
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-fabrication of pigment-capped silver nanoparticles encountering antibiotic-resistant strains and their cytotoxic effect towards human epidermoid larynx carcinoma (HEp-2) cells
Lakshmipathy Muthukrishnana,
Muralidharan Chellappaa,
Anima Nandab,
Sudhakar Thukkaramb,
Gracyfathima Selvarajc,
Bavanilatha Muthiahb,
Suresh Sagadevan *d and
J. Anita Lette
*d and
J. Anita Lette
aLeather Process Technology, Tannery Division, CSIR – Central Leather Research Institute (CLRI), Adyar, Chennai, Tamilnadu 600 020, India. E-mail: mlakshmipathy@gmail.com
bFaculty of Bio & Chemical Engineering, Sathyabama Institute of Science and Technology (Deemed to be University), Rajiv Gandhi Salai, Chennai, Tamilnadu 600119, India
cDepartment of Virology, King Institute of Preventive Medicine and Research, Guindy, Chennai, Tamilnadu 600 032, India
dNanotechnology & Catalysis Research Centre (NANOCAT), University of Malaya, Kuala Lumpur 50603, Malaysia. E-mail: drsureshnano@gmail.com
eDepartment of Physics, Sathyabama Institute of Science and Technology, Chennai-600119, India
First published on 21st May 2019
Abstract
Bacterial biomolecule-mediated nanoparticle (NP) synthesis constitutes a reliable, eco-friendly approach that ameliorates green-chemistry principles. In this study, stable silver nanoparticles were synthesized by exposing aqueous silver ions to an extracellular diffusible pigment produced by Pseudomonas aeruginosa (PA6) under optimized laboratory conditions. Spectroscopic and microscopic analyses showed the typical characteristics of silver with an average size of ∼28.30 nm and spherical shape. The particles were polydispersed and showed no definite agglomeration with a zeta potential of −32.3 mV, conferring stability. Antimicrobial studies were carried out using 5, 15, 25 and 50 μg mL−1 concentrations of pcAgNPs, which showed significant antibacterial activity toward clinically important pathogens at all concentrations compared to with the control sample. The bactericidal effect induced by pcAgNPs associated with cell damage was well demonstrated using electron microscopic studies. ROS production was measured using the DCFH-DA method and the oxidative stress was assessed by measuring the reduced glutathione (GSH) levels. Cytotoxicity studies on HEp-2 (Human Epidermoid Larynx Carcinoma) cells exposed to pcAgNPs showed dose-dependent cytotoxic effect with IC50 of 14.8 μg mL−1 compared to with IC50 of 7.38 μg mL−1 for the Vero cell control. Mechanistically, the pcAgNPs activated p53 that induced catalase, leading to apoptosis and DNA fragmentation via a p53 transcriptional pathway and electron transport arrest, which resulted in cell death. This synergistic efficacy of pigment-AgNPs demonstrated excellent antimicrobial and anti-proliferative activities, providing a potential lead for developing a broad-spectrum antibacterial agent and improving the therapeutic modalities targeting carcinoma cells at the gene level.
1. Introduction
Nanotechnology, a major recent technological development, has experienced tremendous growth in the fabrication of materials at a sub-atomic scale for more precise and unique functionalities.1 Such engineered nanomaterials have extended its potential therapeutic and preventive applications in the biomedical and clinical fields. Specifically, the influence of their size extends their ability to surpass physiological barriers and affinity toward the cellular components of biological origin, allowing them to access the most complicated bio-molecular systems.2 Moreover, nanoparticles are being used as anti-microbial agents and vehicles for targeted delivery,3 gene therapy,4 and cell labeling.5Despite several breakthroughs in the formulation of specialist drugs from leading pharma on par with the increasing incidences of infectious diseases, a push for defending the scope of nanotechnology toward bio-clearance remains vital. As the use of conventional antibiotics rises, antimicrobial resistance patterns develop,6 demanding a continuous need for novel and more effective therapies.7 To date, nanotechnology-driven bio-pharmaceuticals have been the focus of many investigations and remain one of the highly specialized alternative medicines for curing chronic diseases such as cancer.8 Although many studies have focused on nanoparticle-induced cytotoxicity and the corresponding mechanisms,9 the decisive factors and accompanying changes involved in cytotoxicity remain unclear.10 It is noteworthy that several genes participate in the induction of apoptosis, but the role of the p53 pathway that determines the fate of cancerous cells has rarely been studied. Moreover, in more than half of human cancers, the tumor-suppressing p53 gene remains dormant.11 Thus, there is a great interest aimed at developing therapeutic strategies to overcome drug resistance in microbial communities and to anticipate the role of p53 for a prolonged expression through the nanoparticulate system.12
To solve the above-mentioned issues, research studies on synthesizing nanomaterials that exhibit excellent pharmaco-dynamic and pharmacokinetic properties in an eco-friendly fashion and following the green chemistry principle have been proposed.13 Besides chemical and physical methods, the bioprocess-mediated synthesis of nanomaterials is a fascinating approach for the development of a nanoparticle–biomolecule conjugate.14,15 This is achieved using biocatalysts and/or metabolites such as pigments, toxins, and siderophores, exhibiting phenomenal characteristics such as virulence, defense mechanism, and stress.16 In recent years, pigments of microbial origin, especially those produced by Pseudomonads, are gaining much importance in the food and agriculture sector as bio-control agents. The exploitation of such bacteria for their catabolic versatility and broad-spectrum antibiosis can help improve crop production.17,18
In this study, we present an eco-friendly approach for synthesizing silver nanoparticles harnessing a versatile pigment produced by P. aeruginosa in a reliable, stable and cost-effective manner. We investigated the biological role of the bacterial pigment-capped AgNPs toward containing antibiotic-resistant strains and the in vitro antiproliferative effect with respect to the p53 expression using the human epidermoid larynx carcinoma cell line (HEp-2).
2. Materials and methods
2.1. Isolation of Pseudomonas aeruginosa
The bacterium used for the synthesis of silver nanoparticles was isolated from rhizosphere soil samples collected from the paddy fields (Lat. 12°54′40′′N and Long. 79°8′10′′E) in Vellore, Tamilnadu, India. The pseudomonas strains were isolated by performing a dilution plate technique19 on Pseudomonas isolation agar (cat no. 17208, Sigma-Aldrich, USA), and the plates were incubated at 37 ± 2 °C for 24 h. Prior to incubation, the plates were observed for any growth on the selective media. The isolated colonies of the Pseudomonas species were further purified by quadrant streaking on Cetrimide Agar (cat. no. 22470, Sigma-Aldrich, USA) plates and maintained at 4 °C.2.2. Molecular identification of the potential strain
The growth characteristics of the bacterial isolates, viz., lactose fermentation, odour, and pigmentation were observed along with the standard biochemical tests and identified based on Bergey's manual of determinative bacteriology.20 For identification at the molecular level, the total DNA from the potential strain was extracted using a MagAttract Microbial DNA kit (cat. no. 27200.4, Qiagen) and the 16S ribosomal RNA gene was amplified using PCR with suitable primers. The PCR product obtained was sequenced by an automated sequencer ABI PRISM (Model 3700) and the sequences were compared using the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/).2.3. Pigment production
First, 35 pseudomonas strains obtained from various sites were used for the production, extraction, and characterization of pigment, and the conditions were optimized as described in our previous study.21 In brief, the isolates were inoculated into Erlenmeyer flasks containing 100 mL of Pseudomonas broth (cat. no. Z699101, Sigma-Aldrich, USA) and incubated at 37 °C in a rotary shaker for 24 h till the color changed. The pigment produced by the potential strain was quantified based on the absorbance of the pigment in the acidic form at 520 nm and the concentration was determined using the following formula according to Essar et al. (1990):22| concentration (μg mL−1) = O.D520 × 17.07 |
The strain that showed the maximum pigment production prior to incubation was retained for synthesizing AgNPs.
2.4. Bio-synthesis of AgNPs using potential strain
The preliminary screening process was performed by taking 10 mL of the diffusible pigment of the strain and adding it to 90 mL of 10−3 M AgNO3 (CAS No. 7761-88-8, Sigma-Aldrich, USA) in Erlenmeyer flasks till 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentration was achieved. The flasks were examined visually for any colour change prior to the reaction period at 37 °C and 150 rpm, which was indicative of nanoparticle formation. After the formation of nanoparticles, the flasks were removed from the shaker and centrifuged at 8000 rpm for 20 min, freeze-dried and quantified.23
:1 concentration was achieved. The flasks were examined visually for any colour change prior to the reaction period at 37 °C and 150 rpm, which was indicative of nanoparticle formation. After the formation of nanoparticles, the flasks were removed from the shaker and centrifuged at 8000 rpm for 20 min, freeze-dried and quantified.23
2.5. Optimization studies on AgNP synthesis
The ideal conditions for nanoparticle synthesis were optimized by varying the physicochemical parameters such as the temperature (10, 20, 30, 40 and 50 °C), pH (2–9), the concentration of the substrate (0.5–2 mM AgNO3) and the reaction period (0–72 h). Sampling was performed by withdrawing 1 mL aliquots at different time intervals; these aliquots were subjected to spectrophotometric analysis.242.6. Characterization of pcAgNPs
A UV-visible spectrophotometer model UV-1800 (Shimadzu) was used to record the absorption spectra of the reaction mixture. The reduction of silver ions was monitored by the random sampling of a 1 mL aliquot in a quartz cuvette and detected in the scanning range of 800–200 nm at a resolution of 1 nm. The pcAgNPs were separated and saturated by repeated washing and centrifugation at 15000 rpm for 20 min and the final suspension was freeze-dried. Powder X-ray diffraction patterns were measured on a Rigaku multiflex diffractometer that operated at 30 kV and 100 mA at 3 kW with a Cu target Kα radiation using a scintillation counter (λ = 1.5418 Å). For FT-IR analysis, the freeze-dried pcAgNPs were diluted with potassium bromide in a ratio of 1:100 and the spectrum was recorded on a JASCO 4400 model in a diffused reflectance mode in the region of 4000–400 cm−1 and at a resolution of 4 cm−1. HR-TEM (Philips CM200) and SAED were performed to identify the size and shape of the nanoparticles. The pcAgNP samples used for microscopic analyses were prepared on a carbon-coated copper grid.24,25
The particle size and distribution of the bio-synthesized silver nanoparticles were observed using dynamic light scattering (DLS) (Malvern Zetasizer, ver. 6.2). For this experiment, the samples were measured at a fixed angle of 90° at room temperature and the intensity was adjusted in the range of 50–500 kcps by diluting with deionized water. The zeta potential of the silver nanoparticles was also measured by following the above-mentioned protocol and the measurements were analyzed in triplicate.
Atomic force microscopy was employed to study the topography at the nanoscale. The sample was prepared by dissolving pcAgNPs in ethanol and sonicating (Vibra cell, Sonics) to prevent aggregation. Ten μL of the sample was casted onto a silicon slide and the solvent was allowed to evaporate. Then, the thin film of size 1 cm × 1 cm was imaged in the tapping mode with a spring constant of 0.41 N m−1 and resonance frequency 18 kHz via AFM (Ntegra Prima – NTMDT, Ireland).
2.7. Antibacterial activity of pcAgNPs against clinical isolates
Bacterial strains showing resistance toward two different groups of commonly used antibiotics, viz., Methicillin-resistant Staphylococcus aureus (MRSA), Staphylococcus epidermidis (MRSE), Proteus mirabilis, E. coli, Klebsiella pneumoniae, Acinetobacter baumannii, and the vancomycin-resistant Enterococci were used for antibacterial studies. The clinical isolates were procured from M s−1. Sharp Laboratories, Chennai, Tamilnadu, and the pure cultures were maintained in Muller Hinton agar (MHA, HiMedia™, India) slants at 4 °C. The antibacterial activity of pcAgNPs was tested against these drug-resistant bacterial strains using the Kirby Bauer's disk diffusion method.26,27 Sterile disks (HiMedia™, India) containing 5, 15, 25 and 50 μg mL−1 of pcAgNPs were prepared and placed on MHA plates using sterile forceps along with 1 mM AgNO3 as the control. The zone of inhibition (ZOI) was measured after incubation at 37 °C for 18–24 h and compared with that of the control.To detect the membrane damage, different volumes of the MH medium, pcAgNPs and the most susceptible strains (105 CFU mL−1) were added to 5 mL broth with a final concentration of 25 μg mL−1 AgNPs. Control experiments were conducted without AgNPs. The cultures were incubated at 37 ± 2 °C with shaking at 150 rpm. One hundred microlitres of cultures were sampled after being treated for 2 to 4 h. The cultures were centrifuged at 8000 rpm, the supernatant was discarded, and the cells were fixed for observation by scanning electron microscopy.28
2.8. ROS production and glutathione (GSH) measurements
The intracellular localization of stress-induced reactive oxygen species (ROS) was estimated by 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, cat no. D6883, Sigma-Aldrich, USA) as described.29 The strains were selected based on the antibiogram pattern representing Gram-positive and Gram-negative bacteria. These cells at 105 CFU mL−1 were incubated at 37 °C with DCFH-DA (0.5 mM) for 30 min, followed by the addition of pcAgNPs (5, 15, 25, 50 μg mL−1). The bacterial cells were further incubated for 4 h at 37 °C and the fluorescence emission was recorded at 523 nm with excitation at 503 nm on a Jobin Yvon Flurolog-3-11 spectrofluorometer. The ROS production was interpreted from the emission intensity of the treated cells and compared against those of the negative (untreated) and positive (1 mM H2O2) controls.The intracellular GSH level was determined following the protocol of Park et al. with slight modifications.30 In short, the cells (105 CFU mL−1) treated with 5, 15, 25 and 50 μg mL−1 AgNPs were collected and lysed with a 0.6% 5-sulfosalicylic acid solution for 10 min on ice. Fifty μL of the cell lysate was mixed with 450 μL of KH2PO4/EDTA buffer (0.1 M potassium phosphate with 5 mM EDTA, pH 8.0) and 100 μL of 1 mg mL−1 o-phthaldialdehyde solution and incubated for 60 min in dark at room temperature. The fluorescence was measured on a Jobin Yvon Flurolog-3-11 spectrofluorometer with excitation and emission wavelengths of 350 nm and 420 nm, respectively, and compared against that of controls as described. The concentration of GSH was determined from the standard curve and expressed in mM.
2.9. In vitro antiproliferative activity of pcAgNPs
:1) for 1 h at room temperature. After incubation, the cells were stained with AO and EB (1:1) in PBS and incubated for 5 min; then, they were washed with PBS to remove the excess unbinding dye. The stained cells were visualized under UV illumination using the 40× objective (Nikon Eclipse TS100) and the digitized images were captured.2.10. Semi-quantitative RT-PCR analysis
Total RNA was extracted from the experimental cells using the Trizol reagent (TRI, Sigma-Aldrich, St. Louis, USA) kit. The RNA isolated from the cells was reverse-transcribed and amplified using the one-step RT-PCR System (Fermentas, USA). The primer sequences of the forward and reverse p53 were 5′-TTGAGGTGCATGTTTGTGCC-3′ and 5′-AGGAGCTGGTGTTGTTGGAC-3′; for β-actin, they were 5′-TTCCTCCCTGGAGAAGAGCTA-3′ and 5′-CACAGAGTACTTGCGCTCAG-3′, respectively.The PCR conditions were as follows: 94 °C for 5 min; 35 cycles of 94 °C for 1 min, 56–62 °C for 1 min, 72 °C for 1 min; and a final extension step of 72 °C for 10 min. The products were verified by agarose gel electrophoresis and documented using Quantity One software (Bio-Rad, USA). The band intensity of the resolved cDNA fragments of p53 was normalized with that of the internal control β-actin and expressed as OD units relative to β-actin. All of the experiments were performed in two batches and subjected to the individual Student's ‘t’ test to assess the variation p < 0.05 level.
2.11. DNA fragmentation assay
To investigate the internucleosomal DNA fragmentation caused by pcAgNPs, a DNA fragmentation assay was performed according to the standard procedure described by Collins et al. (1997) with slight modifications.33 A total of 1 × 105 cells were treated with pcAgNPs (15 μg mL−1) for 24 h and then collected by centrifugation. Furthermore, the DNA was isolated using a commercially available kit (Genei, Bangalore, India) following the manufacturer's instructions. The DNA was resolved on a 1.5% agarose gel (0.5 μg mL−1 ethidium bromide in the 1× TAE buffer, pH 8.5) at 100 meV for 90 min and the bands were visualized using a UV transilluminator.2.12. Western blotting
First, 1 × 105 cells exposed to pcAgNPs were washed twice with ice-cold 1× PBS solution and centrifuged at 5000 rpm for 5 min. The pellet was lysed in a RIPA buffer containing 150 mM NaCl, Triton X-100, 50 mM Tris–HCl (pH 8), 0.5% sodium deoxycholate, 0.1% SDS, and protease and phosphatase inhibitors. The protein lysates (20 μg) were subjected to protein electrophoresis on 10% NuPAGE® Bis-Tris gels (cat. no. NP0301BOX, ThermoFisher Scientific, CA) according to the manufacturer's instructions. Then, the gels were blotted onto a nitrocellulose membrane (cat. no. 162-0113, Bio-Rad, Italy) for 2 h in Tris–glycine buffer. The membranes were blocked in a PBS 0.1% Tween 20 solution containing 3% of BSA, probed with specific antibodies and developed using the ECL Blotting Substrate (cat. no. K-12045-C20, Advansta, USA).In the western blotting analysis, the following primary antibodies were used: rabbit polyclonal anti-p21 (1:100) (cat. no. TA322940, OriGene, USA), mouse monoclonal anti-p53 (1:500) (cat. no. TA327920, OriGene, USA), and mouse monoclonal anti-catalase (1:100) (cat. no. TA502496, OriGene, USA). Mouse monoclonal anti-β-actin (1:10000) (cat. no. TA355041, OriGene, USA) was used to control loading and to purify the peroxidase-conjugated affinity goat anti-mouse IgG (cat. no. TA130003, OriGene, USA). The primary and secondary antibodies used were diluted in a PBS-0.1% Tween 20 solution containing 3% of BSA (cat. no. P3563, Sigma-Aldrich, USA).
2.13. Densitometric analysis
Protein bands were quantified by densitometric analysis using the Image J software (http://imagej.nih.gov).2.14. Statistical analysis
Experiments conducted in the present study on the antibiogram pattern, measurement of the zone of inhibition, and bactericidal and anti-proliferative activities were performed in triplicate. The results of the experiments were interpreted using ORIGIN 8.0 and expressed as the mean ± SD (standard deviation) of the concentration of AgNPs. The Student's t-test was used to demonstrate the statistical significance. The difference was considered to be statistically significant when the p-value was at least <0.05.3. Results and discussion
3.1. Identification of potential strain
In this study, NPs were synthesized successfully using a rhizosphere-inhabiting environmental isolate Pseudomonas aeruginosa isolated from soil samples collected from paddy fields, and it was designated PA6 for laboratory reference. Of the 51 pigmented strains isolated from six different rhizosphere soils (Table 1), around 68.6% (n = 35) were expected to be Pseudomonas aeruginosa, which were identified based on the biochemical characteristics such as non-lactose fermenting property, being oxidase-positive, pigment production and growth at 40 °C. These strains were then individually screened for the production of pyocyanin; sixteen (45%) of them produced ≤4 μg mL−1, whereas the remaining 55% showed production in the range of 5.3–6.5 μg mL−1. Table 1 reveals that P. aeruginosapa6 isolated from paddy-cultivated soil shows a maximum pigment production of 6.5 μg mL−1, followed by P. aeruginosaw9 with 5.3 μg mL−1. This variation in the production of pyocyanin among the Pseudomonas strains can be attributed to regulators.34 The higher pigment producer P. aeruginosapa6 was preferred and its identification was confirmed as a P. aeruginosa strain; its 16S rRNA partial gene sequence was submitted to the GenBank and we obtained the accession no. KM216846.1.| Source of isolation | No. of pigmented isolates | P. aeruginosa strainsexp | Pyocyanin production (μg mL−1) |
|---|---|---|---|
| a exp expected P. aeruginosa strains based on biochemical characteristics; c2, pa6, pu1, u5, w9, f3 strains of P. aeruginosa isolated from different sites for the production of pyocyanin pigment. | |||
| Corn-cultivated soil | 5 | 3 | 3.2 c2 |
| Paddy-cultivated soil | 13 | 9 | 6.5 pa6 |
| Pulse-cultivated soil | 6 | 4 | 1.9 pu1 |
| Uncultivated soil | 8 | 6 | 3.9 u5 |
| Wastewater irrigated soil | 15 | 10 | 5.3 w9 |
| Fresh water irrigated soil | 4 | 3 | 2.7 f3 |
3.2. Optimization studies on the pcAgNP synthesis
P. aeruginosapa6 exhibited higher pigment production, as evidenced by the change in colour to bluish green and through spectrophotometric analysis. The absorbance maxima (λmax) at ∼278 nm (Fig. 1) revealed the characteristic pyocyanin pigment with bluish green color, which was retained for the synthesis of silver nanoparticles.21,22 A significant change in color (yellowish brown) of the reaction mixture was observed after a thorough examination by varying the physicochemical parameters. The absorbance maxima (λmax) and surface plasmon resonance remained the same, showing a characteristic peak at 420 nm, which confirmed the bio-reduction of silver. It was determined from the spectrophotometric analysis that the yield of AgNPs was found to be maximum at a temperature of 37 ± 2 °C and neutral pH (7.0) with a concentration of 1 mM in a 72 h reaction period. The yield of silver nanoparticles obtained was quantified to be 0.035 mg/100 mL. | ||
| Fig. 1 UV-visible spectroscopic analysis of bluish green pigment (inset) produced by P. aeruginosapa6. | ||
3.3. Characterization of pcAgNPs
The diffusible bluish-green pigment produced by P. aeruginosapa6 was used as a reducing and stabilizing agent in the synthesis protocol. A change in color to yellowish brown indicated the formation of silver nanoparticles. The spectra of pcAgNPs were analyzed periodically using the supernatant obtained by centrifuging the reaction mixture at 8000 rpm for 20 min. The absorption spectra of the silver nanoparticles showed a strong single peak with a maximum at ∼420 nm (Fig. 2) with an increase in the reaction time (72 h) and a maximum yield.35 This is in agreement with the results of a biological method of nanoparticle synthesis.25,36 | ||
| Fig. 2 UV-visible spectrophotometric analysis of pigment-capped AgNPs (pcAgNPs) at different time intervals. | ||
The XRD analysis of the bio-synthesized pcAgNPs (Fig. 3) revealed the face-centered cubic (fcc) symmetry and crystal structure of silver (nanocrystals). Prominent peaks at respective 2θ = 38.02°, 44.24°, 64.35°, and 77.28° representing zero-valent fcc silver indexed for the (1 1 1), (2 0 0), (2 2 0), and (3 1 1) crystal planes, respectively, due to Bragg's reflections were present (JCPDS file no. 04-0783)37 in addition to other peaks, which could have resulted due to the biomolecular interactions with nanoparticles.
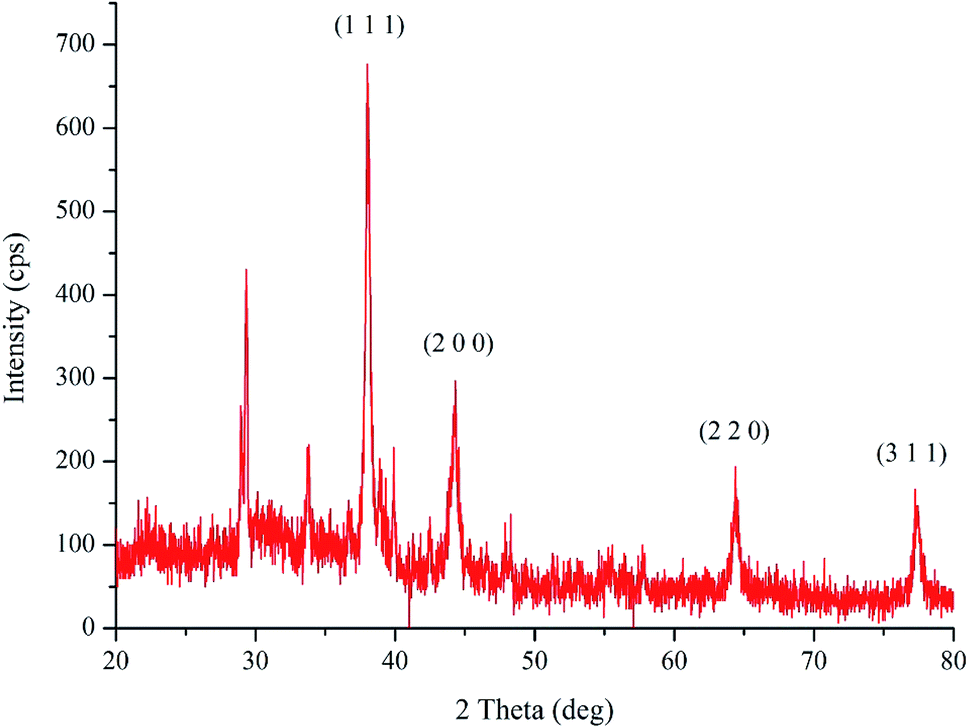 | ||
| Fig. 3 X-ray diffraction pattern of the biosynthesized pcAgNPs. | ||
The topography and size of pcAgNPs were examined by atomic force microscopy (AFM), dynamic light scattering (DLS) and high-resolution transmission electron microscopy (HRTEM). As observed from the atomic microscopy results, the topography of the silver nanoparticles with no definite agglomeration confirms the polydispersity of the nanoparticles (Fig. 4A). Subsequently, the particle size distribution of the silver nanoparticles (Fig. 4B) unveiled the narrow size distribution in the range of 25–55 nm, with the mean diameter of 40 nm and zeta potential of −32.3 mV; this indicated a strong electrostatic attraction between the silver ions and the metabolites, conferring stability.23,38
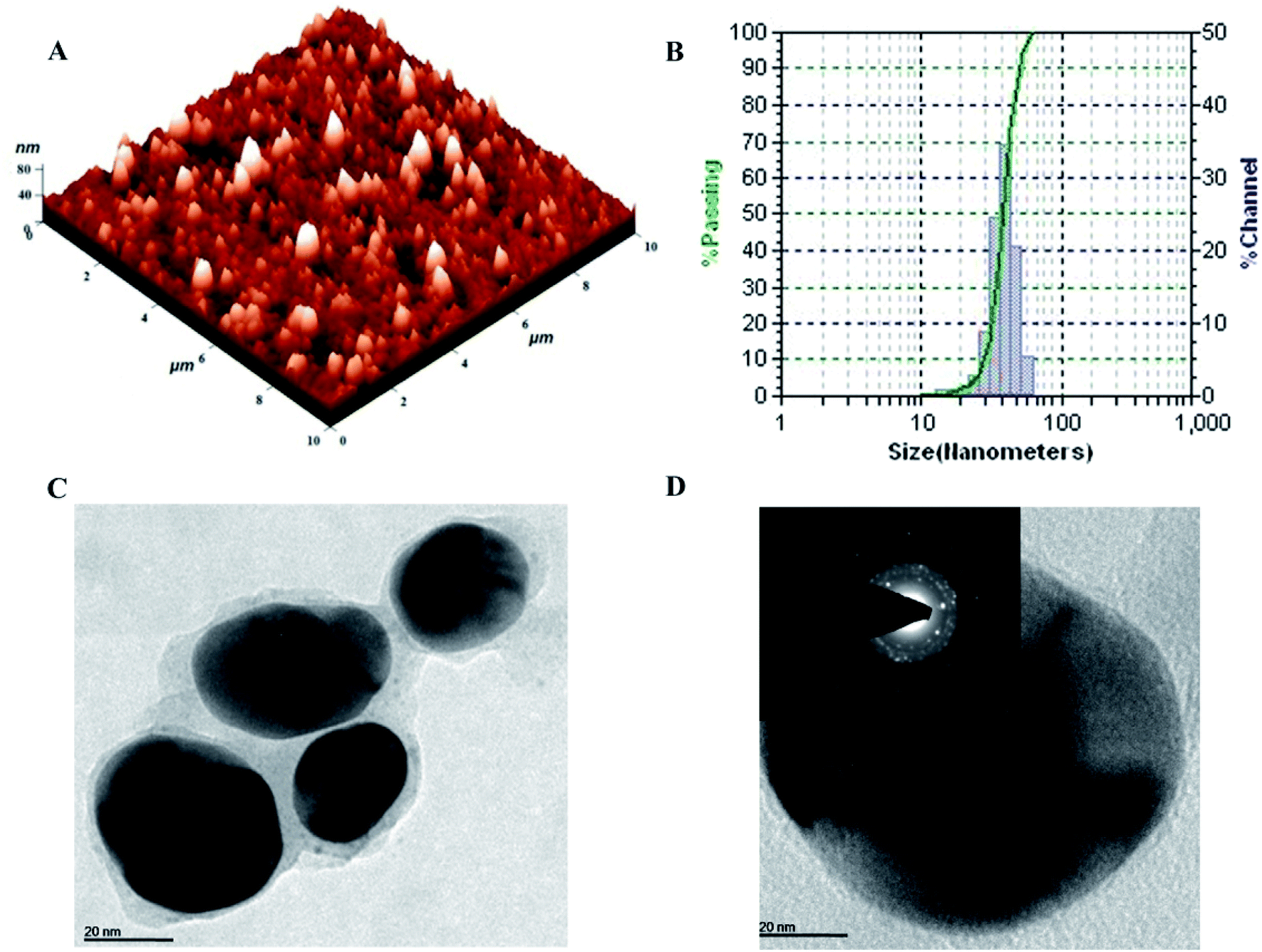 | ||
| Fig. 4 Characterization of pcAgNPs. Atomic Force Microscopic image of size 10 × 10 μm with height profile at 80 nm (A); hydrodynamic diameter in the range of 10–100 nm (B); HR-TEM micrograph at a scale bar of 20 nm (C); and the corresponding SAED pattern showing four diffraction rings, indicating the purity at a scale bar of 20 nm (D). | ||
Further confirmation of the size of the nanoparticles analyzed through HRTEM with SAED patterns (Fig. 4C and D) demonstrated the spherical and nearly spherical shapes of the silver nanoparticles in the size range 25–50 nm, which were found to be consistent with the results obtained from dynamic light scattering. The inset of Fig. 4D shows the selected area diffraction (SAED) pattern of the silver nanoparticles. The smooth surface and ring-like diffraction pattern indicate that the particles are crystalline with the characteristic lattice planes of silver.39
FT-IR spectroscopy was performed to analyze the functional quality of the product and the spectrum was acquired in the range of 4000–400 cm−1 (Fig. 5). The band seen at 3400.9 cm−1 is assigned to the stretching vibrations of O–H and H. The bending vibration of primary amine can be observed in the region of 1650–1580 cm−1 and the C–N stretching vibrations correspond to aliphatic amines, as represented by a peak in the region of 1250–1020 cm−1. This indicated the involvement of a polypeptide, as evidenced from the amide linkages between the amino acid residues. In addition, the stretching vibrations of –C–C– were observed at 1429 cm−1, which accounted for the aromatic compound involved in the reaction; this is probably the role of pyocyanin (pigment), offering stability to the nanoparticles. From the observation, it was confirmed that biomolecules such as protein and pigment moieties were involved in the reduction and stabilization of silver nanoparticles through intermolecular attractions.40,41
 | ||
| Fig. 5 FT-IR spectrum of the biosynthesized pcAgNPs showing the possible biomolecular interactions of the pigment. | ||
3.4. Antimicrobial activity
The antimicrobial study using nanomaterials was performed because of the increase in the multidrug-resistant strains of bacteria, which could provide insights for an alternative drug of choice including AgNPs and silver-based products. pcAgNPs prepared at various concentrations were found to be effective toward a series of antibiotic-resistant bacterial strains on par with those exposed to broad-spectrum antibiotics such as Amikacin, Tetracycline, Vancomycin, Cefepime, Netilmicin, Doxycycline and Chloramphenicol (Table 2) at 25 μg mL−1. The antibiogram pattern of the clinical isolates showed complete resistance toward a series of antibiotics at 25 μg mL−1 as compared with the standard antibiotic susceptibility chart.| Test strains | Antibiotics (25 μg mL−1) | Zone of inhibition (in mm) induced by pcAgNPs | AgNO3 | ||||
|---|---|---|---|---|---|---|---|
| 5 μg mL−1 | 15 μg mL−1 | 25 μg mL−1 | 50 μg mL−1 | ||||
| a AN – Amikacin; TET – Tetracycline; VA – Vancomycin; FEP – Cefepime; NET – Netilmicin; D – Doxycycline; C – Chloramphenicol; NZ – no zone. Data are representative of at least three independent experiments and expressed as mean ± standard deviation (SD) of the concentrations of Antibiotics and pcAgNPs. | |||||||
| S. aureus (MRSA) | AN | 7.7 ± 0.58 | 12.8 ± 0.79 | 15.8 ± 0.62 | 19.1 ± 0.70 | 24 ± 0.82 | 10.5 ± 1.08 |
| TET | 9.3 ± 0.29 | ||||||
| VA | NZ | ||||||
| S. epidermidis (MRSE) | AN | 8.7 ± 0.26 | 11.7 ± 1.25 | 15.2 ± 0.59 | 18 ± 0.82 | 23 ± 2.16 | 10.7 ± 1.25 |
| TET | 9.8 ± 0.21 | ||||||
| VA | NZ | ||||||
| P. mirabilis | AN | 12.3 ± 0.76 | 10.8 ± 1.03 | 15.3 ± 1.25 | 18 ± 1.41 | 24.5 ± 1.23 | 13.5 ± 0.41 |
| FEP | 10.9 ± 0.36 | ||||||
| NET | 10.1 ± 0.51 | ||||||
| E. coli | AN | 12.7 ± 0.58 | 9.8 ± 0.85 | 14 ± 2.16 | 17.3 ± 0.94 | 22.5 ± 1.05 | 11.2 ± 0.85 |
| TET | 9.2 ± 0.59 | ||||||
| NET | 10.8 ± 0.35 | ||||||
| K. pneumoniae | AN | 11.2 ± 0.29 | 10.3 ± 0.47 | 15.7 ± 0.94 | 18.2 ± 0.57 | 24.2 ± 0.85 | 8.7 ± 0.47 |
| TET | 9.3 ± 0.31 | ||||||
| NET | 7.5 ± 0.87 | ||||||
| A. baumannii | AN | 11.5 ± 0.35 | 9.6 ± 0.42 | 14.7 ± 0.47 | 18.3 ± 0.47 | 23.7 ± 0.94 | 9.7 ± 0.38 |
| TET | 10.3 ± 0.29 | ||||||
| D | 7.6 ± 0.23 | ||||||
| Enterococcus sp. (VRE) | C | 10.3 ± 0.25 | 10 ± 0.82 | 16 ± 1.41 | 18.7 ± 0.94 | 22.1 ± 0.98 | 12.3 ± 0.47 |
| TET | 11.8 ± 0.75 | ||||||
| VA | 12 ± 0.15 | ||||||
AgNPs (5, 15, 25 and 50 μg mL−1) exhibited significant antimicrobial activity toward all antibiotic-resistant Gram-positive and Gram-negative strains in a dose-dependent manner. Proteus mirabilis showed maximum susceptibility toward pcAgNPs with ∼25 mm ZOI, followed by K. pneumoniae (24 mm) and Acinetobacter baumannii (24 mm). The isolates that did not respond to the broad-spectrum antibiotics such as MRSA and MRSE showed susceptibility toward pcAgNPs at the same concentration as that of the antibiotics. A similar trend was observed in the case of E. coli and the vancomycin-resistant Enterococcus sp. with a ∼22 mm inhibition zone.
Furthermore, to manifest the antibacterial effect induced by pcAgNPs, the FESEM technique was adopted, for which Enterococcus sp. (VRE) and E. coli models were used. Regular FESEM micrographs (Fig. 6) revealed significant membrane damage on both the strains exposed to 25 μg mL−1 of pcAgNPs in 6 h from their exposure (Fig. 6b and d), whereas the control cells sustained no such damage and retained the coccal and rod-shaped morphologies (Fig. 6a and c). It was assumed that the dissolved silver ions' interaction with the trans-membrane proteins leads to the release of H+ by the influence of the proton-motive force,42 resulting in the formation of ‘pits’ that damage the cell walls.43 Subsequently, the membrane components became disorganized from their intact nature, accounting for the structural changes in the cell walls. This disrupted the permeability, which led to the death of the cells. The antimicrobial susceptibility toward the Gram-negative bacterium sustained extensive structural damage than the observation for the Gram-positive bacterium. It was presumed that the presence of the thick peptidoglycan in the Gram-positive bacterium has the ability to trap the positively charged silver ions. These phenomena suggest a possible antibacterial mechanism exerted by pcAgNPs through membrane damage and inhibition of cellular response.44
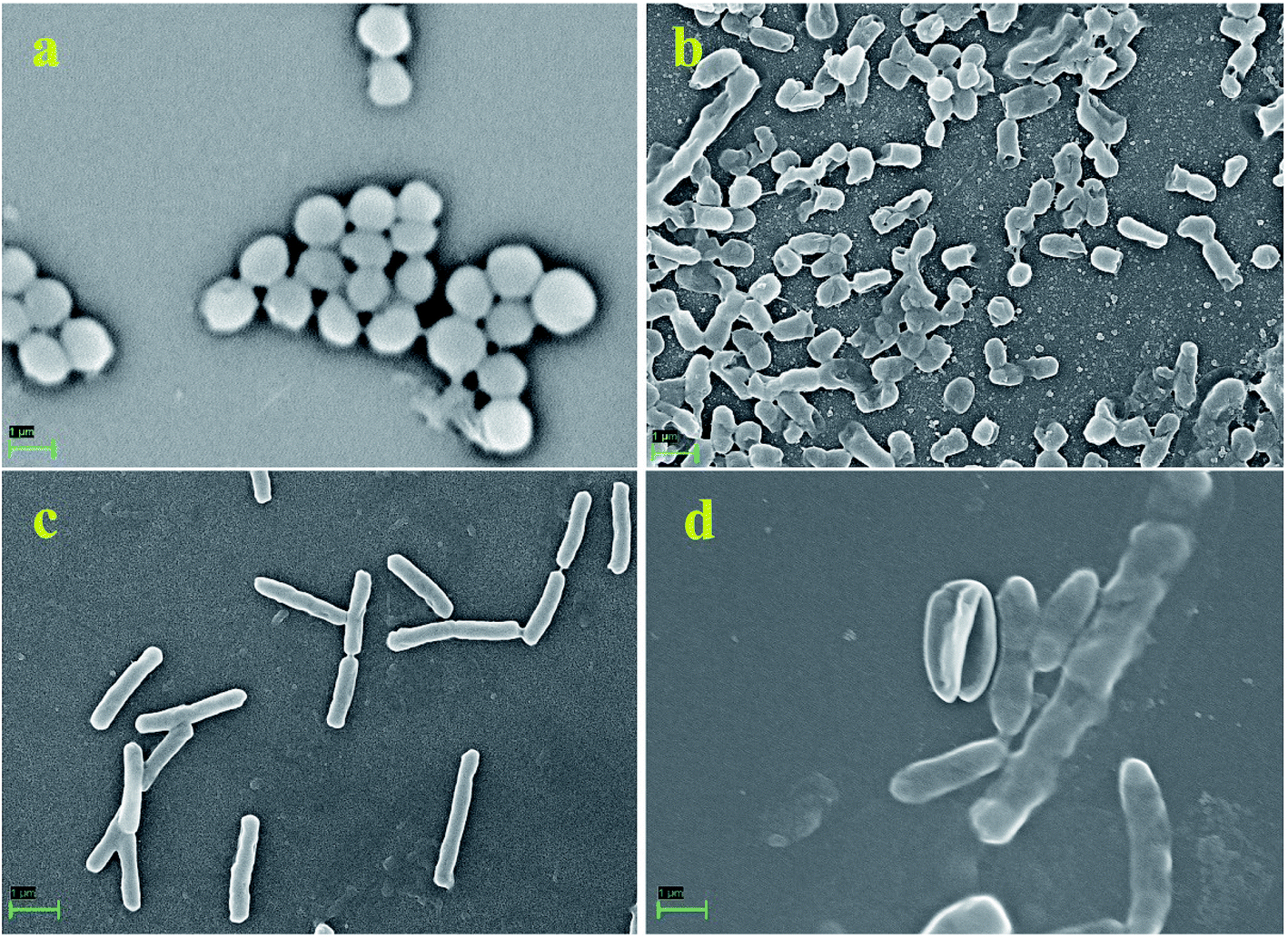 | ||
| Fig. 6 FESEM micrographs of antimicrobial susceptibility testing of pcAgNPs at 25 μg mL−1 toward Gram-positive and Gram-negative strains at a scale bar of 1 μm observed after 24 h of treatment with typical membrane damage. Enterococcus sp. (VRE) control (a); Enterococcus sp. (VRE) treated with pcAgNPs showing cell damage (b); E. coli control (c) and E. coli treated with pcAgNPs (d). | ||
3.5. Mechanistic antibacterial effect
It was evident from the electron microscopic study that pcAgNPs adhere and pass through the plasma membrane, creating pits that result in bacterial cell death. Reports on the metal–microbe interaction induced a significant rise in ROS, bringing about a deleterious effect related to oxidative stress.45 Therefore, the oxidative stress in the Gram-positive Enterococci (VRE) and Gram-negative E. coli after treatment with pcAgNPs was determined through the measurement of intracellular ROS generated using the DCFH-DA assay. This oxidation-sensitive fluorescent probe diffuses passively through the cell membrane and gets deacetylated by esterase to form non-fluorescent DCFH. When it comes in contact with ROS, it reacts to form a fluorescent product DC trapped inside the cell. Thus, the ROS level was directly proportional with the fluorescence intensity at 510 nm and in turn was correlated with the antibacterial effect of pcAgNPs on the bacterial cells. Fig. 7c clearly reveals the ROS generation with increased concentrations of pcAgNPs. The control cells (untreated with pcAgNPs) showed no difference; however, as the concentration of pcAgNPs increased, there was increased ROS generation (75–85%) at 50 μg mL−1 as compared with that for the positive control (100%), pertaining to the ROS-mediated cell death.46 | ||
| Fig. 7 Effect of pcAgNPs on the intracellular GSH concentration (a); DCFH-DA assay (b) for the quantification of ROS generation and the level of ROS production in pcAgNP-treated cells (c). | ||
Furthermore, to guarantee the state of the organisms that succumbed to cell death, the intracellular glutathione (GSH) concentration was measured. Glutathione, the most potent natural antioxidant, influences the physiological and biochemical aspects of both prokaryotic and eukaryotic cells. Eukaryotic cells possess relatively higher concentrations of GSH than prokaryotes, but it is chiefly found in E. coli, Streptococcus and Enterococcus sp. to maintain the cellular redox environment and protect the cells against oxidative stress by scavenging ROS. This tripeptide is present in the cytosol of cells at a concentration of 1–2 mM GSH. When this reduced form of GSH comes in contact with free radicals, it results in the formation of GS* (thiyl radical), which then reacts with another GS* radical to form glutathione disulphide (GSSG), an important regulator of biological processes.47
To decipher the oxidative stress induced by pcAgNPs, the intracellular GSH concentration was measured in pcAgNP-treated cells. As discussed, the initial concentrations of GSH were found to be 1.54 ± 0.41 and 1.26 ± 0.32, respectively, for the Enterococcus sp. and E. coli (control) cells (Fig. 7a). Besides, the pcAgNP-treated cells showed sequential depletion of GSH with a subsequent increase in the concentration. At 25 μg mL−1 concentration, there was three-fold reduction in the concentration of GSH to 0.54 ± 0.14 and 0.41 ± 0.06 mM, respectively, for Enterococci sp. and E. coli. Furthermore, the cells treated with 50 μg mL−1 pcAgNPs showed steady decline in the GSH concentration, i.e., ≤0.2 mM, which was negligible enough to support and protect the cells from cessation. The results from these studies suggested that the oxidative damage caused by ROS associated with reduced glutathione concentration might be responsible for the antibacterial activity of pcAgNPs.48
3.6. In vitro antiproliferative effect of pcAgNPs
 | ||
| Fig. 8 Cytotoxicity of Vero and HEp-2 cells exposed to pcAgNPs and their corresponding IC50 values. The IC50 value of Vero cells was 7.8 μg mL−1 and that of HEp-2 cells was 14.3 μg mL−1. | ||
 | ||
| Fig. 9 Phase contrast microscopic images of Vero and HEp-2 cells exposed to pcAgNPs and their corresponding cells stained with crystal violet observed with a 10× objective. Vero control (a and i); Vero cells at IC50 concentration (b and ii); HEp-2 control (c and iii); HEp-2 cells at IC50 concentration (d and iv). | ||
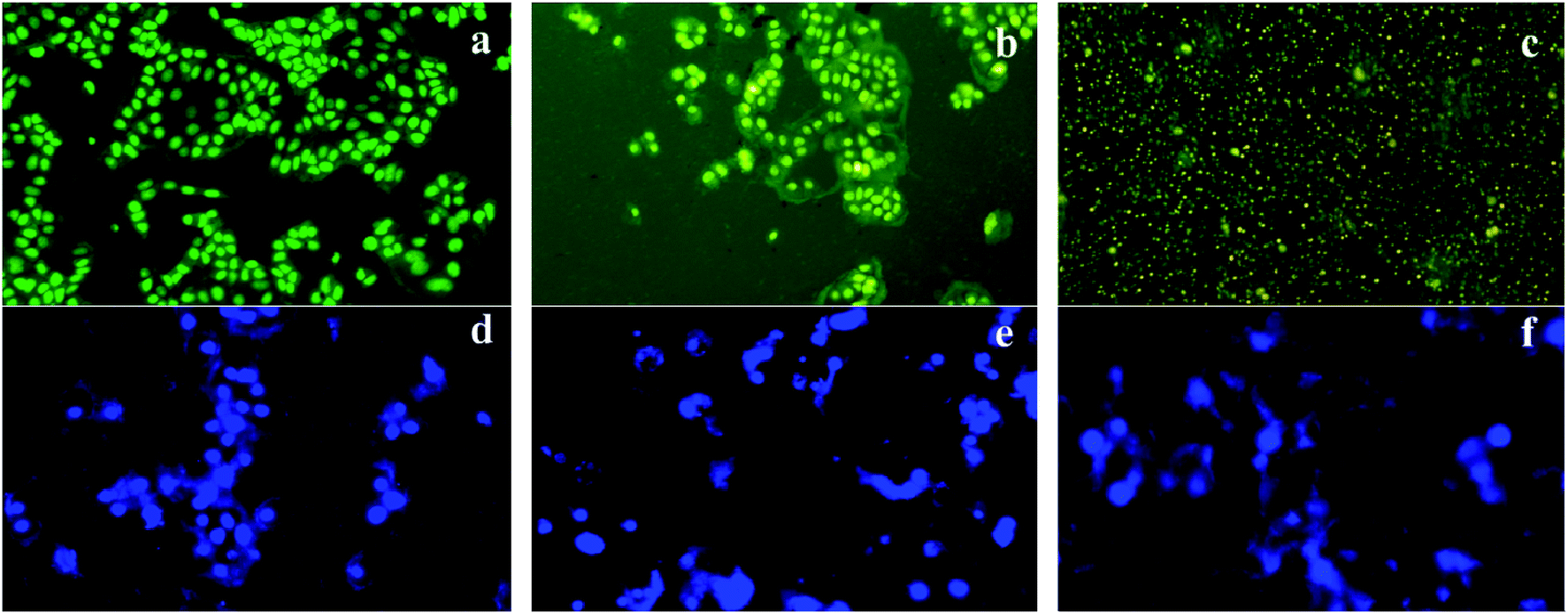 | ||
| Fig. 10 Fluorescence imaging of AO/EB-stained HEp-2 cells (a–c) and DAPI-stained cells (d–f) exposed to pcAgNPs attributing to apoptotic cell death. Green coloration denotes viable cells (a); early apoptotic cells with green fluorescence (b); and late apoptotic cells (c); control cells with intact nuclei (d); chromatin condensation (e) associated with nuclear fragmentation (f). | ||
 | ||
| Fig. 11 Apoptotic mechanism of HEp-2 cells treated with pcAgNPs. DNA fragmentation of HEp-2 cells exposed to pcAgNPs (a). RT-PCR analysis of p53 expression, leading to apoptotic cell death (b and c); pcAgNP activation of p53, leading to increased catalase expression. HEp-2 cells treated with 15 μg mL−1 pcAgNPs for 24 h, showing p53, p21 and catalase expressions assessed using Western blotting analysis. β-actin served as a loading control. Numbers were calculated by quantitative densitometric analysis and indicate the ratio of specific proteins versus β-actin. (d) Data are representative of at least three independent experiments. | ||
The apoptotic regulation mainly depends upon the transcriptional regulation of the target gene(s), possibly reflecting the mechanism of the p53 action in different cell types. Although p53 encodes a nuclear phosphoprotein with cancer-inhibiting properties, most of them remain inactive in more than half of human cancers.54 There is compelling evidence of protein accumulation in the mitochondria in response to abnormal proliferative signals and oxidative stress including DNA damage.
We showed in this study that pcAgNPs induced the expression of p53 as HEp-2 cells possess wild-type p53, which are non-functional and normally kept at low levels. The up-regulation of p53 (Fig. 11c and d) occurred in cells exposed to toxicant(s); in this case, pcAgNPs determined the tumor suppression function and apoptosis.55 We found that the pcAgNP treatment in HEp-2 cells upregulated the levels of p21 (Fig. 11d), the target of p53, when compared with the observation for control cells. Moreover, we also found that pcAgNPs increased the protein levels of catalase, one of the most important scavenger enzymes reported to be activated by p53. Altogether, these results indicate that pcAgNPs activated p53 to induce HEp-2 cell death possibly through the reduction of the intracellular ROS production by catalase.56
Although multiple genes are involved in carcinogenesis, p53 is the most widely studied tumor suppressor gene as most of the wild type-carrying p53 cancer cells need re-activation of its oncosuppressor function. These findings of the pigment-capped AgNPs from P. aeruginosa could provide an insight into gene therapeutic approaches applicable to a wide range of carcinomas. The growing body of research in the area of cancers may bring out a number of possible mechanisms by which p53 regulates tumours and may provide excellent prospects in clinical applications in optimizing targeting strategies.
4. Conclusion
We demonstrated the synthesis of silver nanoparticles using a rhizosphere-inhabiting, stress-tolerant P. aeruginosapa6 and its antimicrobial and anti-proliferative potentials were investigated. The pigment-capped AgNPs showed significant antibacterial activity toward drug-resistant bacteria in a dose-dependent fashion on par with standard antibiotics. The study revealed the possible mechanism by which p53 regulates the cell cycle through apoptosis, thus providing a guide for further optimization of a promising gene therapy system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge CSIR-CLRI and Sathyabama Institute of Science and Technology (Deemed to be University) for funding and providing the infrastructural facility.References
- D. K. Eric, Engines of Creation: The Coming Era of Nanotechnology, Doubleday, 1986 Search PubMed.
- V. Wagner, A. Dullaart, A. K. Bock and A. Zweck, The emerging nanomedicine landscape, Nat. Biotechnol., 2006, 24(10), 1211–1217 CrossRef CAS PubMed.
- D. A. LaVan, T. McGuire and R. Langer, Small-scale systems for in vivo drug delivery, Nat. Biotechnol., 2003, 21(10), 1184–1191 CrossRef CAS PubMed.
- J. Xu, A. Singh and M. M. Amiji, Redox-responsive targeted gelatin nanoparticles for delivery of combination wt-p53 expressing plasmid DNA and gemcitabine in the treatment of pancreatic cancer, BMC Cancer, 2014, 14, 75 CrossRef PubMed.
- O. Salata, Applications of nanoparticles in biology and medicine, J. Nanobiotechnol., 2004, 2(1), 3 CrossRef PubMed.
- D. Anderson, Persistence of antibiotic-resistant bacteria, Curr. Opin. Microbiol., 2003, 6(5), 452–456 CrossRef.
- R. Singh, M. S. Smitha and S. P. Singh, The role of nanotechnology in combating multi-drug resistant bacteria, J. Nanosci. Nanotechnol., 2014, 14(7), 4745–4756 CrossRef CAS PubMed.
- H. R. Kim, Y. Shin da, Y. J. Park, C. W. Park, S. M. Oh and K. H. Chung, Silver nanoparticles induce p53-mediated apoptosis in human bronchial epithelial (BEAS-2B) cells, J. Toxicol. Sci., 2014, 39(3), 401–412 CrossRef CAS PubMed.
- P. V. Asharani, G. L. K. Mun, M. P. Hande and S. Valiyaveettil, Cytotoxicity and genotoxicity of silver nanoparticles in human cells, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- S. Kim and D. Y. Ryu, Silver nanoparticle-induced oxidative stress, genotoxicity and apoptosis in cultured cells and animal tissues, J. Appl. Toxicol., 2013, 33(2), 78–89 CrossRef PubMed.
- B. Vogelstein, D. Lane and A. J. Levine, Surfing the p53 network, Nature, 2000, 408(6810), 307–310 CrossRef CAS PubMed.
- B. Sharma, W. Ma, I. M. Adjei, J. Panyam, S. Dimitrijevic and V. Labhasetwar, Nanoparticle-mediated p53 gene therapy for tumor inhibition, Drug Delivery Transl. Res., 2011, 1(1), 43–52 CrossRef CAS PubMed.
- A. A. Matthew, W. E. Cameron and L. R. Colin, Green chemistry and the health implications of nanoparticles, Green Chem., 2006, 8, 417–432 RSC.
- Q. Wu, H. Cao, Q. Luan, J. Zhang, Z. Wang, J. H. Warner and A. A. R. Watt, Biomolecule-Assisted synthesis of water-soluble silver nanoparticles and their biomedical applications, Inorg. Chem., 2008, 47(13), 5882–5888 CrossRef CAS PubMed.
- S. Su, X. Zuo, D. Pan, H. Pei, L. Wang, C. Fan and W. Huang, Design and applications of gold nanoparticle conjugates by exploiting biomolecule-gold nanoparticle interactions, Nanoscale, 2013, 5(7), 2589–2599 RSC.
- M. Lakshmipathy and A. Nanda, Biocatalysts of silver nanoparticles synthesized using an environmental isolate, Serratia marcescens S01, Int. J. ChemTech Res., 2013, 5(3), 1162–1168 CAS.
- C. Keel, U. Schnider, M. Maurhofer, C. Voisard, J. Laville, U. Burger, P. Wirthner, D. Haas and G. Defago, Suppression of root diseases by Pseudomonas fluorescens CHA0: importance of the bacterial secondary metabolite 2,4-diacetyl phloroglucinol, Mol. Plant-Microbe Interact., 1992, 5(1), 4–13 CrossRef CAS.
- P. Trivedi, A. Pandey and L. M. Palni, In vitro evaluation of antagonistic properties of Pseudomonas corrugata, Microbiol. Res., 2008, 163(3), 329–336 CrossRef PubMed.
- R. F. Harris and L. E. Sommers, Plate-Dilution frequency technique for assay of Microbial ecology, Appl. Microbiol., 1968, 16(2), 330–334 CAS.
- S. B. Robert, E. G. Murray and R. S. Nathan, Bergey's Manual of Determinative Bacteriology, Baltimore, Williams and Wilkins, 1962, p. 1094 Search PubMed.
- S. Karpagam, T. Sudhakar and M. Lakshmipathy, Microbial response of pyocyanin produced by Ps. aeruginosa toward clinical isolates of fungi, Int. J. Pharm. Pharm. Sci., 2013, 5(3), 870–873 Search PubMed.
- D. W. Essar, L. Eberly, A. Hadero and I. P. Crawford, Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications, J. Bacteriol., 1990, 172, 884–900 CrossRef CAS PubMed.
- A. Nanda and M. Saravanan, Biosynthesis of silver nanoparticles from Staphylococcus aureus and its antimicrobial activity against MRSA and MRSE, Nanomedicine, 2009, 5, 452–456 CrossRef CAS PubMed.
- P. Jeevan, K. Ramya and E. A. Rena, Extracellular biosynthesis of silver nanoparticles by culture supernatant of Pseudomonas aeruginosa, Indian J. Biotechnol., 2012, 11, 72–76 CAS.
- L. Muthukrishnan and A. Nanda, Geno-toxic study of silver bio-nanoparticles toward Gram-positive and Gram-negative clinical isolates, J. Pharma Res., 2013, 6, 725–729 CrossRef CAS.
- K. Shameli, M. B. Ahmad, S. D. Jazayeri, P. Shabanzadeh, P. Sangpour, H. Jahangirian and Y. Gharayebi, Investigation of antibacterial properties of silver nanoparticles prepared via green method, Chem. Cent. J., 2012, 6, 73 CAS.
- A. W. Bauer, W. M. M. Kirby, J. C. Sherris and M. Turck, Antibiotic susceptibility testing by a standardized single disk method, Am. J. Clin. Pathol., 1966, 36, 493–496 CrossRef.
- P. Sahoo, P. S. Murthy, S. Dhara, V. P. Venugopalan, A. Das and A. K. Tyagi, Probing the cellular damage in bacteria induced by GaN nanoparticles using confocal laser Raman spectroscopy, J. Nanopart. Res., 2013, 15, 1841 CrossRef.
- S. H. Kim, L. Hyeong-Seon, R. Deok-Seon, C. Soo-Jae and L. Dong-Seok, Antibacterial activity of silver-nanoparticles against Staphylococcus aureus and Escherichia coli, Korean J. Microbiol. Biotechnol., 2011, 39(1), 77–85 CAS.
- E. J. Park, J. Yi, Y. Kim, K. Choi and K. Park, Silver nanoparticles induce cytotoxicity by a Trojan-horse type mechanism, Toxicol. in Vitro, 2010, 24, 872–878 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS.
- Z. Darzynkiewicz, X. Li and J. Gong, Assays of cell viability: discrimination of cells dying by apoptosis, Methods Cell Biol., 1994, 41, 15–38 CrossRef CAS PubMed.
- J. A. Collins, C. A. Schandi, K. K. Young, J. Vesely and M. C. Willingham, Major DNA fragmentation is a late event in apoptosis, J. Histochem. Cytochem., 1997, 45(7), 923–934 CrossRef CAS PubMed.
- H. Liang, J. Duan, C. D. Sibley, M. G. Surette and K. Duan, Identification of mutants with altered phenazine production in Pseudomonas aeruginosa, J. Med. Microbiol., 2011, 60, 22–34 CrossRef CAS PubMed.
- M. Lakshmipathy and A. Nanda, Bio-processing and prophylactic efficacy of silver nanoparticles, Proc. Indian Natl. Sci. Acad., 2013, 79(3), 473–479 CAS.
- N. Duran, P. D. Marcato, O. L. Alves, G. I. H. De Souza and E. Esposito, Mechanistic aspects of biosynthesis of silver nanoparticles by several Fusarium oxysporum strains, J. Nanobiotechnol., 2005, 3, 8 CrossRef PubMed.
- V. Gopinath, D. MubarakAli, S. Priyadarshini, N. M. Priyadharsshini, N. Thajuddin and P. Velusamy, Biosynthesis of silver nanoparticles from Tribulus terrestris and its antimicrobial activity: a novel biological approach, Colloids Surf., B, 2012, 96(1), 69–74 CrossRef CAS PubMed.
- S. Das, J. Das, A. Samadder, S. S. Bhattacharyya, D. Das and A. R. Khuda-Bukhsh, Biosynthesized silver nanoparticles by ethanolic extracts of Phytolacca decandra, Gelsemium sempervirens, Hydrastis Canadensis and Thuja occidentalis induce differential cytotoxicity through G2/M arrest in A375 cells, Colloids Surf., B, 2013, 101, 325–336 CrossRef CAS PubMed.
- T. Shanmugasundaram, M. Radhakrishnan, V. Gopikrishnan, R. Pazhanimurugan and R. Balagurunathan, A study on the bactericidal, anti-biofouling, cytotoxic and antioxidant properties of actinobacterial synthesized silver nanoparticles, Colloids Surf., B, 2013, 111, 680–687 CrossRef CAS PubMed.
- A. Gole, C. Dash, V. Ramakrishnan, S. R. Sainkar and A. B. Mandale, et al., Pepsin-gold colloid conjugates: Preparation, characterization and enzyme activity, Langmuir, 2001, 17, 1674–1679 CrossRef CAS.
- D. Raghunandan, B. D. Mahesh, S. Basavaraja, S. D. Balaji, S. Y. Manjunath and A. Venkataraman, Microwave-assisted rapid extracellular synthesis of stable bio-functionalized silver nanoparticles from guava (Psidium guajava) leaf extract, J. Nanopart. Res., 2011, 13(5), 2021–2028 CrossRef CAS.
- Q. L. Feng, J. Wu, G. Q. Chen, F. Z. Cui, T. N. Kim and J. O. Kim, A mechanistic study of the antibacterial effect of silver ions on Escherichia coli and Staphylococcus aureus, J. Biomed. Mater. Res., 2008, 52, 662–668 CrossRef.
- I. Sondi and B. Salopek-Sondi, Silver nanoparticles as antimicrobial agent: a case study on E. coli as a model for Gram-negative bacteria, J. Colloid Interface Sci., 2004, 275, 177–182 CrossRef CAS PubMed.
- K. B. Holt and A. J. Bard, Interaction of silver (I) ions with the respiratory chain of Escherichia coli: an electrochemical and scanning electrochemical microscopy study of the antimicrobial mechanism of micromolar Ag+, Biochemistry, 2005, 44, 13214–13223 CrossRef CAS PubMed.
- P. V. Vignais, The superoxide-generating NADPH oxidase: structural aspects and activation mechanism, Cell. Mol. Life Sci., 2002, 59(9), 1428–1459 CrossRef CAS PubMed.
- S. Ninganagouda, V. Rathod, D. Singh, J. Hiremath, A. K. Singh, J. Mathew and M. ul-Haq, Growth kinetics and mechanistic action of reactive oxygen species released by silver nanoparticles from Aspergillus niger on Escherichia coli, BioMed Res. Int., 2014, 1–9, DOI:10.1155/2014/753419.
- G. V. Smirnova and O. N. Oktyabrsky, Glutathione in Bacteria, Biochemistry, 2005, 70(11), 1199–1211 CAS.
- H. Xu, F. Qu, H. Xu, W. Lai, Y. Andrew Wang, Z. P. Aguilar and H. Wei, Role of reactive oxygen species in the antibacterial mechanism of silver nanoparticles on Escherichia coli O157:H7, BioMetals, 2012, 25(1), 45–53, DOI:10.1007/s10534-011-9482-x.
- S. M. Hussain, K. L. Hess, J. M. Gearhart, K. T. Geiss and J. J. Schlager, In vitro toxicity of nanoparticles in BRL 3A rat liver cells, Toxicol. In Vitro, 2005, 19(7), 975–983 CrossRef CAS PubMed.
- L. Braydich-Stolle, S. Hussain, J. J. Schlager and M. C. Hofmann, In vitro cytotoxicity of nanoparticles in mammalian germline stem cells, Toxicol. Sci., 2005, 88, 412–419 CrossRef CAS PubMed.
- ATSDR (Agency for toxic substances and Disease Registry), Toxicological profile for Silver, Prepared by Clement international corporation, under Contract 205-88-0608, U.S. Public Health Service, 1990, ATSDR/TP-90-24 Search PubMed.
- C. Carlson, S. M. Hussain, A. M. Schrand, K. Braydich-Stolle, K. L. Hess, R. L. Jones and J. J. Schlager, Unique cellular interaction of silver nanoparticles: size-dependent generation of reactive oxygen species, J. Phys. Chem. B, 2008, 112(43), 13608–13619 CrossRef CAS PubMed.
- M. Schuler, E. Bossy-Wetzel, J. C. Goldstein, P. Fitzgerald and D. R. Green, p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release, J. Biol. Chem., 2000, 275(10), 7337–7342 CrossRef CAS PubMed.
- M. S. Greenblatt, W. P. Bennet, M. Hollstein and C. C. Harris, Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis, Cancer Res., 1994, 54, 4855 CAS.
- K. Oda, H. Arakawa, T. Tanaka, K. Matsuda, C. Tanikawa, T. Mori, H. Nishimori, K. Tamai, T. Tokino, Y. Nakamura and Y. Taya, p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53, Cell, 2000, 102(6), 849–862 CrossRef CAS PubMed.
- G. Y. Liou and P. Storz, Reactive oxygen species in cancer, Free Radic. Res., 2010, 44, 479–496 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |