
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotential role of a disintegrin and metalloproteinase-17 (ADAM17) in age-associated ventricular remodeling of rats†
Hainiang Liu‡
ab,
Haoren Wang‡a,
Dong Chengab,
Qinfu Wangc,
Zuowei Pei a,
Ning Zhud,
Weiyi Fange and
Qin Yu*a
a,
Ning Zhud,
Weiyi Fange and
Qin Yu*a
aDepartment of Cardiology, Affiliated Zhongshan Hospital of Dalian University, Dalian 116001, China. E-mail: yuqin@dlu.edu.cn; dryuqin060621@sina.com; Fax: +86-411-62893555; Tel: +86-411-62887018
bMedical College, Dalian University, Dalian 116622, China
cLife Engineering College, Dalian University, Dalian 116622, China
dDepartment of Cardiology, The Second Affiliated Hospital of Dalian Medical University, Dalian 116023, China
eDepartment of Cardiology, Shanghai Chest Hospital, Shanghai 200030, China
First published on 7th May 2019
Abstract
Excessive tumor necrosis factor-α (TNF-α) could enhance cell death and aggravate left ventricular remodeling and myocardial dysfunction. A disintegrin and metalloproteinase-17 (ADAM17), an important maturation regulator of TNF-α, might be involved in the aging-associated ventricular remodeling. The present study observed myocardial ADAM17 expression in young and aged rats and explored the association between cardiac structure/function and expression of ADAM17 in 6 month-old (n = 10, young group) and 24 month-old SD rats (n = 10, old group). The body, heart weight and heart weight/body weight ratio of rats in the old group were all significantly increased compared to that in the young group (P < 0.05). The left ventricular systolic end-diameter and end-diastolic diameters were significantly enlarged in the old group compared to the young group (P < 0.05), while the systolic function index including the left ventricular ejection fraction and left ventricular fractional shortening were similar between the two groups. The peak mitral flow velocity (E)/peak mitral annulus velocity (E′) ratio was significantly higher in the old group than in the young group (P < 0.05). Histological examination showed more damage of cardiomyocytes, interstitial collagen deposition and inflammatory cell infiltration in the old group. Immunohistochemistry examination showed that myocardial TNF-α expression was mainly located in cardiomyocytes and was significantly higher in the old group than in the young group (P < 0.05). The protein expression of myocardial ADAM17 detected by western blot was significantly higher in the old group than in the young group (P < 0.05), while TIMP-3 expression was similar between the two groups. The present study suggested that ADAM17 and inflammation might play an important role in aging-related myocardial remodeling through regulating TNF-α.
Introduction
Over the past decades, life expectancy has increased in most countries worldwide.1 The prevalence of cardiovascular disease is therefore increasing in aging populations.2 It is known that decreased myocardial reserve and adverse myocardial remodeling contribute to the pathogenesis of cardiovascular disease in aged populations.3 Clinically, aging is related to left ventricular hypertrophy and fibrosis, and cardiac dysfunction, especially impairment of diastolic function.4,5 Exploring the underlying molecular mechanism regulating age-related cardiac remodeling is therefore of clinical importance and helpful to attenuate the prevalence of cardiovascular disease in aged populations.A disintegrin and metalloproteases-17 (ADAM17), also known as tumor necrosis factor-α (TNF-α) converting enzyme (TACE), is widely expressed in human organs, including in the cardiovascular system.6,7 ADAM17 is known to play an important role in regulating valve formation and ventricular remodeling during cardiac development,8 and up-regulated ADAM17 expression was evidenced in patients with myocarditis.9 The structure of ADAM17, its signaling in the cardiovascular system and its participation in certain disorders involving the heart, blood vessels, and neural regulation of autonomic and cardiovascular modulation was summarized in detail previously.10 In animal models of concentric cardiac hypertrophy, application of ADAM17 inhibitors could reduce left ventricular hypertrophy, perhaps through the preservation of TNF-α on the surface of cardiomyocytes.11 Until now, it remains largely elusive whether the aging process is linked with myocardial expression changes of ADAM17 or not. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a native inhibitor of ADAM-17.12 Increased ADAM17 activity and soluble TNF-α abundance were evidenced in TIMP-3 deficient animals.13,14 Zheng et al. demonstrated that enhanced ADAM17 expression, along with a decreased TIMP-3 and increased TNF-α expression, is correlated with cardiac remodeling after acute myocardial infarction (AMI).15
The present study aimed to compare the myocardial expression of ADAM17 and TIMP-3, as well as TNF-α between young and aged rats, to explore a potential role of the above parameters on aging and related cardiac remodeling.
Experimental
Animal model
Sprague-Dawley (SD) rats (SPF grade) were purchased from Experimental Animal Center of Dalian Medical University, rats were divided into two groups: the young group (6 months old, n = 10) and the old group (24 months old, n = 10). The age of experimental animals is a critical variable in biological science research.16 It was shown that 6 month-old rats were equivalent to 18 years for humans and 24 month-old rats were equivalent to 60 years for humans.17 Therefore, rats at the age of 6 months and 24 months were selected in this study, representing the young and aged groups. Rats were housed in separate boxes (n = 10 per cage) at a constant temperature (23 ± 2 °C) with free access to food and tap water under a 12 h light/dark cycle prior to the experiments for two weeks. The animal experiments were performed in accordance with the European Community Guidelines for Care and Use of Animals, and approved by the Institutional Animal Research and Ethics Committee of Dalian Medical University and efforts were made to minimize the number and suffering of the experimental animals (The Animal Ethic Clearance Approval Number: 201719).Echocardiographic measurements
After adaption to the environment for 2 weeks, all the rats were anesthetized by 10% chloral hydrate (3 ml kg−1) and depilated with 5% sodium sulfide depilation. The rats were placed in a supine position. All images were acquired with a 12 MHz transducer connected to a Vivid E9 echocardiography machine (GE Vingmed Ultrasound, Horten, Norway). M-mode ultrasound was used to measure left ventricular end-systolic diameter (LVESD), left ventricular end-diastolic diameter (LVEDD), and interventricular septal thickness (IVST) from a parasternal long-axis view at the papillary muscle level, and left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) values were calculated as LVFS = (LVEDD − LVESD)/LVEDD × 100%, and LVEF was calculated according to the Teichholz formula.18 Pulsed-wave Doppler echocardiography was performed on an apical four-chamber view to measure peak mitral flow velocity (E). Tissue Doppler echocardiography was performed on an apical four-chamber view to measure the peak mitral annulus velocity (E′). The E/E′ ratio, a preload-independent index of LV filling pressure, was then calculated. The above echocardiographic parameters were obtained by a professional echocardiographer. All parameters were evaluated on an average of three consecutive cardiac circles.Samples preparation
Blood was collected from an angular vein after echocardiographic examination. Blood was immediately centrifuged at 1000g at 4 °C for 15 min and then serum was stored at −20 °C for further use. All rats were sacrificed under deep anesthesia (intramuscular ketamine hydrochloride injection, 100 mg kg−1), hearts were excised and weighed. The heart weight (HW) to body weight (BW) ratio was then calculated. The atria and right ventricle were then separated from the heart, the left ventricle was cut into three sections (each 3 mm in thickness) along the long axis and processed for histological (middle section), immunohistological (basal section), and biochemical analysis (apical section). For the biochemical tests, samples were rapidly harvested and immediately put into liquid nitrogen and stored at −80 °C for further analysis. For histological analysis, a part of the myocardial tissue was fixed in 4% paraformaldehyde, embedded in paraffin. The other part was cut into tissue pieces of about 1 mm × 1 mm × 1 mm with a double-sided blade, which were then fixed in 2.5% glutaraldehyde and stored at 4 °C for transmission electron microscope analysis.Histopathological examination
The previously embedded myocardial tissue paraffin blocks were cut into 5 μm thick sections. Histopathological changes were evaluated by H&E staining. Fibrosis was evaluated by Sirius red staining. After Sirius red staining, a single slide was taken and the total field area for ten randomly selected fields were analyzed in a blind fashion using Image Analysis System (Media Cybernetics, Inc. Rockville, MD USA). The collagen volume fraction (CVF = area of the collagen/area of field of vision × 100%) was measured and ten separate areas of high power fields (200×) in each section were visualized under a light microscope. Ten sections from each rat were observed and the results were averaged.The ultrastructural changes of rat myocardium were observed by transmission electron microscopy. All reagents of electron microscopy were provided by the Electron Microscope Laboratory of Dalian Medical University. The specific operation was as follows: the tissue specimen was fixed, washed with 0.01 M PBS solution for 15 min; 1% osmic acid was added, soaked for 2 hours at room temperature; washed in 0.01 M PBS for 15 min; and dehydrated by 50% ethanol, 70% ethanol, 80% ethanol, 90% ethanol, and 100% ethanol. The rest of the steps were performed by technicians in the electron microscopy room: acetone gradient dehydration; Epon 812 infiltration, embedding; made ultra-thin slices; and uranium lead double staining. Myocardial ultrastructure was observed under an electron microscope (JEM-2000EX, JEOL, Japan).
Real-time PCR analysis
The levels of ADAM17, TIMP-3, collagen I, collagen III, and TNF-α mRNA in myocardial tissue were detected by a Real-Time Quantitative Polymerase Chain Reaction (qPCR). The total RNA was isolated from 100 mg left ventricular tissue using the High Pure RNA Isolation Kit according to the manufacturer's instructions (Takara Bio Inc., Dalian, China). All primer sequences were designed and synthesized by (Shanghai Shanjing Bio Inc., Shanghai, China) as follows, primers specific for rat ADAM17 (5′-GTGCTGACACCGACAACTCGT-3′ and 5′-CAGCTGGTCAATGAAATCCCAAA-3′), TIMP-3 (5′-GCTACTACTTGCCTTGCTTTG-3′ and 5′-ATTGCTGATGCTCTTGTCTG-3′), collagen I (5′-TGCCGTGACCTCAAGATGTG-3′ and 5′-CACAAGCGTGCTGTAGGTGA-3′), collagen III (5′-CTGGACCAAAAGGTGATGCTG-3′ and 5′-TGCCAGGGAATCCTCGATGTC-3′), TNF-α (5′-TGGCCCAGACCCTACACA-3′ and 5′-TGCCCGGACTCCGTGAT-3′) and GADPH (5′-TCCGCCCCTTCCGCTGATG-3′ and 5′-CACGGAAGGCCATGCCAGTGA-3′). Reverse transcription was performed with a PrimeScript™ RT reagent Kit with gDNA Eraser (Takara Bio Inc, Dalian, China) according to the manufacturer's instructions. Real-time PCR was carried out in a 20 μl reaction system containing 10 μl SYBR®Premix Ex TaqTMII (Tli RNaseH Plus), 1.6 μl each primer, 0.4 μl ROX Reference Dye II, 2 μl cDNA, and 6 μl dH2O. Real-time PCR was performed with the following protocol: initial predenaturation at 95 °C for 30 s, 40 cycles of denaturation at 95 °C for 5 s, annealing at 60 °C for 34 s. Housekeeping gene GAPDH was used as a positive control and internal standard. The result was the average of three times of the Real-time PCR. ΔCt was calculated as ΔCt = target genes Ct value − the GAPDH genes Ct value. Different groups of the genes on the internal change ratio = 2−ΔCt.19Measurement of serum TNF-α by ELISA
The previously collected serum samples were taken out from the −20 °C refrigerator and thawed at room temperature. Detection of serum TNF-α concentration was performed as described by the instructions of ELISA kit (Boster Biological Technology co. Ltd, California, USA). 100 μl of the test specimen was added to the reaction plate to establish a standard curve. 100 μl of test specimen was added to the reaction hole and incubated at 37 °C for 90 minutes. The prepared biotin anti-rat TNF-α antibody working solution was sequentially added at 100 μl per hole, and the reaction was incubated for 60 minutes at 37 °C and then washed 3 times with 1× wash buffer. The prepared ABC working solution was added in order of 100 μl per hole, and reacted at 37 °C for 30 minutes and then was washed 5 times with 1× washing buffer. The treated TMB coloring solution was added in order of 90 μl per hole, and reacted at 37 °C for 25–30 minutes in darkness. The optical density (OD) value of each pilot hole was read on a microplate reader (SpectraMax Plus, USA) at a wavelength of 450 nm within 3 minutes. According to the OD, a standard curve was drawn to help determine the serum levels of TNF-α.Detection of TNF-α in rat heart tissue by immunohistochemistry
We used immunohistochemistry to detect the expression and distribution of the inflammatory factor marker TNF-α in rat myocardial tissue. Left ventricle paraffin sections (5 μm) were stained according to the streptavidin–biotin complex method. The procedure involved the following steps: (1) the paraffin sections were de-waxed and then dehydrated; (2) antigen retrieval; (3) incubation with hydrogen peroxide (3% H2O2); (4) goat serum (SL038, Solarbio Inc., Beijing, China) was added, incubated at room temperature for 15 min, and the serum was removed; (5) the primary antibody, TNF-α antibody (WL01896, wanlei Inc., Shenyang, China), was diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 with PBS, which was dropped onto the tissue sections, and then put in a wet box 4 °C overnight. Sections were soaked in PBS for 5 min, repeated 3 times; (6) the secondary antibody, biotinylate goat anti rabbit IgG (A0277, Beyotime Inc., Beijing, China), was diluted 1:200 with PBS, which was dropped onto the tissue sections, and was incubated at 37 °C for 30 min in a wet box. Sections were soaked in PBS for 5 min, repeated 3 times; (7) HRP-labeled streptavidin (A0303, Beyotime Inc., Beijing, China) was dropped onto the tissue sections and then was incubated at 37 °C for 30 min. The sections were soaked in PBS for 5 min, repeated 3 times; (8) DAB chromogenic reagent (DA1010, Beyotime Inc., Beijing, China) was dropped onto the tissue sections; (9) hematoxylin counter staining; (10) dehydrated, transparent and sealed.
:200 with PBS, which was dropped onto the tissue sections, and then put in a wet box 4 °C overnight. Sections were soaked in PBS for 5 min, repeated 3 times; (6) the secondary antibody, biotinylate goat anti rabbit IgG (A0277, Beyotime Inc., Beijing, China), was diluted 1:200 with PBS, which was dropped onto the tissue sections, and was incubated at 37 °C for 30 min in a wet box. Sections were soaked in PBS for 5 min, repeated 3 times; (7) HRP-labeled streptavidin (A0303, Beyotime Inc., Beijing, China) was dropped onto the tissue sections and then was incubated at 37 °C for 30 min. The sections were soaked in PBS for 5 min, repeated 3 times; (8) DAB chromogenic reagent (DA1010, Beyotime Inc., Beijing, China) was dropped onto the tissue sections; (9) hematoxylin counter staining; (10) dehydrated, transparent and sealed.
The expression levels were estimated by the integrated OD of the positive cells. Integrated OD was the average cumulative OD of the positive staining area of each group determined by Image Pro Plus 6.0 software (Media Cybernetics, USA).
Western blotting analysis
The proteins were extracted from the myocardial tissue samples and analyzed by western blotting, as described previously.20 Extracted protein lysates from the left ventricle were separated on SDS-PAGE and then transferred to the PVDF (polyvinylidene fluoride) membrane. After sealing with 5% (m/v) skimmed milk powder, the membranes were then incubated with primary antibodies, including ADAM17 (Abcam, AB39162, 1:2000), TIMP-3 (Abcam, AB3984, 1:2000) and β-actin (Wanleibio, WL01845, 1:1000) at 4 °C overnight and subsequently with sheep anti-rabbit IgG-HRP (Wanleibio, WLA023, 1:5000) at room temperature for 45 minutes. The blotting band density was quantified by densitometric analysis using Image pro-Plus 6.0 software (Media Cybernetics, USA) and then normalized to the values of β-actin.
Statistical analyses
Experimental data were analyzed using SPSS 20. The experimental data is represented as mean ± SD. For data that conform to the normal distribution, a Student's t test is used. For data that does not conform to a normal distribution, a nonparametric test is used. A P value < 0.05 was regarded as statistically significant.Results
Body, heart weight, and heart weight to body weight ratio in young and old rats
Table 1 showed that body weight, heart weight, and heart weight to body weight ratio were all significantly higher in the old group than in young group.| Young group | Old group | |
|---|---|---|
| n = 10 | n = 10 | |
| a All data are expressed by the mean ± SD. *P < 0.05 vs. young group. BW, body weight and HW, heart weight. | ||
| Body weight (g) | 623.90 ± 39.25 | 795.60 ± 36.07* |
| Heart weight (mg) | 2160.0 ± 289.84 | 3198.0 ± 397.93* |
| HW/BW (mg g−1) | 3.46 ± 0.40 | 4.2 ± 0.51* |
Changes of left ventricular structure and function in old rats
Table 2 and Fig. 1 show that LVESD, LVEDD, and E/E′ were significantly higher in old rats than in young rats.| Young group | Old group | |
|---|---|---|
| n = 10 | n = 10 | |
| a All data are expressed by the mean ± SD. *P < 0.05 vs. young group. LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; IVST, interventricular septal thickness; LVESD, left ventricular systolic end-diameter; LVEDD, left ventricular end-diastolic diameter; and E/E′, peak mitral flow velocity (E) to mitral annulus velocity (E′) ratio. | ||
| LVEF (%) | 93.30 ± 4.57 | 89.80 ± 6.44 |
| LVFS (%) | 65.10 ± 7.61 | 61.70 ± 6.46 |
| IVST (mm) | 1.94 ± 0.27 | 2.31 ± 0.50 |
| LVESD (mm) | 1.81 ± 0.22 | 3.20 ± 0.34* |
| LVEDD (mm) | 4.12 ± 0.26 | 5.03 ± 0.28* |
| E/E′ | 15.12 ± 2.71 | 21.39 ± 5.18* |
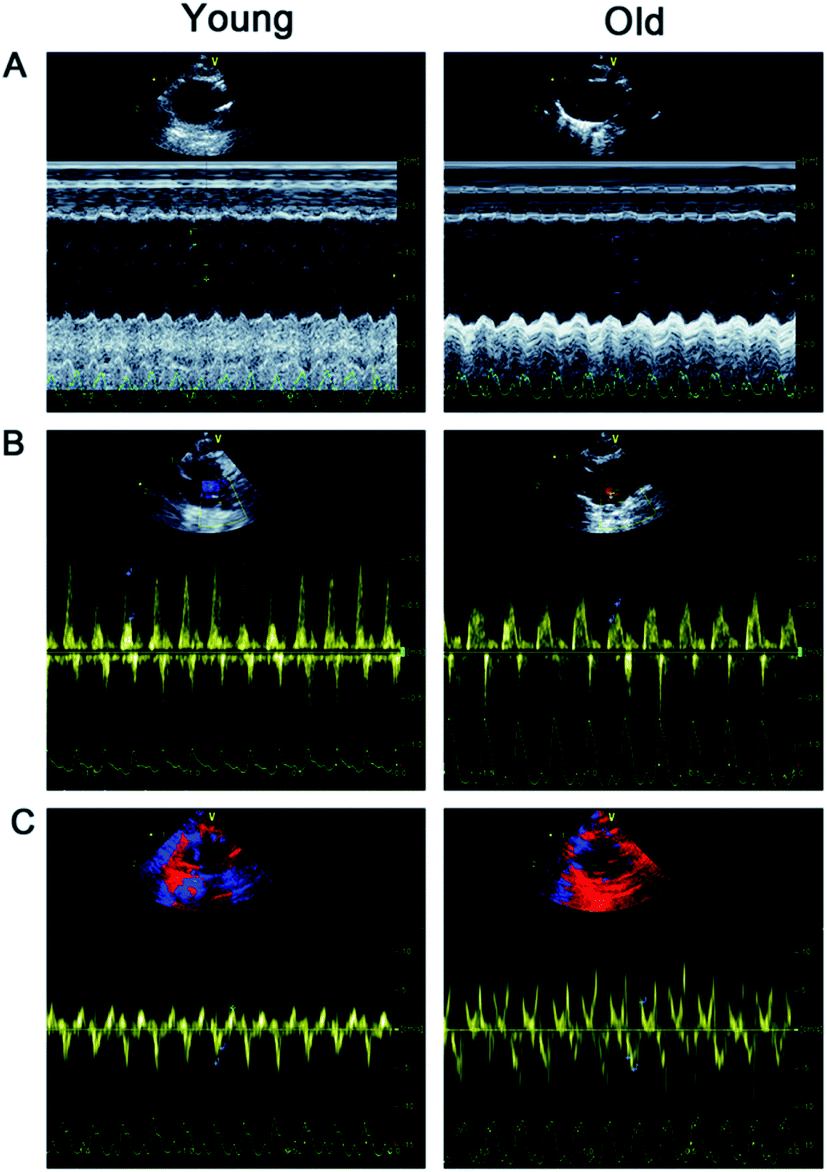 | ||
| Fig. 1 Hemodynamic parameters and left ventricular function were analyzed by echocardiography. (A) Representative M-mode ultrasound images; (B) representative pulsed-wave Doppler images; (C) representative tissue Doppler images. | ||
Histopathological analysis
Fig. 2 shows representative results of H&E staining (Fig. 2A), Sirius red staining (Fig. 2B) and quantitative analysis of the collagen volume fraction (CVF) (Fig. 2C) in the young and old groups. H&E staining revealed that myocardial fibers were displayed uniform and neat. The nucleus structure of the cardiomyocytes was clear, size was uniform, only a small amount of collagen fibers in the myocardial fibers or around the blood vessels was detected in the young group. In the old group, the cell gap widened and myocardial collagen fiber was increased. The cell gap is filled with a large number of inflammatory factors. Sirius red staining dictated a large amount of red collagen fibers in myocardial fibers or around the blood vessels in old group. The mean positive staining area (expressed as collagen volume fraction, CVF, %) was significantly higher in the old group than in the young group.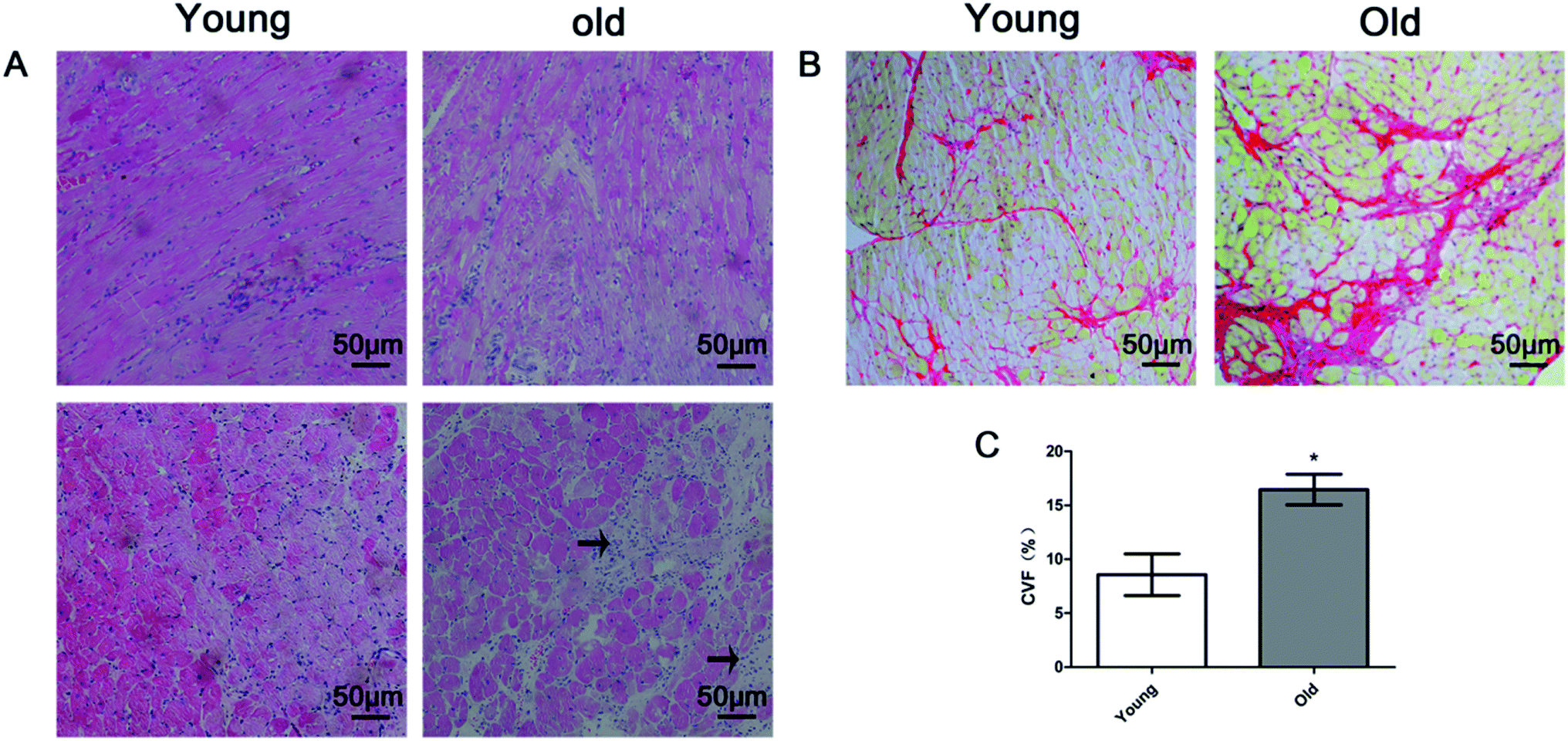 | ||
| Fig. 2 (A) Representative examples of HE staining (magnification 200×) in young and old rats. Interstitial inflammatory cell infiltration in the old group (arrow); (B) representative examples of Sirius red staining (magnification 200×) in the young and old group; (C) bar plots of collagen volume fraction (CVF) in the young and old groups (mean ± SD, n = 10 in each group). *P < 0.05 vs. young group. | ||
Fig. 3 presents the transmission electron microscopy results of the ultrastructure of the myocardium. In the young group, the myocardium observed few autophagosomes (Fig. 3A, left), the myocardial fibers arranged neatly (Fig. 3B, left), mitochondria were dense, no edema (Fig. 3B, left) and the intercalary disc were clear and continuous (Fig. 3C, left). In the old group, the myocardium observed a large number of autophagosomes (Fig. 3A, right), the myocardial fibers arranged loosely (Fig. 3B, right), dropsy mitochondria (Fig. 3B, right) and the intercalary disc gap widening (Fig. 3B, right).
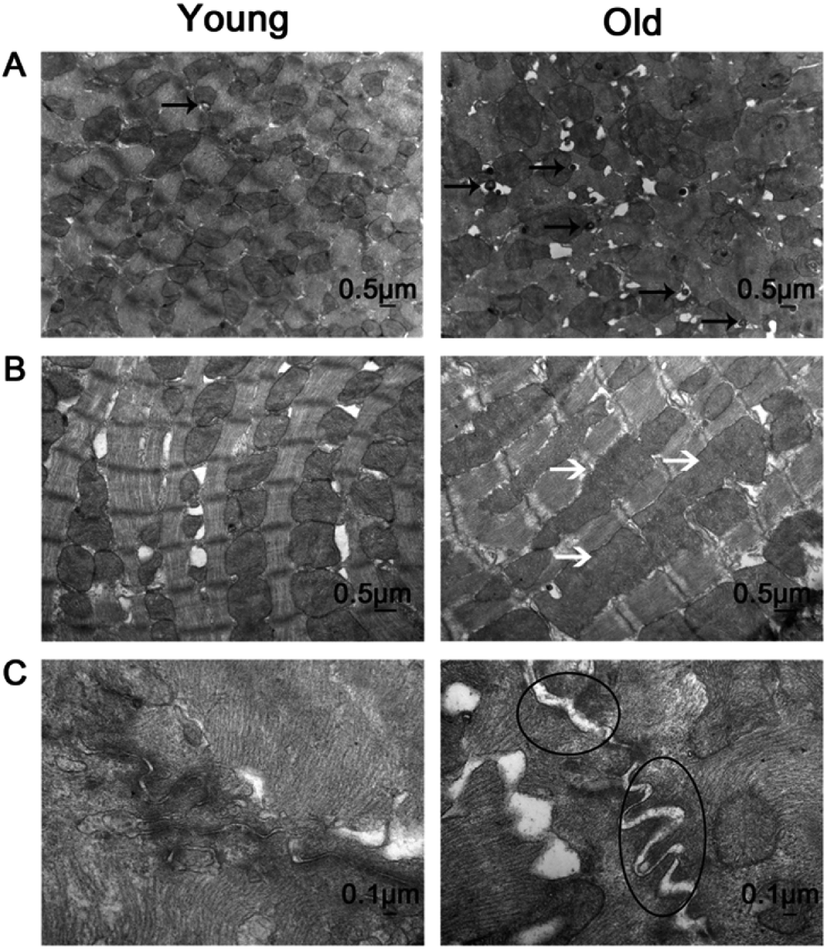 | ||
| Fig. 3 Representative examples of transmission electron microscopy in the young and old group. (A) Magnification 15000×: autophage corpuscle (black arrows); (B) magnification 20000×: swollen mitochondrias (white arrows); (C) magnification 60000×: widening of the intercalary disc gap (circles). | ||
Myocardial mRNA expression of ADAM17 and cardiac remodeling molecules
Fig. 4 shows that the myocardial mRNA expression of collagen I and collagen III (Fig. 4A), TNF-α (Fig. 4B), ADAM17 (Fig. 4C) and TIMP-3 (Fig. 4D) of the young group and old group. Compared with the young group, the mRNA expressions of TNF-α were significantly upregulated and collagen I and collagen III were significantly down-regulated while the mRNA expressions of ADAM17, TIMP-3, were similar between the young and old groups.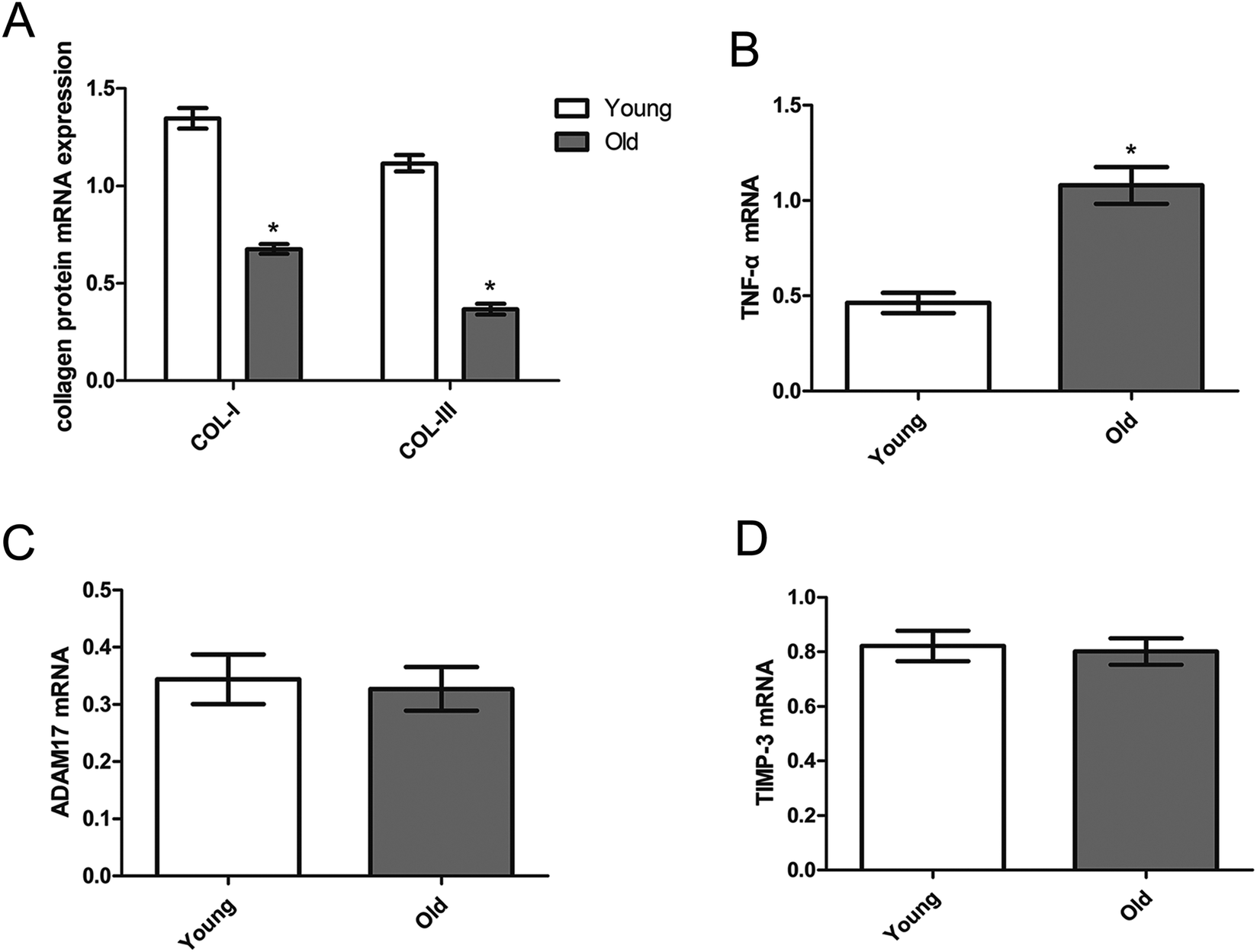 | ||
| Fig. 4 Myocardial mRNA expression of collagen I and III (A), TNF-α (B), ADAM17 (C) and TIMP-3 (D) in the young (open) and old (dark) group. Data are expressed by the mean ± SD (n = 10 in each group). *P < 0.05 vs. the young group. | ||
Myocardial TNF-α expression detected by immunohistochemistry analysis and serum TNF-α level detected by Elisa analysis
Fig. 5A and B show increased myocardial TNF-α expression in the old group compared to the young group. Serum TNF-α level was detected by Elisa analysis. Fig. 5C shows that serum TNF-α was significantly higher in the old group than in the young group.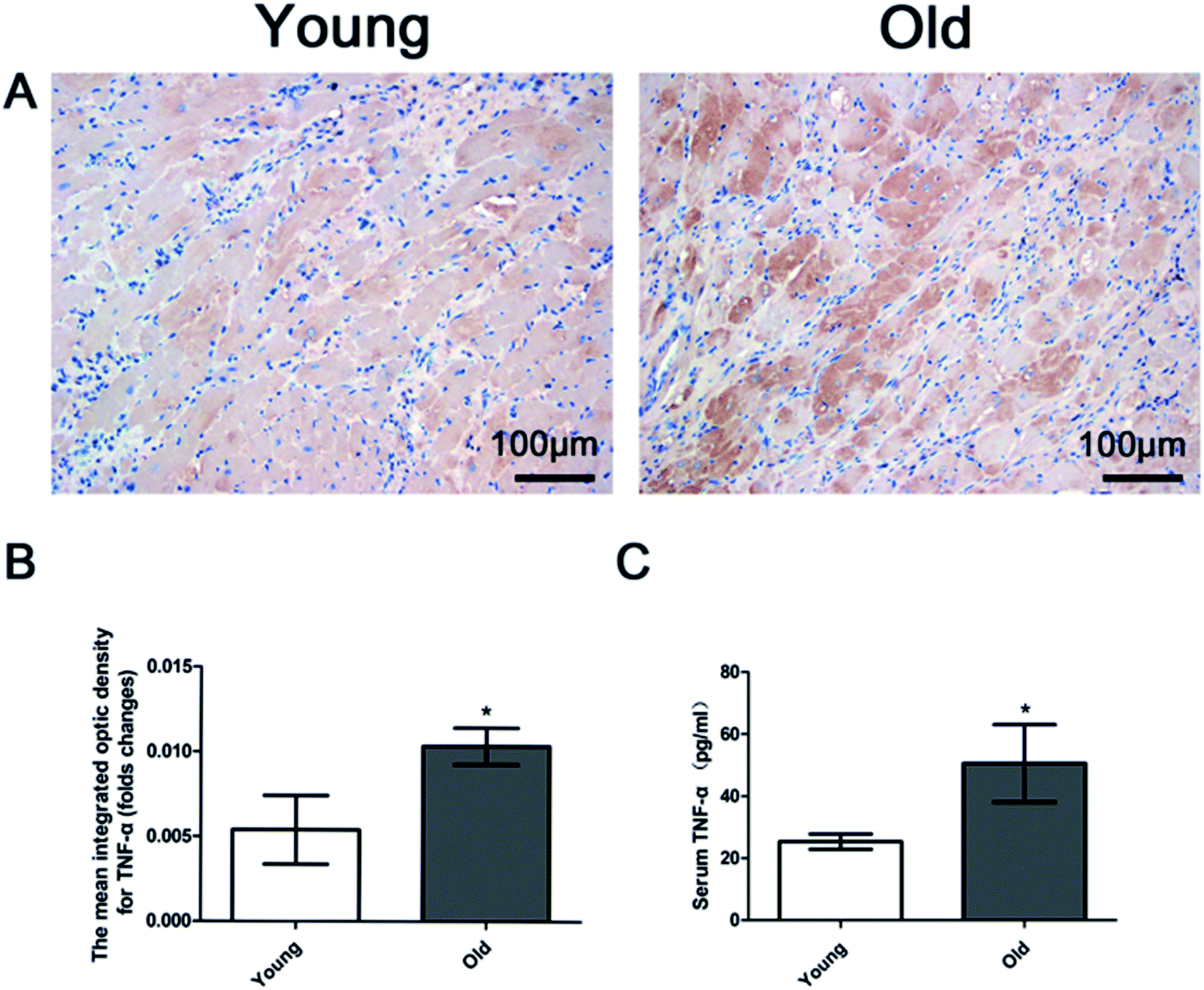 | ||
| Fig. 5 (A) Representative immunohistochemistry images (magnification 200×); (B) bar graph of myocardial TNF-α expression; (C) bar graph showing quantification of serum TNF-α level. Data are expressed by the mean ± SD (n = 10 in each group). *P < 0.05 vs. young group. | ||
Protein expression of ADAM17 and TIMP-3 in cardiac tissues
Fig. 6 shows the protein expression of ADAM17 and TIMP-3 in the cardiac tissue of the young group and old group. Protein expression of ADAM17 was significantly upregulated in the old group compared with the young group, but protein expression of TIMP-3 was similar between the young and old group.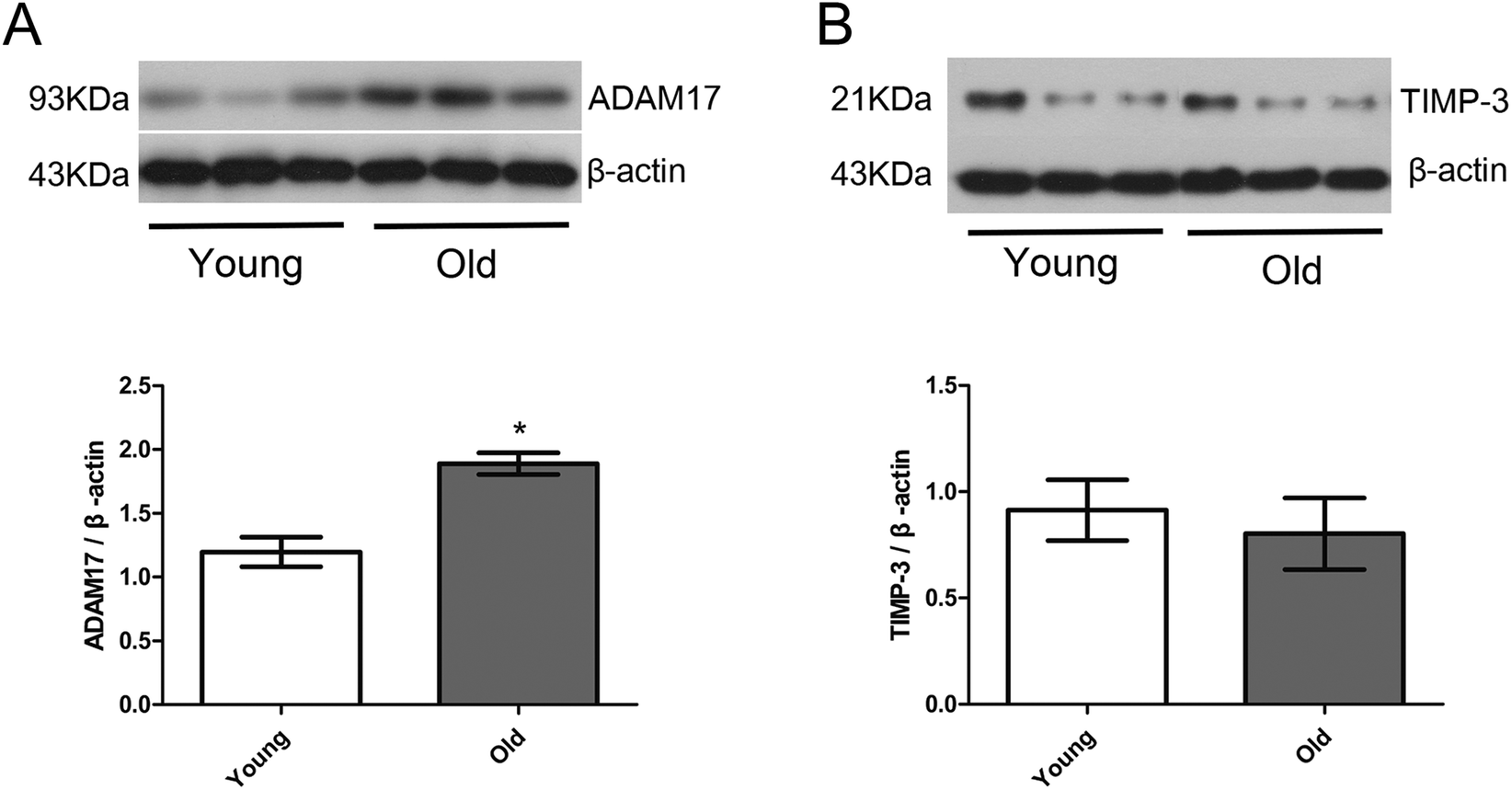 | ||
| Fig. 6 Protein expression of ADAM17 (A) and TIMP-3 (B) in cardiac tissues analyzed by western blotting. Upper panels show representative images of the corresponding protein and the lower graphs are quantitative analysis of each protein expression. Data are expressed by the mean ± SD (n = 10 in each group). *P < 0.05 vs. young group. | ||
Discussion
Natural aging is closely related to the increased prevalence of cardiovascular diseases. A large number of experimental and clinical studies have shown that aging itself is an independent risk factor for cardiac structural changes and dysfunction. In healthy individuals without cardiovascular disease, aging can lead to left ventricular remodeling, diastolic function decline, while systolic function is preserved at rest, but exercise capacity might decline.21 In this study, 24 month-old rats were used to represent the natural aging process in rats. The impact of aging on cardiac structure and function were assessed. We found that the BW, HW, and HW/BW ratio were all significantly increased in old rats compared to young rats, indicating the presence of cardiac remodeling in aged rats. Echocardiographic data also indicated ventricular cavity enlargement and diastolic dysfunction in aged rats. This finding is in line with a previous report showing a close relationship between aging and diastolic dysfunction.22,23The results of transmission electron microscopy showed that the aging myocardium was damaged, mainly manifested as mitochondrial edema, myocardial disorders, myofibrillar lysis and rupture, interstitial edema and fusion of autophagy bodies. These results were consistent with previous studies.24 It is well known that aging is associated with increased content of extracellular matrix protein (ECM) in addition to the myocardial degeneration process.25–27
By observing and quantifying the pathological feature using Image-ProPlus software on Sirius red stained sections, we evidenced significant myocardial fibrosis in the aged group, as expressed by enhanced ECM deposition. The deposition of myocardial collagen in aged rats might increase myocardial stiffness, which might thus be an important reason for the increase of diastolic index E/E′. In addition, by observing HE staining pathological sections, we also found a large number of inflammatory cell infiltration between myocardial cells, suggesting that inflammation may play an important role in the occurrence and development of myocardial remodeling during aging.28–31
Collagen protein is the main component of ECM. However, the mRNA expression level of collagen I and collagen III did not significantly change during myocardial aging in our study. Previous studies have shown that some fibrosis markers are imbalanced expressed at mRNA and protein levels in elderly patients with myocardial fibrosis. Mays et al. also found that the collagen synthesis rate of 24 month-old rats was 1/3 of that in 2 month old rats.32 Collagen cross-linking between cells was significantly enhanced during aging, and the resistance of the collagen network structure to degradation was then enhanced.33
It can be seen that collagen biosynthesis in myocardium does not increase with age, and the degradation pathway of ECM may play an important role in collagen accumulation. In addition, phenomenons of chronic proinflammatory reactions emerging in the organism progressively increased in the process of natural aging, also known as inflammatory senescence,34 which can also cause damage to cardiac tissue cells and imbalance in the production and degradation of the cardiac extracellular matrix.29,30 In line with this hypothesis, an enhanced infiltration of inflammatory cells in aging reconstructed myocardium was evidenced in HE stained myocardial tissues. TNF-α is one of the most important proinflammatory cytokines. In recent years, more and more studies have found that serum TNF-α, IL-6, IL-1 and other inflammatory factors increase with age, and participate in the pathogenesis of most age-related diseases.35 In this experiment, we also observed increased TNF-α expression in the myocardium, and a higher serum TNF-α level in aged rats. Our results are in line with the previous studies showing that TNF-α is crucial in the early stage of left ventricular remodeling,36–38 over-expression of TNF-α and chronic infusion of TNF-α may lead to cardiomyocyte and extracellular matrix remodeling, resulting in increased mean cross-sectional area of ventricular myocytes, myocardial necrosis, myocardial interstitial fibrosis, ventricular dilatation, and eventually ventricular remodeling.39–42
ADAM17, also known as tumor necrosis factor-α converting enzyme, is an important regulator of TNF maturation. It promotes the production of soluble TNF-α by specific cleavage of pro-TNF-α to mediate the activation of TNF-α after translation.6,7 Activation of TNF-α after translation is an important cause of myocardial remodeling induced by over expression of TNF-α in myocardium. Studies have shown that the use of ADAM17 inhibitors could reduce the production of soluble TNF-α, which can prevent ventricular dilatation in transgenic mice.9
Therefore, in cardiovascular diseases, ADAM17 can be used as a potential therapeutic target to block the production of soluble TNF-α. In addition, ADAM17 can reduce the degradation of Angiotensin II and aggravate Angiotensin II-induced myocardial remodeling by mediating the sheddases of ACE2.43,44 Knockout of ADAM17 significantly reduced endothelial cell migration and collateral circulation formation,45 decreased cardiac enlargement and reversed ventricular remodeling.8,40,44 Targeted ADAM17 siRNA therapy for 30 days effectively prevented ventricular hypertrophy in spontaneously hypertensive rats and in mice continuously infused with Angiotensin-II.44 Previous clinical and animal studies have shown that ADAM17 expression in myocardial tissue of pathological myocardial remodeling such as dilated cardiomyopathy, Angiotensin-II-induced myocardial remodeling and post-infarction myocardial remodeling is increased at both mRNA and protein levels.8,40,44 However, the expression of ADAM17 in aging myocardial remodeling has not been explored. In this study, we found that the expression of ADAM17 protein in the myocardium of aged rats was significantly higher than that of young rats, which suggested the possibility of ADAM17 participating in the myocardial fibrosis of aged rats.
Activated ADAM17 itself has been shown to affect the synthesis and secretion of collagen in the cell matrix and the interaction between the cell matrix through the sheddases of transmembrane glycoproteins of extracellular cytokine receptors and integrins.46,47 TIMP-3 is the only endogenous inhibitor that inhibits ADAM17 activity.11 Studies have found that ADAM17 and TIMP3 are abundant in the heart. Under stationary conditions, ADAM17 dimerizes on the cell surface through its cytoplasmic tail, and TIMP3 inhibits the cleavage activity of ADAM17 by binding to the ADAM17 homologous dimer on the cell surface. When the ERK or P38 MAPK pathway is activated under hypoxia, inflammatory factors and stress, the intracellular segment of ADAM17 is phosphorylated, and the ADAM17 homologous dimer on the cell surface is converted into a monomer. At the same time, the TIMP3 group is released from ADAM17. At this time, ADAM17 is activated and its protein hydrolysis activity is enhanced.48 TIMP-3 can inhibit the sheddases of cytokines and their receptors such as TNF, TNFR, IL-6R by ADAM17, which has been confirmed in animal experiments. The activity of ADAM17 in TIMP3 deficient mice increased significantly, resulting in the existence of a large quantity of soluble TNF-α.13,14 In addition, it was also found that down-regulation of TIMP-3 resulted in elevated levels of soluble TNF-α in rats, leading to psoriasis-like inflammatory dermatosis.14 Zheng et al. declared that within one week after acute myocardial infarction, the expression of ADAM17 in myocardial tissue increased, accompanied by a decrease in TIMP3 expression and an increase in TNF-alpha expression, suggesting that these changes were associated with myocardial remodeling after myocardial infarction.15 The novel finding of the present study is as follows: aging is linked with increased myocardial protein expression of ADAM17 but not accompanied by simultaneous reduction of TIMP-3 (the endogenous inhibitor of ADAM17) in this model, which is quite different to that seen in the previous pathological myocardial remodeling mentioned above. It is suggested that up-regulation of TIMP-3 in an aged heart could inhibit ADAM17 activity and slow down aging-related myocardial remodeling. In addition, activated ADAM17 not only promotes the release of soluble TNF-α, but also mediates myocardial remodeling through other pathways in myocardial tissue. Therefore, direct down-regulation of ADAM17 in the heart may also improve age-related myocardial remodeling. The present study was an observational study and future more in-depth mechanism studies are warranted to explore the role of ADAM17 and TIMP-3 in age-related myocardial remodeling.
Theoretically, the use of ADAM17 inhibitors or specific ADAM17 knockout animal models will help elucidate the mechanism of ADAM17 during myocardial remodeling. However, recent studies have shown that the use of nonspecific ADAM17 inhibitors is limited due to their strong side effects.49 The mice with ADAM17 specific knockout established by Fan50 and others are not yet available in our laboratory, and future studies with these knockout mice might facilitate related mechanistic research.
Conclusions
Our results show that the remodeling of aged myocardium is mainly manifested by myocardial cell injury, interstitial collagen deposition and invasion of a large number of inflammatory cells, resulting in increased heart mass, ventricular enlargement and diastolic dysfunction, but this has little effect on the contractile function of rats. There is an unbalanced expression of the ADAM17 gene level and protein level during aging. Aging increased the expression of ADAM17 protein in rat myocardium, accompanied by increased expression of TNF-α in myocardium and increased levels of TNF-α in serum. The aging myocardium remodeling is accompanied by a high inflammatory state. The expression of TIMP-3 was not changed in the aging process. ADAM17 may also play an important role in myocardial remodeling with increasing age.Author contributions
Qin Yu designed this study; Hainiang Liu drafted the manuscript; Hainiang Liu and Haoren Wang analyzed data and interpreted the results of experiments; Hainiang Liu and Dong Cheng prepared the figures; Haoren Wang and Qinfu Wang guided the experiment technology; and Zuowei Pei, Weiyi Fang and Ning Zhu supervised the experimental process. All authors have read and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by Scientific Research Fund from Dalian Science and Technology Bureau (grant number: 2007E21SF203; 2014E14SF149); the Specialized Research Fund of Higher Education of Liaoning Provincial Education Department (grant number: L2010023), Grant to promote Dalian's medical soft power from Dalian Municipal Government, Natural Science Funds of Liaoning Province (grant number: 201602033) and National Natural Science Funds of China (grant number: 81770405).References
- K. Christensen, G. Doblhammer, R. Rau and J. W. Vaupel, Lancet, 2009, 374, 1196–1208 CrossRef.
- P. A. Heidenreich, J. G. Trogdon, O. A. Khavjou, J. Butler, K. Dracup, M. D. Ezekowitz, E. A. Finkelstein, Y. Hong, S. C. Johnston, A. Khera, D. M. Lloyd-Jones, S. A. Nelson, G. Nichol, D. Orenstein, P. W. Wilson and Y. J. Woo, Circulation, 2011, 123, 933–944 CrossRef.
- D. F. Dai, Y. A. Chiao, R. J. Wessells, R. Bodmer, H. H. Szeto and P. S. Rabinovitch, in Handbook of the Biology of Aging, ed. M. R. Kaeberlein and G. M. Martin, Academic Press, San Diego, 8th edn, 2016, DOI:10.1016/C2012-0-07319-X, ch. 17, pp. 459–494.
- J. Eng, R. L. McClelland, A. S. Gomes, W. G. Hundley, S. Cheng, C. O. Wu, J. J. Carr, S. Shea, D. A. Bluemke and J. A. Lima, Radiology, 2016, 278, 714–722 CrossRef.
- P. S. Hees, J. L. Fleg, E. G. Lakatta and E. P. Shapiro, Am. J. Cardiol., 2002, 90, 1231–1236 CrossRef.
- R. A. Black, C. T. Rauch, C. J. Kozlosky, J. J. Peschon, J. L. Slack, M. F. Wolfson, B. J. Castner, K. L. Stocking, P. Reddy, S. Srinivasan, N. Nelson, N. Boiani, K. A. Schooley, M. Gerhart, R. Davis, J. N. Fitzner, R. S. Johnson, R. J. Paxton, C. J. March and D. P. Cerretti, Nature, 1997, 385, 729–733 CrossRef CAS.
- M. L. Moss, S. L. Jin, M. E. Milla, D. M. Bickett, W. Burkhart, H. L. Carter, W. J. Chen, W. C. Clay, J. R. Didsbury, D. Hassler, C. R. Hoffman, T. A. Kost, M. H. Lambert, M. A. Leesnitzer, P. McCauley, G. McGeehan, J. Mitchell, M. Moyer, G. Pahel, W. Rocque, L. K. Overton, F. Schoenen, T. Seaton, J. L. Su and J. D. Becherer, et al., Nature, 1997, 385, 733–736 CrossRef CAS PubMed.
- W. Shi, H. Chen, J. Sun, S. Buckley, J. Zhao, K. D. Anderson, R. G. Williams and D. Warburton, Dev. Biol., 2003, 261, 371–380 CrossRef CAS.
- M. Satoh, M. Nakamura, H. Satoh, H. Saitoh, I. Segawa and K. Hiramori, J. Am. Coll. Cardiol., 2000, 36, 1288–1294 CrossRef CAS.
- J. Xu, S. Mukerjee, C. R. Silva-Alves, A. Carvalho-Galvao, J. C. Cruz, C. M. Balarini, V. A. Braga, E. Lazartigues and M. S. Franca-Silva, Front. Physiol., 2016, 7, 469 Search PubMed.
- Z. I. Dibbs, A. Diwan, S. Nemoto, G. DeFreitas, M. Abdellatif, B. A. Carabello, F. G. Spinale, G. Feuerstein, N. Sivasubramanian and D. L. Mann, Circulation, 2003, 108, 1002–1008 CrossRef CAS.
- A. Amour, P. M. Slocombe, A. Webster, M. Butler, C. G. Knight, B. J. Smith, P. E. Stephens, C. Shelley, M. Hutton, V. Knauper, A. J. Docherty and G. Murphy, FEBS Lett., 1998, 435, 39–44 CrossRef CAS.
- M. Federici, M. L. Hribal, R. Menghini, H. Kanno, V. Marchetti, O. Porzio, S. W. Sunnarborg, S. Rizza, M. Serino, V. Cunsolo, D. Lauro, A. Mauriello, D. S. Smookler, P. Sbraccia, G. Sesti, D. C. Lee, R. Khokha, D. Accili and R. Lauro, J. Clin. Invest., 2005, 115, 3494–3505 CrossRef CAS PubMed.
- J. Guinea-Viniegra, R. Zenz, H. Scheuch, D. Hnisz, M. Holcmann, L. Bakiri, H. B. Schonthaler, M. Sibilia and E. F. Wagner, Genes Dev., 2009, 23, 2663–2674 CrossRef CAS.
- D. Y. Zheng, J. Zhao, J. M. Yang, M. Wang and X. T. Zhang, Life Sci., 2016, 151, 61–69 CrossRef CAS.
- J. E. McCutcheon and M. Marinelli, Eur. J. Neurosci., 2009, 29, 997–1014 CrossRef.
- N. A. Andreollo, E. F. Santos, M. R. Araujo and L. R. Lopes, Arq. Bras. Cir. Dig., 2012, 25, 49–51 CrossRef.
- J. G. Dumesnil, D. Dion, K. Yvorchuk, R. A. Davies and K. Chan, Can. J. Cardiol., 1995, 11, 1007–1014 CAS.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- L. Ye, H. Haider, S. Jiang, R. Ge, P. K. Law and E. K. Sim, J. Heart Lung Transplant., 2005, 24, 1393–1402 CrossRef PubMed.
- E. G. Lakatta and D. Levy, Circulation, 2003, 107, 139–146 CrossRef PubMed.
- A. S. Balcioglu, M. E. Durakoglugil, K. Okyay, Y. Tavil and A. Abaci, Echocardiography, 2014, 31, 1062–1070 CrossRef.
- F. A. Zouein, L. E. de Castro Bras, D. V. da Costa, M. L. Lindsey, M. Kurdi and G. W. Booz, J. Cardiovasc. Pharmacol., 2013, 62, 13–21 CrossRef CAS PubMed.
- H. Frenzel and J. Feimann, Mech. Ageing Dev., 1984, 27, 29–41 CrossRef CAS.
- A. Sheydina, D. R. Riordon and K. R. Boheler, Clin. Sci., 2011, 121, 315–329 CrossRef CAS.
- J. H. Kim, H. E. Choy, K. H. Nam and S. C. Park, Ann. N. Y. Acad. Sci., 2001, 928, 65–70 CrossRef CAS.
- J. Zhao, R. Randive and J. A. Stewart, World J. Diabetes, 2014, 5, 860–867 CrossRef PubMed.
- B. E. Aboulhoda, Ann. Anat., 2017, 214, 21–30 CrossRef PubMed.
- D. L. Mann, Annu. Rev. Physiol., 2003, 65, 81–101 CrossRef CAS PubMed.
- B. M. Meador, C. P. Krzyszton, R. W. Johnson and K. A. Huey, J. Appl. Physiol., 2008, 104, 991–997 CrossRef CAS PubMed.
- M. Wang and A. M. Shah, J. Mol. Cell. Cardiol., 2015, 83, 101–111 CrossRef CAS PubMed.
- P. K. Mays, R. J. McAnulty and G. J. Laurent, Mech. Ageing Dev., 1991, 59, 229–241 CrossRef CAS PubMed.
- D. P. Thomas, R. J. McCormick, S. D. Zimmerman, R. K. Vadlamudi and L. E. Gosselin, Am. J. Physiol., 1992, 263, H778–H783 CAS.
- D. P. Shanley, D. Aw, N. R. Manley and D. B. Palmer, Trends Immunol., 2009, 30, 374–381 CrossRef CAS.
- M. De Martinis, C. Franceschi, D. Monti and L. Ginaldi, Exp. Mol. Pathol., 2006, 80, 219–227 CrossRef CAS PubMed.
- J. Taieb, C. Delarche, F. Ethuin, S. Selloum, T. Poynard, M. A. Gougerot-Pocidalo and S. Chollet-Martin, J. Leukocyte Biol., 2002, 72, 1142–1147 CAS.
- R. Menghini, L. Fiorentino, V. Casagrande, R. Lauro and M. Federici, Atherosclerosis, 2013, 228, 12–17 CrossRef CAS PubMed.
- M. Kveiborg, R. Instrell, C. Rowlands, M. Howell and P. J. Parker, PLoS One, 2011, 6, e17168 CrossRef CAS.
- B. Bozkurt, S. B. Kribbs, F. J. Clubb Jr, L. H. Michael, V. V. Didenko, P. J. Hornsby, Y. Seta, H. Oral, F. G. Spinale and D. L. Mann, Circulation, 1998, 97, 1382–1391 CrossRef CAS.
- T. Kubota, C. F. McTiernan, C. S. Frye, S. E. Slawson, B. H. Lemster, A. P. Koretsky, A. J. Demetris and A. M. Feldman, Circ. Res., 1997, 81, 627–635 CrossRef CAS PubMed.
- F. D. Pagani, L. S. Baker, C. Hsi, M. Knox, M. P. Fink and M. S. Visner, J. Clin. Invest., 1992, 90, 389–398 CrossRef CAS PubMed.
- T. Yokoyama, L. Vaca, R. D. Rossen, W. Durante, P. Hazarika and D. L. Mann, J. Clin. Invest., 1993, 92, 2303–2312 CrossRef CAS PubMed.
- D. W. Lambert, M. Yarski, F. J. Warner, P. Thornhill, E. T. Parkin, A. I. Smith, N. M. Hooper and A. J. Turner, J. Biol. Chem., 2005, 280, 30113–30119 CrossRef CAS.
- X. Wang, T. Oka, F. L. Chow, S. B. Cooper, J. Odenbach, G. D. Lopaschuk, Z. Kassiri and C. Fernandez-Patron, Hypertension, 2009, 54, 575–582 CrossRef CAS PubMed.
- J. L. Lucitti, J. K. Mackey, J. C. Morrison, J. J. Haigh, R. H. Adams and J. E. Faber, Circ. Res., 2012, 111, 1539–1550 CrossRef CAS PubMed.
- A. M. Manso, L. Elsherif, S. M. Kang and R. S. Ross, Cardiovasc. Res., 2006, 69, 574–584 CrossRef CAS.
- M. Sun, F. Dawood, W. H. Wen, M. Chen, I. Dixon, L. A. Kirshenbaum and P. P. Liu, Circulation, 2004, 110, 3221–3228 CrossRef CAS PubMed.
- P. Xu, J. Liu, M. Sakaki-Yumoto and R. Derynck, Sci. Signaling, 2012, 5, ra34 CrossRef PubMed.
- P. R. Murumkar, S. DasGupta, S. R. Chandani, R. Giridhar and M. R. Yadav, Expert Opin. Ther. Pat., 2010, 20, 31–57 CrossRef CAS PubMed.
- D. Fan, A. Takawale, M. Shen, W. Wang, X. Wang, R. Basu, G. Y. Oudit and Z. Kassiri, Circ.: Heart Failure, 2015, 8, 970–979 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01190k |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |