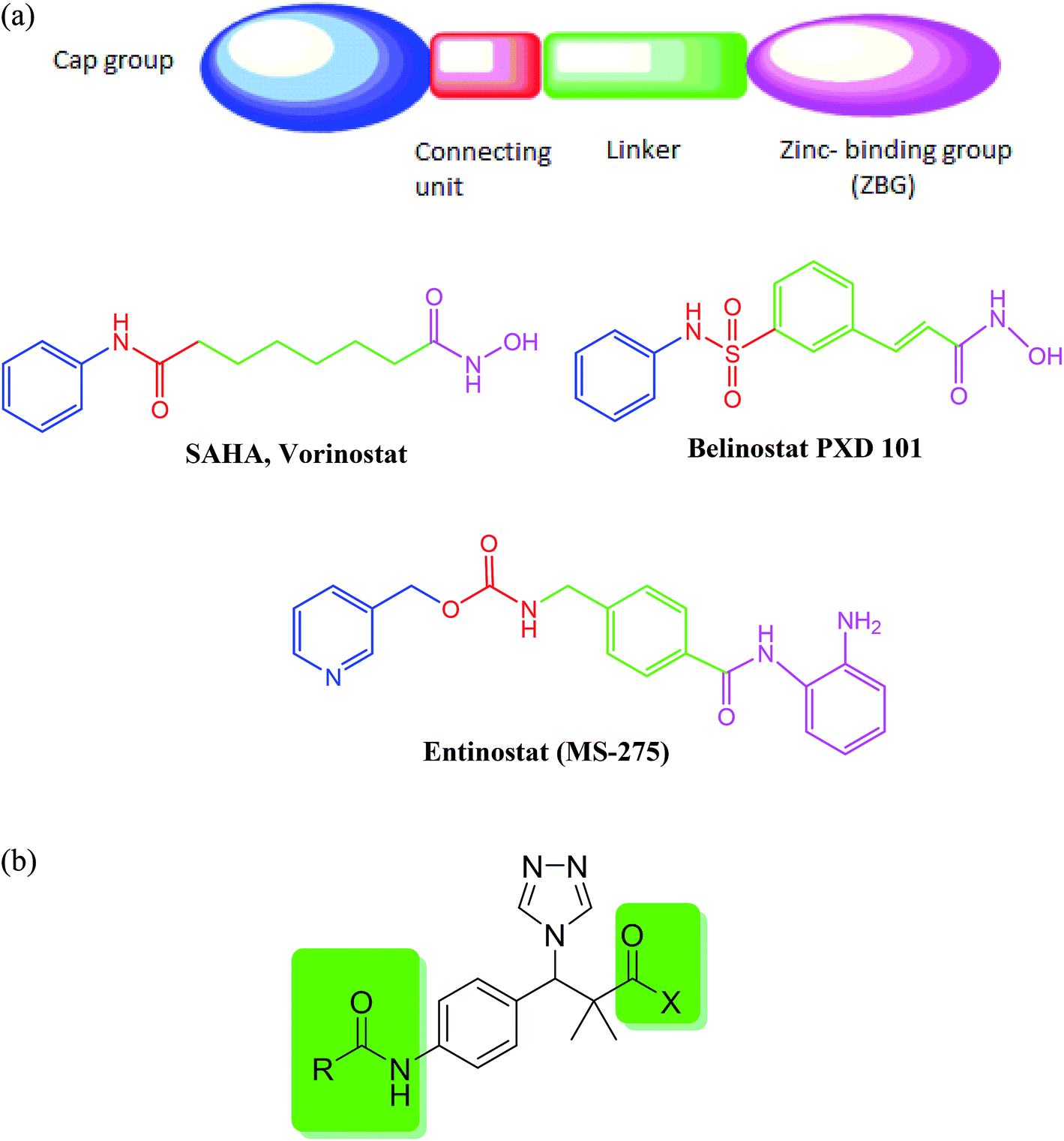 | ||
| Fig. 1 (a) Classical histone deacetylase (HDAC) inhibitors. (b) Structure design of our proposed HDAC inhibitors. | ||
Until now, four HDACIs are approved for cancer treatment including three synthetic hydroxamate HDACIs (SAHA, Belinostat PXD 101, and Panobinostat) and one natural product cyclic depsipeptide (romidepsin).12 Other compounds in clinical trials include hydroxamic acid derivatives such as panobinostat (LBH589) and pracinostat (SB-939) with an N-hydroxyacrylamide moiety exhibiting excellent HDAC inhibitory activity13,14 and benzamides such as entinostat (MS-275) and mocetinostat (MGCD-0103).15,16 Therefore, developing and identification of potent HDACIs is a promising therapeutic strategy in cancer treatment.
Our proposed HDAC inhibitors were designed to combine the most important structural features reported for HDAC in a simple chemical structure (Fig. 1b). The compounds contain a sterically hindered amide, ester or hydroxamate on one side of the molecule. A triazole moiety was incorporated for steric effect as well as to optimize the binding interaction and improve the pharmacokinetic properties. A central aromatic ring connects the amide group with another acetamide moiety to mimic the well-known benzamide inhibitors.
Discussion
We now report the synthesis of the targeted structures; N-hydroxy-2,2-dimethyl-3-(4-(2-(phenylamino)acetamido)phenyl)-3-(4H-1,2,4-triazol-4-yl)propanamide (8), N-hydroxy-3-(4-(2-((2-(hydroxyamino)-2-oxoethyl)amino)acetamido)phenyl)-2,2-dimetthyl-3-(4H-1,2,4-riazol-4-yl)propanamide (11) and their analogues 16a–d, 18 as HDAC inhibitors.Methyl 3-hydroxy-2,2-dimethyl-3-(4-nitrophenyl)propanoate (3) was prepared by the reaction of 4-nitrobenzaldehyde 1 with trimethylsilyl ketene acetal 2 in the presence of pyridine-N-oxide and LiCl in DMF at room temperature under nitrogen atmosphere to afford the ester 3 in 69% yield.17,18 Reduction of 3 by using hydrogen atmosphere in the presence of Pd/C catalyst in ethanol at room temperature for 3 h afforded the desired ester 4 in 87% yield. The acylation reaction of the ester 4 with chloroacetyl chloride in the presence of triethyl amine and catalytic amount of DMAP in dichloromethane for 12 h. Afforded methyl 3-(4-(2-chloroacetamido)phenyl)-3-hydroxy-2,2-dimethylpropanoate 5 in 70% yield, Scheme 1.
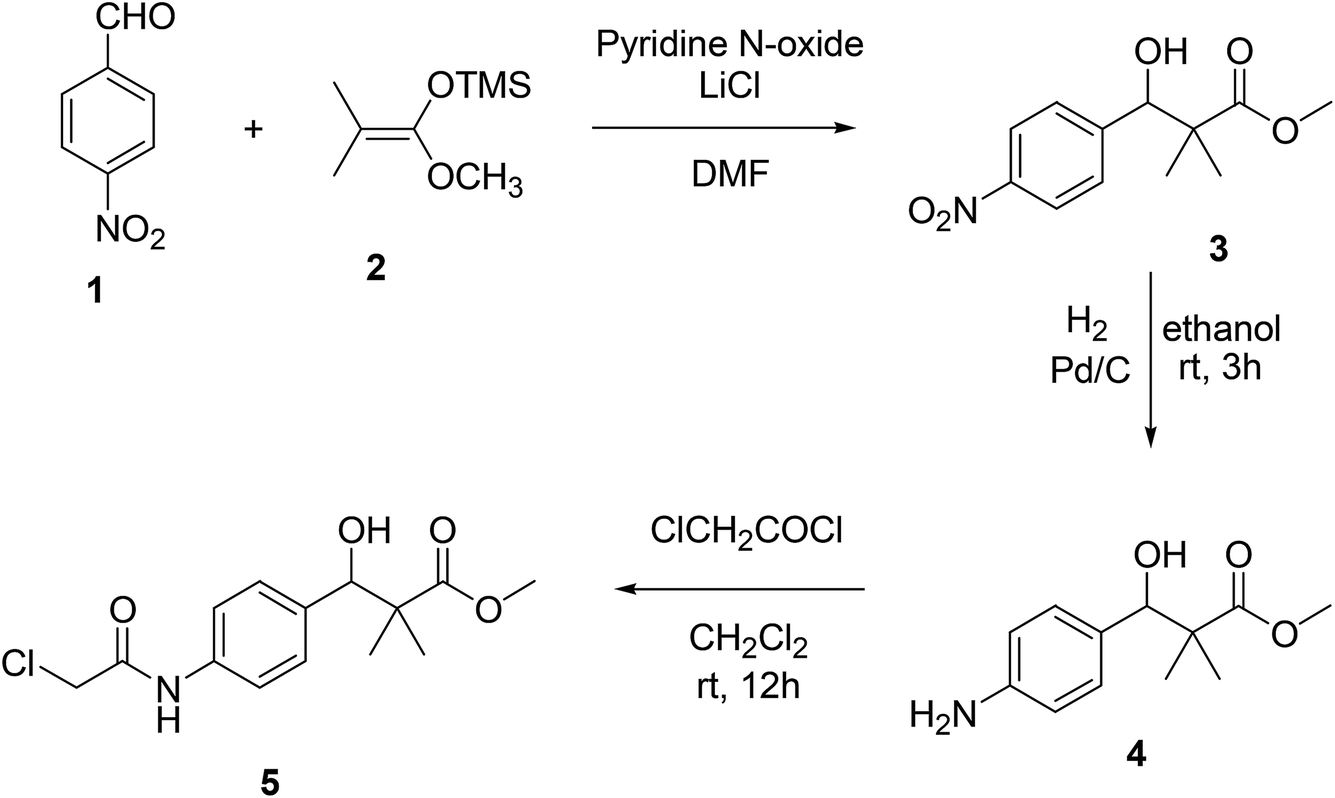 | ||
| Scheme 1 Sequential synthesis of methyl 3-(4-(2-chloroacetamido)phenyl)-3-hydroxy-2,2-dimethylpropanoate 5. | ||
Structure modification of chloroacetamido derivative 5 could simply be achieved by the reaction with nucleophiles and the attachment of triazole moiety to produce both targeted structures N-hydroxy-2,2-dimethyl-3-(4-(2-(phenylamino)acetamido)phenyl)-3-(4H-1,2,4-triazol-4-yl)propanamide (8) and N-hydroxy-3-(4-(2-((2-(hydroxyamino)-2-oxoethyl)amino)acetamido)phenyl)-2,2-dimethyl-3-(4H-1,2,4-triazol-4-yl)propanamide (11). Thus, the reaction of chloroacetamide derivative 5 with aniline in pyridine under reflux condition for 10 h. Afforded methyl 3-hydroxy-2,2-dimethyl-3-(4-(2-(phenylamino)acetamido)phenyl)propanoate (6) in 84% yield. Successful attachment of triazole residue was achieved by the reaction of 6 with triazole in the presence of CDI in acetonitrile under reflux condition for 48 h. To afford the triazole derivative 7 in 67% yield. The first targeted compound N-hydroxy-2,2-dimethyl-3-(4-(2-(phenylamino)acetamido)phenyl)-3-(4H-1,2,4-triazol-4-yl)propanamide (8) was finally produced in 58% yield by the reaction of 7 with hydroxyl amine hydrochloride in the presence of KOH in ethanol under reflux condition, Scheme 2.
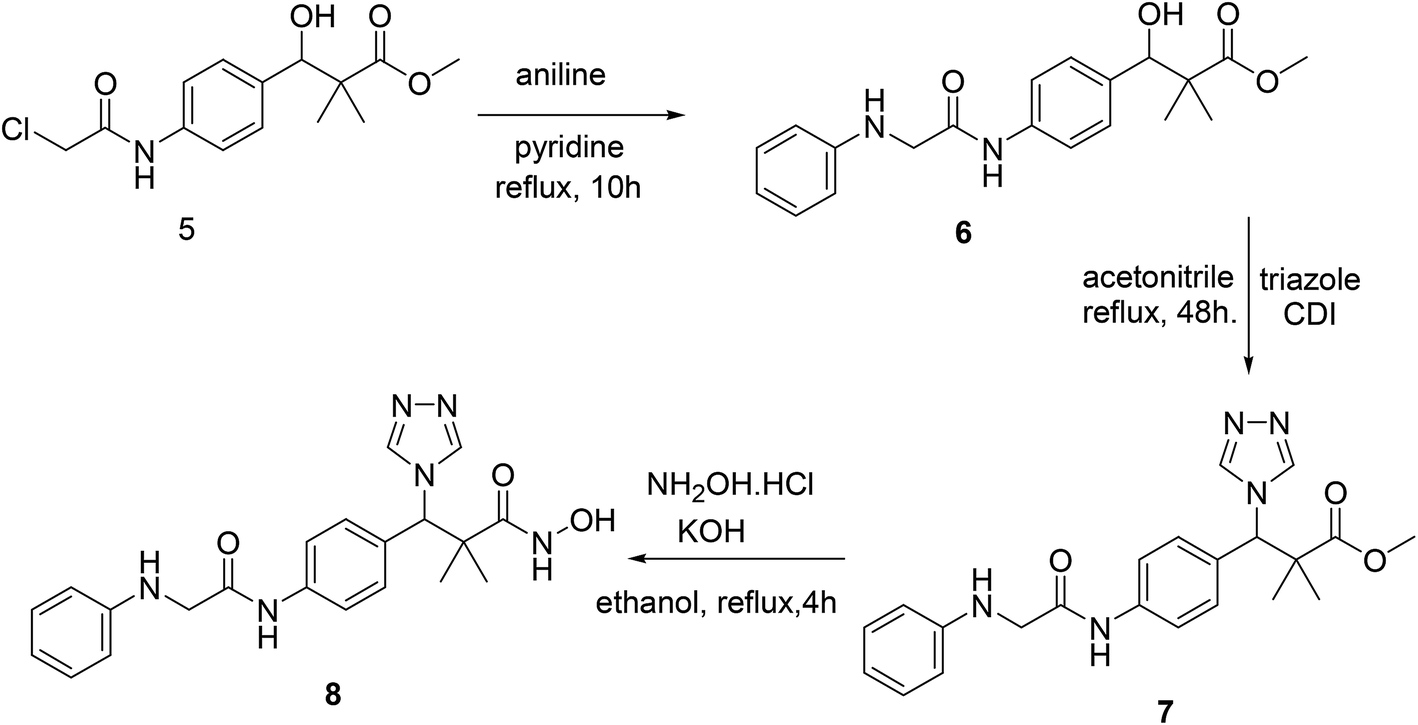 | ||
| Scheme 2 Sequential synthesis of the targeted molecule N-hydroxy-2,2-dimethyl-3-(4-(2-(phenylamino)acetamido)phenyl)-3-(4H-1,2,4-triazol-4-yl)propanamide (8). | ||
Similarly, the second targeted molecule N-hydroxy-3-(4-(2-((2-(hydroxyamino)-2-oxoethyl)amino)acetamido)phenyl)-2,2-dimethyl-3-(4H-1,2,4-triazol-4-yl)propan-amide (11) is prepared from chloroacetamido derivative 5 via three step sequential reactions. Thus, 5 reacted with glycine methyl ester hydrochloride in pyridine under reflux condition for 10 h. Afforded methyl 3-hydroxy-3-(4-(2-((2-methoxy-2-oxoethyl)amino)acetamido)phenyl)-2,2-dimethylpropanoate (9) in 71% yield. Next, the reaction of 9 with triazole in the presence of CDI in acetonitrile under reflux condition for 48 h. Afforded 10 in 71% yield and finally 11 was produced in 74% yield by the reaction of 10 with hydroxyl amine hydrochloride in the presence of KOH in ethanol under reflux condition, Scheme 3.
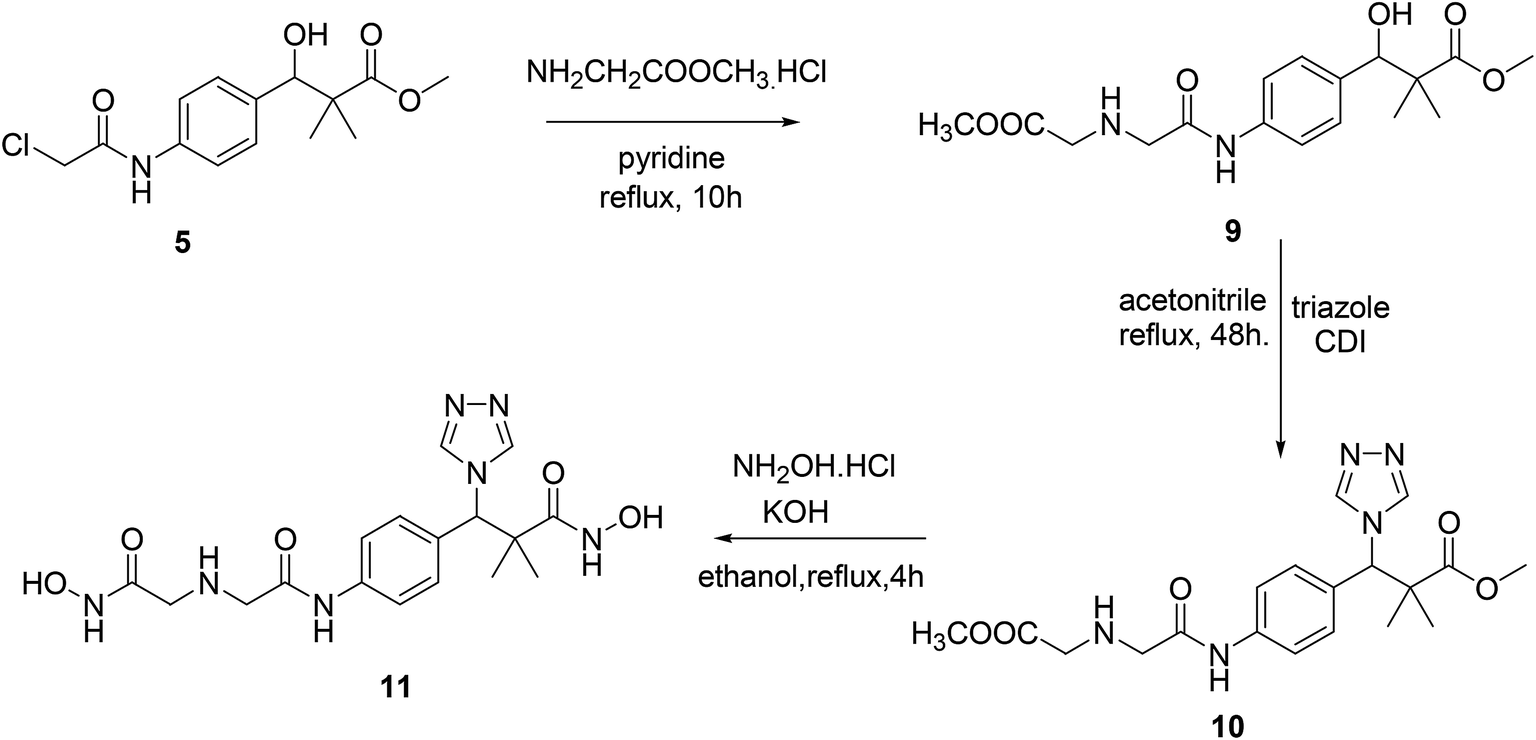 | ||
| Scheme 3 Sequential synthesis of the targeted molecule N-hydroxy-3-(4-(2-((2-(hydroxyamino)-2-oxoethyl)amino)acetamido)phenyl)-2,2-dimethyl-3-(4H-1,2,4-triazol-4-yl)propanamide (11). | ||
The structure assignment of both targeted structures N-hydroxy-2,2-dimethyl-3-(4-(2-(phenylamino)acetamido)phenyl)-3-(4H-1,2,4-triazol-4-yl)propanamide (8) and N-hydroxy-3-(4-(2-((2-(hydroxyamino)-2-oxoethyl)amino)acetamido)phenyl)-2,2-dimethyl-3-(4H-1,2,4-triazol-4-yl)propanamide (11) is based on 1H and 13C NMR spectroscopy and physicochemical analysis. Thus, the 1H NMR spectrum of 11 exhibit signals at δ 6.10 and 9.00 ppm are typically associated with two NH groups. The 1H NMR spectrum also presented an important signal at 10.15 ppm that is typically associated with 2 OH oximes. The 13C NMR spectrum displayed signals at δ = 49.4, 52.0, 173.8 and 174.9 ppm, associated with two CH2 and 2 CO groups, respectively (Fig. 2).
 | ||
| Fig. 2 Selected 1H and 13C NMR spectral data of N-hydroxy-2,2-dimethyl-3-(4-(2-(phenylamino)acetamido)phenyl)-3-(4H-1,2,4-triazol-4-yl)propanamide (8) and N-hydroxy-3-(4-(2-((2-(hydroxyamino)-2-oxoethyl)amino)acetamido)phenyl)-2,2-dimethyl-3-(4H-1,2,4-triazol-4-yl)propanamide (11). | ||
Our next target was the preparation of a number of 3-(4-chlorophenyl)-N-arylidene-2,2-dimethylpropanehydrazide 16a–d and N-benzyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (18) as structure analogues of compounds 8 and 11 for the CYP26 inhibitors evaluation. The reaction of 4-chlorobenzaldehyde (12) with methyl 2-bromo-2-methylpropanoate (13) in benzene under reflux condition afforded 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanoate (14) in 79% yield. Hydrazinolysis of ester 14 afforded the hydrazide 15. The reaction of hydrazide with aldehyde in ethanol under reflux condition for 6 h. Afforded the hydrazone 16a–d, Scheme 4.
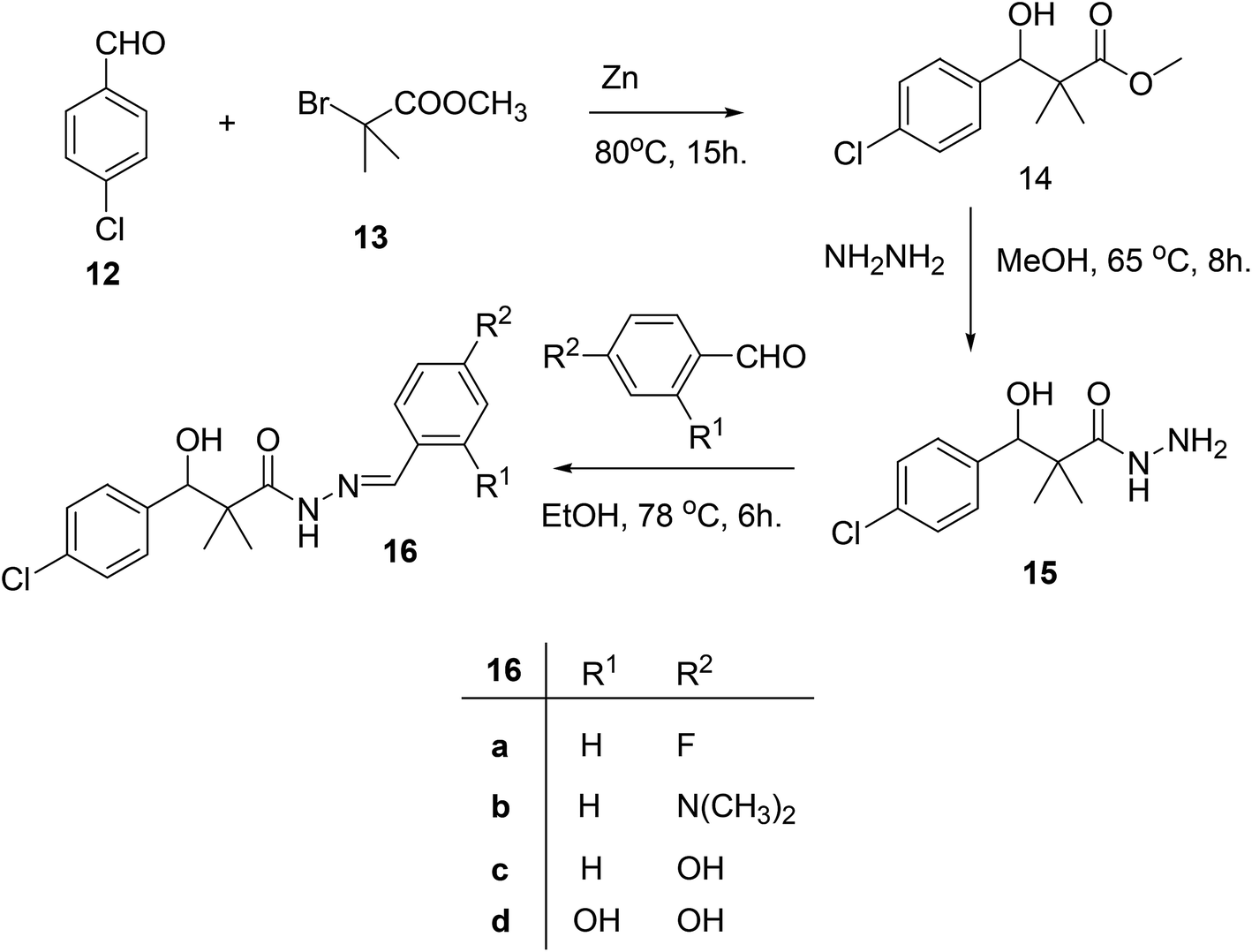 | ||
| Scheme 4 Sequential synthesis of 3-(4-chlorophenyl)-N-arylidene-2,2-dimethylpropanehydrazide 16a–d. | ||
On the other hand, the hydrazide reacted with benzyl amine under azide coupling condition to afford N-benzyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (18) in 77% yield, Scheme 5.
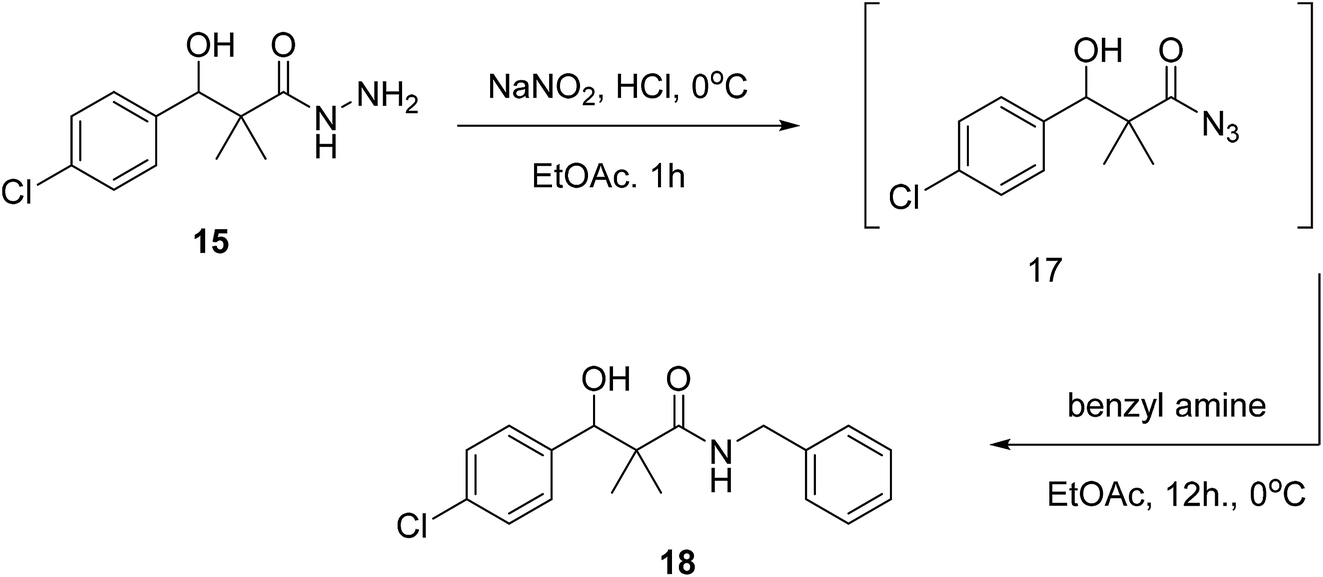 | ||
| Scheme 5 Sequential synthesis of N-benzyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (18). | ||
The structure assignment of 3-(4-chlorophenyl)-N-arylidene-2,2-dimethylpropanehydrazide 16a–d and N-benzyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (18) is based on 1H and 13C NMR spectroscopy and physicochemical analysis. Thus, the 1H NMR spectrum of 16a showed newly introduced salicylaldehyde signals at 8.59 and 11.40 for CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) and OH groups. N-Benzyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (18) was identified by 1H and 13C NMR spectroscopy. The 1H NMR spectrum showed newly introduced benzyl group signals at 4.35 and 6.08 for CH2 and NH groups. The 13C NMR spectrum displays signals at 46.2 corresponding to CH2 group.
and OH groups. N-Benzyl-3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropanamide (18) was identified by 1H and 13C NMR spectroscopy. The 1H NMR spectrum showed newly introduced benzyl group signals at 4.35 and 6.08 for CH2 and NH groups. The 13C NMR spectrum displays signals at 46.2 corresponding to CH2 group.
The cytotoxicity of each compound was investigated against different cancer cell lines namely; HCT-116, MCF-7, PC3, A549 at concentrations (0.1, 1, 10, and 100 μM) using MTT assay colorimetric assay. Data illustrated in (Table 1) shows the percentage of viability of HCT-116 cells after 48 h from treatment with different concentrations of the compounds versus control. The results revealed that the compound 16c had the lowest IC50 value (0.69 μM), followed by compounds 16b and 18 with IC50 values equal 11.6 μM, 3.39 μM, respectively compared to the standard drug doxorubicin (IC50; 2.29 μM) (Table 2).
| % of cell growth treated with different (conc. μM± SEM) of cpds. | ||||
|---|---|---|---|---|
| Cpds. | 0.1 | 1 | 10 | 100 |
| 3 | 61.47361 | 64.73607 | 64.99267 | 48.42375 |
| 4 | 57.14809 | 61.58358 | 51.6129 | 44.17155 |
| 5 | 79.98534 | 90.72581 | 100.2933 | 71.66422 |
| 6 | 62.86657 | 68.58504 | 75.80645 | 50.62317 |
| 7 | 94.72141 | 86.91349 | 70.96774 | 49.41349 |
| 8 | 54.80205 | 54.72874 | 59.12757 | 49.81672 |
| 9 | 64.03959 | 73.05718 | 62.79326 | 49.59677 |
| 10 | 94.72141 | 86.91349 | 70.96774 | 49.41349 |
| 11 | 92.33871 | 93.8783 | 86.36364 | 48.86364 |
| 14 | 79.98534 | 90.72581 | 100.2933 | 71.66422 |
| 15 | 62.86657 | 68.58504 | 75.80645 | 50.62317 |
| 16a | 65.9824 | 61.10704 | 58.5044 | 50.73314 |
| 16b | 51.46628 | 53.07918 | 50.18328 | 47.83724 |
| 16c | 52.84311 | 49.85924 | 51.64956 | 53.00587 |
| 16d | 67.88757 | 63.57404 | 71.86435 | 50.3476 |
| 18 | 53.55572 | 53.81232 | 47.54399 | 86.40029 |
| 5-FU | 98.05718 | 77.52933 | 78.95894 | 80.42522 |
| Dox | 73.24047 | 70.01466 | 16.31232 | 21.04106 |
| Compd. | IC50 (μM) |
|---|---|
| 3 | ND |
| 4 | 13.2 |
| 5 | 100 |
| 6 | 77.6 |
| 7 | 77.5 |
| 8 | 81.3 |
| 9 | 85.1 |
| 10 | ND |
| 11 | ND |
| 14 | ND |
| 15 | ND |
| 16a | ND |
| 16b | 11.6 |
| 16c | 0.69 |
| 16d | ND |
| 18 | 3.39 |
| 5-FU | ND |
| Dox | 2.29 |
Interestingly, the results of the different concentrations antiproliferative assay conducted on other cancer cell lines were consistent with the HeLa cells results, with compounds 16b, 16c, 18 on top of the activity list and also showing close activity to the standard drugs doxorubicin and 5-fluoro uracil (Tables 3–6).
| % of cell growth treated with different (conc. μM± SEM) of cpds. | ||||
|---|---|---|---|---|
| Cpds. | 0.1 | 1 | 10 | 100 |
| 3 | 74.25979 | 70.00955 | 73.87775 | 69.91404 |
| 4 | 86.67622 | 75.45368 | 80.70678 | 65.52053 |
| 5 | 80.09531 | 68.47507 | 66.93548 | 54.43548 |
| 6 | 92.33871 | 93.8783 | 86.36364 | 48.86364 |
| 7 | 74.70833 | 85.58333 | 88.41667 | 86.875 |
| 8 | 71.82426 | 73.20917 | 69.532 | 64.51767 |
| 9 | 74.88061 | 74.11652 | 78.08023 | 62.89398 |
| 10 | 75.02388 | 72.34957 | 77.31614 | 57.06781 |
| 11 | 76.69532 | 71.58548 | 69.81853 | 73.68672 |
| 14 | 73.35244 | 69.67526 | 67.95606 | 68.33811 |
| 15 | 72.6361 | 69.62751 | 71.48997 | 70.63037 |
| 16a | 78.12798 | 83.52436 | 81.27985 | 79.36963 |
| 16b | 58.11843 | 61.7001 | 49.37918 | 47.94651 |
| 16c | 64.89971 | 62.89398 | 57.02006 | 55.44413 |
| 16d | 60.875 | 64.41667 | 64.79167 | 60.08333 |
| 18 | 63.32378 | 65.85482 | 68.62464 | 65.42502 |
| 5-FU | 73.35244 | 77.93696 | 69.43649 | 62.98949 |
| Dox | 82.56925 | 81.3276 | 65.234 | 82.52149 |
| % of cell growth treated with different (conc. μM± SEM) of cpds. | ||||
|---|---|---|---|---|
| Cpds. | 0.1 | 1 | 10 | 100 |
| 3 | 89.1342 | 78.44568 | 99.3658 | 97.3465 |
| 4 | 98.1562 | 94.49858 | 105.4714 | 93.839 |
| 5 | 105.6513 | 109.1291 | 90.90091 | 91.14076 |
| 6 | 77.58333 | 76.16667 | 72.91667 | 46.20833 |
| 7 | 88.04348 | 108.8969 | 112.4396 | 90.49919 |
| 8 | 95.57787 | 93.839 | 91.3806 | 78.48898 |
| 9 | 98.39604 | 98.69585 | 104.3322 | 82.44641 |
| 10 | 114.7054 | 113.806 | 116.6242 | 79.50832 |
| 11 | 108.4695 | 106.8505 | 102.1736 | 109.3089 |
| 14 | 113.4463 | 102.1736 | 104.0923 | 99.77515 |
| 15 | 103.9125 | 104.5121 | 98.51596 | 98.33608 |
| 16a | 111.5875 | 113.0265 | 109.5488 | 93.89897 |
| 16b | 83.2259 | 85.56438 | 77.10988 | 60.32079 |
| 16c | 78.66887 | 80.70754 | 73.09249 | 70.63409 |
| 16d | 60.875 | 64.41667 | 64.79167 | 60.08333 |
| 18 | 102.6533 | 105.6513 | 102.4134 | 106.9105 |
| 5-FU | 104.632 | 103.013 | 92.81967 | 68.53545 |
| Dox | 108.9492 | 115.1851 | 84.06536 | 84.42512 |
| % of cell growth treated with different (conc. μM± SEM) of cpds. | ||||
|---|---|---|---|---|
| Cpds. | 0.1 | 1 | 10 | 100 |
| 3 | 75.70833 | 70.70833 | 71.29167 | 67.91667 |
| 4 | 83.20833 | 69.66667 | 69 | 149.3333 |
| 5 | 78.08333 | 71.54167 | 73.58333 | 123.8333 |
| 6 | 98.39604 | 98.69585 | 104.3322 | 82.44641 |
| 7 | 85.74879 | 84.46055 | 87.39936 | 87.56039 |
| 8 | 66.75 | 65.66667 | 67.08333 | 57.70833 |
| 9 | 77.58333 | 76.16667 | 72.91667 | 46.20833 |
| 10 | 77.33333 | 80.83333 | 72.20833 | 47.25 |
| 11 | 72.41667 | 77.91667 | 71.58333 | 66.58333 |
| 14 | 74.70833 | 85.58333 | 88.41667 | 86.875 |
| 15 | 62.125 | 66.5 | 67.375 | 61.29167 |
| 16a | 89.1342 | 78.44568 | 99.3658 | 97.3465 |
| 16b | 66.20833 | 66.5 | 53.83333 | 44.08333 |
| 16c | 70.5 | 63.54167 | 55.08333 | 46.29167 |
| 16d | 81.11916 | 77.89855 | 75.60386 | 75.36232 |
| 18 | 59.45833 | 59.66667 | 51.20833 | 45.16667 |
| 5-FU | 66 | 62.25 | 49 | 50.16667 |
| Dox | 63.25 | 65.16667 | 49.875 | 49.04167 |
| % of cell growth treated with different (conc. μM± SEM) of cpds. | ||||
|---|---|---|---|---|
| Cpds. | 0.1 | 1 | 10 | 100 |
| 3 | 78.55572 | 82.36804 | 70.41789 | 59.45748 |
| 4 | 80.43478 | 95.04831 | 90.05636 | 72.98712 |
| 5 | 84.29952 | 83.49436 | 85.82931 | 80.47504 |
| 6 | 88.88889 | 91.38486 | 78.7037 | 66.86795 |
| 7 | 60.875 | 64.41667 | 64.79167 | 60.08333 |
| 8 | 88.04348 | 108.8969 | 112.4396 | 90.49919 |
| 9 | 100.7246 | 94.40419 | 93.67955 | 75.60386 |
| 10 | 88.88889 | 91.38486 | 78.7037 | 66.86795 |
| 13 | 93.84058 | 87.39936 | 97.70531 | 65.29791 |
| 14 | 88.28502 | 87.56039 | 94.68599 | 92.3913 |
| 15 | 85.74879 | 84.46055 | 87.39936 | 87.56039 |
| 16a | 89.1342 | 78.44568 | 99.3658 | 97.3465 |
| 16b | 98.59098 | 86.95652 | 96.53784 | 46.61836 |
| 16c | 91.10306 | 97.26248 | 110.1449 | 46.37681 |
| 16d | 85.74879 | 84.46055 | 87.39936 | 87.56039 |
| 18 | 97.38325 | 93.27697 | 94.36393 | 43.19646 |
| 5-FU | 79.99195 | 76.24799 | 73.59098 | 66.74718 |
| Dox | 79.99195 | 62.72142 | 65.25765 | 73.79227 |
The accurate IC50 results on A549 cells again proved compounds 16b and 16c as superior antiproliferative agents with IC50 equal to 79.4 μM and 85.1 μM respectively compared to an IC50 of 72.4 μM for the standard drug 5-fluoro uracil.
The consistent comparable activity of compounds 16b and 16c on all tested cell lines suggested that the triazole ring is not of great importance compared to the hydroxyl group. Moreover, shifting the benzyl amide group next to the hydroxy isobutyl group resulted in a highly active compound (compound 18) possibly due to an increase in the stabilization of the amide bond that is essential for HDAC inhibition. Surprisingly, compound 18 although is the most different in structure among the designed and synthesized compounds as it lacks the triazole, the ester or the hydroxamic acid groups and the benzamide substituents, is one of the most active compounds against almost all tested cancer cell lines. This implies that for our compounds the structurally hindered hydroxyisobutyl acetamide is more important than the benzamide and the hydroxamate moieties. Therefore, compound 18 is considered as a potential lead for further optimization and development followed by compounds 16b and 16c which are basically aminohydrazone derivatives of compound 18 (Table 7).
| Compd. | IC50 (μM) |
|---|---|
| 3 | ND |
| 4 | ND |
| 5 | ND |
| 6 | ND |
| 7 | ND |
| 8 | ND |
| 9 | ND |
| 10 | ND |
| 11 | ND |
| 14 | ND |
| 15 | ND |
| 16A | ND |
| 16b | 79.4 |
| 16c | 85.1 |
| 16D | ND |
| 18 | 89.2 |
| 5-FU | 72.4 |
| Dox | ND |
The activity of the docked compounds was investigated by comparison of their ligand–receptor interactions with the amino acids of the receptor binding site. As shown in Table 8 and Fig. 3, the original ligand forms three hydrogen bonds with Tyr 308, His145, and Gly154. Moreover, it forms two van der Waals interactions with His 183 and Phe 155. Compound 15 forms six hydrogen bonds with Tyr 308, Asp 269, His 183, His 145 and Asp 181, and it forms one van der Waals interaction with Arg 39. Compound 16a forms three hydrogen bonds with His 145 and His 183, and it forms two van der Waals interactions with Phe 155 and Arg 39. Compound 16b forms five hydrogen bonds with Tyr 308, Asp 269, His 146, Asp 181 and His 145, and it forms three van der Waals interactions with Phe 155, His 183 and Arg 39. Compound 16c forms three hydrogen bonds with Ala 141 and Gly 154, and it forms two van der Waals interactions with Phe 155 and His 183. Compound 16d forms three hydrogen bonds with Asp 104, Asp 269, and Tyr 308 and it forms two van der Waals interactions with His 145 and Gly 154. Compound 18 forms four hydrogen bonds with Gly 154, Tyr 308, His 146 and His 145, and it forms two van der Waals interactions with Phe 155 and Arg 39.
| Molecular target (PDB code) | Original ligand | Docked compounds | ||||
|---|---|---|---|---|---|---|
| Co-crystallized ligand | Hydrogen bond no. | van der Waals interactiona | Compound | Hydrogen bond no. | van der Waals interactiona | |
| a van der Waals interactions are arene–arene interactions or arene–cation interaction. | ||||||
| 3MAX | N-(4-Aminobiphenyl-3-yl)benzamide | 3 Hydrogen bonds with Tyr 308 His145 Gly154 | His 183 and Phe 155 | 15 | 6 Hydrogen bonds with Tyr 308, Asp 269, His 183, His 145 and Asp 181 | Arg 39 |
| 16A | 3 Hydrogen bonds with His 145 and His 183 | Phe 155 and Arg 39 | ||||
| 16B | 5 Hydrogen bonds with Tyr 308, Asp 269, His 146, Asp 181 and His 145 | Phe 155, His 183 and Arg 39 | ||||
| 16C | 3 Hydrogen bonds with Gly 154, and Ala 141 | Phe 155 and His 183 | ||||
| 16D | 3 Hydrogen bonds with Tyr 308, Asp 269 and Asp 104 | His 145 and Gly 154 | ||||
| 18 | 4 Hydrogen bonds with Gly 154, Tyr 308, His 146 and His 145 | Phe 155 and Arg 39 | ||||
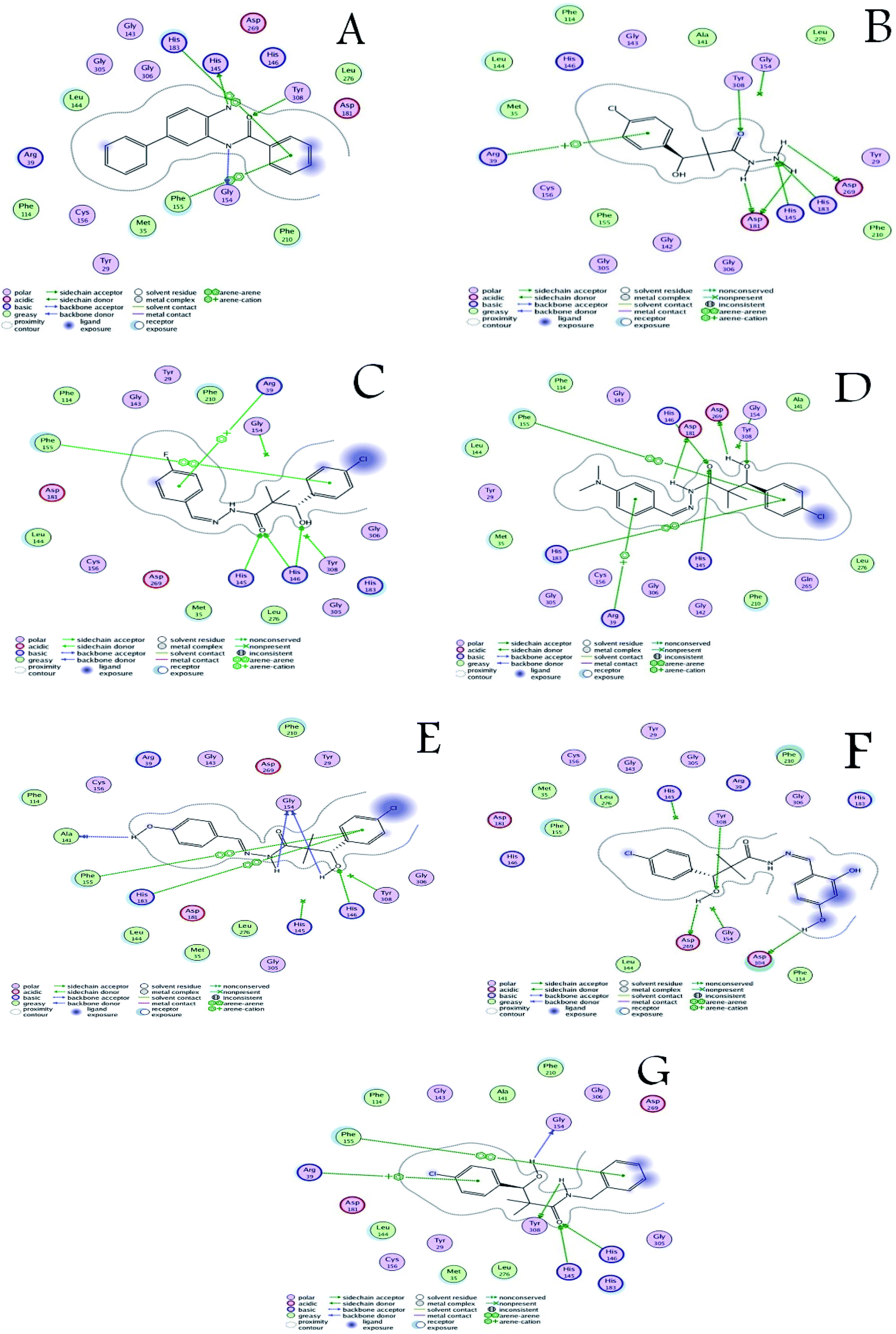 | ||
| Fig. 3 (A) Ligand–receptor interactions of the original ligand, and (B–G) ligand–receptor interactions of the docked compounds 15, 16a–d and 18 respectively. | ||
From the above discussion, compound 18 is the only compound that was able to bind the three key residues that are involved in binding the co-crystallized ligand through hydrogen bonding namely; His 145, Gly 154, and Tyr 308. Compound 16B formed hydrogen bonding with only two of the key residues namely; His 145, and Tyr 308. This is in close agreement with the antiproliferative assay results that identified compounds 18, 16B, 16C as the most active compounds. Therefore, based on the comparative docking with the co-crystallized ligand compound 18 is considered a potential hit for HDAC inhibition followed by compound 16B with rooms for pharmacokinetic and target interaction optimization.
Conclusion
In conclusion, efficient and very simple methods for the synthesis of various HDAC inhibitors were executed. Some of the synthesized compounds exhibited promising antiproliferative activity against various cancer cell lines namely; HCT-116, MCF-7, PC3, A549. Compounds 16b, 16c, 18 were consistently the most active on all tested cells and showed comparable activities to the standard drugs doxorubicin and 5-fluoro uracil. Compound 18 was selected as a potential lead for further optimization and development due to its interesting structural features that differs from other members of the series. The docking experiments performed substantiated the biological testing results and identified compound 18 as a potential HDAC inhibitor.Experimental
General
Solvent were purified and dried by standard procedures. The boiling range of the petroleum ether used was 40–60 °C. Thin layer chromatography (TLC): silica gel 60 F254 plastic plates (E. Merck, layer thickness 0.2 mm) detected by UV absorption. Elemental analyses were performed on a Flash EA-1112 instrument at the Microanalytical laboratory, Faculty of Science, Suez Canal University, Ismailia, Egypt. Melting points were determined on a Buchi 510 melting-point apparatus and the values are uncorrected. 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively (Bruker AC 400) in CDCl3 and DMSO solution with tetramethylsilane as an internal standard. The NMR analyses were performed at Faculty of Science, Sohag University. The mass spectra were measured with a KRATOS analytical compact; on MALDI-MS the spectrometer was using 2,5-dihydroxy benzoic acid (DHB) as matrix. The starting compound 3 were obtained as described in literature.17,18General procedure for synthesis of 3-(4-chlorophenyl)-N-arylidene-2,2-dimethylpropane hydrazide 16a–d.
A mixture of 3-(4-chlorophenyl)-3-hydroxy-2,2-dimethylpropane hydrazide (15) (2.1 g, 0.90 mmol) and aldehydes (1.0 mmol) were refluxed in ethanol (30 ml) for 6 hours. After cooling to room temperature, the resulting solid was filtered, washed with ethanol and recrystallized from ethanol.Antiproliferative assay
Materials and methods
Data were calculated as percent of cell viability by the following formula: % cell viability = (mean absorbance in test wells/mean absorbance in control wells) × 100.23
For the docking studies, crystal structure of human HDAC2 complexed with an N-(2-aminophenyl)benzamide was obtained from the freely accessible protein data bank (PDB code: 3max), verification process was performed by redocking of the co-crystallized ligand into the active site using the default settings. The compounds under study were constructed 2D using ChemBio-office 2015, converted to 3D by builder interface of MOE program, and then were subjected to energy minimization with MMFF94X force and the partial charges were automatically calculated. Different conformers for each compound are imported by systematic conformational of the MOE and saved in an mdb database file to be docked into the active site of the receptor. Each complex was analyzed for interaction, 2D images were taken by using MOE visualizing tool.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Science & Technology Development Fund in Egypt STDF Project ID: 22909 for funding this research proposal also our appreciation extended to Ass. Lect. Mohamed S. Nafie, Bioorganic Chemistry, Faculty of science, Suez Canal University for the computational chemistry work.References
- M. Rahman and M. R. Hasan, Metabolites, 2015, 5(4), 571 CrossRef CAS PubMed.
- Q. Wu, Z. Yang, Y. Nie, Y. Shi and D. Fan, Cancer Lett., 2014, 347, 159–166 CrossRef CAS PubMed.
- L. Gianni, G. Grasselli, S. Cresta, A. Locatelli, L. Vigano and G. Minotti, Cancer Chemother. Biol. Response Modif., 2003, 21, 29–40 CAS.
- G. Marzaro, A. Guiotto and A. Chilin, Expert Opin. Ther. Pat., 2012, 22(3), 223 CrossRef CAS PubMed.
- S. Poorirani, S. Sadeghian-Rizi, G. Khodarahmi, M. R. Khajouei and F. Hassanzadeh, Results Pharma Sci., 2018, 13(5), 450 CrossRef.
- B. D. Strahl and C. D. Allis, Nature, 2000, 403, 41–45 CrossRef CAS PubMed.
- S. E. Rundlett, A. A. Carmen, R. Kobayashi, S. Bavykin, B. M. Turner and M. Grunstein, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14503–14508 CrossRef CAS.
- A. Petrella, B. Fontanella, A. Carratu, V. Bizzarro, M. Rodriquez and L. Parente, Mini-Rev. Med. Chem., 2011, 11, 519–527 CrossRef CAS.
- D. R. Walkinshaw and X. J. Yang, Curr. Oncol., 2008, 15(5), 237–243 CAS.
- J. E. Bolden, M. J. Peart and R. W. Johnstone, Nat. Rev. Drug Discovery, 2006, 5, 769–784 CrossRef CAS.
- Y. Dai, Y. Guo, M. L. Curtin, J. Li, L. J. Pease, J. Guo, P. A. Marcotte, K. B. Glaser, S. K. Davidsen and M. R. Michaelides, Bioorg. Med. Chem. Lett., 2003, 13, 3817–3820 CrossRef CAS.
- D. Son, C. S. Kim, K. R. Lee and H. J. Park, Bioorg. Med. Chem. Lett., 2016, 26, 2365–2369 CrossRef CAS PubMed.
- A. P. Zorzi, M. Bernstein, Y. Samson, D. A. Wall, S. Desai, D. Nicksy, N. Wainman, E. Eisenhauer and S. Baruchel, Pediatr. Blood Cancer, 2013, 60, 1868–1874 CrossRef CAS.
- P. Atadja, Cancer Lett., 2009, 280, 233–241 CrossRef CAS PubMed.
- K. Rao-Bindal, N. V. Koshkina, J. Stewart and E. S. Kleinerman, Curr. Cancer Drug Targets, 2013, 13, 411–422 CrossRef CAS PubMed.
- Y. Boumber, A. Younes and G. Garcia-Manero, Expert Opin. Invest. Drugs, 2011, 20, 823–829 CrossRef CAS PubMed.
- R. H. Kalita, A. J. Borah and P. Phukan, Indian J. Chem., 2013, 52(B), 289 Search PubMed.
- F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Green Chem., 2010, 12, 1301 RSC.
- D. S. El-Kady, A. A. Abd Rabou, M. A. Tantawy, A. A.-H. Abdel-Rahman, A. A.-S. Abdel-Megeed, M. M. AbdElhalim and G. A. Elmegeed, Appl. Biochem. Biotechnol., 2019 DOI:10.1007/s12010-018-02943-6.
- D. K. Maurya, N. Nandakumar and T. P. Devasagayam, J. Clin. Biochem. Nutr., 2011, 48(1), 85 CrossRef CAS.
- A. A.-H. Sebeka, A. M. A. Osman, I. E.-T. El Sayed, M. El Bahanasawy and M. A. Tantawy, J. Appl. Pharm. Sci., 2017, 7(10), 9 CAS.
- S. Ranganathan, D. Halagowder and N. D. Sivasithambaram, PLoS One, 2015, 10(10), e0141370 CrossRef PubMed.
- M. A. Tantawy, M. S. Nafie, G. A. Elmegeed and I. A. I. Ali, Bioorg. Chem., 2017, 73, 128–146 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |