
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-doped TiO2 nanotube arrays with uniformly embedded CoxP nanoparticles for high-efficiency hydrogen evolution reaction†
Yong Weia,
Jijiang Fua,
Hao Songab,
Ben Zhanga,
Chaoran Pia,
Lu Xiaa,
Xuming Zhang *a,
Biao Gaoab,
Yang Zheng*a and
Paul K. Chub
*a,
Biao Gaoab,
Yang Zheng*a and
Paul K. Chub
aThe State Key Laboratory of Refractories and Metallurgy, Institute of Advanced Materials and Nanotechnology, Wuhan University of Science and Technology, Wuhan 430081, China. E-mail: xumzhang@wust.edu.cn; yzheng@wust.edu.cn
bDepartment of Physics, Department of Materials Science and Engineering, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, China
First published on 15th April 2019
Abstract
Efficient and stable non-precious metal based electrocatalysts are crucial to the hydrogen evolution reaction (HER) in renewable energy conversion. Herein, CoxP nanoparticles (NPs) are uniformly embedded in N-doped TiO2 nanotube arrays (CoxP/N-TiO2 NTAs) by low-temperature phosphorization of the precursor of metallic cobalt NPs embedded in N-doped TiO2 NTAs (Co/N-TiO2 NTAs) which were fabricated by phase separation of CoTiO3 NTAs in ammonia. Owing to the abundant exposed surface active sites of CoxP NPs, tight contact between the CoxP NPs and TiO2 NTAs, fast electron transfer in N-doped TiO2, and channels for effective diffusion of ions and H2 bubbles in the tubular structure, the CoxP/N-TiO2 NTAs have excellent electrocatalytic activity in HER exemplified by a low overpotential of 180 mV at 10 mA cm−2 and small Tafel slope of 51 mV dec−1 in 0.5 M H2SO4. The catalyst also shows long-term cycling stability and is a promising non-precious metal catalyst for HER.
Introduction
Because of environmental pollution and greenhouse gas emission derived from the consumption of fossil fuels, alternative green energy is attracting tremendous interest.1,2 For example, hydrogen energy is very promising due to the harmless byproducts, high energy density, abundant resource, and recyclability, and electrochemical water splitting is an important technology.3 Electrocatalysts play key roles in electrochemical water splitting which includes two half reactions: hydrogen evolution reaction (HER) on the cathode and oxygen evolution reaction (OER) on the anode.4 It is well known that platinum (Pt)-based electrocatalysts are ideal for HER because of the low overpotentials and large exchange current densities,5 but the high cost and terrestrial scarcity of Pt are hurdles for large-scale industrial application.6 Therefore, noble metal-free electrocatalysts having good activity and stability,7 such as transition metal chalcogenides,8–10 nitrides,11,12 and phosphides,13–15 are attractive alternatives.Transition metal phosphides (TMPs) are attractive non-precious metal based electrocatalysts for HER in acidic media, for instance, cobalt phosphide,16 nickel phosphide,17 and molybdenum phosphide,18 have been studied intensively and exhibited excellent HER performance,19–22 because the negatively charged P atoms in TMPs could trap positively charged protons during HER.18 However the poor conductivity of TMP-based catalysts is a crucial obstacle to limit the HER performance. Hybrid structure composed of TMP nanoparticles anchored on conductive substrate is desirable, for example, CoP nanoparticles grown on graphene oxide23 and carbon nanotubes decorated with CoP nanocrystals24 exhibit excellent HER activity in acid solution. Although the hybrid electrocatalysts shows improved HER activity,25 the random distribution of nanosize TMP nanoparticles on conductive agent without good adhesion and coupling compromised their HER activity and stability. Meanwhile, the fast electron transport pathways as well as effective channels for ions and gas bubbles diffusion are still challenge. Titanium oxide nanotube arrays (TiO2 NTAs) have been proposed as a support substrate for superior electrochemical electrodes in many field because of the unique hollow structure, effective electron transfer pathways, and excellent physical and chemical stability under a wide variety of conditions.26–32 More important, the highly ordered TiO2 NTAs prepared by anodization of Ti substrate exhibits large surface areas, and they do not require additional binders to adhere to Ti substrates. Recently, there are many literatures on the incorporation of noble metal or economic electrocatalysts on TiO2 NTAs to decrease their overpotential for water splitting, such as RuO2 doped TiO2 NT for OER application, amorphous MoSx-coated TiO2 NTAs and Ru0.33Se nanoparticles decorated TiO2 NTAs for enhanced HER activity.33–35
Herein, we describe a novel electrocatalyst, CoxP NPs uniformly embedded in N-doped TiO2 NTAs (CoxP/N-TiO2 NTAs). First, the CoTiO3 NTAs was fabricated by hydrothermal treatment of amorphous TiO2 NTAs in cobalt acetate solution.36 Second, metallic cobalt is in situ segregated and anchored in/on the wall of the N-doped TiO2 nanotubes (Co/N-TiO2 NTAs) after thermal reduction of the CoTiO3 NTAs under ammonia by phase separation. Then, the CoxP/N-TiO2 NTAs with original morphology was obtained after phosphorization of Co/N-TiO2 NTAs. In this unique structure, the N-doped TiO2 NTAs provide the fast electron transfer pathways and the original hollow channels is in favor of fast diffusion of ions and generated H2 bubbles. More important, the formed CoxP NPs are confined and tightly anchored in/on the wall of TiO2 NTAs resulting in low contact resistance and excellent structural stability. The catalyst shows a low overpotential of 180 mV at 10 mA cm−2 with a small Tafel slope of 51 mV dec−1 under acidic conditions and a long lifetime suggesting large potential in water splitting and related applications.
Experimental methods
Ti plates (purity: 99.6%) with dimensions 10 mm × 10 mm × 1 mm were used as the substrates. Cobaltous acetate (Co(Ac)2), sodium hypophosphite (NaH2PO2) were obtained from Aladdin Company and nitric acid, hydrofluoric acid and other reagents were purchased from Sinopharm Chemical Reagent Co. Ltd., China. All the chemicals were analytical reagent grade and used without further purification. Deionized water (DIW) was used throughout the experiments.Materials preparation
Synthesis of TiO2 nanotube arrays (TiO2 NTAs)
The TiO2 NTAs were prepared according to the process described in the literature.37,38 The Ti plates were ultrasonically cleaned successively with acetone, ethanol, and DIW. Electrochemical anodization was carried out using a two-electrode configuration with a graphite plate being the cathode and Ti plate being the anode. After anodization in an ethylene glycol solution containing 5 vol% DI water and 0.5 wt% ammonium fluoride (NH4F) at 60 V for 1 h using a power supply (IT6834, ITECH, China) at room temperature, the samples were washed with DIW and dried in air.Synthesis of CoTiO2 nanotube arrays (CoTiO3 NTAs)
The CoTiO3 NTAs were prepared by a hydrothermal method.36 The as-prepared amorphous TiO2 NTAs were placed vertically and immersed in 30 ml of a 0.2 M cobalt acetate (Co(Ac)2) aqueous solution in a 40 mL Teflon-lined stainless steel autoclave. The autoclave was sealed and heated in an oven to 200 °C for 6 h. Afterwards, the product was removed from the autoclave and ultrasonically washed with DIW for 5 minutes. The sample was then annealed at 450 °C for 2 h in air to improve the crystallinity of the CoTiO3 NTAs.Synthesis of Co/N-doped TiO2 nanotube arrays (Co/N-TiO2 NTAs)
The as-prepared CoTiO3 NTAs were calcined in an ammonia ambient at 600 °C (5 °C min−1) for 3 hours in a tube furnace to prepare Co NPs decorated N doped-TiO2 NTAs. After natural cooling, black Co/N-TiO2 NTAs were obtained.Synthesis of CoxP/N doped-TiO2 nanotube arrays (CoxP/N-TiO2 NTAs)
The as-prepared Co/N-TiO2NTAs were placed downstream from NaH2PO2 (0.2 g) in a furnace tube at a distance of about 20 cm and treated at different temperature for 3 h under flowing Ar. After phosphorization, the CoxP/N-TiO2 NTAs were collected after the furnace cooled down to room temperature. The samples were denoted as CoxP/N-TiO2-300, CoxP/N-TiO2-450, CoxP/N-TiO2-550, respectively.Materials characterization
The morphology and microstructure of the samples were characterized by field-emission scanning electron microscopy (FE-SEM, FEI Nova 400 Nano), transmission electron microscopy (TEM, JEOL 2010, Tokyo, Japan), X-ray diffraction (XRD, Philips X'Pert Pro) with Cu Kα radiation, X-ray photoemission spectroscopy (XPS, ESCALAB MK-II, VG Instruments, London, UK, calibrated with the C 1s peak at 284.6 eV).Electrochemical measurements
The electrochemical measurements were carried out using a three-electrode system in 0.5 M H2SO4 with carbon as the counter electrode, saturated calomel electrode (SCE) as the reference electrode, and the sample with an exposed surface area of 1 cm2 as the working electrode (CHI 760e, Shanghai CHI Company, China). The HER activity was evaluated by linear sweep voltammetry (LSV) at a scanning rate of 5 mV s−1 and the potentials were referenced to the reversible hydrogen electrode (RHE) according ERHE = ESCE + 0.2415 + 0.0591 × pH. The electrochemical impedance spectra (EIS) were acquired at an overpotential of 100 mV in the frequency range between 100 kHz and 0.01 Hz with an AC perturbation of 5 mV. The stability of the electrocatalyst was assessed by cyclic voltammetry (CV) at a scanning rate of 200 mV s−1. All the potentials in the polarization curves and Tafel plots were iR corrected using Rs from EIS.Results and discussion
Fig. 1a schematically illustrates the synthesis of the CoxP/N-TiO2 NTAs. Uniform TiO2 NTAs are fabricated on the Ti plates by electrochemical anodization and the CoTiO3 NTAs are prepared by a simple hydrothermal treatment of the as-prepared amorphous TiO2 NTAs and Co(Ac)2. The Co/N-TiO2 NTAs are produced by thermal reduction of CoTiO3 NTAs under ammonia and the CoxP/N-TiO2 NTAs are obtained after phosphorization. Fig. 1b–e display the morphology and microstructure of the TiO2 NTAs after different steps. After anodic oxidation, the ordered TiO2 NTAs with a uniform diameter of 90 nm are vertically aligned on the Ti plate. After the hydrothermal treatment in 0.2 M Co(Ac)2 at 200 °C for 6 h, Fig. 1c shows that the diameter of CoTiO3 NTAs decreases because of the thick wall of the NTAs. It is due to self-crystallization of amorphous TiO2 to titanate in the presence of corresponding salts as previously reported.39 After the thermal treatment at 600 °C in NH3 (Fig. 1d), NPs with a diameter of about 80 nm are formed uniformly on the surface of the NTAs by the phase separation effect.40–42 Subsequently, the morphology is maintained after phosphorization, as shown in Fig. 1e.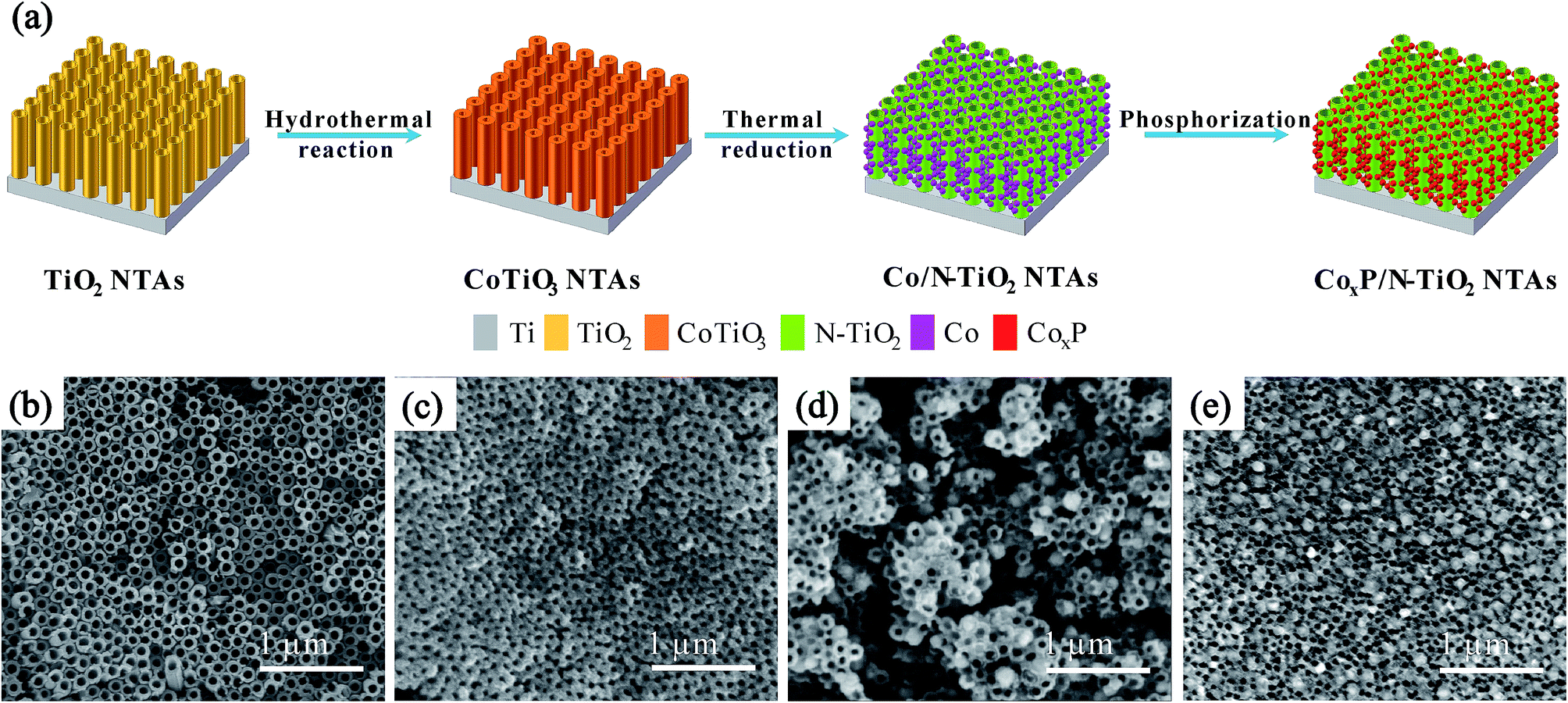 | ||
| Fig. 1 (a) Illustration of the synthesis of CoxP/N-TiO2. Corresponding SEM images: (b) TiO2 NTAs, (c) CoTiO3 NTAs, (d) Co/N-TiO2 NTAs, and (e) CoxP/N-TiO2 NTAs. | ||
The structural evolution of the products is monitored by XRD as in Fig. 2. The XRD pattern of as-anodized TiO2 NTAs shows a series of peaks (green line) at 38.4°, 40.2°, 53.0°, 62.9°, 70.6°, 76.1°, and 82.2°assigned to the substrate Ti (JCPDS no. 89-2762) reflecting the amorphous structure of the as-anodized TiO2 NTAs (Fig. 2a).40 However, there is no peak from as-hydrothermal cobalt titanate can be observed in Fig. 2b suggesting the amorphous nature and the obvious phase assigned to CoTiO3 (JCPDS no. 77-1373) can be formed at a high annealing temperature (Fig. S1†). After nitridation, several new XRD peaks at 44.2°, 51.5°, 75.8° corresponding to the (111), (200), and (220) planes of metallic Co (JCPDS no. 89-7093) are observed, as shown Fig. 2c (purple line), confirming segregation of metallic Co from the hydrothermal product. After phosphorization (Fig. 2d), the XRD pattern shows characteristic peaks of cobalt phosphide (CoP, JCPDS no. 89-2747 and Co2P, JCPDS no. 89-3030) in addition to the TiO2 and Ti substrate indicating that metallic Co is converted to cobalt phosphide.43
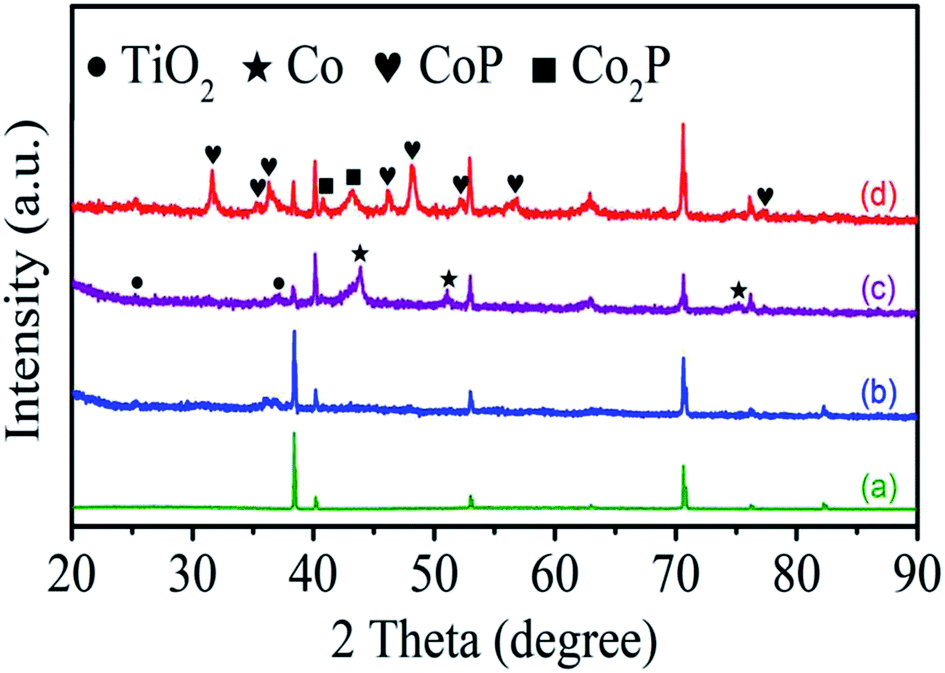 | ||
| Fig. 2 XRD patterns of (a) TiO2 NTAs, (b) CoTiO3 NTAs, (c) Co/N-TiO2 NTAs, and (d) CoxP/N-TiO2 NTAs. | ||
Fig. 3 presents the microstructure and morphology of CoxP/N-TiO2-300 examined by SEM and TEM. The cross-sectional SEM image of CoxP/N-TiO2-300 in Fig. 3a discloses a large amount of CoxP NPs with a large exposed surface and uniformly embedded in the NTAs. The tubular channels allow for fast diffusion of ions and gas bubbles in a stable structure benefiting the electrochemical activity and durability during HER. Fig. 3b depicts the cross-sectional TEM image of CoxP/N-TiO2-300 clearly showing the tubular structure after phosphorization. Fig. 3c shows a well assembled and porous structure and the high-resolution TEM image (Fig. 3d) reveals lattice spacings of 0.351 nm, 0.206 nm, and 0.171 nm corresponding to the (101) plane of anatase TiO2, (211) plane of Co2P, and (103) plane of CoP, respectively. Fig. 3e shows the energy-dispersive X-ray spectroscopy (EDS) maps of Ti, O, N, Co, and P disclosing that Ti, N and O are uniformly distributed through the entire NTAs benefiting the conductivity and electrochemical properties.40 In contrast, Co and P are scattered at the same positions demonstrating that the CoxP NPs are embedded in the N-doped TiO2 NTAs and there is no doped P in the NTAs after phosphorization.
 | ||
| Fig. 3 (a) Cross-sectional SEM images of CoxP/N-TiO2-300; (b) and (c) low-resolution TEM images of CoxP/N-TiO2-300; (d) high-resolution TEM image of CoxP/N-TiO2-300; (e) EDS maps. | ||
X-ray photoelectron spectroscopy (XPS) is conducted to characterize the chemical state of the phosphatized sample and shown in Fig. 4. The survey spectrum shows Co, P, Ti, N, and O (Fig. 4a) consistent with EDS mapping. A fitted weak peaks at 458.8 and 464.8 eV in the Ti 2p spectrum (Fig. 4b) and that at 398.3 eV in the N 1s spectrum (Fig. 4c) are related to N–Ti–O confirming formation of N-doped TiO2.40,44,45 The high-resolution Co 2p spectrum shows two pairs of double peaks and two satellite peaks (Fig. 4e). The peaks at 777.8 and 792.7 eV can be assigned to Co in cobalt phosphide46 and those at 781.4 eV and 797.3 eV are associated with Co2+. The peaks at 785.5 eV and 802.4 eV are satellite peaks.46 The P 2p spectrum can be deconvolved into three peaks (Fig. 4f). The ones at 129.1 eV and 129.9 eV are low valence P in CoxP NPs and that at 133.5 eV can be attributed to PO43− or P2O5 because of oxidation.47 The XPS results confirm that the hybrid composite composed of CoxP and N-doped TiO2, in consist with the XRD and TEM analysis.
 | ||
| Fig. 4 (a) The survey spectra of CoxP/N-TiO2 NTAs-300 and the fine XPS spectra of (b) Ti 2p, (c) N 1s, (d) O 1s, (e) Co 2p and (f) P 2p acquired from the CoxP/N-TiO2 NTAs-300. | ||
The electrocatalytic activity of CoxP/N-TiO2 NTAs in HER is evaluated using a three-electrode system in 0.5 M H2SO4. Fig. 5a shows the polarization curves after removing the effect of solution resistance by iR compensation (Fig. S2†). As expected, the Pt sheet exhibits excellent HER activity with a very low onset overpotential of −34 mV for 1 mA cm−2 and only needs −59 mV to reach the benchmark current density of 10 mA cm−2. The bare Ti plate has no electrocatalytic activity. After nitridation, the Co/N-TiO2 NTAs show poor electrocatalytic properties with a current density of 10 mA cm−2 at a large overpotential of −380 mV. After phosphorization at 300 °C for 3 h, the optimal HER activity is achieved. A small onset potential of −123 mV for 1 mA cm−2 is observed and only −180 mV is needed to reach the current density of 10 mA cm−2. By further increasing the phosphorization temperature, the HER activity of the CoxP/N-TiO2 NTAs decline dramatically due to insufficient phosphorization as a result of quick decomposition of NaH2PO2 as well as fast segregation of Co NPs on the surface blocking the mouth of the NTs (Fig. S3 and S4†). Fig. 5b presents the Tafel slopes derived from the polarization curves by fitting the Tafel equation η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj + a, where j is the current density and b is the Tafel slope, in different overpotential ranges. The Pt sheet shows a low Tafel slope of 30 mV dec−1 close to that reported previously.48 In comparison, the Tafel slopes of the CoxP/N-TiO2 NTAs are 51, 57, and 70 mV dec−1 when phosphorization is carried out at 300 °C, 450 °C, and 550 °C, respectively. CoxP/N-TiO2 NTAs-300 has better HER properties than other transition metal phosphides based electrocatalysts, as shown in Fig. S5.† Electrochemical impedance spectroscopy (EIS) is employed to determine the charge transfer resistance as shown in Fig. 5c. The Nyquist plots are fitted with an equivalent circuit (inset in Fig. 5c), where the Rs is the solution resistance of the electrochemical system, Rct is the charge transfer resistance, and CPE is the constant phase element. CoxP/N-TiO2 NTAs-300 has a lowest resistance of 8 Ω implying efficient electron transport. The stability which is also a critical factor in HER catalyst is evaluated by continuous CV cycling at 200 mV s−1. After 3000 cycles (Fig. 5d), the polarization curve exhibits negligible degradation, demonstrating superior stability under acidic conditions. The excellent HER activity of CoxP/N-TiO2 NTAs-300 with uniformly dispersed CoxP NPs stems from the unique nanostructure which provides not only fast charge transfer pathways and low contact resistance between the catalyst and electrode, but also effective hollow channels for diffusion of ions and H2 bubbles.
logj + a, where j is the current density and b is the Tafel slope, in different overpotential ranges. The Pt sheet shows a low Tafel slope of 30 mV dec−1 close to that reported previously.48 In comparison, the Tafel slopes of the CoxP/N-TiO2 NTAs are 51, 57, and 70 mV dec−1 when phosphorization is carried out at 300 °C, 450 °C, and 550 °C, respectively. CoxP/N-TiO2 NTAs-300 has better HER properties than other transition metal phosphides based electrocatalysts, as shown in Fig. S5.† Electrochemical impedance spectroscopy (EIS) is employed to determine the charge transfer resistance as shown in Fig. 5c. The Nyquist plots are fitted with an equivalent circuit (inset in Fig. 5c), where the Rs is the solution resistance of the electrochemical system, Rct is the charge transfer resistance, and CPE is the constant phase element. CoxP/N-TiO2 NTAs-300 has a lowest resistance of 8 Ω implying efficient electron transport. The stability which is also a critical factor in HER catalyst is evaluated by continuous CV cycling at 200 mV s−1. After 3000 cycles (Fig. 5d), the polarization curve exhibits negligible degradation, demonstrating superior stability under acidic conditions. The excellent HER activity of CoxP/N-TiO2 NTAs-300 with uniformly dispersed CoxP NPs stems from the unique nanostructure which provides not only fast charge transfer pathways and low contact resistance between the catalyst and electrode, but also effective hollow channels for diffusion of ions and H2 bubbles.
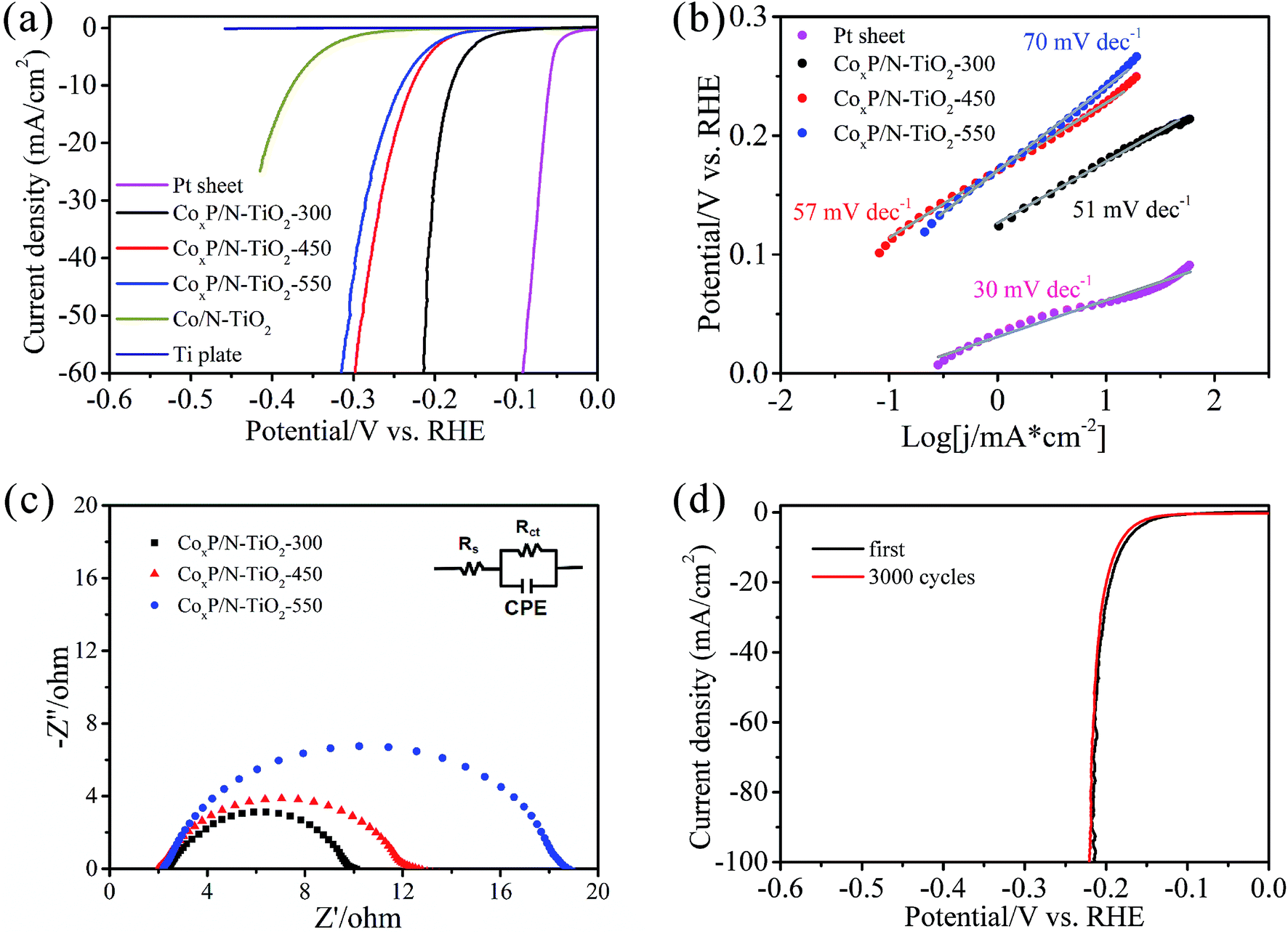 | ||
| Fig. 5 (a) HER polarization curves acquired in 0.5 M H2SO4 at 5 mV s−1 after iR calibration; (b) Tafel plots; (c) Nyquist plots together with the electrical equivalent circuit; (d) LSV curves of CoxP/N-TiO2-300 before and after 3000 cycles. | ||
Conclusion
CoxP NPs are uniformly embedded in N-doped TiO2 NTAs by nitridation and phosphorization of CoTiO3 NTAs. CoxP/N-TiO2 NTAs-300 delivers excellent electrocatalytic performance in HER in 0.5 M H2SO4, for instance, a low overpotential of 180 mV at a current density of 10 mA cm−2, small Tafel slope of 51 mV dec−1, and long-term durability. The CoxP NPs confined in the N-doped TiO2 NTs provide high structural stability and low contact resistance between the catalyst and electrode. The N-doped TiO2 NTs provide fast electron transfer pathways as well as channels for effective diffusion of ions and H2 bubbles. The novel electrocatalyst has excellent HER activity and our results reveal a different approach to design and synthesize non-noble-metal electrocatalysts.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 31500783, 51572100, 51504171 and 61434001), Major Project of Technology Innovation of Hubei Province (2018AAA011), HUST Key Interdisciplinary Team Project (2016JCTD101), Fundamental Research Funds for the Central Universities (HUST: 2015QN071), Wuhan Yellow Crane Talents Program, and City University of Hong Kong Strategic Research Grant (SRG) No. 7005105, and Hong Kong Research Grants Council (RGC) General Research Funds (GRF) No. CityU 11205617.References
- L. Chen, J. Zang, X. Liu, Y. Zhang, S. Jia, P. Tian and Y. Wang, Electrochim. Acta, 2018, 281, 386–393 CrossRef CAS.
- L. Lin, W. Lei, S. Zhang, Y. Liu, G. G. Wallace and J. Chen, Energy Storage Materials, 2019 DOI:10.1016/j.ensm.2019.02.023.
- S. Deng, Y. Zhong, Y. Zeng, Y. Wang, X. Wang, X. Lu, X. Xia and J. Tu, Adv. Sci., 2017, 5, 1700772 CrossRef PubMed.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, L. Zou and Z. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- M. Kuang, Q. Wang, P. Han and G. Zheng, Adv. Energy Mater., 2017, 7, 1700193 CrossRef.
- F. H. Saadi, A. I. Carim, E. Verlage, J. C. Hemminger, N. S. Lewis and M. P. Soriaga, J. Phys. Chem. C, 2015, 118, 29294–29300 CrossRef.
- Z. Zhou, N. Mahmood, Y. Zhang, L. Pan, L. Wang, X. Zhang and J. Zou, J. Energy Chem., 2017, 26, 1223–1230 CrossRef.
- D. Wang, Z. Wang, C. Wang, P. Zhou, Z. Wu and Z. Liu, Electrochem. Commun., 2013, 34, 219–222 CrossRef.
- F. Li, L. Zhang, J. Li, X. Lin, X. Li, Y. Fang, J. Huang, W. Li, M. Tian and J. Jin, J. Power Sources, 2015, 292, 15–22 CrossRef CAS.
- N. Kornienko, J. Resasco, N. Becknell, C. Jiang, Y. Liu, K. Nie, X. Sun, J. Guo, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2015, 137, 7448–7455 CrossRef CAS PubMed.
- J. Xie, S. Li, X. Zhang, J. Zhang, R. Wang, H. Zhang, B. Pan and Y. Xie, Chem. Sci., 2014, 5, 4615–4620 RSC.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 54, 14710–14714 CrossRef PubMed.
- Z. Pu, I. S. Amiinu, X. Liu, M. Wang and S. Mu, Nanoscale, 2016, 8, 17256–17261 RSC.
- M. Zhuang, X. Ou, Y. Dou, L. Zhang, Q. Zhang, R. Wu, Y. Ding, M. Shao and Z. Luo, Nano Lett., 2016, 16, 4691–4698 CrossRef CAS PubMed.
- Z. Xing, Q. Liu, A. M. Asiri and X. Sun, ACS Catal., 2015, 5, 145–149 CrossRef CAS.
- J. F. Callejas, C. G. Read, E. J. Popczun, J. M. Mcenaney and R. E. Schaak, Chem. Mater., 2015, 27, 3769–3774 CrossRef CAS.
- Y. Pan, Y. Liu, J. Zhao, K. Yang, J. Liang, D. Liu, W. Hu, D. Liu, Y. Liu and C. Liu, J. Mater. Chem. A, 2014, 3, 1656–1665 RSC.
- C. Huang, C. Pi, X. Zhang, K. Ding, P. Qin, J. Fu, X. Peng, B. Gao, P. K. Chu and K. Huo, Small, 2018, 14, 1800667 CrossRef PubMed.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1500985 CrossRef.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- Z. Cai, Y. Shi, S. Bao, Y. Shen, X. Xia and L. Zheng, ACS Catal., 2018, 8, 3895–3902 CrossRef CAS.
- L. Jiao, Y. Zhou and H. Jiang, Chem. Sci., 2016, 7, 1690–1695 RSC.
- Q. Liu, J. Tian, W. Cui, P. Jiang, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2015, 45, 6710–6714 Search PubMed.
- Z. Pu, Q. Liu, P. Jiang, A. M. Asiri, A. Y. Obaid and X. Sun, Chem. Mater., 2014, 26, 4326–4329 CrossRef CAS.
- R. Vadakkekara, R. Illathvalappil and S. Kurungot, ChemElectroChem, 2018, 5, 4000–4007 CrossRef CAS.
- M. M. Momeni, Y. Ghayeb and F. Ezati, Surf. Eng., 2018, 1–7 CrossRef.
- D. E. Schipper, Z. Zhao, A. P. Leitner, L. Xie, F. Qin, M. K. Alam, S. Chen, D. Wang, Z. Ren, Z. Wang, J. Bao and K. H. Whitmire, ACS Nano, 2017, 11, 4051–4059 CrossRef CAS PubMed.
- W. Wang, J. Dong, X. Ye, Y. Li, Y. Ma and L. Qi, Small, 2016, 12, 1469–1478 CrossRef CAS PubMed.
- P. A. Gross, N. Javahiraly, N. G. Sabat, T. Cottineau, E. R. Savinova and V. Keller, Appl. Phys. Lett., 2016, 109, 153903 CrossRef.
- M. Kan, X. Qian, T. Zhang, D. Yue and Y. Zhao, ACS Sustainable Chem. Eng., 2017, 5, 10940–10946 CrossRef CAS.
- W. Chen, T. Wang, J. Xue, S. Li, Z. Wang and S. Sun, Small, 2017, 13, 1602420 CrossRef PubMed.
- Z. Liu, X. Zhang, B. Wang, M. Xia, S. Gao, X. Liu, A. Zavabeti, J. Z. Ou, K. K. Zadeh and Y. Wang, J. Phys. Chem. C, 2018, 122, 12589–12597 CrossRef CAS.
- K. Wang, Q. Chen, Y. Hu, W. Wei, S. Wang, Q. Shen and P. Qu, Small, 2018, 14, 1802132 CrossRef PubMed.
- H. Yoo, K. Oh, Y. R. Lee, K. H. Row, G. Lee and J. Choi, Int. J. Hydrogen Energy, 2017, 42, 6657–6664 CrossRef CAS.
- X. Zhang, B. Gao, L. Hu, L. Li, W. Jin, K. Huo and P. K. Chu, CrystEngComm, 2014, 16, 10280–10285 RSC.
- X. Zhang, K. Huo, L. Hu, Z. Wu and P. K. Chu, J. Am. Ceram. Soc., 2010, 93, 2771–2778 CrossRef CAS.
- X. Peng, J. Fu, X. Zhang, Y. Li, M. Huang, K. Huo and P. K. Chu, Nanosci. Nanotechnol. Lett., 2013, 5, 1251–1257 CrossRef CAS.
- K. Huo, H. Wang, X. Zhang, Y. Cao and P. K. Chu, ChemPlusChem, 2012, 77, 323–329 CrossRef CAS.
- J. Xu, N. Xu, X. Zhang, B. Gao, B. Zhang, X. Peng, J. Fu, P. K. Chu and K. Huo, J. Mater. Chem. B, 2017, 5, 1779–1786 RSC.
- J. Xu, N. Xu, X. Zhang, P. Xu, B. Gao, X. Peng, S. Mooni, Y. Li, J. Fu and K. Huo, Sens. Actuators, B, 2017, 244, 38–46 CrossRef CAS.
- X. Peng, L. Wang, L. Hu, Y. Li, B. Gao, H. Song, C. Huang, X. Zhang, J. Fu, K. Huo and P. K. Chu, Nano Energy, 2017, 34, 1–7 CrossRef CAS.
- Z. H. Xue, H. Su, Q.-Y. Yu, B. Zhang, H.-H. Wang, X.-H. Li and J.-S. Chen, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- M. Sathish, B. Viswanathan, R. P. Viswanath and C. S. Gopinath, Chem. Mater., 2015, 17, 6349–6353 CrossRef.
- T. Yu, X. Tan, L. Zhao, Y. Yin, P. Chen and J. Wei, Chem. Eng. J., 2010, 157, 86–92 CrossRef CAS.
- L. Tian, X. Yan, X. Chen, L. Liu and X. Chen, J. Mater. Chem. A, 2016, 4, 13011–13016 RSC.
- H. Song, M. Dai, H. Song, X. Wan, X. Xu, C. Zhang and H. Wang, Catal. Commun., 2014, 43, 151–154 CrossRef CAS.
- X. Yang, A. Lu, Y. Zhu, M. N. Hedhili, S. Min, K. Huang, Y. Han and L. Li, Nano Energy, 2015, 15, 634–641 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01184f |
| This journal is © The Royal Society of Chemistry 2019 |