
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and biological activities of petrosiols B and D†
Jialin Gengab,
Qidong Renab,
Caizhu Changac,
Xinni Xiea,
Jun Liu *ab and
Yuguo Du*abd
*ab and
Yuguo Du*abd
aState Key Laboratory of Environmental Chemistry and Eco-Toxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Science, Beijing 100085, China. E-mail: junliu@rcees.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Chemistry and Environmental Engineering, Wuhan Institute of Technology, Wuhan 430205, China
dNational Engineering Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, China
First published on 1st April 2019
Abstract
A divergent total synthesis of natural diacetylenic tetraols, petrosiol B and petrosiol D, was accomplished by taking advantage of a carbohydrate chiral template. In particular, petrosiol B, which is the first total synthesis so far, was achieved in 13 linear steps with a 10% overall yield applying Ohira–Bestmann homologation, NaH-mediated dehydrobromination, and Cu(I)-catalyzed Cadiot–Chodkiewicz coupling as the key reaction steps. The synthetic petrosiols B and D were subjected to the study on differentiation activities toward neuronal progenitor PC12 cells. Our results suggested that both petrosiol B and petrosiol D could induce the differentiation of neuronal progenitor PC12 cells via the enhancement of Nrf2 activity. By comparing petrosiols B, D and their natural homologue E, petrosiol B displayed the most intensive cell differentiation activity and the highest Nrf2 activity enhancement as well.
Introduction
Polyacetylene compounds are a class of natural product with two or more conjugated acetylenic and olefinic bonds. Not surprisingly, marine organisms have become increasingly important and thoroughly investigated sources of polyacetylenes over the past few years.1 It is believed that these polyacetylenes are biosynthetically derived from their fatty acid progenitors via desaturase-promoted dehydrogenation, leading to double bonds at specific sites of the carbon chain in normally high Z-selectivity.2 Further dehydrogenation of some double bonds in the presence of acetylenases generates alkynes which play crucial roles in their biological activities.1a Results from human nutrition research reveals that some of the polyacetylene compounds exhibit health chemopreventive effects, while some of their structural analogues are toxins with strong neurotoxicity.3,4 Due to their unique structural characteristics and impressive pharmacological properties such as anti-inflammatory, antimicrobial, antitumor, antiviral and neuritogenic activities, polyacetylenes have attracted significant interest in terms of the isolation, structure determination, bioactivity screening, and synthetic attempt.1 Recent examples of this polyacetylene family include debilisones A–F,5 strongylodiols A–I,6 ivorenolide A–B,7 and atractylodemaynes A–G.8Petrosiols A–E (1-5) (Fig. 1) are polyacetylene-containing natural products isolated from the Okinawan marine sponge Petrosia strongylata by Ojika's group in 2013.9 The structure and absolute stereochemistry of the petrosiols was determined by derivatization of petrosiol C and a modified Mosher's method as well, sharing a common structural feature of unusual triyne–triyne inserted tetraol skeleton with different side chain residues.
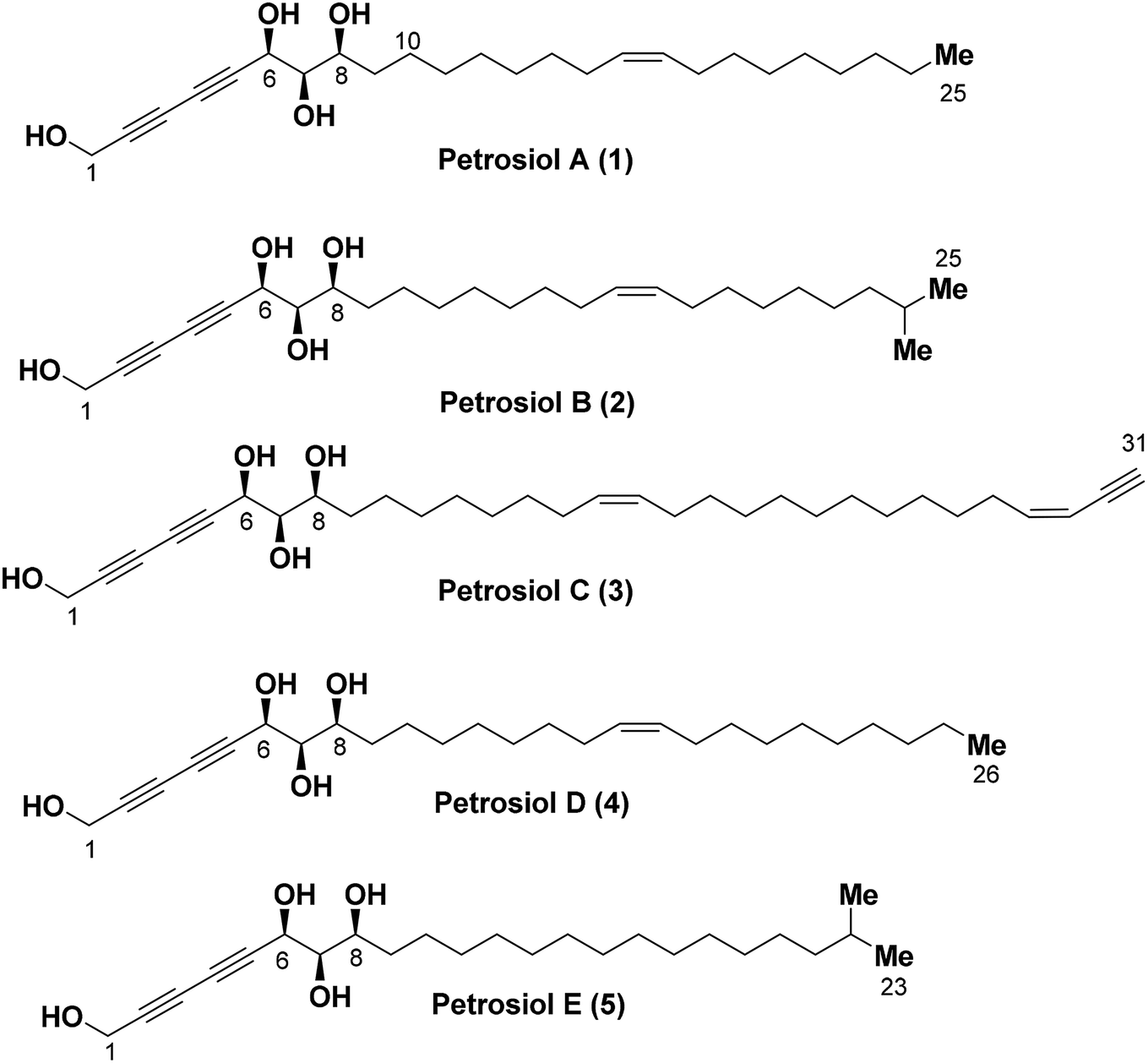 | ||
| Fig. 1 Structures of petrosiols A–E (1–5). | ||
Because of their novel structure feature, impressive broad-band biological activities, and most importantly the limited availability, petrosiols have attracted the attention toward the total synthesis. Srihari and co-workers reported the first total synthesis of petrosiol D using (+)-diethyl L-tartrate as starting material and Cadiot–Chodkiewicz coupling as the key step in 2013.10 The same group also reported the total synthesis of petrosiols A and E using a similar strategy in 2016.11 As part of our interests in the total synthesis of various complex natural products based on carbohydrate skeletons, we have reported the first total synthesis of petrosiol E from D-xylose in 2014.12 So far, there is no report regarding the syntheses of petrosiols B and C.
With the synthetic petrosiol E in hand, we studied its molecular mechanism in stimulating neuronal differentiation and antioxidative stress in PC12 neuronal progenitor cells.13 We found that petrosiol E could stimulate the mitogen-activated protein kinase and serine/tyrosine kinase signaling to enhance the activity of Nrf2. We also identified a dual role of petrosiol E in potentiating the differentiation of neuronal progenitors and in protecting them against arsenic-induced oxidative stress in PC12 cells. The impressive biological activity and the lack of structure–activity relationship (SAR) studies on petrosiols encouraged us to undertake a flexible and scalable synthesis of these novel natural products. Herein, we disclose the first total synthesis of petrosiol B and total synthesis of petrosiol D using readily available carbohydrate D-mannose as the chiral pool.
Results and discussion
Based on the results from our previous biological study,13 petrosiols could induce nerve growth factor (NGF)-like neuronal differentiation of PC12 cells in a dose-dependent manner. It thus could provide a potential agent for the prevention and treatment of neurodegenerative diseases such as Parkinson's disease (PD) and Alzheimer disease (AD). In this regard, it is highly demanding a method to reach the common crucial tetraol skeleton of petrosiols in an efficient and easy scaling up way, although we have realized that the structure sensitivity toward oxidation and light makes them challenging synthetic target molecules.Our retrosynthetic analysis for the total syntheses of petrosiols B and D is presented in Scheme 1.
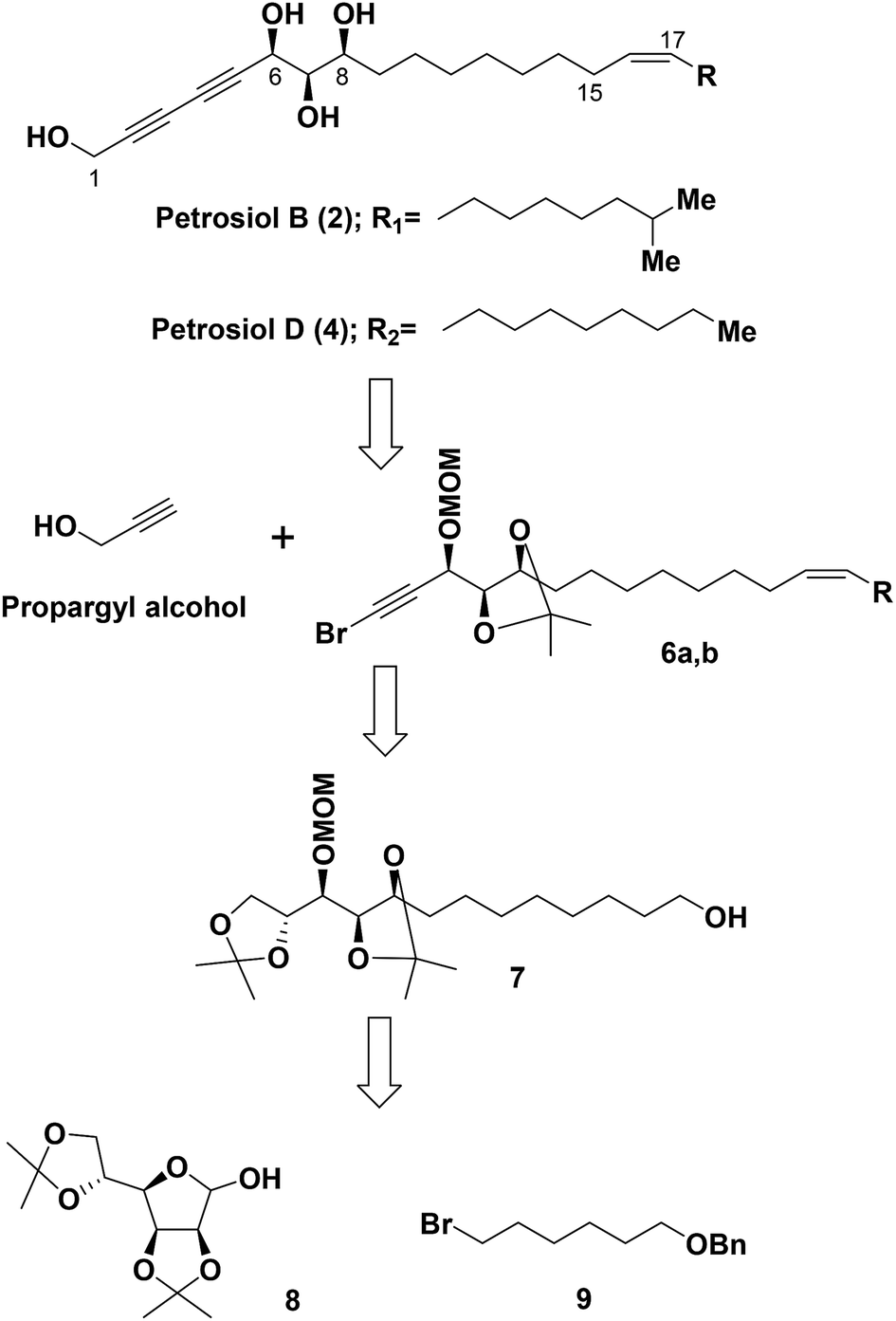 | ||
| Scheme 1 Retrosynthetic analysis of petrosiols B and D. | ||
Accordingly, a divergent approach was designed taking advantage of the same triyne–triyne tetraol framework (C1–C17) which can be accessed by coupling of the precursor bromo-alkyne 6 with propargyl alcohol via Cadiot–Chodkiewicz reaction for both petrosiol B and petrosiol D. The requisite precursor 6 could be generated from the primary alcohol 7 followed by functional group transformation via sequential alkylation of the terminal alkyne, Lindlar catalyst promoted cis-reduction, acetonide cleavage, and Corey–Fuchs reaction. As for the key intermediate 7, it could be obtained by coupling of bromide 9 and hemiacetal 8 via a multi-step sequential modification. Details of the studies thus undertaken are described below.
The synthetic sequence commenced with the formation of the terminal alkyne 10 from commercially available 2,3:5,6-di-O-isopropylidene-α-D-mannofuranose (8) as the starting material following a modified procedure from Lievre's report (Scheme 2).14 We thought that a total synthesis of the target petrosiols from a chiral carbohydrate material with a reliably assigned absolute configuration might substantiate the reported structural assignment. Thus, the masked carbonyl group of hemiacetal 8 was converted into the corresponding terminal alkyne by addition of Ohira–Bestmann reagent and K2CO3 in refluxing methanol.15,16 The desired epimerization at the propargylic stereocenter (C-5) occurred smoothly and confirmed by an up-field shifting of H5 signal (4.66 ppm, J = 7.6, 2.0 Hz).14 The full inversion of stereocenter could be attributed to the greater thermodynamic stability of the trans over the cis C4–C5 acetonide structure.16
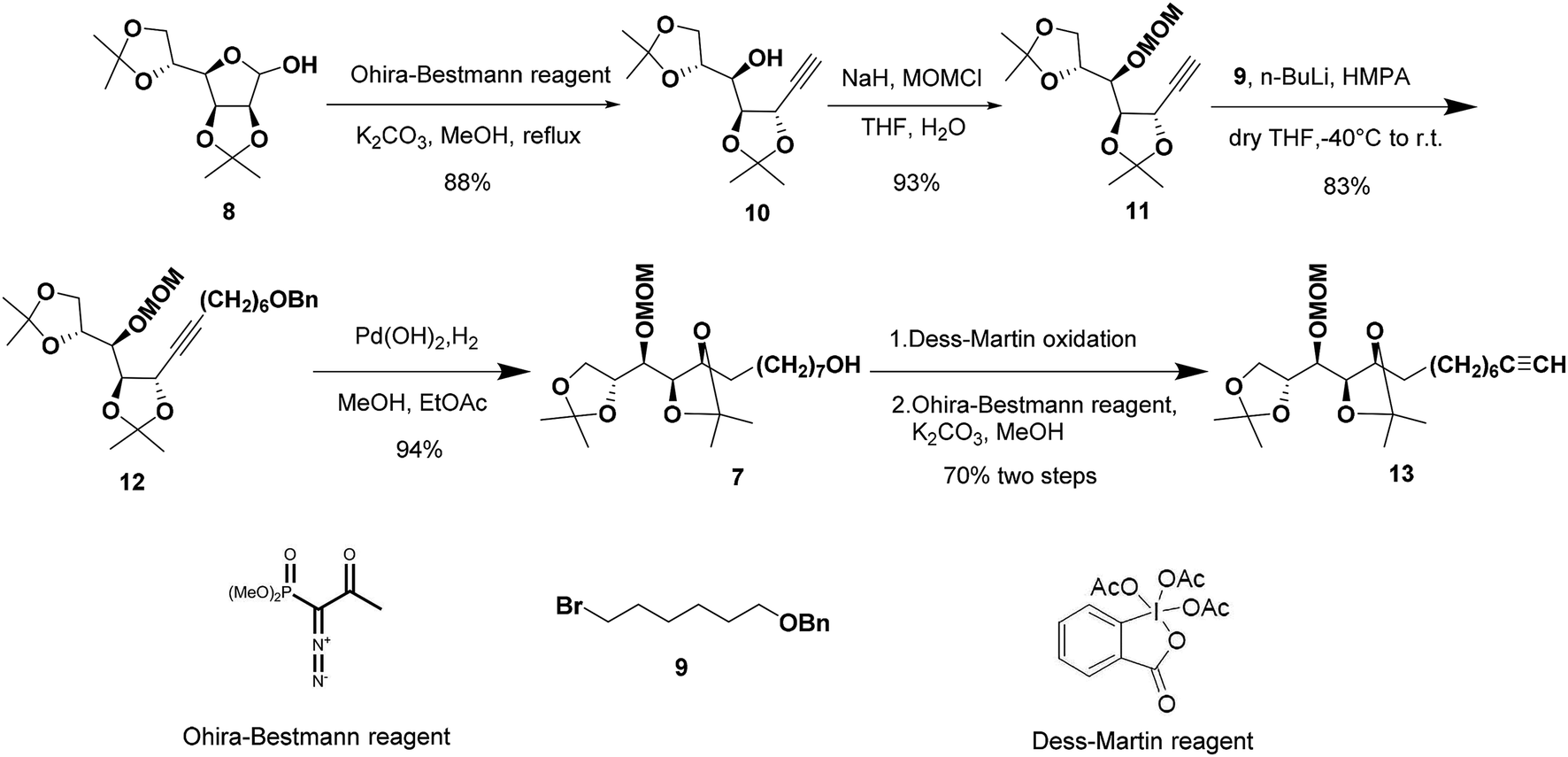 | ||
| Scheme 2 Synthesis of the C5–C16 fragment. | ||
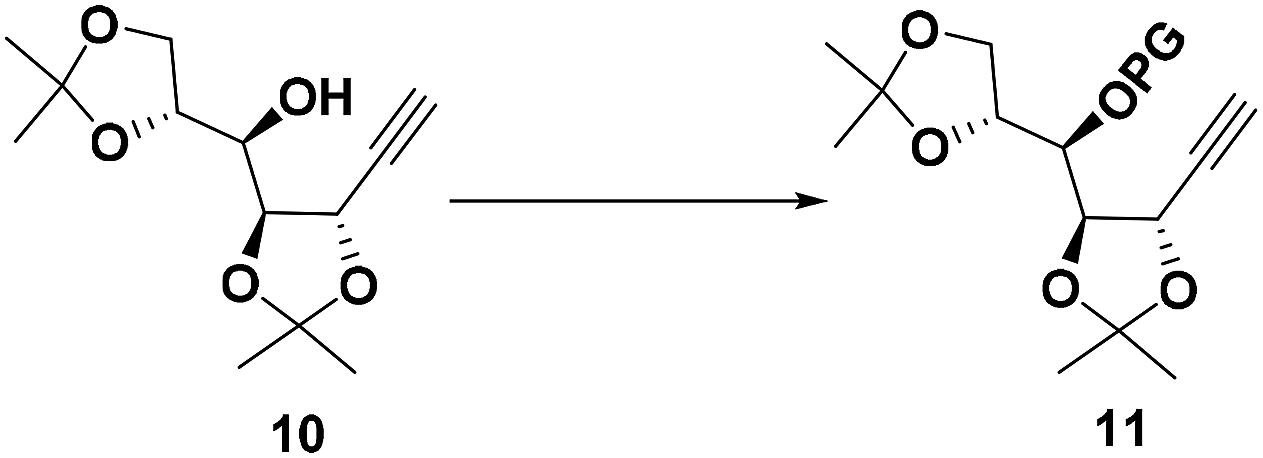
To differentiate the secondary alcohol of 10 from its latent primary alkyl hydroxyl group introduced later, alkyne 10 was exposed to excess MOMCl in dichloromethane at rt for 48 h with DIPEA as the base. However, the MOM-protected product 11 was isolated in only 10% yield together with 71% of recovered 10. Attempts to improve the yield of this protection by changing solvents or elevating reaction temperature were fruitless (Table 1, entries 1–4). Other attempts to protect the secondary alcohol using TBDPS as protection group also failed (entries 5–6). Using stronger base such as DMAP or NaH did not improve the reaction yield at all. To our surprise, the desired MOM-protected product 11 was eventually obtained in 93% yield in the presence of NaH in THF containing two drops of water at room temperature (entry 8). After reviewing the literature, we recognize that catalytic amounts of water and NaH produce highly active NaOH, which is the real base to promote the reaction.17 We assumed that the exothermic process when adding water into the reaction mixture also facilitated the reaction in some extent.18
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | Solvent | T (°C) | Time (h) | Yield (%) |
| 1 | MOMCl (2 eq.), DIPEA (3 eq.) | DCM | 20 | 48 | <10 |
| 2 | MOMCl (5 eq.), DIPEA (3 eq.) | DCM | Reflux | 48 | 18 |
| 3 | MOMCl (2 eq.), DIPEA (3 eq.), DMAP (cat) | THF | 20 | 48 | <10 |
| 4 | MOMCl (2 eq.), DIPEA (3 eq.), DMAP (cat) | DCM | Reflux | 48 | 20 |
| 5 | TBDPSCl (1.5 eq.), imidazole (4 eq.) | DMF | 20 | 48 | <5 |
| 6 | TBDPSCl (3 eq.), imidazole (4 eq.) | DMF | 80 | 48 | <5 |
| 7 | MOMCl (3 eq.), NaH (5 eq.) | THF | 20 | 7 | <5 |
| 8 | MOMCl (3 eq.), NaH (5 eq.), with few drops of water | THF | 20 | 5 | 93 |
Terminal alkyne 11 was then alkylated with bromide 9 in the presence of HMPA leading to the desired alkyne 12 in a yield of 83%. Hydrogenation of 12 in methanol/ethyl acetate removed benzyl group and saturated the triple bond at the same time gave 7 in excellent yield. The primary alcohol 7 was oxidized to the corresponding aldehyde by Dess–Martin oxidation19 and the crude aldehyde was used directly for the Ohira–Bestmann homologation20 afforded 13 in 70% yield over two steps.
Alkylation of the terminal alkyne 13 with iodoalkanes afforded compounds 14a,b, corresponding to petrosiols B and D respectively, in good yields (Scheme 3). Partial hydrogenation of 14a,b with Lindlar catalyst (5% Pd on CaCO3 poisoned with lead) under H2 atmosphere afforded the cis-olefins 15a,b with excellent Z/E selectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Regioselective oxidative cleavage of the terminal isopropylidene group in 15a,b was performed with periodic acid in dry ethyl acetate affording one-carbon diminished aldehyde,20 which was immediately treated with CBr4/PPh3 in DCM21,22 to generate 1,L-dibromoalkenes 16a,b in about 51–52% yield. NaH-mediated dehydrobromination12,23 of dibromoalkenes in wet THF at room temperature provided monobromoalkynes 6a,b in excellent yield. Cu(I)-catalyzed Cadiot–Chodkiewicz cross coupling of propargyl alcohol with monobromoalkynes 6a,b afforded the corresponding diynes 17a,b.24 Finally, one-pot global deprotection of 17a,b were achieved with 3 N HCl in refluxing ethanol, delivering petrosiol B (2) in 97% yield and petrosiol D (4) in 98% yield, respectively. The spectroscopic data (1H, 13C NMR, and HRMS) and specific rotation data of both synthetic petrosiol B and petrosiol D were in good agreement with those of the natural products (see the ESI†).
:1). Regioselective oxidative cleavage of the terminal isopropylidene group in 15a,b was performed with periodic acid in dry ethyl acetate affording one-carbon diminished aldehyde,20 which was immediately treated with CBr4/PPh3 in DCM21,22 to generate 1,L-dibromoalkenes 16a,b in about 51–52% yield. NaH-mediated dehydrobromination12,23 of dibromoalkenes in wet THF at room temperature provided monobromoalkynes 6a,b in excellent yield. Cu(I)-catalyzed Cadiot–Chodkiewicz cross coupling of propargyl alcohol with monobromoalkynes 6a,b afforded the corresponding diynes 17a,b.24 Finally, one-pot global deprotection of 17a,b were achieved with 3 N HCl in refluxing ethanol, delivering petrosiol B (2) in 97% yield and petrosiol D (4) in 98% yield, respectively. The spectroscopic data (1H, 13C NMR, and HRMS) and specific rotation data of both synthetic petrosiol B and petrosiol D were in good agreement with those of the natural products (see the ESI†).
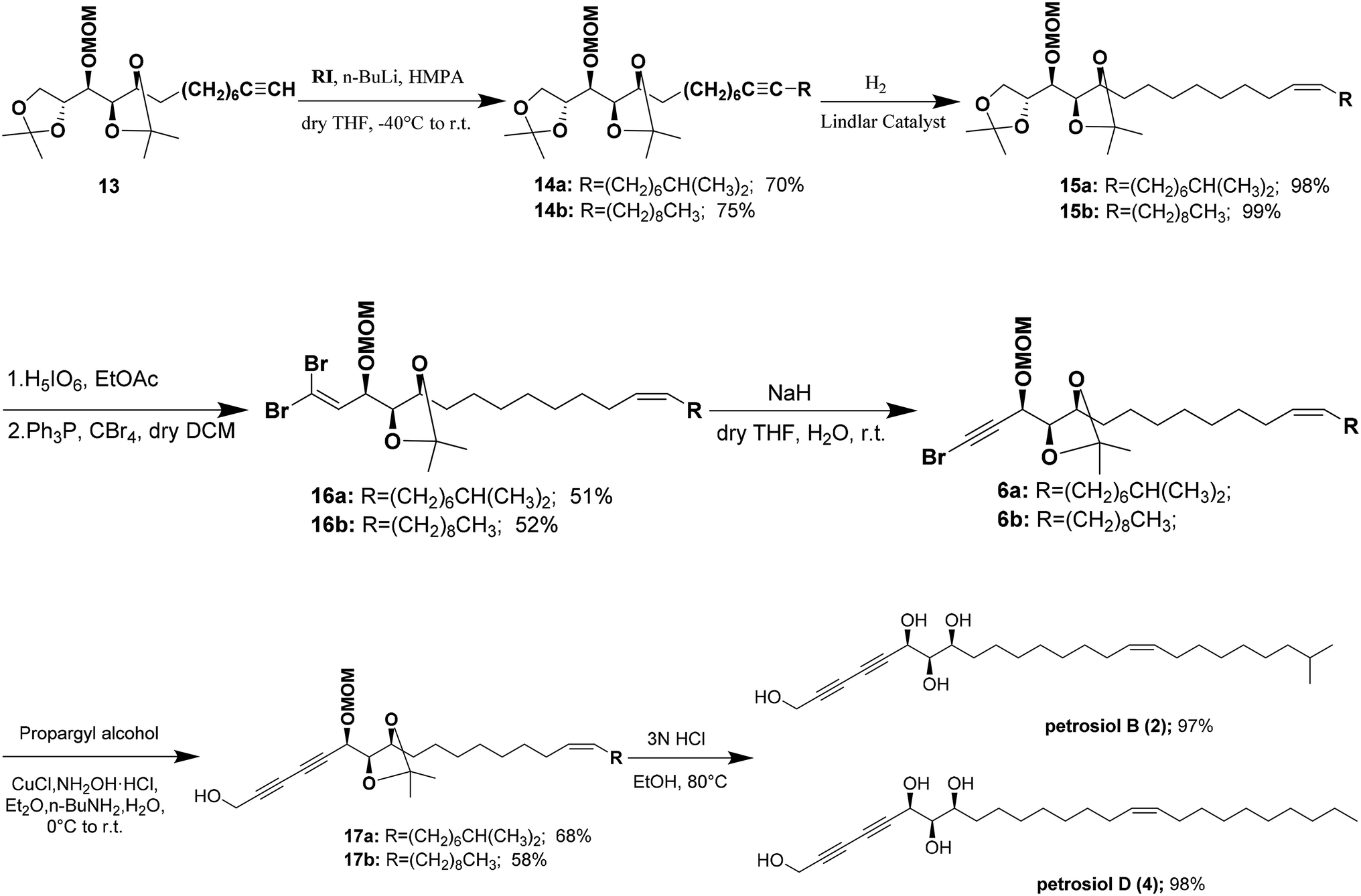 | ||
| Scheme 3 Total synthesis of petrosiols B and D. | ||
Bioactivity section
Biological activity of the synthetic petrosiols B and D were investigated following our previous evidences on homologue E induced PC12 cells differentiation.13 To find out the feasible concentrations in differentiation experiment, petrosiols ranging from 0.15 μg mL−1 to 10 μg mL−1 were subjected to PC12 cell to evaluate the cell viability using the Cell Counting Kit-8 (CCK-8). As shown in Fig. 2, petrosiol displayed no obvious effects on cell viability from day 1 to day 5 when the cell was treated with petrosiol (B/D) below the concentration of 5 μg mL−1. In particular, petrosiol B inhibited PC12 cell viability by about 74% (P < 0.001) after 1 day treatment and further repressed by about 80% (P < 0.001) after 5 day treatment at 10 μg mL−1 (Fig. 2A), while petrosiol D inhibited cell viability by about 54% (P < 0.001) after 1 day and about 75% (P < 0.001) after 5 day at 10 μg mL−1 (Fig. 2B). Therefore, concentrations lower than 5 μg mL−1 were chosen for the synthetic petrosiols in the following biological experiments.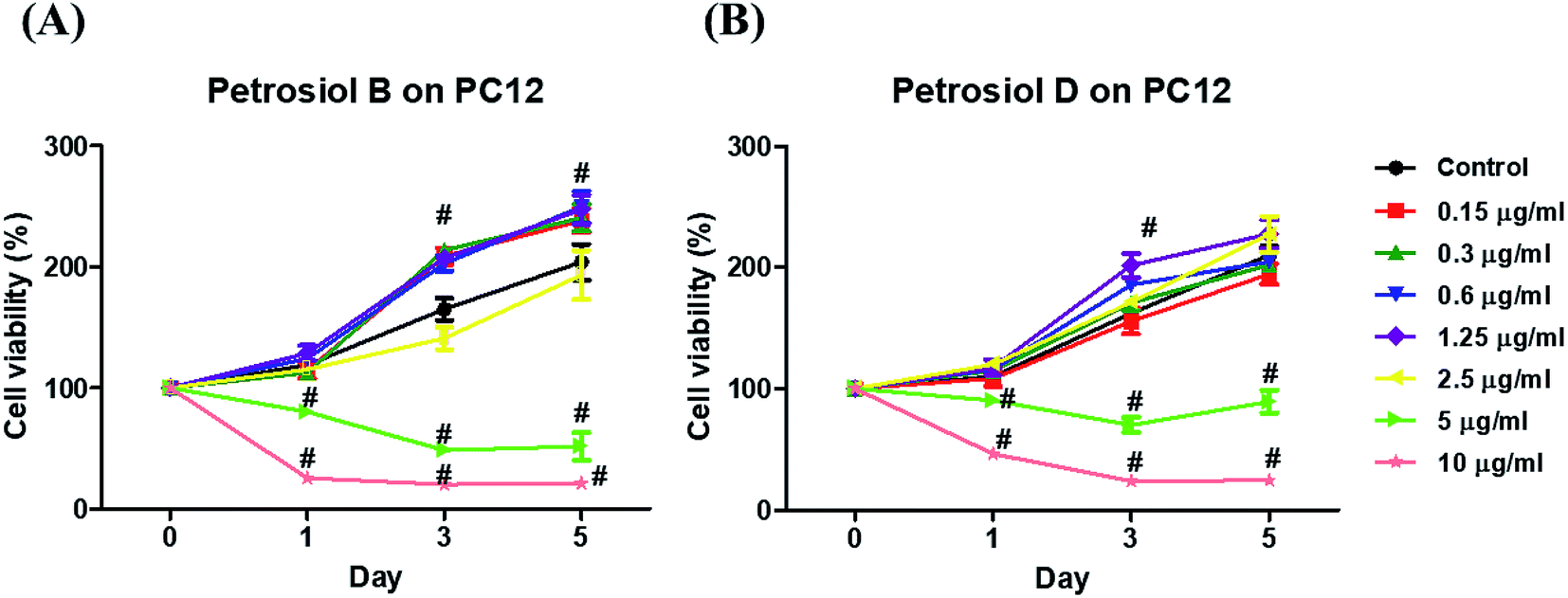 | ||
| Fig. 2 Screening of sub-lethal concentrations of petrosiols B (A) and D (B) on PC12 cells. Cell viability was determined through CCK-8 assay at different concentrations of petrosiols B and D for 1, 3, and 5 d. #P < 0.001, relative to untreated control. | ||
To evaluate the effect of petrosiol on neuronal progenitors differentiation, PC12 cells were treated with petrosiols at different concentrations and under varying exposure time. As shown in Fig. 3 and 4, treatment of PC12 cells with petrosiols B, D and E all significantly increased the number of neurites in cells in a dose- and time-dependent manner compared to control treatment, and petrosiol B showed strongest activity on neuronal differentiation. Specifically on day 5 at 2.5 μg mL−1, petrosiol B induced about 35% (P < 0.001) of cells generating neurite outgrowth, while 20% (P < 0.001) petrosiol D, 19% (P < 0.001) for petrosiol E under the same incubation conditions (Fig. 3C). Interestingly, petrosiols D and E induced a comparative differentiating cells on day 5 (Fig. 3C) though D showed stronger inducing effect than E on day 1 and 3 (Fig. 3A and B). The current result suggested that petrosiol compounds triggered neuronal differentiation of PC12 cells, though petrosiol B exhibited stronger effect than its homologues D and E.
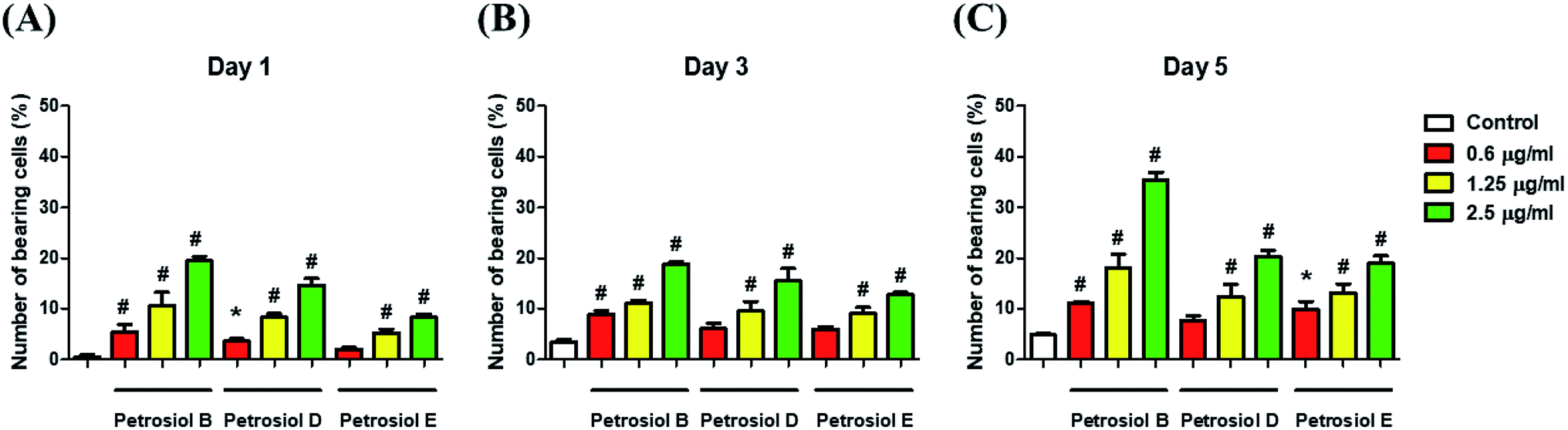 | ||
| Fig. 3 Petrosiols B and D promoted neurite outgrowth and activity of Nrf2 in PC12 cells. Petrosiol B, D and E promoted neurite outgrowth in PC12 cells on day 1 (A), 3 (B), and 5 (C). *P < 0.05, relative to control. #P < 0.001, relative to control. | ||
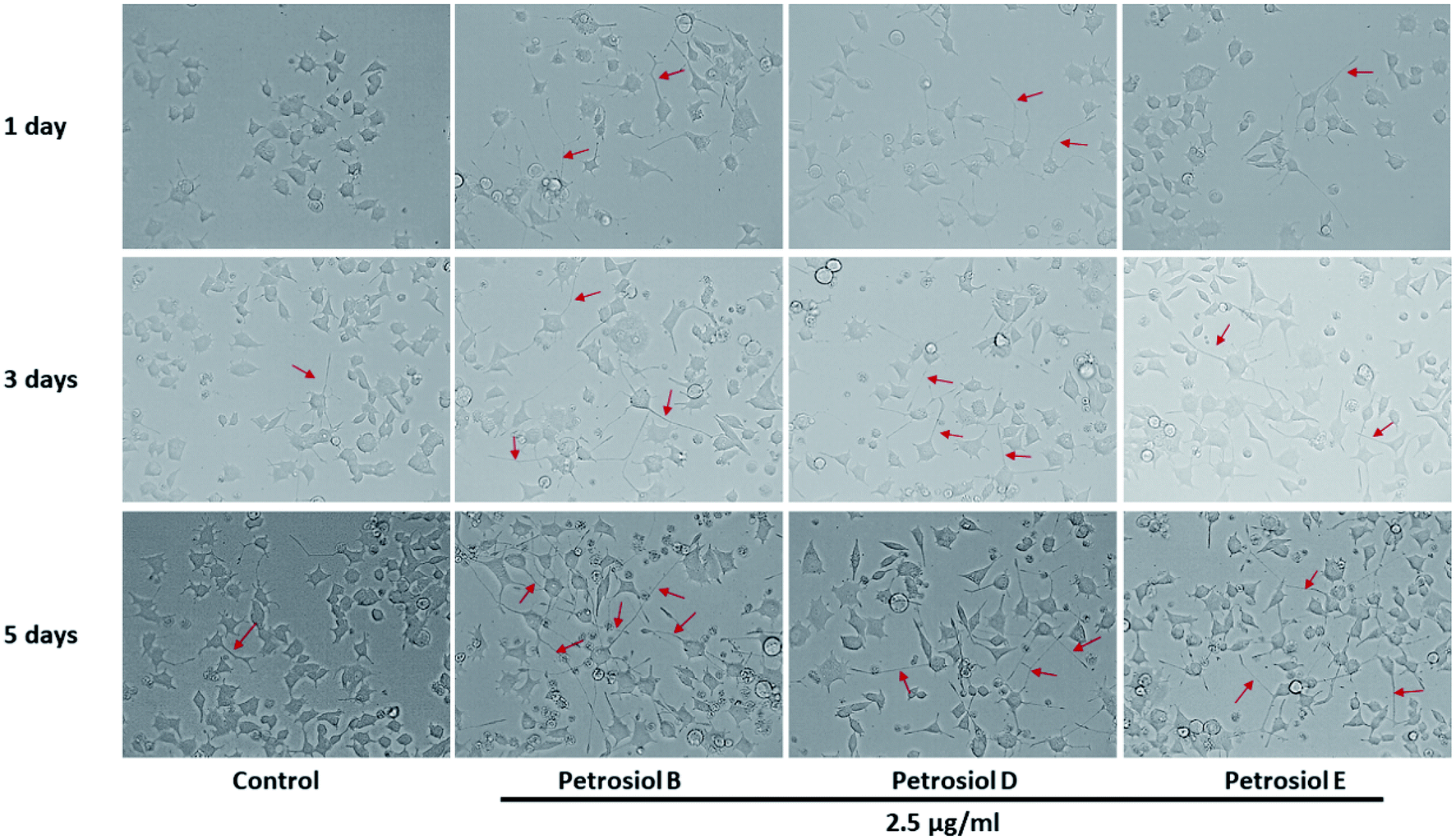 | ||
| Fig. 4 The morphology changes of PC12 cells after petrosiols B, D, and E induction at 2.5 μg mL−1 at day 3 and day 5. | ||
We also investigated the molecular mechanism concealing the petrosiol-induced neuronal differentiation. Our previous study indicated that petrosiol E induced PC12 differentiation by enhancing the activity of Nrf2 which is indispensable in an augmented differentiation process. We thus explored Nrf2 activity of PC12 cells in the presence of petrosiols B and D by detecting the contents of Nrf2 protein in nucleus, respectively. As shown in Fig. 5, petrosiols (at 1.25 and 2.5 μg mL−1) significantly upregulated the contents of Nrf2 in nuclear portion after treatment for 24 h, relative to untreated control cells. After 24 h treatment with petrosiols B, D and E at 2.5 μg mL−1, the nuclear Nrf2 level was increased by 3.5- (P < 0.001), 3.7- (P < 0.001) and 2.6-fold (P < 0.05) compared to the untreated control (Fig. 5B), respectively. To further confirm the petrosiols induced Nrf2 activation, downstream target gene heme oxygenase-1 (HO-1)25 of Nrf2 was also checked at mRNA level upon petrosiol exposure. It was found that petrosiols B, D and E at 2.5 μg mL−1 increased mRNA level of HO-1 by 10.4- (P < 0.001), 6.7- (P < 0.001) and 2.9-fold (P < 0.05) relative to the untreated control, respectively, which were consistent with the enhancement of nuclear Nrf2 protein (Fig. 5C). These findings strongly indicated that petrosiols could enhance neuronal differentiation through activating Nrf2.
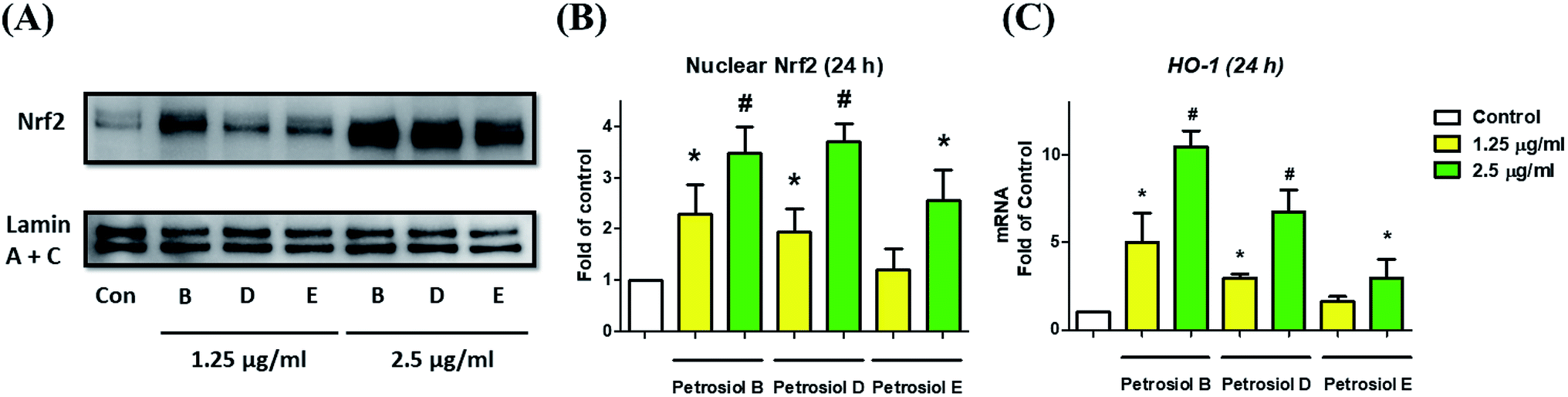 | ||
| Fig. 5 Petrosiols upregulated Nrf2 protein level in nuclear. (A) Protein levels in PC12 cell nuclear were determined via western blot. (B) Quantitative analysis of Nrf2 protein abundance was conducted with ImageJ software. Lamin A + C served as loading control. (C) The mRNA levels of HO-1 expression upon various concentrations of petrosiols B, D and E for 24 h. *P < 0.05, relative to control. #P < 0.001, relative to control. | ||
Conclusions
In conclusion, we have achieved the first total synthesis of petrosiol B and a total synthesis of petrosiol D at the same time using readily available carbohydrate D-mannose as the chiral template. The synthetic petrosiols (B, D and E) were found to elicit the differentiation of neuronal progenitor PC12 cells through an enhanced Nrf2 activity in cell nuclear. Among them, petrosiol B presented the best potential in this regards. Further efforts for the divergent syntheses and SAR studies of other petrosiol-derived polyacetylenes1,26 are currently underway in our laboratory.Experimental
General methods
Unless noted otherwise, commercially available materials were used without further purification. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. All solvents were dried according to the established procedures ahead of use. Flash chromatography (FC) was performed using silica gel (200–300 meshes) according to the standard protocol. All reactions under standard conditions were monitored by thin-layer chromatography (TLC) on gel F254 plates. Optical rotations were measured using a polarimeter with a thermally jacketed 2.5 cm cell at approximately 25 °C. High-resolution mass spectrometry data (HRMS) were acquired using a Q-TOF analyzer in methanol as solvent. 1H NMR, 13C NMR were measured on 400 MHz or 100 MHz spectrometers. Chemicals shifts (δ) were expressed in ppm relative to residual CHCl3, CD3OD or TMS. Multiplicity is tabulated as s for singlet, d for doublet, t for triplet, q for quadruplet, and m for multiplet and br when the signal in question is broadened.Synthesis of compound 10
MeOH (10 mL) was added to a mixture of 8 (3.0 g, 11.5 mmol) and K2CO3 (4.0 g, 28.9 mmol). The mixture was stirred under an argon atmosphere and allowed to reflux. Dimethyl(1-diazo-2-oxopropyl)phosphonate (4.33 mL, 28.8 mmol) was added dropwise within 6 h, at the end of which time the mixture was cooled to room temperature, then it was filtered on glass-frit and concentrated. After an extraction with EtOAc/H2O, the combined organic extracts was dried over Na2SO4 and concentrated. The crude residue was purified by flash chromatography (petroleum ether/EtOAc 4:1) to afford 10 as a syrup. Yield: 2.6 g (88%): [α]28D = −25.7 (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.66 (dd, J = 7.6, 2.0 Hz, 1H), 4.28 (d, J = 7.6 Hz, 1H), 4.13–3.98 (m, 3H), 3.56 (dd, J = 7.4, 2.3 Hz, 1H), 2.54 (d, J = 2.1 Hz, 1H), 1.51 (s, 3H), 1.43 (d, J = 2.5 Hz, 6H), 1.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 111.2, 109.7, 81.0, 80.7, 76.3, 75.1, 70.0, 67.0, 66.8, 27.0, 26.8, 26.3, 25.5; HRMS (ESI): calcd. for C13H20O5K+ [M + K]+, 295.0942; found 295.0983. The 1H and 13C NMR spectra were identical to those reported in ref. 14.
Synthesis of compound 11
To a stirred solution of 10 (889 mg, 3.47 mmol) in dry THF (50 mL) were added NaH (694 mg, 17.4 mmol) and methoxymethyl chloride (0.79 mL, 10.4 mmol). After 5 min, deionized water (50 μL) was added slowly into the mixture via syringe at room temperature and it will be exothermic. The reaction mixture was stirred for 5 h and quenched with saturated NH4Cl aqueous solution. The organic phase in the resulting solution mixture was extracted with EtOAc (3 times, 50 mL each), dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 7:1) to afford 11 as a colorless oil. Yield: 967 mg (93%): [α]25D = −25.0 (c 2.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.83 (d, J = 6.8 Hz, 1H), 4.74 (d, J = 6.7 Hz, 1H), 4.65 (dd, J = 7.5, 2.0 Hz, 1H), 4.24–4.17 (m, 1H), 4.15 (dd, J = 7.5, 3.7 Hz, 1H), 4.09–3.99 (m, 2H), 3.85 (t, J = 4.3 Hz, 1H), 3.42 (s, 3H), 2.54 (d, J = 2.0 Hz, 1H), 1.48 (s, 3H), 1.42 (d, J = 3.0 Hz, 6H), 1.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 110.9, 109.0, 98.3, 82.2, 81.0, 76.7, 75.8, 74.9, 66.8, 65.8, 56.4, 26.7, 26.6, 26.4, 25.5; HRMS (ESI): calcd. for C15H24O6Na+ [M + Na]+, 323.1465; found 323.1457.
Synthesis of compound 12
n-Butyllithium (2.5 M in hexanes, 1.33 mL, 3.32 mmol) was added dropwise to a stirred solution of terminal alkyne 11 (830 mg, 2.77 mmol) and HMPA (0.73 mL, 4.15 mmol) in dry THF (30 mL) at −40 °C. The reaction mixture was stirred at −40 °C for 20 minutes, then added 9 gave an orange colored solution. The reaction mixture was stirred at room temperature overnight and quenched with saturated NH4Cl aqueous solution. The organic phase in the resulting solution mixture was extracted with EtOAc (3 times, 50 mL each), dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 7:1) to afford 12 as a colorless oil. Yield: 967 mg (83%): [α]25D = −21.6 (c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.36–7.26 (m, 5H), 4.84 (d, J = 6.6 Hz, 1H), 4.73 (d, J = 6.6 Hz, 1H), 4.60 (dt, J = 7.7, 1.9 Hz, 1H), 4.49 (s, 2H), 4.24–4.16 (m, 1H), 4.06–3.97 (m, 3H), 3.86 (t, J = 4.0 Hz, 1H), 3.45 (t, J = 6.6 Hz, 2H), 3.41 (s, 3H), 2.21 (td, J = 7.1, 1.9 Hz, 2H), 1.65–1.57 (m, 2H), 1.56–1.44 (m, 5H), 1.43–1.34 (m, 13H); 13C NMR (100 MHz, CDCl3) δ 138.8, 128.5, 127.8, 127.7, 110.2, 108.9, 98.2, 87.9, 82.2, 76.8, 76.6, 75.3, 73.1, 70.5, 67.4, 65.6, 56.4, 29.8, 28.9, 28.6, 26.8, 26.7, 26.6, 25.9, 25.5, 18.9; HRMS (ESI): calcd. for C28H43O7+[M + H]+, 491.3003, found 491.3004.
Synthesis of compound 7
To a solution of 12 (900 mg, 1.83 mmol) in 30 mL of methanol and ethyl acetate (v/v = 1:2) was added palladium hydroxide (moisture ca. 60%, 20% Pd, 51 mg) at room temperature under H2 atmosphere. The mixture was stirred at room temperature for 1.5 h, filtered through a plug of silica gel, washed with ethyl acetate, and concentrated to afford crude compound 7 as a colorless oil which was used without further purification. Yield: 697 mg (94%): [α]25D = −30.4 (c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.83 (d, J = 6.7 Hz, 1H), 4.69 (d, J = 6.7 Hz, 1H), 4.20–4.13 (m, 1H), 4.06–3.98 (m, 3H), 3.73–3.69 (m, 1H), 3.66 (dd, J = 8.1, 2.8 Hz, 1H), 3.60 (t, J = 6.6 Hz, 2H), 3.39 (s, 3H), 1.59–1.42 (m, 5H), 1.40–1.25 (m, 21H); 13C NMR (100 MHz, CDCl3) δ 108.7, 108.6, 98.4, 81.5, 77.3, 76.8, 76.1, 65.9, 63.1, 56.4, 33.2, 32.9, 29.8, 29.6, 29.5, 27.6, 26.8, 26.6, 26.3, 25.9, 25.4; HRMS (ESI): calcd. for C21H40O7Na+ [M + Na]+, 427.2666, found 427.2656.
Synthesis of compound 13
To a solution of 7 (160 mg, 0.40 mmol) in 10 mL of DCM was added NaHCO3 (66 mg, 0.80 mmol) and Dess–Martin periodinane (251 mg, 0.40 mmol) at 0 °C. The mixture was stirred at room temperature for 0.5 h and quenched with saturated NaHCO3 aqueous solution. The organic phase in the resulting solution mixture was extracted with DCM (3 times, 50 mL each), dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The colorless oil was extracted with petroleum ether and DCM, concentrate the supernatant to afford crude aldehyde which was used without further purification.Dimethyl-2-oxopropylphosphonate (152 mg, 119 μL, 0.80 mmol) was added to a suspension of K2CO3 (164 mg, 1.20 mmol) and the crude aldehyde in MeOH (10 mL) under N2 atmosphere. The mixture was stirred at room temperature for 4.5 h. The blue mixture was concentrated, extracted with EtOAc, dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 7:1) to afford 13 as a colorless oil. Yield: 119 mg (66% for three steps): [α]25D = −26.4 (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.85 (d, J = 6.7 Hz, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.24–4.13 (m, 1H), 4.08–3.97 (m, 3H), 3.76–3.70 (m, 1H), 3.68 (dd, J = 8.0, 2.8 Hz, 1H), 3.40 (s, 3H), 2.17 (td, J = 7.0, 2.6 Hz, 2H), 1.93 (t, J = 2.6 Hz, 1H), 1.63–1.45 (m, 5H), 1.43–1.24 (m, 19H); 13C NMR (100 MHz, CDCl3) δ 108.69, 108.66, 98.4, 84.9, 81.6, 77.3, 76.8, 76.1, 68.3, 66.0, 56.5, 33.3, 29.7, 29.2, 28.8, 28.6, 27.7, 26.8, 26.7, 26.3, 25.5, 18.6; HRMS (ESI): calcd. for C22H38O6Na+ [M + Na]+, 421.2561, found 421.2569.
Synthesis of compound 14a
n-Butyllithium (2.5 M in hexanes, 0.14 mL, 0.36 mmol) was added dropwise to a stirred solution of terminal alkyne 13 (110 mg, 0.28 mmol) and HMPA (72 μL, 0.41 mmol) in dry THF (10 mL) at −40 °C. The reaction mixture was stirred at room temperature and added 1-iodo-7-methyloctane gave an orange colored solution. Quenched with saturated NH4Cl aqueous solution after 4.5 h. The organic phase in the resulting solution mixture was extracted with EtOAc (3 times, 50 mL each), dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 12:1) to afford 14a as a colorless oil. Yield: 101 mg (70%): [α]25D = −20.5 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.84 (d, J = 6.6 Hz, 1H), 4.70 (d, J = 6.6 Hz, 1H), 4.23–4.12 (m, 1H), 4.07–3.98 (m, 3H), 3.75–3.70 (m, 1H), 3.67 (dd, J = 8.0, 2.8 Hz, 1H), 3.40 (s, 3H), 2.18–2.07 (m, 4H), 1.60–1.42 (m, 8H), 1.41–1.29 (m, 21H), 1.28–1.23 (m, 4H), 1.18–1.09 (m, 2H), 0.85 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 108.68, 108.65, 98.4, 81.6, 80.5, 80.3, 77.4, 76.8, 76.1, 66.0, 56.4, 39.2, 33.3, 29.8, 29.6, 29.4, 29.3, 29.3, 29.1, 29.0, 28.1, 27.7, 27.5, 26.8, 26.7, 26.4, 25.5, 22.8, 19.0, 18.9; HRMS (ESI): calcd. for C31H56O6Na+ [M + Na]+, 547.3969, found 547.3969.
Synthesis of compound 14b
n-Butyllithium (2.5 M in hexanes, 91 μL, 0.23 mmol) was added dropwise to a stirred solution of terminal alkyne 13 (70 mg, 0.18 mmol) and HMPA (46 μL, 0.26 mmol) in dry THF (8 mL) at −40 °C. The reaction mixture was stirred at room temperature and added 1-iodononane gave an orange colored solution. Quenched with saturated NH4Cl aqueous solution after 20 h. The organic phase in the resulting solution mixture was extracted with EtOAc (3 times, 30 mL each), dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 7:1) to afford 14b as a colorless oil. Yield: 67 mg (75%): [α]25D = −18.4 (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.85 (d, J = 6.7 Hz, 1H), 4.71 (d, J = 6.7 Hz, 1H), 4.22–4.13 (m, 1H), 4.11–3.91 (m, 3H), 3.77–3.71 (m, 1H), 3.68 (dd, J = 8.0, 2.8 Hz, 1H), 3.41 (s, 3H), 2.16–2.09 (m, 4H), 1.62–1.42 (m, 9H), 1.41–1.12 (m, 29H), 0.87 (t, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 108.70, 108.66, 98.4, 81.6, 80.5, 80.3, 77.4, 76.9, 76.1, 66.0, 56.5, 33.3, 32.1, 29.8, 29.7, 29.5, 29.4, 29.3, 29.1, 29.0, 27.7, 26.9, 26.7, 26.4, 25.5, 22.9, 19.0, 14.3; HRMS (ESI) calcd for C31H56O6Na+ [M + Na]+ 547.3969, found 547.3975.
Synthesis of compound 15a
To a solution of 14a (40 mg, 0.076 mmol) in 6 mL of methanol and ethyl acetate was added Lindlar catalyst (12 mg, 0.023 mmol) at room temperature under H2 atmosphere. The mixture was stirred at room temperature for 1.5 h, filtered through a plug of silica gel, washed with ethyl acetate, and concentrated to afford 15a as a colorless oil which was used without further purification. Yield: 39 mg (98%): [α]25D = −15.0 (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.43–5.25 (m, 2H), 4.84 (d, J = 6.8 Hz, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.25–4.13 (m, 1H), 4.09–3.97 (m, 3H), 3.75–3.71 (m, 1H), 3.68 (dd, J = 8.0, 2.9 Hz, 1H), 3.40 (s, 3H), 2.07–1.92 (m, 4H), 1.61–1.45 (m, 4H), 1.43–1.22 (m, 29H), 1.20–1.09 (m, 2H), 0.85 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 130.1, 130.0, 108.7, 108.6, 98.4, 81.6, 77.4, 76.1, 66.0, 56.4, 39.2, 33.3, 30.0, 29.95, 29.94, 29.90, 29.6, 29.5, 29.4, 28.2, 27.7, 27.6, 27.40, 27.39, 26.8, 26.7, 26.4, 25.5, 22.8; HRMS (ESI) calcd for C31H58O6Na+ [M + Na]+ 549.4126, found 549.4125.Synthesis of compound 15b
To a solution of 14b (300 mg, 0.59 mmol) in 40 mL of methanol and ethyl acetate was added Lindlar catalyst (30 mg, 0.059 mmol) at room temperature under H2 atmosphere. The mixture was stirred at room temperature for 1.5 h, filtered through a plug of silica gel, washed with ethyl acetate, and concentrated to afford 15b as a colorless oil which was used without further purification. Yield: 298 mg (99%): [α]25D = −23.9 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.41–5.28 (m, 2H), 4.85 (d, J = 6.8 Hz, 1H), 4.71 (d, J = 6.8 Hz, 1H), 4.24–4.14 (m, 1H), 4.09–3.96 (m, 3H), 3.76–3.71 (m, 1H), 3.68 (dd, J = 8.0, 2.8 Hz, 1H), 3.40 (s, 3H), 2.09–1.88 (m, 4H), 1.64–1.44 (m, 3H), 1.43–1.20 (m, 35H), 0.87 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 130.1, 130.0, 108.69, 108.65, 98.4, 81.6, 77.4, 76.1, 66.0, 56.5, 33.3, 32.1, 29.96, 29.95, 29.9, 29.80, 29.76, 29.7, 29.53, 29.51, 29.4, 27.7, 27.41, 27.39, 26.8, 26.7, 26.4, 25.5, 22.9, 14.3; HRMS (ESI) calcd for C31H58O6Na+ [M + Na]+ 549.4126, found 549.4128.Synthesis of compound 16a
A solution of compound 15a (32 mg, 0.061 mmol) and H5IO6 (28 mg, 0.12 mmol) in 2 mL EtOAc was stirred for 1 h. The reaction mixture was filtered, and the filtrate was evaporated to afford aldehyde as an orange-red oil.To a solution of triphenylphosphine (127 mg, 0.49 mmol) in 2 mL dry DCM was added carbon tetrabromide (81 mg, 0.24 mmol) at 0 °C. The resulting mixture was stirred for 5 min, and then aldehyde was added slowly via syringe. Quenched with saturated NaHCO3 aqueous solution after 2 hours. The organic phase in the resulting solution mixture was extracted with DCM (3 times, 20 mL each), dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 20:1) to afford 16a as a colorless oil. Yield: 19 mg (51% for three steps): [α]25D = −42.7 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.44 (d, J = 9.1 Hz, 1H), 5.42–5.28 (m, 2H), 4.71 (d, J = 6.8 Hz, 1H), 4.61 (d, J = 6.8 Hz, 1H), 4.40 (dd, J = 9.1, 5.3 Hz, 1H), 4.03–3.95 (m, 1H), 3.73 (dd, J = 7.8, 5.4 Hz, 1H), 3.40 (s, 3H), 2.12–1.89 (m, 4H), 1.61–1.45 (m, 4H), 1.42 (d, J = 1.6 Hz, 6H), 1.38–1.21 (m, 17H), 1.18–1.10 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 135.8, 130.2, 130.0, 109.4, 94.7, 93.8, 82.1, 77.1, 76.3, 56.0, 39.3, 33.9, 30.0, 29.97, 29.96, 29.7, 29.59, 29.56, 29.4, 28.2, 27.7, 27.6, 27.43, 27.42, 26.9, 26.2, 22.9; HRMS (ESI) calcd for C28H50Br2O4K+ [M + K]+ 647.1707, found 647.1713.
Synthesis of compound 16b
A solution of compound 15b (262 mg, 0.51 mmol) and H5IO6 (234 mg, 1.02 mmol) in 20 mL EtOAc was stirred for 1 h. The reaction mixture was filtered, and the filtrate was evaporated to afford aldehyde as a orange red oil.To a solution of triphenylphosphine (1.08 g, 4.12 mmol) in 20 mL dry DCM was added carbon tetrabromide (680 mg, 2.06 mmol) at 0 °C. The resulting mixture was stirred for 5 min, and then aldehyde was added slowly via syringe. Quenched with saturated NaHCO3 aqueous solution after 2 hours. The organic phase in the resulting solution mixture was extracted with DCM (3 times, 100 mL each), dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 20:1) to afford 16b as a colorless oil. Yield: 166 mg (52% for three steps): [α]25D = −64.9 (c 0.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.44 (d, J = 9.0 Hz, 1H), 5.49–5.27 (m, 2H), 4.71 (d, J = 6.8 Hz, 1H), 4.61 (d, J = 6.8 Hz, 1H), 4.40 (dd, J = 9.1, 5.3 Hz, 1H), 4.04–3.91 (m, 1H), 3.72 (dd, J = 7.8, 5.3 Hz, 1H), 3.40 (s, 3H), 2.16–1.85 (m, 4H), 1.65–1.45 (m, 3H), 1.44–1.18 (m, 29H), 0.87 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 135.7, 130.1, 130.0, 109.4, 94.6, 93.8, 82.0, 77.1, 76.2, 56.0, 33.9, 32.1, 30.0, 29.9, 29.81, 29.76, 29.7, 29.6, 29.54, 29.51, 29.4, 27.7, 27.4, 26.9, 26.2, 22.9, 14.3; HRMS (ESI) calcd for C28H50Br2O4K+ [M + K]+ 647.1707, found 647.1710.
Synthesis of compound 17a
To a stirred solution of the dibromoalkene 16a (65 mg, 0.10 mmol) in 6 mL anhydrous THF was added 60% sodium hydride (42 mg, 1.05 mmol) as a solid at 0 °C. The resulting mixture was warmed to room temperature, added two drops of water slowly and stirred overnight. After the reaction mixture was quenched with saturated aq. NH4Cl, it was extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and finally evaporated under a reduced pressure to give the crude 1-bromoalkyne 6a, which was used for the next reaction without further purification.To a stirred aqueous solution of 30% n-BuNH4 (4 mL) was added crystal CuCl at room temperature, resulted in the formation of a blue solution immediately. A few crystals of hydroxylamine hydrochloride were added until the blue color disappeared. The resulting colorless solution indicated the present of Cu(I) salt. A solution of propargyl alcohol (0.12 mL, 2.09 mmol) in Et2O (0.5 mL) was added to the solution at room temperature, yielding a yellow acetylide suspension, which was immediately cooled to 0 °C. A solution of 1-bromoalkyne 6a in Et2O (0.5 mL) was added dropwise. The resulting mixture was warmed to room temperature and stirred for 4 h. More crystals of hydroxylamine hydrochloride were added throughout the reaction to prevent the reaction mixture from turning blue or green. The reaction mixture was extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 6:1) to afford 17a as a colorless oil. Yield: 37 mg (69% for two steps): [α]25D = −79.5 (c 0.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.45–5.29 (m, 2H), 4.92 (d, J = 6.8 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 4.52 (d, J = 6.3 Hz, 1H), 4.34 (s, 2H), 4.03 (td, J = 7.9, 3.2 Hz, 1H), 3.80 (t, J = 6.9 Hz, 1H), 3.40 (s, 3H), 2.09–1.85 (m, 4H), 1.82–1.68 (m, 1H), 1.63–1.46 (m, 5H), 1.45–1.22 (m, 21H), 1.21–1.10 (m, 2H), 0.86 (d, J = 6.5 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 130.2, 130.0, 109.7, 94.5, 81.6, 78.6, 77.7, 75.4, 71.5, 69.8, 67.7, 56.0, 51.6, 39.2, 34.2, 30.01, 29.99, 29.97, 29.7, 29.62, 29.56, 29.5, 28.2, 27.8, 27.6, 27.4, 27.2, 26.1, 22.9; HRMS m/z (ESI) calcd for C31H53O5+ [M + H]+ 505.3888, found 505.3877.
Synthesis of compound 17b
To a stirred solution of dibromoalkene 16b (110 mg, 0.18 mmol) in 20 mL anhydrous THF was added 60% sodium hydride (211 mg, 3.53 mmol) as a solid at 0 °C. The resulting mixture was warmed to room temperature, added two drops of water slowly and stirred overnight. After the reaction mixture was quenched with saturated aq. NH4Cl, it was extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and finally evaporated under a reduced pressure to give the crude 1-bromoalkyne 6b, which was used for the next reaction without further purification.To a stirred 30% n-BuNH4 aqueous solution (6 mL) was added crystal of CuCl at room temperature, resulted in the formation of a blue solution immediately. A few crystal of hydroxylamine hydrochloride were added until the blue color disappeared. The resulting colorless solution indicated the present of Cu(I) salt. A solution of propargyl alcohol (0.17 mL, 3.53 mmol) in Et2O (0.5 mL) was added to the solution at room temperature, yielding a yellow acetylide suspension, which was immediately cooled to 0 °C. A solution of 1-bromoalkyne 6b in Et2O (0.5 mL) was added dropwise. The resulting mixture was warmed to room temperature and stirred for 4 h. More crystals of hydroxylamine hydrochloride were added throughout the reaction to prevent the reaction mixture from turning blue or green. The reaction mixture was extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and finally evaporated under a reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc 6:1) to afford 17b as a colorless oil. Yield: 53 mg (58% for two steps): [α]25D = −114.0 (c 0.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.45–5.27 (m, 2H), 4.92 (d, J = 6.8 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 4.52 (d, J = 6.3 Hz, 1H), 4.33 (s, 2H), 4.03 (td, J = 7.9, 3.3 Hz, 1H), 3.79 (dd, J = 7.4, 6.4 Hz, 1H), 3.39 (s, 3H), 2.12–1.87 (m, 4H), 1.83–1.68 (m, 2H), 1.65–1.46 (m, 2H), 1.44–1.23 (m, 28H), 0.93–0.82 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 130.2, 130.0, 109.7, 94.5, 81.6, 78.5, 77.7, 75.3, 71.5, 69.7, 67.7, 56.0, 51.6, 34.2, 32.1, 29.97, 29.95, 29.80, 29.75, 29.7, 29.6, 29.53, 29.51, 29.45, 27.8, 27.4, 27.1, 26.1, 22.9, 14.3; HRMS m/z (ESI) calcd for C31H52O5Na+ [M + Na]+ 527.3707, found 527.3706.
Synthesis of petrosiol B (2)
To a stirred solution of diyne 17a (25 mg, 0.048 mmol) in 3 mL EtOH was added 3 N HCl (0.5 mL) aqueous solution at room temperature. The mixture was stirred at 70 °C for 1.5 h. The reaction mixture was cooled to room temperature and evaporated under a reduced pressure to afford 2 as a white powder. Yield: 20 mg (98%): [α]25D = −4.2 (c 0.25, MeOH); 1H NMR (400 MHz, CDCl3/MeOD = 4:1) δ 5.45–5.27 (m, 2H), 4.46 (d, J = 6.9 Hz, 1H), 4.27 (s, 2H), 3.82–3.68 (m, 1H), 3.44 (dd, J = 6.9, 2.2 Hz, 1H), 2.13–1.88 (m, 4H), 1.69–1.40 (m, 4H), 1.38–1.23 (m, 17H), 1.21–1.09 (m, 2H), 0.87 (d, J = 6.6 Hz, 6H); 1H NMR (400 MHz, MeOD) δ 5.46–5.26 (m, 2H), 4.43 (d, J = 7.6 Hz, 1H), 4.24 (s, 2H), 3.83–3.67 (m, 1H), 3.37 (dd, J = 7.6, 2.3 Hz, 1H), 2.14–1.91 (m, 4H), 1.66–1.45 (m, 4H), 1.41–1.24 (m, 17H), 1.23–1.13 (m, 2H), 0.89 (d, J = 6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3/MeOD = 4:1) δ 130.2, 130.1, 78.4, 77.6, 76.2, 71.3, 70.2, 69.0, 64.5, 50.7, 39.3, 34.3, 30.07, 30.06, 30.0, 29.9, 29.8, 29.59, 29.58, 28.2, 27.6, 27.47, 27.46, 26.0, 22.8; 13C NMR (100 MHz, MeOD) δ 130.9, 130.8, 79.3, 79.2, 77.7, 72.1, 70.6, 69.4, 65.4, 51.0, 40.2, 35.0, 30.9, 30.84, 30.77, 30.7, 30.39, 30.35, 29.2, 28.5, 28.2, 28.1, 26.9, 23.1; HRMS (ESI) calcd for C26H44O4K+ [M + K]+ 459.2871, found 459.2865.
Synthesis of petrosiol D (4)
To a stirred solution of diyne 17b (19 mg, 0.037 mmol) in 4 mL EtOH was added 3 N HCl (0.6 mL) aqueous solution at room temperature. The mixture was stirred at 70 °C for 2 h. The reaction mixture was cooled to room temperature and evaporated under a reduced pressure to afford 4 as a white powder. Yield: 15 mg (98%): [α]25D = −4.8 (c 0.18, MeOH); 1H NMR (400 MHz, CDCl3/MeOD = 4:1) δ 5.44–5.27 (m, 2H), 4.46 (d, J = 6.7 Hz, 1H), 4.27 (s, 2H), 3.82–3.71 (m, 1H), 3.45 (d, J = 6.3 Hz, 1H), 2.08–1.92 (m, 4H), 1.68–1.42 (m, 3H), 1.37–1.18 (m, 23H), 0.93–0.82 (d, J = 6.7 Hz, 3H); 1H NMR (400 MHz, MeOD) δ 5.55–5.25 (m, 2H), 4.44 (d, J = 7.6 Hz, 1H), 4.24 (s, 2H), 3.82–3.70 (m, 1H), 3.38 (dd, J = 7.6, 2.3 Hz, 1H), 2.14–1.93 (m, 4H), 1.67–1.42 (m, 3H), 1.41–1.24 (m, 23H), 0.95–0.85 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3/MeOD = 4:1) δ 130.1, 130.0, 78.3, 77.6, 76.1, 71.3, 70.1, 68.9, 64.4, 50.6, 34.2, 32.1, 30.0, 29.9, 29.81, 29.76, 29.7, 29.50, 29.47, 29.46, 27.37, 27.35, 25.9, 22.8, 14.2; 13C NMR (100 MHz, MeOD) δ 130.8 (2C), 79.3, 79.2, 77.6, 72.1, 70.6, 69.4, 65.3, 51.0, 35.0, 33.1, 30.9, 30.83, 30.75, 30.73, 30.65, 30.6, 30.5, 30.4, 30.3, 28.2, 28.1, 26.9, 23.8, 14.5; HRMS (ESI) calcd for C26H44O4Na+ [M + Na]+ 443.3132, found 443.3134.
Bioactivity section
:1000; Proteintech) and anti-Lamin A + C (ab169532, 1:5000; Abcam). Anti-Lamin A + C antibodies were used for equal loading of nuclear proteins.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2015CB931903 and 2016YFA0203102) and Natural Science Foundation of China (Projects 21672255 and 21621064). We thank Xinyi Zhang and Miao Chen for HRMS measurements.Notes and references
- (a) Z. Zhou, M. Menna, Y. Cai and Y. Guo, Chem. Rev., 2015, 115, 1543–1596 CrossRef CAS PubMed; (b) R. Negri, Fitoterapia, 2015, 106, 92–109 CrossRef CAS PubMed.
- R. E. Minto and B. J. Blacklock, Prog. Lipid Res., 2008, 47, 233–306 CrossRef CAS PubMed.
- U. Wittstock, K.-H. Lichtnow and E. Teuscher, Planta Med., 1997, 63, 120–124 CrossRef CAS PubMed.
- K. Xu, J. Jiang, Z. Feng, Y. Yang, L. Li, C. Zang and P. Zhang, J. Nat. Prod., 2016, 79, 1567–1575 CrossRef CAS PubMed.
- (a) N. Panthama, S. Kanokmedhakul and K. Kanokmedhakul, J. Nat. Prod., 2010, 73, 1366–1369 CrossRef CAS PubMed; (b) T. K. Kuilya and R. K. Goswami, Org. Biomol. Chem., 2016, 14, 8789–8799 RSC.
- (a) K. Watanabe, Y. Tsuda, Y. Yamane, H. Takahashi, K. Iguchi, H. Naoki, T. Fujita and R. W. M. Van Soest, Tetrahedron Lett., 2000, 41, 9271–9276 CrossRef CAS; (b) K. Watanabe, Y. Tsuda, M. Hamada, M. Omori, G. Mori, K. Iguchi, H. Naoki, T. Fujita and R. W. M. Van Soest, J. Nat. Prod., 2005, 68, 1001–1005 CrossRef CAS PubMed.
- (a) B. Zhang, Y. Wang, S. P. Yang, Y. Zhou, W. B. Wu, W. Tang, J. P. Zuo, Y. Li and J. M. Yue, J. Am. Chem. Soc., 2012, 134, 20605–20608 CrossRef CAS PubMed; (b) Y. Wang, Q. F. Liu, J. J. Xue, Y. Zhou, H. C. Yu, S. P. Yang, B. Zhang, J. P. Zuo, Y. Li and J. M. Yue, Org. Lett., 2014, 16, 2062–2065 CrossRef CAS PubMed.
- C. M. Yao and X. W. Yang, J. Ethnopharmacol., 2014, 151, 791–799 CrossRef CAS PubMed.
- (a) K. Horikawa, T. Yagyu, Y. Yoshioka, T. Fujiwara, A. Kanamoto, T. Okamoto and M. Ojika, Tetrahedron, 2013, 69, 101–106 CrossRef CAS; (b) B. K. Choi, B. Y. Cha, T. Yagyu, J. T. Woo and M. Ojika, Bioorg. Med. Chem., 2013, 21, 1804–1810 CrossRef CAS PubMed.
- A. Sathish Reddy and P. Srihari, Tetrahedron Lett., 2013, 54, 6370–6372 CrossRef CAS.
- P. Gangadhar, A. Sathish Reddy and P. Srihari, Tetrahedron, 2016, 72, 5807–5817 CrossRef CAS.
- L. Wang, X. Zhang, J. Liu and Y. Du, Tetrahedron, 2014, 70, 8231–8235 CrossRef CAS.
- J. Liu, L. Wang, Y. Du and S. Liu, Adv. Sci., 2017, 4, 1700089 CrossRef PubMed.
- F. Dolhem, C. Lievre and G. Demailly, Tetrahedron, 2003, 59, 155–164 CrossRef CAS.
- G. Pandey, K. C. Bharadwaj, M. I. Khan, K. S. Shashidhara and V. G. Puranik, Org. Biomol. Chem., 2008, 6, 2587–2595 RSC.
- J. K. Chahal, D. Craig, C. A. Smethurst and A. J. P. White, ARKIVOC, 2011, 4, 127–139 Search PubMed.
- (a) K. M. Pietrusiewicz and J. Monkiewicz, J. Org. Chem., 1983, 48, 788–790 CrossRef CAS; (b) K. M. Pietrusiewicz and J. Monkiewicz, Tetrahedron Lett., 1986, 27, 739–742 CrossRef CAS.
- (a) G. V. M. Sharma, T. R. Prasad, P. R. Krishna, K. Krishnudu, M. R. Rao and A. C. Kunwar, Tetrahedron: Asymmetry, 2000, 11, 4499–4507 CrossRef CAS; (b) G. V. M. Sharma, A. S. Chander, P. R. Krishna, K. Krishnudu, M. R. Rao and A. C. Kunwar, Tetrahedron: Asymmetry, 2000, 11, 2643–2646 CrossRef CAS.
- (a) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS; (b) Y. G. Gao, J. Liu, L. L. Wang, M. Xiao and Y. G. Du, Eur. J. Org. Chem., 2014, 10, 2092–2098 CrossRef.
- (a) S. Müller, B. Liepold, G. J. Roth and H. J. Bestmann, Synlett, 1996, 6, 521–522 CrossRef; (b) M. V. Mijangos and L. D. Miranda, Org. Biomol. Chem., 2018, 16, 9409–9419 RSC.
- (a) S. Moura, C. Thomassigny, C. Ligeour, C. Greck, D. Joseph, E. Drège and F. Dumas, Green Chem., 2011, 13, 1812–1818 RSC; (b) F. Sartillo-Piscil, M. Vargas, C. Anaya de Parrodi and L. Quintero, Tetrahedron Lett., 2003, 44, 3919–3921 CrossRef CAS.
- (a) E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769–3772 CrossRef; (b) P. Michel, D. Gennet and A. Rassat, Tetrahedron Lett., 1999, 40, 8575–8578 CrossRef CAS.
- (a) D. Grandjean, P. Pale and J. Chuche, Tetrahedron Lett., 1994, 35, 3529–3530 CrossRef CAS; (b) T. Durand-Reville, L. B. Gobbi, B. L. Gray, S. V. Ley and J. S. Scott, Org. Lett., 2002, 4, 3847–3850 CrossRef CAS PubMed.
- (a) K. S. Sindhu, A. P. Thankachan, P. S. Sajitha and G. Anilkumar, Org. Biomol. Chem., 2015, 13, 6891–6905 RSC; (b) J. P. Marino and H. N. Nguyen, J. Org. Chem., 2002, 67, 6841–6844 CrossRef CAS PubMed.
- A. Loboda, M. Damulewicz, E. Pyza, A. Jozkowicz and J. Dulak, Cell. Mol. Life Sci., 2016, 73, 3221–3247 CrossRef CAS PubMed.
- K. Xu, P. Yang, Y. Yang, Z. Feng, J. Jiang and P. Zhang, Org. Lett., 2017, 19, 686–689 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra01166h |
| This journal is © The Royal Society of Chemistry 2019 |