DOI:
10.1039/C9RA01019J
(Paper)
RSC Adv., 2019,
9, 7432-7439
Structural exploration of AuxM− (M = Si, Ge, Sn; x = 9–12) clusters with a revised genetic algorithm†
Received
8th February 2019
, Accepted 27th February 2019
First published on 6th March 2019
Abstract
We used a revised genetic algorithm (GA) to explore the potential energy surface (PES) of AuxM− (x = 9–12; M = Si, Ge, Sn) clusters. The most interesting finding in the structural study of AuxSi− (x = 9–12) is the 3D (Au9Si− and Au10Si−) → quasi-planar 2D (Au11Si− and Au12Si−) structural evolution of the Si-doped clusters, which reflects the competition of Au–Au interactions (forming a 2D structure) and Au–Si interactions (forming a 3D structure). The AuxM− (x = 9–12; M = Ge, Sn) clusters have quasi-planar structures, which suggests a lower tendency of sp3 hybridization and a similarity of electronic structure for the Ge or Sn atom. Au9Si− and Au10Si− have a 3D structure, which can be viewed as being built from Au8Si− and Au9Si− with an extra Au atom bonded to a terminal gold atom, respectively. In contrast, the quasi-planar structures of AuxM− (x = 9–12; M = Ge, Sn) reflect the domination of the Au–Au interactions. Including the spin–orbit (SO) effects is very important to calculate the simulated spectrum (structural fingerprint information) in order to obtain quantitative agreement between theoretical and future experimental PES spectra.
I. Introduction
The properties of nanoclusters with several hundreds of atoms and molecules strongly depend on both their size and shape. The discovery of catalytic activities of supported gold nanoparticles has attracted intense research interest in the structures and properties of gaseous gold clusters,1 which provides an atomic-level understanding of catalytic mechanisms of gold nanoparticles. Various techniques have been used to investigate the structures of gold nanoclusters, such as photoelectron spectroscopy (PES),2 ion mobility,3,4 infrared multiphoton dissociation spectroscopy,5,6 and trapped ion electron diffraction.7 All of these techniques are quite powerful, especially when combined with ab initio calculations in obtaining structural information. Many of the unusual properties of the gold clusters derive from the strong relativistic effects that reduce the Au 5d–6s energy gap and enhance s–d hybridization.8–10 Small-sized anion gold clusters (Aun−) up to 12 atoms have been found to have 2D planar structures and are understood in terms of the s–d hybridization.3,10,11 In the medium-sized gold clusters, Au16− to Au18− clusters have been found to possess highly stable hollow-cage structures,7,12 and both Au20− and Au20 clusters have tetrahedral structures.6,13 Au24− possibly possesses tubular structure,14,15 and other large gold clusters, such as Au25−, Au32−, Au34−, and Au55− to Au64−,15–23 show core shell structures.
In comparison to pure gold clusters, doped gold clusters have received increasing attention because that the properties of gold clusters can be greatly influenced by the presence of impurities.24–32 For example, both W and V atoms can change the structure of the pure Au12− cluster to form an endohedral structure.33–35 The pure gold clusters doped with transition-metal atom also have been studied through experiment and theory.36–38 However, doping an isoelectronic substitution atom, such as Ag and Cu, in a pure gold cluster has a little effect on the electronic and geometrical structure of gold cluster anions.39–42
Few studies have been undertaken on the gold clusters doped with group-14 atoms.26,43–62 Small Si-doped gold clusters, such as Au4Si−1/0, Au4Si2−1/0, Au2Si2−1/0, and Au3Si3−1/0/+1, have been investigated by Wang et al.26,43,44 The Au/H analogy has been found in the tetrahedral Au4Si−1/0 cluster. For Ge or Sn dopant, they also affect the structure of gold clusters by different ways relative to the Si dopant. For example, the theoretical calculation shows that tetrahedral structure does not exist in Au4Ge− and Au4Sn−, and they have square-pyramidal structures.46 The tetrahedral structure derives from the sp3 hybridization in the Au4Si−. The results suggest that Si atom has stronger tendency to form directional bonding by sp3 hybridization than Ge and Sn. In order to obtain different isomers of atom cluster system, the genetic algorithm (GA) combined with density functional theory has been used to search the potential energy surface (PES). This method has been proven as an effective stochastic global search algorithm.63,64 To compare with the future experimental data, we computed the density of state (DOS) spectra by including the relativistic and spin–orbit effects for several lowest-lying isomers. The simulated DOS spectra for the isomers of different systems will be compared with future experimental data, providing considerable credence for the identified isomers for these clusters.
In this paper, the revised genetic algorithm was used to explore the possible low-lying cluster structures of AuxM− (x = 9–12; M = Si, Ge, Sn) systems. We want to explore the structure evolution of different Si-, Ge-, and Sn-doped gold anion clusters and find out the different effect of Si, Ge, and Sn atoms on pure gold clusters (Aux; x = 9–12).
II. Theoretical methods
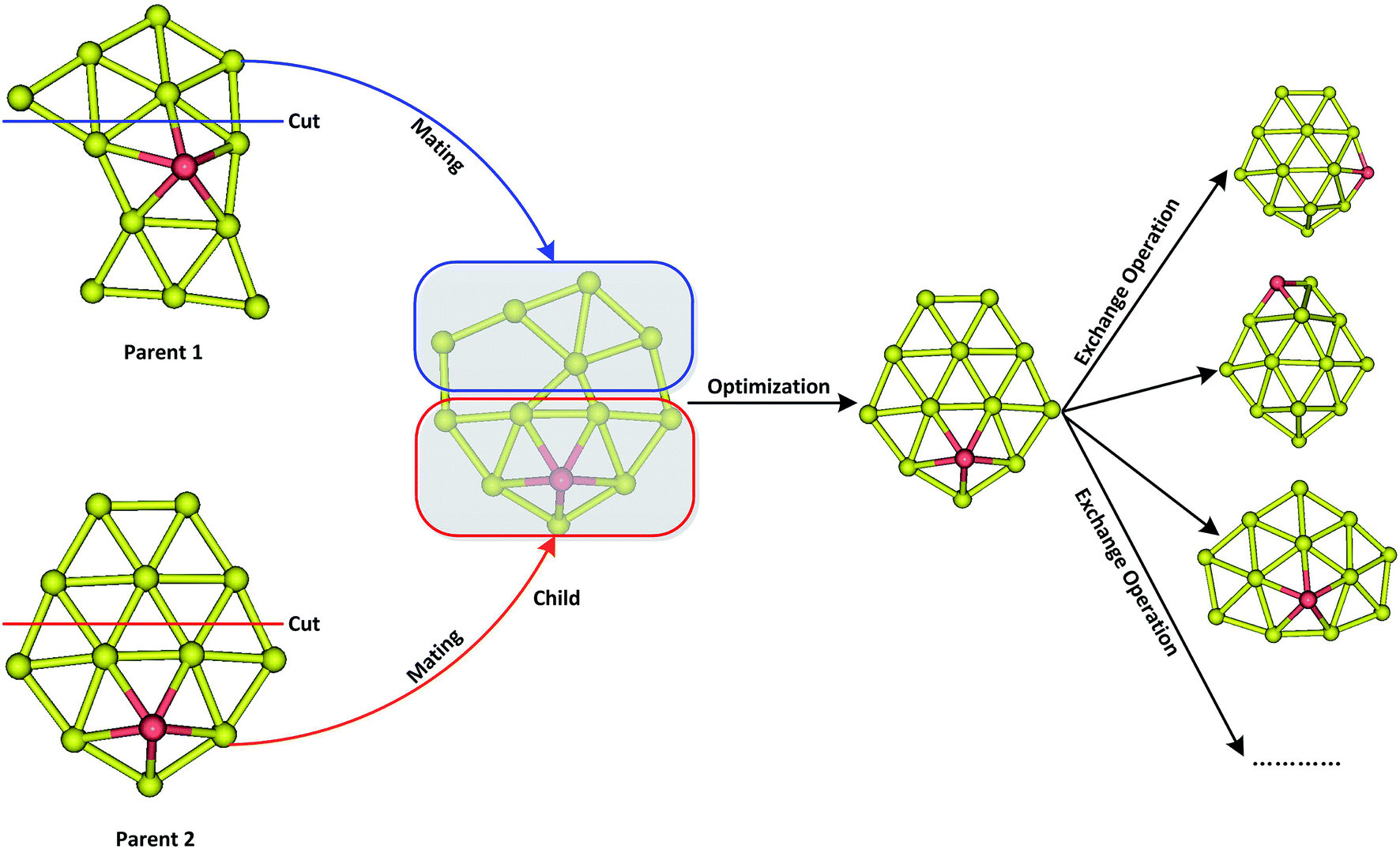
A revised genetic algorithm (GA), which is a global searching method based on biological evolution theory, was used to search global minimum of AuxM− (x = 9–12; M = Si, Ge, Sn) systems.64 The GA method contains several similar procedures as Deaven used: initial population, fitness function evaluation, selection, crossover, mutation and selecting new population.63 A new subprocess was added after crossover by our GA. The new process was called “exchange operation”, switching Cartesian coordinates of “M” and “Au” element in cluster (M = Si, Ge, Sn). Fig. 1 shows the basic operator of mating by cut and swap. Due to the complex PES of multicomponent cluster, the swap operator is very important for multicomponent searching to effectively explore the local minimum around PES basin of a fixed cluster architecture. The initial population containing 20 individuals were generated by random process, and their coordinates were optimized by quasi-Newton L-BFGS routine.65 The fitness was calculated by individual energy in population, and the fitness functions can be represented as below:
| P = (Ei − Emin)/(Emax − Emin) |
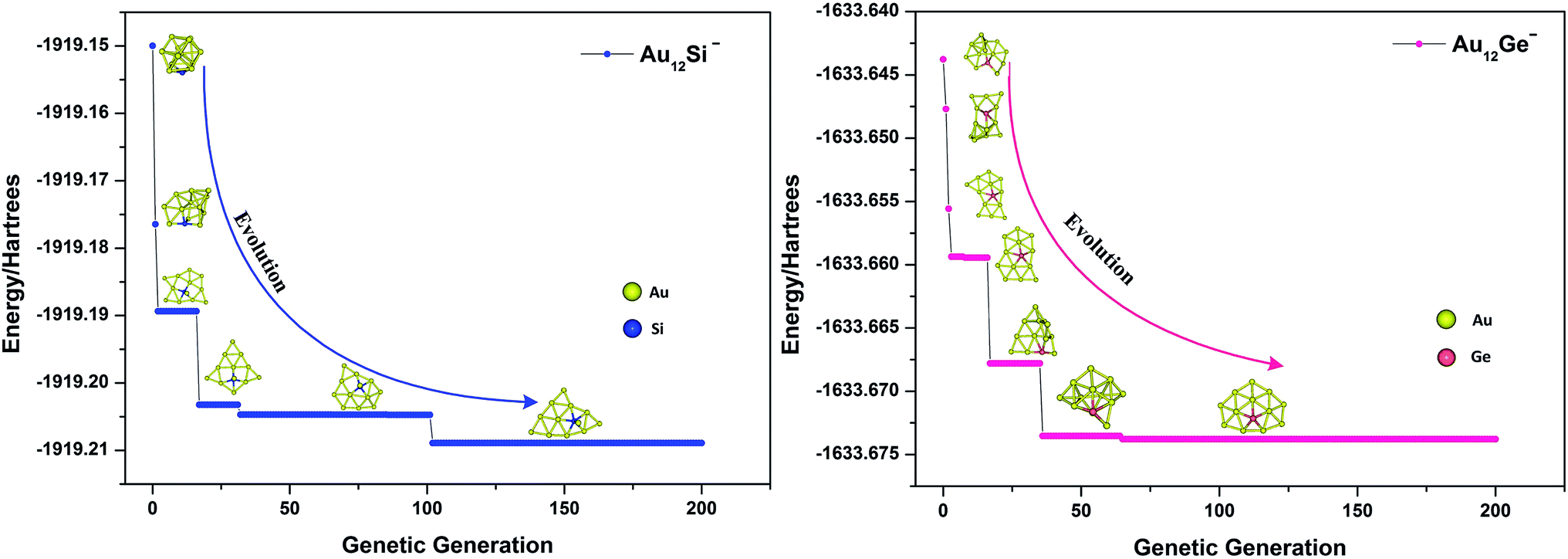
where Emax and Emin are the highest and lowest energy clusters in the current population, respectively. The selection of two parent individuals was accomplished by roulette wheel, and the low energy individual will have high probability to be selected for crossover. The crossover was achieved by a random plane passing through the center of mass of parent clusters, and the child was generated by collecting two slices atoms which lay above and below the plane of parent 1 and parent 2. The swap operation, which has been executed by exchanging coordinates between different elements based on local optimization structure, was performed for each new produced child. This operation was carried out by a fixed number of times set up by user (10 times for current research), and the lowest energy structure was selected as a new child to replace the child generated by crossover. The mutation is very important for keeping population diversity and also prevents searching trapped in local basin on PES. Ten percent of the offspring were selected for mutation, and the mutation was accomplished by the perturbation of each atomic coordinates with randomly values ranging from −1 to 1. Relaxation to local minimum was performed using L-BFGS method after each operation.65 All processes of crossover, mutation and selection were then repeated for a specified number of generations. Fig. 2 shows the total energy of Au12Ge− and Au12Sn− clusters against genetic generation by our revised GA.
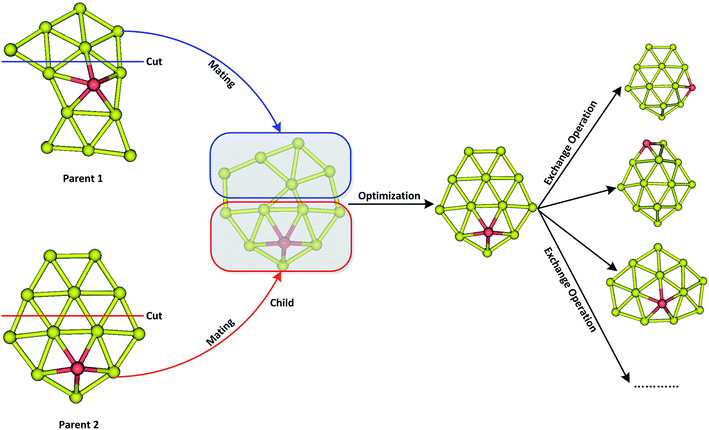 |
| | Fig. 1 The schematic diagram of mating operator by cut and swap operator via exchanging two different elements after mating. | |
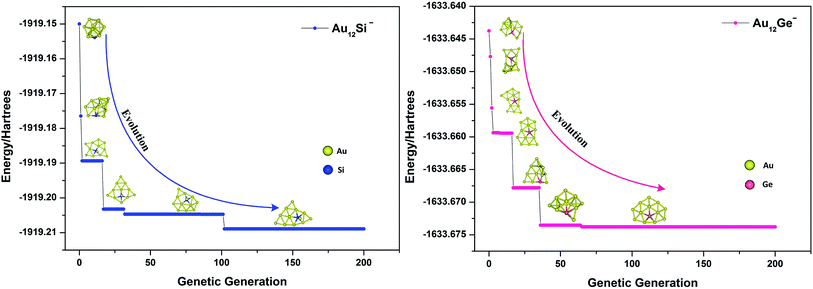 |
| | Fig. 2 The total energy against genetic generation of Au12Si− and Au12Ge− clusters using revised genetic algorithm. | |
To search for low-lying structures of AuxM− (x = 9–12; M = Si, Ge, Sn) systems, we used the revised GA global optimization technique combined with DFT for geometry optimization. Generalized gradient approximation in the Perdew–Burke–Ernzerhof (PBE)66 functional and the double-numerical polarized (DNP) basis set with effective core potentials (ECPs), implemented in the DMol3 code,67 were chosen for structure optimization of AuxM− (x = 9–12; M = Si, Ge, Sn) systems. To generate enough isomers in GA searches, a medium level convergence criterion was chosen such that the optimization gradient convergence was less than 4 × 10−3 hartree per Å and the optimization energy convergence was 2 × 10−5 hartree in the DFT calculations. When more than 200 generations were collected, the top-5 lowest-energy structures for last generation were reoptimized using the hybrid functional PBE0 and CRBNBL basis set for all of elements (PBE0/CRBNBL) in the NWChem software package.68 The isomers were ranked according to their relative energies. The relative energies of the top-5 isomers of the AuxM− (x = 9–12; M = Si, Ge, Sn) systems were further calculated using the PBEPBE/Def2-TZVPPD functional/basis set, PBE1PBE/Def2-TZVPPD functional/basis set, B3LYP/Def2-TZVPPD functional/basis set, and MP2/Def2-TZVPPD second-order Møller–Plesset perturbation theory/basis set, implemented in Gaussian 09 software package.69 In order to further confirm cluster structure information and compare with future experiment data, we simulated the photoelectron spectroscopy of AuxM− (x = 9–12; M = Si, Ge, Sn) systems. It is very important to include the spin–orbit (SO) effects in the calculation of the density of states in order to obtain quantitative agreement between theoretical and experimental PES spectra.58 We calculated simulated spectra of all candidate isomers at the PBE0/CRBNBL(SO) level using NWChem software package.68 Photoelectron spectra were calculated using PBE0 functional and CRBNBL basis set for Si, Ge, and Sn, CRBNBL basis set for Au with spin–orbit effects included. The first vertical detachment energies (VDEs) of the anion clusters were calculated at PBE0/CRENBL(SO) level as the energy difference between the optimization anion isomer and the neutral at the corresponding anion geometry. The binding energies of deeper orbitals were then added to the first VDE to approximate the higher binding energy detachment features. Each peak was then fitted with a 35 meV-wide Gaussian curve to simulate the DOS spectra.
III. Theoretical results and discussions
The top-5 lowest-energy structures of the AuxM− (x = 9–12; M = Si, Ge, Sn) systems are depicted in Fig. S1–S4 in the ESI,† respectively, together with their simulated PES spectra and coordinates. Tables 1–4 list the relative energies of the corresponding isomers at several levels of theory (see table titles). The simulated spectra of the primary structures of the AuxM− (x = 9–12; M = Si, Ge, Sn) systems are depicted in Fig. 3–6.
Table 1 Relative energies of five low-lying isomers of Au9M− (M = Si, Ge, Sn) at PBE0/CRENBL (NWChem) and PBEPBE/Def2-TZVPPD (G09), PBE1PBE/Def2-TZVPPD (G09), B3LYP/Def2-TZVPPD (G09), and MP2/Def2-TZVPPD (G09) levels of theory and basis seta
| Relative energies (eV) |
| Isomer |
|
PBE0 |
PBEPBE |
PBE1PBE |
B3LYP |
MP2 |
| Isomers are ranked according to their relative energies at the PBE0/CRENBL level of theory. |
| Au9Si− |
1 |
0.000 |
0.000 |
0.000 |
0.040 |
0.448 |
| 2 |
0.182 |
0.174 |
0.265 |
0.315 |
0.972 |
| 3 |
0.186 |
0.141 |
0.252 |
0.195 |
0.908 |
| 4 |
0.186 |
0.072 |
0.171 |
0.160 |
0.802 |
| 5 |
0.379 |
0.170 |
0.127 |
0.000 |
0.000 |
| Au9Ge− |
1 |
0.000 |
0.034 |
0.122 |
0.150 |
0.422 |
| 2 |
0.005 |
0.000 |
0.000 |
0.000 |
0.019 |
| 3 |
0.047 |
0.001 |
0.104 |
0.097 |
0.409 |
| 4 |
0.069 |
0.123 |
0.205 |
0.336 |
0.828 |
| 5 |
0.104 |
0.069 |
0.074 |
0.069 |
0.000 |
| Au9Sn− |
1 |
0.000 |
0.000 |
0.000 |
0.000 |
0.034 |
| 2 |
0.090 |
0.140 |
0.015 |
0.147 |
0.000 |
| 3 |
0.270 |
0.295 |
0.206 |
0.330 |
0.322 |
| 4 |
0.271 |
0.232 |
0.129 |
0.229 |
0.112 |
| 5 |
0.273 |
0.324 |
0.233 |
0.379 |
0.372 |
Table 2 Relative energies of five low-lying isomers of Au10M− (M = Si, Ge, Sn) at PBE0/CRENBL (NWChem) and PBEPBE/Def2-TZVPPD (G09), PBE1PBE/Def2-TZVPPD (G09), B3LYP/Def2-TZVPPD (G09), and MP2/Def2-TZVPPD (G09) levels of theory and basis seta
| Relative energies (eV) |
| Isomer |
|
PBE0 |
PBEPBE |
PBE1PBE |
B3LYP |
MP2 |
| Isomers are ranked according to their relative energies at the PBE0/CRENBL level of theory. |
| Au10Si− |
1 |
0.000 |
0.000 |
0.000 |
0.000 |
0.017 |
| 2 |
0.010 |
0.113 |
0.249 |
0.281 |
0.789 |
| 3 |
0.032 |
0.011 |
0.048 |
0.044 |
0.182 |
| 4 |
0.069 |
0.075 |
0.084 |
0.152 |
0.185 |
| 5 |
0.072 |
0.098 |
0.144 |
0.045 |
0.000 |
| Au10Ge− |
1 |
0.000 |
0.133 |
0.082 |
0.321 |
0.647 |
| 2 |
0.015 |
0.000 |
0.019 |
0.144 |
0.526 |
| 3 |
0.066 |
0.099 |
0.069 |
0.196 |
0.388 |
| 4 |
0.101 |
0.060 |
0.000 |
0.000 |
0.000 |
| 5 |
0.140 |
0.176 |
0.147 |
0.291 |
0.483 |
| Au10Sn− |
1 |
0.000 |
0.000 |
0.000 |
0.000 |
0.098 |
| 2 |
0.078 |
0.103 |
0.060 |
0.063 |
0.000 |
| 3 |
0.112 |
0.161 |
0.146 |
0.187 |
0.291 |
| 4 |
0.149 |
0.151 |
0.185 |
0.190 |
0.353 |
| 5 |
0.175 |
0.213 |
0.186 |
0.205 |
0.192 |
Table 3 Relative energies of five low-lying isomers of Au11M− (M = Si, Ge, Sn) at PBE0/CRENBL (NWChem) and PBEPBE/Def2-TZVPPD (G09), PBE1PBE/Def2-TZVPPD (G09), B3LYP/Def2-TZVPPD (G09), and MP2/Def2-TZVPPD (G09) levels of theory and basis seta
| Relative energies (eV) |
| Isomer |
|
PBE0 |
PBEPBE |
PBE1PBE |
B3LYP |
MP2 |
| Isomers are ranked according to their relative energies at the PBE0/CRENBL level of theory. |
| Au11Si− |
1 |
0.000 |
0.001 |
0.031 |
0.022 |
0.243 |
| 2 |
0.082 |
0.000 |
0.000 |
0.000 |
0.000 |
| 3 |
0.106 |
0.074 |
0.089 |
0.259 |
0.506 |
| 4 |
0.114 |
0.136 |
0.159 |
0.188 |
0.394 |
| 5 |
0.130 |
0.415 |
0.459 |
0.951 |
1.658 |
| Au11Ge− |
1 |
0.000 |
0.009 |
0.065 |
0.186 |
0.470 |
| 2 |
0.035 |
0.238 |
0.161 |
0.828 |
1.264 |
| 3 |
0.051 |
0.043 |
0.112 |
0.247 |
0.643 |
| 4 |
0.125 |
0.000 |
0.000 |
0.000 |
0.000 |
| 5 |
0.134 |
0.047 |
0.116 |
0.122 |
0.210 |
| Au11Sn− |
1 |
0.000 |
0.000 |
0.000 |
0.032 |
0.226 |
| 2 |
0.009 |
0.037 |
0.050 |
0.106 |
0.410 |
| 3 |
0.100 |
0.022 |
0.143 |
0.071 |
0.474 |
| 4 |
0.144 |
0.075 |
0.088 |
0.000 |
0.000 |
| 5 |
0.196 |
0.183 |
0.257 |
0.200 |
0.452 |
Table 4 Relative energies of five low-lying isomers of Au12M− (M = Si, Ge, Sn) at PBE0/CRENBL (NWChem) and PBEPBE/Def2-TZVPPD (G09), PBE1PBE/Def2-TZVPPD (G09), B3LYP/Def2-TZVPPD (G09), and MP2/Def2-TZVPPD (G09) levels of theory and basis seta
| Relative energies (eV) |
| Isomer |
|
PBE0 |
PBEPBE |
PBE1PBE |
B3LYP |
MP2 |
| Isomers are ranked according to their relative energies at the PBE0/CRENBL level of theory. |
| Au12Si− |
1 |
0.000 |
0.000 |
0.021 |
0.150 |
0.640 |
| 2 |
0.084 |
0.069 |
0.000 |
0.192 |
0.343 |
| 3 |
0.117 |
0.075 |
0.003 |
0.000 |
0.000 |
| 4 |
0.131 |
0.268 |
0.249 |
0.942 |
2.037 |
| 5 |
0.142 |
0.099 |
0.137 |
0.000 |
0.741 |
| Au12Ge− |
1 |
0.000 |
0.016 |
0.031 |
0.147 |
0.511 |
| 2 |
0.005 |
0.000 |
0.000 |
0.000 |
0.102 |
| 3 |
0.135 |
0.175 |
0.127 |
0.124 |
0.000 |
| 4 |
0.157 |
0.151 |
0.152 |
0.142 |
0.217 |
| 5 |
0.182 |
0.357 |
0.241 |
0.680 |
0.764 |
| Au12Sn− |
1 |
0.000 |
0.000 |
0.000 |
0.092 |
0.332 |
| 2 |
0.089 |
0.111 |
0.062 |
0.138 |
0.167 |
| 3 |
0.090 |
0.038 |
0.033 |
0.000 |
0.000 |
| 4 |
0.163 |
0.324 |
0.204 |
0.821 |
1.140 |
| 5 |
0.216 |
0.323 |
0.185 |
0.735 |
0.856 |
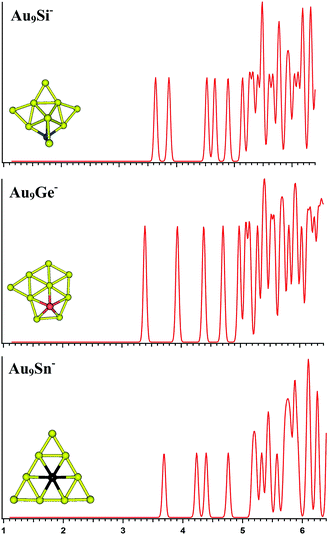 |
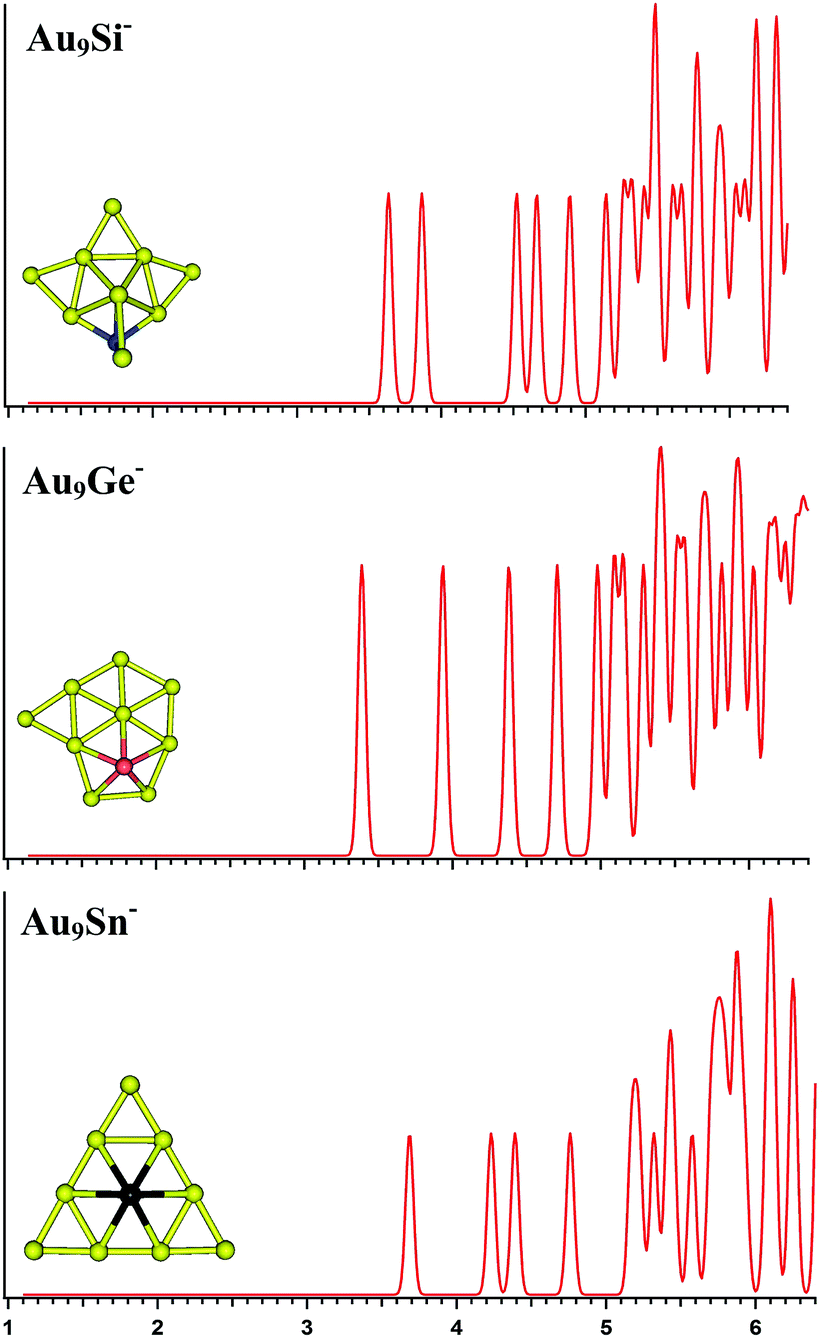
| | Fig. 3 The simulated photoelectron spectra of Au9M− (M = Si, Ge, Sn) using PBE0/CRENBL (NWChem) level of theory and basis set including the relativistic and spin–orbit effects. The insets show the corresponding structures. The dopant atoms are shown in color (Si in blue, Ge in red, and Sn in black). | |
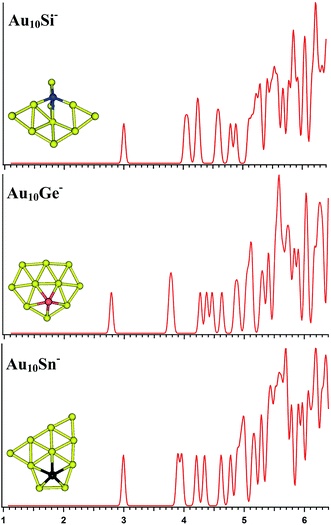 |
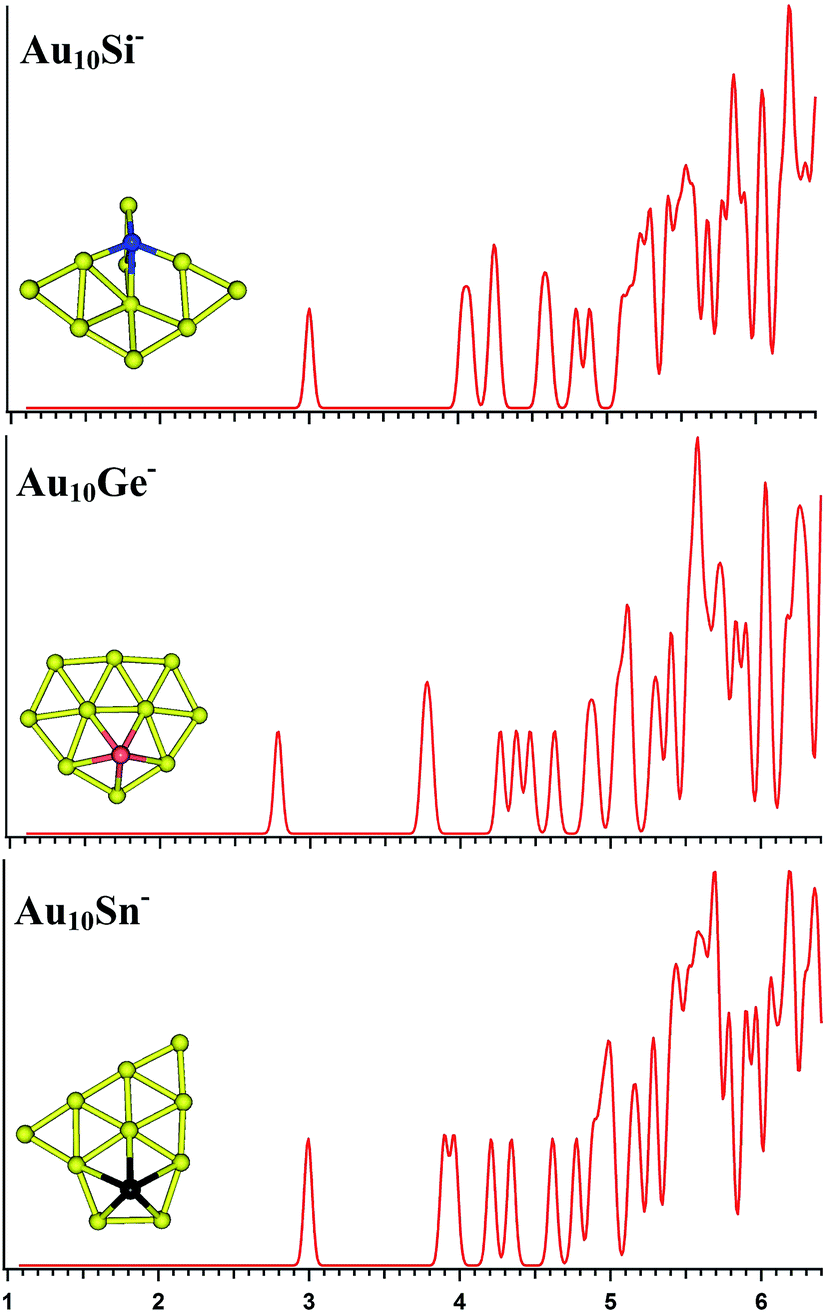
| | Fig. 4 The simulated photoelectron spectra of Au10M− (M = Si, Ge, Sn) using PBE0/CRENBL (NWChem) level of theory and basis set including the relativistic and spin–orbit effects. The insets show the corresponding structures. The dopant atoms are shown in color (Si in blue, Ge in red, and Sn in black). | |
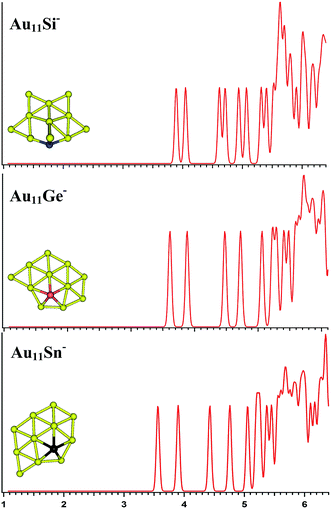 |
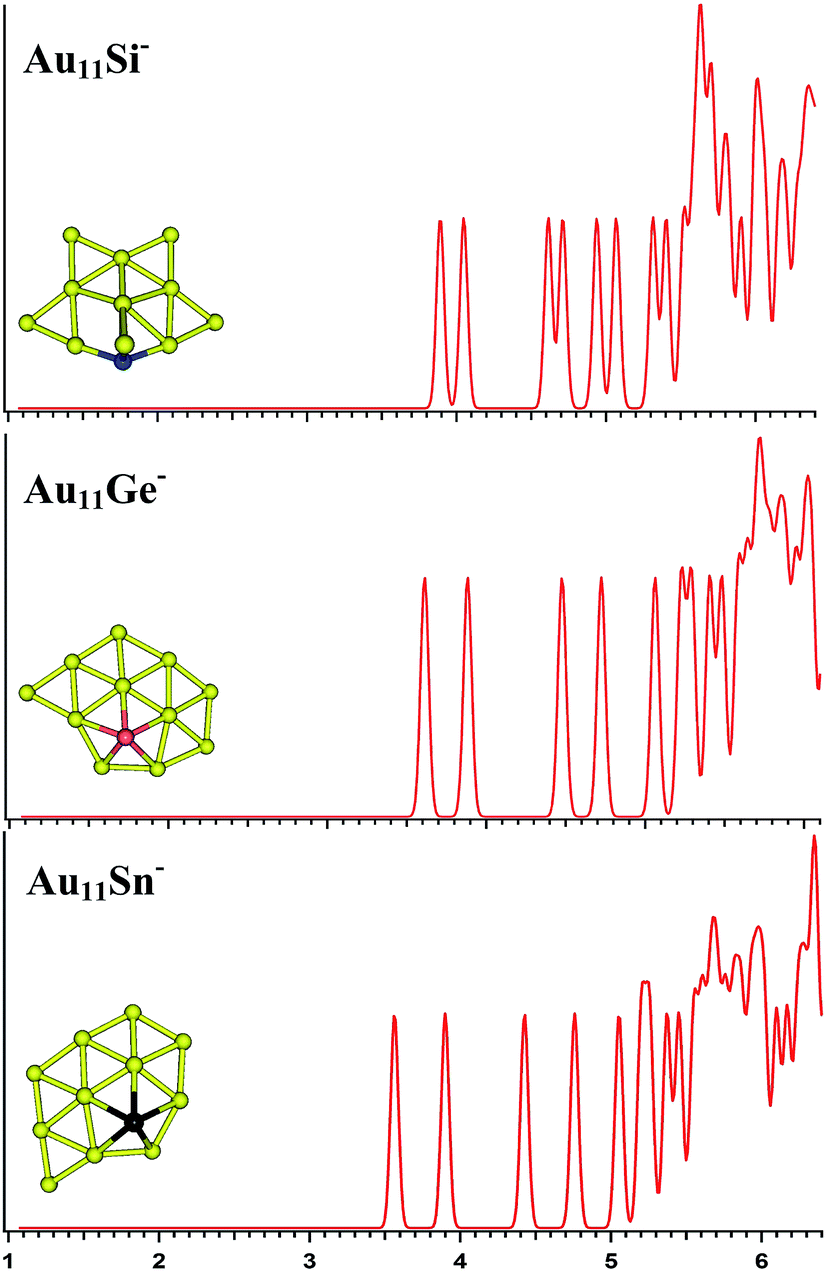
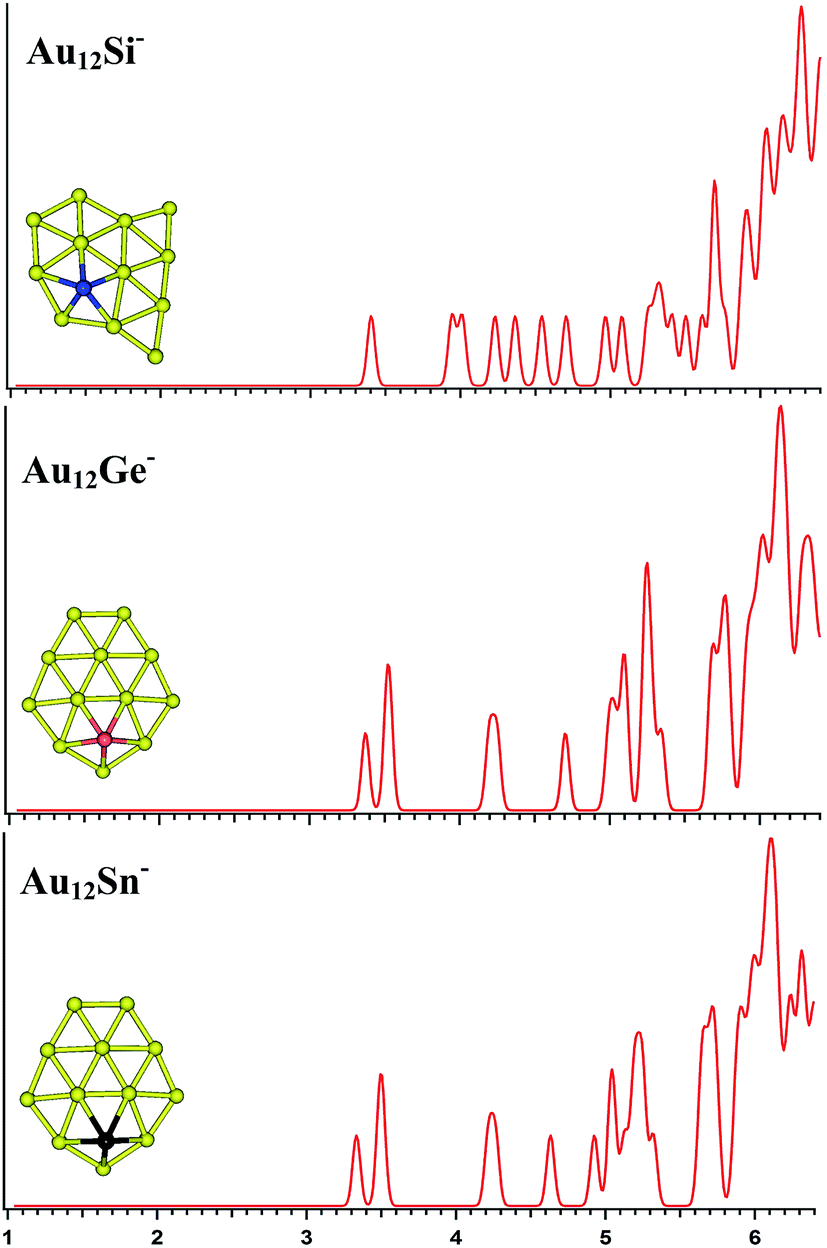
| | Fig. 5 The simulated photoelectron spectra of Au11M− (M = Si, Ge, Sn) using PBE0/CRENBL (NWChem) level of theory and basis set including the relativistic and spin–orbit effects. The insets show the corresponding structures. The dopant atoms are shown in color (Si in blue, Ge in red, and Sn in black). | |
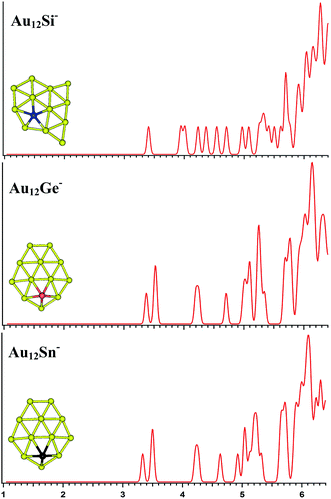 |
| | Fig. 6 The simulated photoelectron spectra of Au12M− (M = Si, Ge, Sn) using PBE0/CRENBL (NWChem) level of theory and basis set including the relativistic and spin–orbit effects. The insets show the corresponding structures. The dopant atoms are shown in color (Si in blue, Ge in red, and Sn in black). | |
Au9M−
For Au9Si−, PBE0 (NWChem), PBEPBE, and PBE1PBE computations all predict isomer 1 as the global minimum structure (Fig. S1la in the ESI†), which can be viewed as the lowest-energy structure of Au8Si− cluster bonded by another Au atom through a terminal gold atom.58 The B3LYP and MP2 calculations seem to prefer isomer 5 (Fig. S1le in the ESI†) as the lowest energy structure. In order to further identify the primary structure information for future PES experiment spectrum of Au9Si− cluster, the simulated spectra of the top-5 lowest-energy structures of Au9Si− cluster (Fig. S1la–e in the ESI†) have been calculated at the PBE0/CRENBL(SO) level. It is very important for theoretical spectrum simulation of Au atom to consider spin-orbital interaction by previous study.58 For Au9Ge−, PBE0 (NWChem) computation predicts isomer 1 (Fig. S1ma in the ESI†) as the global minimum structure, while the PBEPBE, PBE1PBE and B3LYP computations suggest isomer 2 (Fig. S1mb in the ESI†) as the global minimum structure. The MP2 calculations seem to prefer isomer 5 (Fig. S1me in the ESI†) as the lowest energy structure. For Au9Sn−, PBE0 (NWChem), PBEPBE, PBE1PBE, and B3LYP computations all predict isomer 1 as the global minimum structure (Fig. S1ra in the ESI†), while the MP2 computation suggests isomer 2 (Fig. S1rb in the ESI†) as the global minimum structure. Due to the similar chemical properties of Ge and Sn atom, they should have similar lowest-energy structure as previous study. But the Au9Ge− and Au9Sn− have different lowest energy structures by our calculation. Fig. 3 shows the simulated spectra of the primary structures of Au9M− (M = Si, Ge, Sn) clusters. The primary structure of Au9Si− cluster was dominated by Au8Si− cluster, reflecting that the electronic structure of Au9Si− cluster should be similar with Au8Si− cluster. The result shows that the lowest-energy structure of Au9Si− cluster has a strong tendency of forming tetrahedral bonding structure by sp3 hybridization for Si atom. However, Ge- and Sn-doped gold anion cluster have a quasi-planar structure differing from Au9Si− cluster. It suggests the lower tendency of sp3 hybridization for Ge and Sn atoms. The primary structures of Au9Ge− and Au9Sn− are mainly dominated by Au–Au interactions.
Au10M−
The PBE0 (NWChem), PBEPBE, PBE1PBE, and B3LYP calculations predict isomers 1 (Fig. S2la in the ESI†) as the global minimum structure for Au10Si−. Isomer 1 can be viewed based on the isomer 1 (Fig. S1la in the ESI†) of Au9Si− cluster by adding an Au atom. However, the MP2 calculation reveals isomer 5 as the lowest energy structure, which has a quasi-planar structure differing from isomer 1 (Fig. S2la in the ESI†). For Au10Ge−, The PBE0 (NWChem) computation (Table 2) indicates that isomer 1 (Fig. S2ma in the ESI†) has the global minimum, while PBEPBE calculation seems to prefer isomer 2 (Fig. S2mb in the ESI†) as the lowest energy structure, and PBE1PBE, B3LYP, and MP2 calculations reveal isomer 4 as the lowest energy structure. The isomer 1 of Au10Ge− is also a quasi-planar structure and can be viewed as being derived from isomer 1 of Au9Ge− by adding one Au atom. For Au10Sn−, PBE0 (NWChem), PBE1PBE, PBEPBE and B3LYP calculations reveal that isomer 1 is the lowest-energy structure, while MP2 calculation prefers isomer 2 (Fig. S2rb in the ESI†) as the lowest energy structure. Different theory level computations produce different energy order (Table 2). The isomer 1 and isomer 2 are the primary structures of Au10Sn− and can be viewed as being derived from isomer 2 of Au9Sn−. Fig. 4 shows the simulated spectra of the primary structures of Au10M− (M = Si, Ge, Sn) clusters. Au10Si− has a 3D structure, and shows a strong tendency of forming tetrahedral bonding structure by sp3 hybridization for Si atom. Au10M− (M = Ge, Sn) systems show the quasi-planar structures, reflecting that the structure is mainly dominated by Au–Au interactions.
Au11M−
For Au11Si−, the PBE0 (NWChem) calculation (Table 3) reveals isomer 1 (Fig. S3la in the ESI†) as the ground-state structure. Isomer 1 can be viewed as being derived from isomer 5 (Fig. S2le in the ESI†) of Au10Si− by attaching one Au atom to form triangle structure, and has a quasi-planar structure differing from the 3D structures of Au9Si− and Au10Si− because of sp3 hybridization for Si atom. Other computations suggest that isomer 2 (Fig. S3lb in the ESI†) is the lowest energy structure. The simulated spectrums of 5-top isomers of Au11Si− cluster can be found in ESI† (Fig. S3la–e in the ESI†). For Au11Ge−, PBE0 (NWChem) calculation (Table 3) indicates that isomer 1 (Fig. S3ma in the ESI†) is the global minimum structure and can be viewed as being derived from isomer 3 (Fig. S2mc in the ESI†) of Au10Ge− by adding one Au atom to form triangle structure. Other computations suggest isomer 2 (Fig. S3mb in the ESI†) as the lowest energy structure. For Au11Sn−, the lowest-energy structure is found to be isomer 1 (Fig. S3ra in the ESI†), which has the same structure as Au11Ge− (Fig. S3ma in the ESI†) for PBE0 (NWChem) calculation. B3LYP and MP2 calculations reveal that isomer 4 is the lowest-energy structure, and can be viewed as being derived from isomer 2 (Fig. S2rb in the ESI†) of Au10Sn− by attaching one Au atom to form triangle structure. Fig. 5 shows the simulated spectra of the primary structures of Au11M− (M = Si, Ge, Sn) clusters. All of the Au11M− (M = Si, Ge, Sn) systems show the same quasi-planar structure, reflecting that the structure is mainly dominated by Au–Au interactions.
Au12M−
The PBE0 (NWChem) and PBEPBE calculations indicate that isomer 1 (Fig. S4la in the ESI,† a quasi-planar structure) is the lowest-energy structure for Au12Si− cluster, and other calculations reveal isomer 2 and isomer 3 as the ground state structure. For Au12Ge−, PBE0 (NWChem) calculation (Table 4) reveals isomer 1 (Fig. S4ma in the ESI†) as the lowest energy structure, while PBE1PBE, PBEPBE, and B3LYP computations predict isomer 2 (Fig. S4mb in the ESI†) as the global minimum structure. Isomer 2 has very similar quasi-planar structure with isomer 1 of Au12Si− cluster. The MP2 calculations reveal isomer 3 has the global minimum, which can be viewed evolved from the isomer 1 of Au11Ge− by attaching one Au atom and subsequent structural relaxation. For Au12Sn−, PBE0 (NWChem), PBEPBE, and PBE1PBE calculations predict isomer 1 (Fig. S4ra in the ESI†) as the global minimum, which has a similar structure with isomer 1 of Au12Ge− cluster. However, the B3LYP and MP2 calculation reveals isomer 3 (Fig. S4rc in the ESI†) as the lowest energy structure. Fig. 6 shows the simulated spectra of the primary structures of Au12M− (M = Si, Ge, Sn) clusters. The Au12M− (M = Si, Ge, Sn) clusters have quasi-planar structures, reflecting that their structures are dominated by Au–Au interactions.
To summarize, the structure evolutions of AuxM− (x = 9–12, M = Si, Ge, Sn) systems have been explored by revised GA method. The most intriguing finding is the structure change of 3D to quasi-planar for AuxSi− (x = 9–12). The result suggests the competition between the tendency of forming tetrahedral bonding structure by sp3 hybridization for Si atom (Au–M interaction) and the tendency of forming planar structures by Au–Au interactions for small anion gold clusters. The Si-doped gold clusters easily form local tetrahedral bonding structures because of the strong sp3 hybridization of the Si atom. But the structure changing from 3D to quasi-planar is found at x = 11 for AuxSi− (x = 9–12) systems, which reflects that their structures are dominated by Au–Au interactions when the cluster size increases for AuxSi− (x = 9–12) systems. However, the Ge- or Sn-doped gold clusters exhibit different structural images from pure gold anion clusters, which reflects its lower tendency of sp3 hybridization in the doped clusters. The structural evolutions of AuxM− (x = 9–12; M = Si, Ge, Sn) systems reflect that the Au–Au interactions become competitive in deciding the cluster structures when the cluster size increases for dopant clusters of gold. Including the spin–orbit (SO) effects are very important to calculate the density of states in order to obtain quantitative agreement between theoretical and future experimental PES spectra.
IV. Conclusion
In conclusion, a detailed investigation of the structures and isomers of AuxM− (x = 9–12; M = Si, Ge, Sn) clusters using revised genetic algorithm combined with density functional theory were reported. The subprocess of exchange operation was added to explore local basin on PES for multicomponent systems in GA method. The Au9Si− and Au10Si− clusters have a tetrahedral-based 3D structure, and Au11Si− and Au12Si− clusters have a quasi-planar structure. The results show that the Si-doped gold clusters, which often form 3D direction bonding structure owing to the strong sp3 hybridization of Si atom, will be replaced by Au–Au interactions to form quasi-planar structure when the cluster size increases for dopant clusters of gold. However, the quasi-planar structures of Ge- or Sn-doped gold clusters reflect that the Ge and Sn atoms have the lower tendency of s–p hybridization in the doped clusters. For AuxM− (x = 9–12; M = Ge, Sn) systems, quasi-planar structures tend to be formed due to the domination of Au–Au interactions, suggesting that the Ge and Sn atoms have the lower tendency of s–p hybridization in the doped clusters.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The theoretical work was supported by the National Natural Science Foundation of China (Grant No. 51601189), and the foundation from Sichuan science and technology program (Grant No. 2017JY0117). Some of the computations were performed at the Supercomputing Center of the Chinese Academy of Sciences.
References
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- K. J. Taylor, C. L. Pettiettehall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 96, 3319–3329 CrossRef CAS.
- F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Gilb, T. Bierweiler and M. M. Kappes, J. Chem. Phys., 2002, 117, 6982–6990 CrossRef CAS.
- P. Weis, Int. J. Mass Spectrom., 2005, 245, 1–13 CrossRef CAS.
- A. Fielicke, A. Kirilyuk, C. Ratsch, J. Behler, M. Scheffler, G. von Helden and G. Meijer, Phys. Rev. Lett., 2004, 93, 023401–023405 CrossRef PubMed.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed.
- X. Xing, B. Yoon, U. Landman and J. H. Parks, Phys. Rev. B, 2006, 74, 165423 CrossRef.
- P. Pyykko, Chem. Rev., 1988, 88, 563–594 CrossRef CAS.
- P. Pyykko, Inorg. Chim. Acta, 2005, 358, 4113–4130 CrossRef.
- H. Hakkinen, M. Moseler and U. Landman, Phys. Rev. Lett., 2002, 89, 033401–033405 CrossRef PubMed.
- H. Hakkinen, B. Yoon, U. Landman, X. Li, H. J. Zhai and L. S. Wang, J. Phys. Chem. A, 2003, 107, 6168–6175 CrossRef.
- S. Bulusu, X. Li, L.-S. Wang and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8326–8330 CrossRef CAS PubMed.
- J. Li, X. Li, H. J. Zhai and L. S. Wang, Science, 2003, 299, 864–867 CrossRef CAS PubMed.
- B. Yoon, P. Koskinen, B. Huber, O. Kostko, B. von Issendorff, H. Hakkinen, M. Moseler and U. Landman, ChemPhysChem, 2007, 8, 157–161 CrossRef CAS PubMed.
- S. Bulusu, X. Li, L.-S. Wang and X. C. Zeng, J. Phys. Chem. C, 2007, 111, 4190–4198 CrossRef CAS.
- M. Ji, X. Gu, X. Li, X. G. Gong, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2005, 44, 7119–7123 CrossRef CAS PubMed.
- A. F. Jalbout, F. F. Contreras-Torres, L. A. Perez and I. L. Garzon, J. Phys. Chem. A, 2008, 112, 353–357 CrossRef CAS PubMed.
- A. Lechtken, D. Schooss, J. R. Stairs, M. N. Blom, F. Furche, N. Morgner, O. Kostko, B. von Issendorff and M. M. Kappes, Angew. Chem., Int. Ed., 2007, 46, 2944–2948 CrossRef CAS PubMed.
- X. Gu, S. Bulusu, X. Li, X. C. Zeng, J. Li, X. G. Gong and L.-S. Wang, J. Phys. Chem. C, 2007, 111, 8228–8232 CrossRef CAS.
- I. E. Santizo, F. Hidalgo, L. A. Perez, C. Noguez and I. L. Garzon, J. Phys. Chem. C, 2008, 112, 17533–17539 CrossRef CAS.
- I. L. Garzon, K. Michaelian, M. R. Beltran, A. Posada-Amarillas, P. Ordejon, E. Artacho, D. Sanchez-Portal and J. M. Soler, Phys. Rev. Lett., 1998, 81, 1600–1603 CrossRef CAS.
- H. Hakkinen, M. Moseler, O. Kostko, N. Morgner, M. A. Hoffmann and B. von Issendorff, Phys. Rev. Lett., 2004, 93, 093401–093405 CrossRef PubMed.
- W. Huang, M. Ji, C.-D. Dong, X. Gu, L.-M. Wang, X. G. Gong and L.-S. Wang, ACS Nano, 2008, 2, 897–904 CrossRef CAS PubMed.
- T. K. Ghanty, A. Banerjee and A. Chakrabarti, J. Phys. Chem. C, 2010, 114, 20–27 CrossRef CAS.
- D. Yuan, X. Gong and R. Wu, Phys. Rev. B, 2008, 78, 035441 CrossRef.
- X. Li, B. Kiran, L. F. Cui and L. S. Wang, Phys. Rev. Lett., 2005, 95, 253401 CrossRef PubMed.
- K. Koszinowski, D. Schroder and H. Schwarz, ChemPhysChem, 2003, 4, 1233–1237 CrossRef CAS PubMed.
- A. M. Joshi, W. N. Delgass and K. T. Thomson, J. Phys. Chem. B, 2006, 110, 23373–23387 CrossRef CAS PubMed.
- S. Pande, T. Jian, N. S. Khetrapal, L. S. Wang and X. C. Zeng, J. Phys. Chem. C, 2018, 122, 6947–6954 CrossRef CAS.
- N. S. Khetrapal, T. Jian, G. V. Lopez, S. Pande, L. S. Wang and X. C. Zeng, J. Phys. Chem. C, 2017, 121, 18234–18243 CrossRef CAS.
- N. S. Khetrapal, T. Jian, R. Pal, G. V. Lopez, S. Pande, L. S. Wang and X. C. Zeng, Nanoscale, 2016, 8, 9805–9814 RSC.
- Y. R. Zhao, Y. Qian, M. G. Zhang and Y. F. Hu, Mol. Phys., 2015, 113, 3598–3605 CrossRef CAS.
- P. Pyykko and N. Runeberg, Angew. Chem., Int. Ed., 2002, 41, 2174–2176 CrossRef CAS PubMed.
- X. Li, B. Kiran, J. Li, H. J. Zhai and L. S. Wang, Angew. Chem., Int. Ed., 2002, 41, 4786–4789 CrossRef CAS PubMed.
- H. J. Zhai, J. Li and L. S. Wang, J. Chem. Phys., 2004, 121, 8369–8374 CrossRef CAS PubMed.
- Y. Gao, S. Bulusu and X. C. Zeng, J. Am. Chem. Soc., 2005, 127, 15680–15681 CrossRef CAS PubMed.
- Y. Gao, S. Bulusu and X. C. Zeng, ChemPhysChem, 2006, 7, 2275–2278 CrossRef CAS PubMed.
- S. Neukermans, E. Janssens, H. Tanaka, R. E. Silverans and P. Lievens, Phys. Rev. Lett., 2003, 90, 033401–033405 CrossRef CAS PubMed.
- L.-M. Wang, S. Bulusu, H.-J. Zhai, X.-C. Zeng and L.-S. Wang, Angew. Chem., Int. Ed., 2007, 46, 2915–2918 CrossRef CAS PubMed.
- L.-M. Wang, R. Pal, W. Huang, X. C. Zeng and L.-S. Wang, J. Chem. Phys., 2009, 130, 051101–051105 CrossRef PubMed.
- L.-M. Wang, R. Pal, W. Huang, X. C. Zeng and L.-S. Wang, J. Chem. Phys., 2010, 132, 114306–114314 CrossRef PubMed.
- R. Pal, L.-M. Wang, W. Huang, L.-S. Wang and X. C. Zeng, J. Chem. Phys., 2011, 134, 054306–054307 CrossRef PubMed.
- B. Kiran, X. Li, H. J. Zhai, L. F. Cui and L. S. Wang, Angew. Chem., Int. Ed., 2004, 43, 2125–2129 CrossRef CAS PubMed.
- B. Kiran, X. Li, H.-J. Zhai and L.-S. Wang, J. Chem. Phys., 2006, 125, 133204 CrossRef PubMed.
- P. Pyykko and Y. F. Zhao, Chem. Phys. Lett., 1991, 177, 103–106 CrossRef CAS.
- R. Pal, S. Bulusu and X. C. Zeng, J. Comput. Methods Sci. Eng., 2007, 7, 185–193 CAS.
- C. Majumder, A. K. Kandalam and P. Jena, Phys. Rev. B, 2006, 74, 205437 CrossRef.
- C. Majumder, Phys. Rev. B, 2007, 75, 235409 CrossRef.
- M. Walter and H. Hakkinen, Phys. Chem. Chem. Phys., 2006, 8, 5407–5411 RSC.
- Q. Sun, Q. Wang, G. Chen and P. Jena, J. Chem. Phys., 2007, 127, 214706 CrossRef PubMed.
- L.-M. Wang, S. Bulusu, W. Huang, R. Pal, L.-S. Wang and X. C. Zeng, J. Am. Chem. Soc., 2007, 129, 15136 CrossRef CAS PubMed.
- M. Abe, T. Nakajima and K. Hirao, J. Chem. Phys., 2002, 117, 7960–7967 CrossRef CAS.
- R. Pal, L.-M. Wang, W. Huang, L.-S. Wang and X. C. Zeng, J. Am. Chem. Soc., 2009, 131, 3396–3404 CrossRef CAS PubMed.
- S. J. Lu, X. L. Xu, H. G. Xu and W. J. Zheng, J. Chem. Phys., 2018, 148, 244306 CrossRef PubMed.
- Y. Zhao, F. Zhao and L. H. Kang, J. Mol. Model., 2018, 24, 61–68 CrossRef PubMed.
- K. Joshi and S. Krishnamurty, J. Phys. Chem. C, 2017, 121, 17514–17522 CrossRef CAS.
- E. M. Dore and J. T. Lyon, J. Cluster Sci., 2016, 27, 1365–1381 CrossRef CAS.
- Y. R. Liu, T. Huang, Y. B. Gai, Y. Zhang, Y. J. Feng and W. Huang, Sci. Rep., 2015, 5, 17738–17745 CrossRef CAS PubMed.
- S. Gautam, N. Goel and K. Dharamvir, RSC Adv., 2014, 4, 13927–13933 RSC.
- T. Quoc Tri, S.-J. Lu, L.-J. Zhao, X.-L. Xu, H.-G. Xu, T. Van Tan, J. Li and W. J. Zheng, J. Phys. Chem. A, 2018, 122, 3374–3382 CrossRef PubMed.
- S.-J. Lu, X.-L. Xu, G. Feng, H.-G. Xu and W.-J. Zheng, J. Phys. Chem. C, 2016, 120, 25628–25637 CrossRef CAS.
- S. J. Lu, L. R. Hu, X. L. Xu, H. G. Xu, H. Chen and W. J. Zheng, Phys. Chem. Chem. Phys., 2016, 18, 20321–20329 RSC.
- D. M. Deaven and K. M. Ho, Phys. Rev. Lett., 1995, 75, 288–291 CrossRef CAS PubMed.
- R. L. Johnston, Dalton Trans., 2003, 4193–4207 RSC.
- R. H. Byrd, P. H. Lu, J. Nocedal and C. Y. Zhu, SIAM J. Sci. Comput., 1995, 16, 1190–1208 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- R. A. Kendall, E. Apra, D. E. Bernholdt, E. J. Bylaska, M. Dupuis, G. I. Fann, R. J. Harrison, J. L. Ju, J. A. Nichols, J. Nieplocha, T. P. Straatsma, T. L. Windus and A. T. Wong, Comput. Phys. Commun., 2000, 128, 260–283 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Predicted low-energy structures of the AuxM− (x = 9–12; M = Si, Ge, Sn) clusters using the revised genetic algorithm combined with density functional theory. See DOI: 10.1039/c9ra01019j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Tianquan Liangb,
Enhui Wua,
Jun Lia and
Jing Houc
*a,
Tianquan Liangb,
Enhui Wua,
Jun Lia and
Jing Houc