
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction, structural characterization, and thermal and biomedical properties of sulfated polysaccharides from razor clam Solen marginatus
Nabil Souissi *a,
Soumaya Boughribab,
Ola Abdelhedib,
Marwa Hamdib,
Mourad Jridib,
Suming Lic and
Moncef Nasrib
*a,
Soumaya Boughribab,
Ola Abdelhedib,
Marwa Hamdib,
Mourad Jridib,
Suming Lic and
Moncef Nasrib
aLaboratoire de Biodiversité Marine, Institut National des Sciences et Technologies de la Mer, Centre de Sfax, Avenue Madagascar BP, 1035-3018, Sfax, Tunisia. E-mail: nabil.souissi@gmail.com; Tel: +216 22 95 08 54
bLaboratoire de Génie Enzymatique et de Microbiologie, Université de Sfax, Ecole Nationale d’Ingénieurs de Sfax, B. P. 1173-3038 Sfax, Tunisia
cInstitut Européen des Membranes, UMR CNRS 5635, Université de Montpellier, Place Eugene Bataillon, 34095 Montpellier Cedex 5, France
First published on 12th April 2019
Abstract
In this study, the antioxidant, antibacterial and anticoagulant activities of sulfated polysaccharides extracted from Solen marginatus flesh were investigated via physicochemical characterization of the crude polysaccharide SM-CP and its deproteinized fraction (SM-DP); their total sugar contents were 47.15% and 66.01%. The results obtained via molecular weight evaluation showed that SM-CP mainly had a high molecular weight (1075 kDa), whereas SM-DP had a lower molecular weight (almost 237.9 kDa); in addition, thermal analysis (differential scanning calorimetry and thermogravimetry) was conducted; the results indicated that SM-CP was thermally more stable as its degradation temperature was 307 °C, whereas SM-DP was thermally less stable, with the degradation temperature of 288 °C. Moreover, the results obtained via the investigation of biological properties revealed that the extracted polysaccharides exhibited strong antioxidant and anticoagulant activities. Subsequently, SM-CP was fractionated using the DEAE-cellulose column. The peak (FII) eluted at high NaCl concentrations indicated highest anticoagulant activity as designated by the prolongation of the activated partial thromboplastin time (over 120 s), prothrombin time (28 s) and low level of fibrinogen (0.7 g l−1). The overall data demonstrated the significant therapeutic potential of the polysaccharides extracted from razor clam flesh.
1 Introduction
Due to its diversity, the marine world can offer an infinite number of original molecules that have not been discovered yet. Nowadays, one of the research challenges is the exploration of marine biodiversity for possible future applications. The interest in marine sources is significantly increasing, especially, for the exploration of bioactive compounds, such as pigments, fatty acids, proteins and carbohydrates, since natural products can be applied in numerous industries; among these molecules, polysaccharides represent the third class of molecules extracted from marine bivalves, occupying 27% of all bioactive substances after protein and protein hydrolysates.1 Marine polysaccharides are exploited in therapeutic, food and cosmetic products. For instance, carrageenan extracted from red algae is widely used in the food industry to control the texture of dairy desserts as well as in the treatment of breast cancer.2Specifically, sulfated polysaccharides (SP) represent a considerable challenge for research because of their great structural variety. In fact, in addition to their functional properties such as gelation, texturing and emulsification,2 marine SP exhibit antiviral,3 antithrombotic,4 antibacterial,5,6 angiotensin I-converting enzyme inhibition,7 antioxidant8 and anti-obesity activities.9
Actually, free radicals, oxidative stress, reactive oxygen species and antioxidants have become extensively familiar terms for health professionals, industrialists and even for the general public. For instance, during food processing, various synthetic antioxidant compounds, such as BHA, BHT and TBHQ, are commonly employed. However, due to safety concerns related to the use of these synthetic antioxidants, significant interest has been paid towards the search of alternative natural antioxidants;10 moreover, according to the medical community, cardiovascular diseases, such as heart diseases and stroke, associated with thrombosis are the major cause of death;11 therefore, to provide protection against these pathologies, it is important to have adequate natural antioxidant and anticoagulant defenses that replace the synthetic products.
Solen marginatus is a widely distributed marine bivalve in the Mediterranean Sea, particularly in the North of Africa including Tunisian coasts.12 Moreover, S. marginatus is available in international markets at fairly high prices,13 and it is commercially exploited in several European countries such as Spain, Portugal, Italy and Ireland.14 However, in Tunisia, among seafood, S. marginatus has a low commercial value, and only three bivalves (i.e. Ruditapes decussatus, Mytilus galloprovincialis and Crassostrea gigas) have been mainly exploited.12
To date, only few studies have been reported on the reproductive cycle of S. marginatus, its genetic characterization and pathological alterations, whereas no study has been reported on the biotechnological use and applications of S. marginatus; hence, herein, we have emphasized on the biochemical study of this razor clam, its valorization by the extraction and fractionation of SP, and the investigation of the antioxidant, anticoagulant and antibacterial activities of these SP.
2 Materials and methods
2.1. Sample collection
Fresh samples were obtained from the gulf of Gabes coastal area named Smara in Tunisia during the period between March and April; the obtained samples were then packed in polyethylene bags filled with ice to be transported to the laboratory within an hour. Initially, the samples were cleaned with tap water; then, fleshes were separated from the shells and kept in sealed plastic bags at −20 °C until further use within one week for the characterization and extraction of polysaccharides.2.2. Polysaccharide extraction
Crude polysaccharides were obtained from the fleshes of S. marginatus by ethanol precipitation according to a previously reported procedure.7 Herein, 100 g of fleshes was homogenized in 100 ml of distilled water and then ground (Moulinex R62). Thereafter, the mixture was treated for 20 min at 95 °C to inactivate endogenous enzymes. After this, Alcalase® (pH = 8.0 at 50 °C) was added to the abovementioned mixture with the enzyme–substrate ratio E/S equal to 10 U mg−1 of protein. The mixture was then kept at 50 °C for 12 h at an adequate pH (pH = 8.0), subsequently boiled for 10 min to inactivate the enzyme and centrifuged at 6000g for 20 min at 4 °C. The supernatant was separated from the pellet and precipitated twice with absolute ethanol (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) at 4 °C for 12 h; then, it was centrifuged at 6000g for 20 min at 4 °C. The recovered pellet was dissolved in distilled water and then freeze-dried (Modulyo Freeze dryer, Thermo Fisher, USA) to obtain the Solen marginatus crude polysaccharide (SM-CP) powder.
:2, v/v) at 4 °C for 12 h; then, it was centrifuged at 6000g for 20 min at 4 °C. The recovered pellet was dissolved in distilled water and then freeze-dried (Modulyo Freeze dryer, Thermo Fisher, USA) to obtain the Solen marginatus crude polysaccharide (SM-CP) powder.
The deproteinized polysaccharide SM-DP was extracted using the method reported by Huang et al.15 The SM-CP was incubated at 4 °C for 4 h in the presence of 20% (w/v) of a 15% trichloroacetic acid (TCA) solution and then centrifuged at 6000g for 20 min at 4 °C. After this, the supernatant was precipitated with absolute ethanol at the ratio of 1:2 (v/v) and kept at 4 °C for 12 h. Finally, the resulting precipitate was dissolved in distilled water and then freeze-dried to obtain the SM-DP.
2.3. Physicochemical analysis of the S. marginatus flesh and its polysaccharide extracts
Initially, the physicochemical properties, such as protein, lipid, and ash contents, of the S. marginatus flesh were determined according to the methods described in AOAC.16For polysaccharide extracts, the protein content was determined using the Folin–Ciocalteu's phenol reagent method as reported by Lowry et al.17 The total carbohydrates were estimated by the phenol-sulfuric acid method.18 The total uronic acid content was calorimetrically evaluated according to the method described by Bitter & Muir19 using glucuronic acid as a standard. The determination of the sulfate content in polysaccharides was performed using the barium-chloride-gelatin method described by Ji et al.20
2.4. Fourier transform infrared (FTIR) spectroscopy analysis
The structural analysis of samples was conducted via Fourier-transform infrared spectroscopy (FTIR) (PerkinElmer, Spectrum 65, France) using approximately 10 mg of each sample mixed with 100 mg of dried KBr. The spectra of the polysaccharide extracts were obtained between 450 and 4000 cm−1 using 32 scans at the resolution of 4 cm−1.2.5. Molecular weights of the polysaccharide extracts
The molecular weights (MW) of the SM-CP and SM-DP were estimated by an instrumental setup consisting of a set of a size exclusion chromatography (SEC) system coupled with a multi-detector analyzer (Wyatt Dawn DSP-F) composed of a differential refractometer and a multi-angle laser light scattering detector. The samples were injected as solutions in sodium nitrate (10 mM, pH 7.0) at the concentration of 2 mg ml−1 into the TSKgel® (GMPWXL) column and eluted at the flow gradient of 0.4 ml min−1.The molecular weight distribution (MWD) was determined using high-performance liquid chromatography (Thermo Scientific Dionex UltiMate 3000). A volume of 20 μl of each sample (50 mg ml−1) was injected onto the column (BioBasic SEC-300) and eluted using 0.1 M of phosphate buffer (pH = 7.4), already filtered, at the flow rate of 1 ml min−1. A series of reference Dextrans was used to calibrate the column.
2.6. 1H nuclear magnetic resonance (NMR) spectroscopy
1H-NMR spectroscopy provides structural and conformational information about the studied samples. Both the SM-CP and the SM-DP obtained from S. marginatus were structurally characterized using the Bruker 400 spectrometer (Bruker Biospin AG, Fallanden, Switzerland) at 25 °C. The SM-CP and SM-DP were dried in vacuum over P2O5 for 48 h, and then, 20 mg of the sample was dissolved in 1 ml of 99% deuterium oxide (D2O). The data obtained was analyzed using the MestRe Nova 5.3.0 (Mesterlab Research S.L.) software. Moreover, chemical shifts are provided in ppm.2.7. Anion exchange chromatography
The fractionation of polysaccharides was performed using anion-exchange chromatography. Briefly, two hundred mg of polysaccharides was dissolved in 2 ml of sodium acetate (0.1 M, pH 6) and then placed on the surface of a DEAE-cellulose column (1 cm × 15 cm), already equilibrated with sodium acetate buffer. At first, the elution was accomplished with sodium acetate (0.1 M, pH = 6.0) and then with NaCl at various concentrations (1 M, 2 M and 3 M) at the flow rate of 0.5 ml min−1. The sub-fractions were obtained, and the contents of total carbohydrates and uronic acids were determined in them, as previously described. Then, sub-fractions related to the same peak were mixed and desalted through an ultrafiltration membrane of 1 kDa cut-off to be finally freeze-dried.2.8. Thermal analyses
2.9. In vitro antioxidant activity evaluation
where ODblank, ODsample and ODcontrol are the absorbance of the blank, sample and control DPPH tubes, respectively. Absorbance was determined at 517 nm. The test was repeated three times for each sample.

After this, the absorbance of all the test tubes was measured twice at 470 nm before and after incubation for 1 h at 50 °C. The tests were performed in triplicate, and the antioxidant activity was calculated as follows:
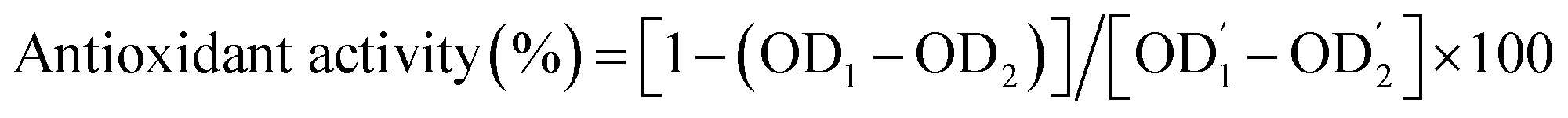
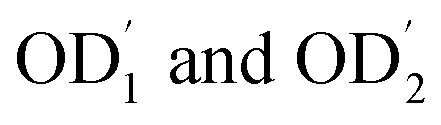 are the absorbances of the control measured at the beginning and the end of incubation, respectively.
are the absorbances of the control measured at the beginning and the end of incubation, respectively.
| Metal chelating activity (%) = [(ODcontol + ODblank − ODsample)/ODcontrol] × 100 |
| A = 0.001B + 0.0049; R2 = 0.987 |
2.10. In vitro anticoagulant activity evaluation
The anticoagulant activity of polysaccharides was evaluated using the blood samples obtained from healthy volunteers, and then, these samples were mixed with sodium citrate (3.2%) at the ratio of 9:1. Thereafter, the mixture was centrifuged at 3000g for 15 min at ambient temperature. Then, the pellet was discarded, and the supernatant, regarded as the citrated pool of plasma, was recovered. The citrated pool of plasma was used to determine the three anti-coagulation mechanisms of S. marginatus polysaccharide extracts and their DEAE-c fractions at various concentrations using the STart® analyzer (Diagnostica Stago, France).
2.11. Antibacterial activity evaluation
3 Results and discussion
3.1. Biochemical composition of the S. marginatus flesh and its polysaccharide extracts
The physicochemical characterization of the S. marginatus flesh showed that the muscle contained 3.53 ± 0.58% of total sugars, 18.39 ± 0.15% of proteins, 0.89 ± 0.07 of lipids and 3.43 ± 0.27 of ash. These values are in the same range as those reported in ref. 27 and 28 wherein it has been found that Ruditapes decussatus and Cerastoderma glaucum species have 3.84% and 3.72% of total sugar and 9.24% and 12% of proteins, respectively. The present results reveal that S. marginatus would be an effective source of proteins and carbohydrates. Hence, the extraction of polysaccharides was carried out.The extraction of crude polysaccharides from S. marginatus flesh was performed by a hydrolysis step conducted using Alcalase® followed by ethanol precipitation, and the deproteinized fraction was treated with TCA. The results of the extraction yield, protein, total sugar, sulfated group and uronic acid contents in the SM-CP and SM-DP are presented in Table 1. As shown, SM-DP revealed the highest content of sugar and uronic acids reaching 66.01% and 44.04% vs. 47.15% and 10.12% of the crude sample, respectively. On the other hand, SM-CP contained the greatest amount of sulfate groups (5.04%) and protein content (11.08%). Similarly, the characterization of the polysaccharide extract obtained from the viscera of smooth hound revealed a high sugar content (58.9%) and medium levels of proteins, uronic acids and sulfate groups (5.5%, 3.5% and 7.9%, respectively).7
| Extraction yield (%) | Proteins (%) | Total sugars (%) | Sulfated groups (%) | Uronic acids (%) | |
|---|---|---|---|---|---|
| a Results are expressed in percentages (%) based on the wet weight matter. SM-CP and SM-DP extraction yields are expressed in percentages based on the wet weight of the raw material and the polysaccharide crude, respectively. a,b Different letters in the same column indicate significant differences (P ≤ 0.05). | |||||
| SM-CP | 4.26 ± 0.5 | 11.08 ± 0.44a | 47.15 ± 5.18b | 5.04 ± 0.34a | 10.12 ± 1.41b |
| SM-DP | 59.5 ± 4.95 | 1.87 ± 0.62b | 66.01 ± 5.26a | 1.58 ± 0.35b | 44.04 ± 3.76a |
Moreover, the results presented in Table 1 prove the efficiency of the deproteinization step in reducing the protein content in the SM-CP to 1.87% vs. 11.08% for SM-DP without affecting the carbohydrate level, which remains constant. These results were further confirmed by the UV-visible spectra of both samples (data not shown herein); the SM-DP had smaller absorbance peaks at 260 and 280 nm as compared to its corresponding crude polysaccharide; this indicated the presence of nucleic acids and proteins in very small amounts. In addition, the largest absorbance peaks observed at 200 nm and those related to the specific characteristics of sulfated polysaccharides29 were clearly observed for the extracted polysaccharides. All these data confirmed that the SM-DP had high degree of purity as compared to the SM-CP.
As is commonly known, polysaccharides are structurally associated with proteins in the form of glycoproteins;30 thus, a deproteinization step for the extraction of SM-CP is required. In our study, a temperature of 4 °C in the presence of TCA was applied for polysaccharide deproteinization to guarantee highest deproteinization yield (59%) from the SM-CP powder. In fact, De Kruif et al.31 reported that low temperatures facilitated the separation of two polymers (proteins and carbohydrates) into two separate phases. In the same context, Zhu et al.32 showed that the deproteinization of abalone viscera polysaccharide led to the reduction of the protein level from 28.51% to 16.93%. In addition, in the ref. 6, it was found that the total carbohydrate and protein contents of the SP obtained from Sepioteuthis lessoniana were 61.3% and 0.8%, respectively.
3.2. Molecular weight of the polysaccharide extracts
The MW distribution of the polysaccharides obtained from the S. marginatus flesh was investigated using size exclusion chromatography (SEC), and the corresponding spectra are shown in Fig. 1. The profiles indicated similar MW repartition, which ranged from 2 to 9 min. Moreover, a slight shift and longer retention time were observed for SM-CP as compared to the case of SM-DP; this suggested higher MW of the crude polysaccharide sample. In addition, upon observing the spectra of both polysaccharides, it was found that the last eluting peak, representing the lowest MW, was completely missing in the case of SM-CP, whereas it was slightly present in the case of the deproteinized sample.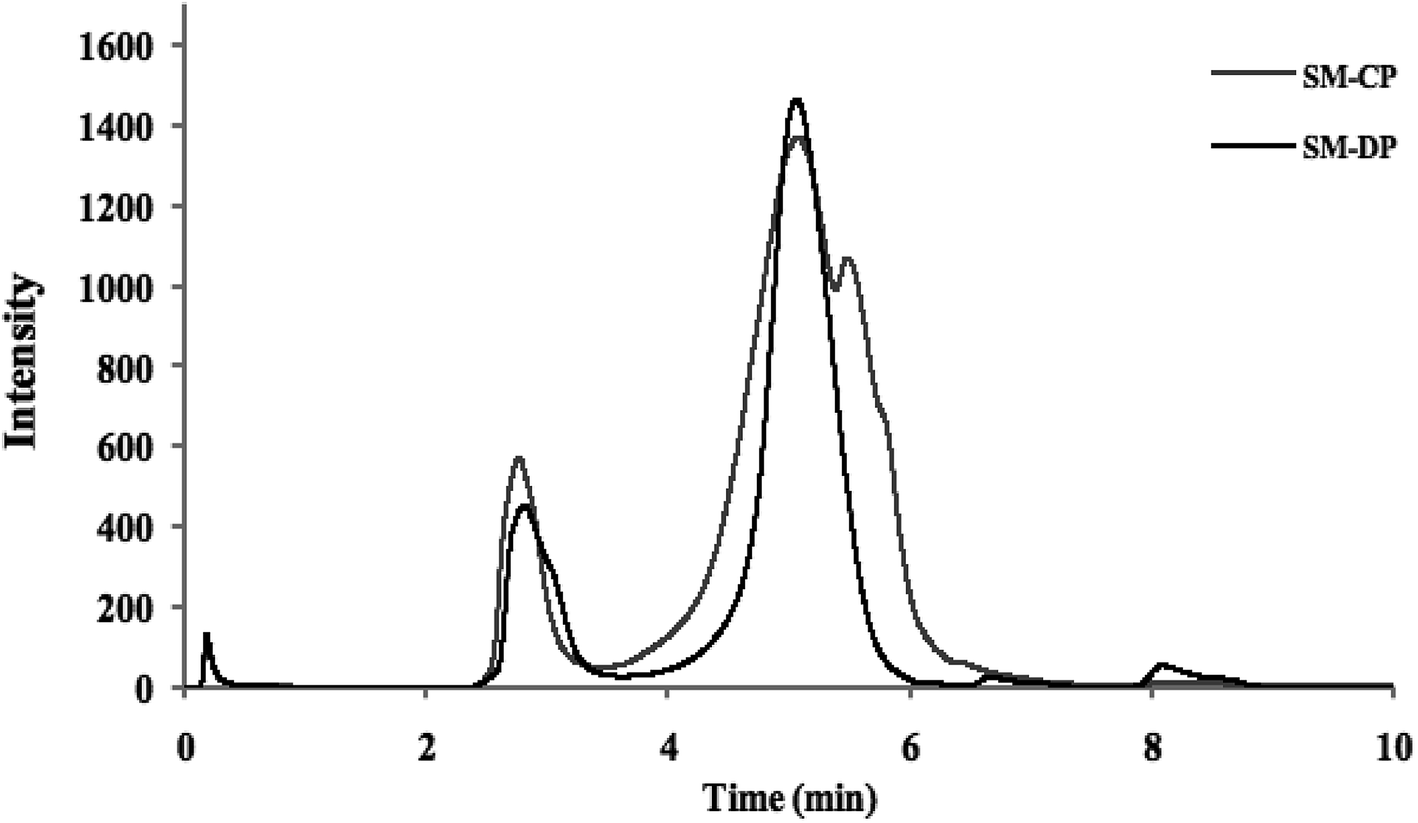 | ||
| Fig. 1 Molecular weight distribution of SM-DP and SM-CP. | ||
These data were confirmed by molecular weight values. The results obtained showed that the SM-CP constituted a high molecular weight fraction (1075 kDa), whereas the SM-DP constituted a lower molecular weight fraction (almost 237.9 kDa). In the same context, Wang et al.8 extracted polysaccharides from a bivalve species, Corbicula fluminea, and found that the obtained fractions were composed of two major peaks at 2113.4 and 61.5 kDa for the crude and at 1980.9 and 248.4 kDa for the Sevag-deproteinized polysaccharide fraction. Hence, note that the molecular weight depends on the steps in the extraction method and the deproteinization treatment method that affect the molecular weight of the carbohydrate chains.
3.3. FTIR spectroscopy analysis
Fourier transform infrared spectroscopy, based on the analysis of the absorption frequencies of chemical groups, was used to characterize the extracted polysaccharides. The obtained FTIR spectra are shown in Fig. 2. | ||
| Fig. 2 Infrared spectra of SM-CP and SM-DP. | ||
Both spectra are similar and showed the characteristic bands of the commercial SP, the glycosaminoglycanes reported by Mansour et al.,33 and the fingerprint range for sulfated polysaccharides (from 500 cm−1 to 2000 cm−1) was conserved, as indicated by Shanura Fernando et al.34 In fact, the presence of amide A and B bands at 3346 cm−1 and 2946 cm−1 is the characteristic of the hydroxyl and (CH) bond groups, respectively. In addition, both polysaccharides exhibited characteristic bands at 944 cm−1, 885 cm−1 and 830 cm−1, which were attributed to the anomeric C–H deformation, vibration of α-L-guluronic acids and β-D-mannuronic acids, respectively.35 The bands at 1620 cm−1 and 1423 cm−1 corresponded to the COO (asymmetric) and COO (symmetric), respectively.36 In addition, the signals observed in the ranges of 1082–1085 cm−1 and 1155–1160 cm−1 for the SM-CP and SM-DP are assigned to the symmetric and asymmetric stretching of an ether sulfate group (RO–SO−3), respectively.37 The bands located at 1250 cm−1 and 792 cm−1 were ascribed to the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and C–O–S bands,38 respectively, which confirmed the presence of sulfated groups. Specific uronic acid bands were observed between 1383.5 cm−1 and 1470 cm−1.39
O stretching vibration and C–O–S bands,38 respectively, which confirmed the presence of sulfated groups. Specific uronic acid bands were observed between 1383.5 cm−1 and 1470 cm−1.39
Similar spectra have been reported for the sulfated polysaccharides extracted from different marine sources such as abalone (Haliotis discus hannai Ino) gonad,40 bullet tuna (Auxis rochei) by-products41 and Asian clam (Corbicula fluminea).8
3.4. 1H-NMR spectroscopy analysis
NMR analysis was performed on polysaccharides extracted from S. marginatus, and the spectra are displayed in Fig. 3. The signals obtained in the SM-CP and SM-DP spectra were assembled in four clusters. The first cluster located between 0.5 and 0.9 ppm in SM-CP was attributed to the stretching of RCH3 without any attached functional groups. The second cluster extended from 2.4 to 3.5 ppm in the SM-CP and between 2.5 and 3.5 ppm in the SM-DP, corresponding to the H3–H6 chemical shifts of different sugar residues. In this region, the SM-CP and SM-DP showed 3 main chemical shifts located at 2.6, 2.8 and 3.2 ppm and 2.7, 3.1 and 3.2 ppm, confirming the opulence of H2 and H3 of the β-linked residues in glucuronic acid and iduronic acid, respectively. | ||
| Fig. 3 1H NMR spectra of SM-CP (A) and SM-DP (B). | ||
In addition, both spectra showed various resonances perceived between 4.0 and 4.9 ppm, indicating the presence of sulfated groups with a major chemical shift at 4.5 ppm that confirmed the abundance of glucuronic acid in the SM-CP and SM-DP.37
The final cluster detected between 4.2 and 4.7 ppm refers to α-linked osidic units.42 On the other hand, the intensity of the resonance peak at 4.9 ppm was significantly lower for SM-CP as compared to that for SM-DP; this suggested the greatest efficiency of the TCA treatment that resulted in the abundance of osidic units in SM-DP.
3.5. Thermal analysis
The DSC and TGA results of the studied polysaccharides are presented in Table 2. The DSC curves of the SM-CP and SM-DP display similar profiles, characterized by two endothermic peaks. The first peak was attributed to the glass transition temperature value (Tg), whereas the second peak was assigned to the melting of the sample. As shown in Table 2, the Tg value was similar for both samples, and a less pronounced reduction was observed in the SM-CP (57 °C) as compared to that in the SM-DP (53 °C).| Tg (°C) | Δw (%) 1st transformation region | Δw (%) 2nd transformation region | Tmax (°C) | Residue (%) | |
|---|---|---|---|---|---|
| a Tmax = Temperature of maximum degradation for the second transformation. SM-CP and SM-DP represent S. marginatus crude polysaccharide and deproteinized polysaccharide, respectively. | |||||
| SM-CP | 57.9 | 6.741 | 66.9 | 307.1 | 19.91 |
| SM-DP | 53.5 | 11.06 | 63.67 | 288.1 | 21.49 |
The TGA analysis was used to evaluate the thermal stability of the polysaccharides extracted from S. marginatus as well as their maximum degradation temperature through determination of their weight loss (Δw) as a function of the temperature rise under a controlled nitrogen atmosphere. The TGA results and the first derivative of the TGA curves obtained for the SM-CP and SM-DP are depicted in Table 2 and Fig. 4, respectively. The weight loss derivation profiles of both polysaccharides are similar, showing two major peaks. The first peak, corresponding to moisture loss, was observed when the temperature was increased up to 200 °C. The second peak, which was more extended, was detected between 200 and 600 °C with the maximum degradation temperature of 307 °C and 288 °C for the SM-CP and SM-DP, respectively (Fig. 4). The second transformation region resulted in the sample weight loss reaching 66.9% for SM-CP and 63.7% for SM-DP. Based on the temperature of maximum degradation and the weight loss values, it may be suggested that the deproteinization step decreases the temperature of polysaccharide degradation and the glass transition value. These data confirm that SM-CP is thermally more stable than SM-DP; this may be associated with the dissociation of bonds existing between the proteins and the polysaccharides before deproteinization.
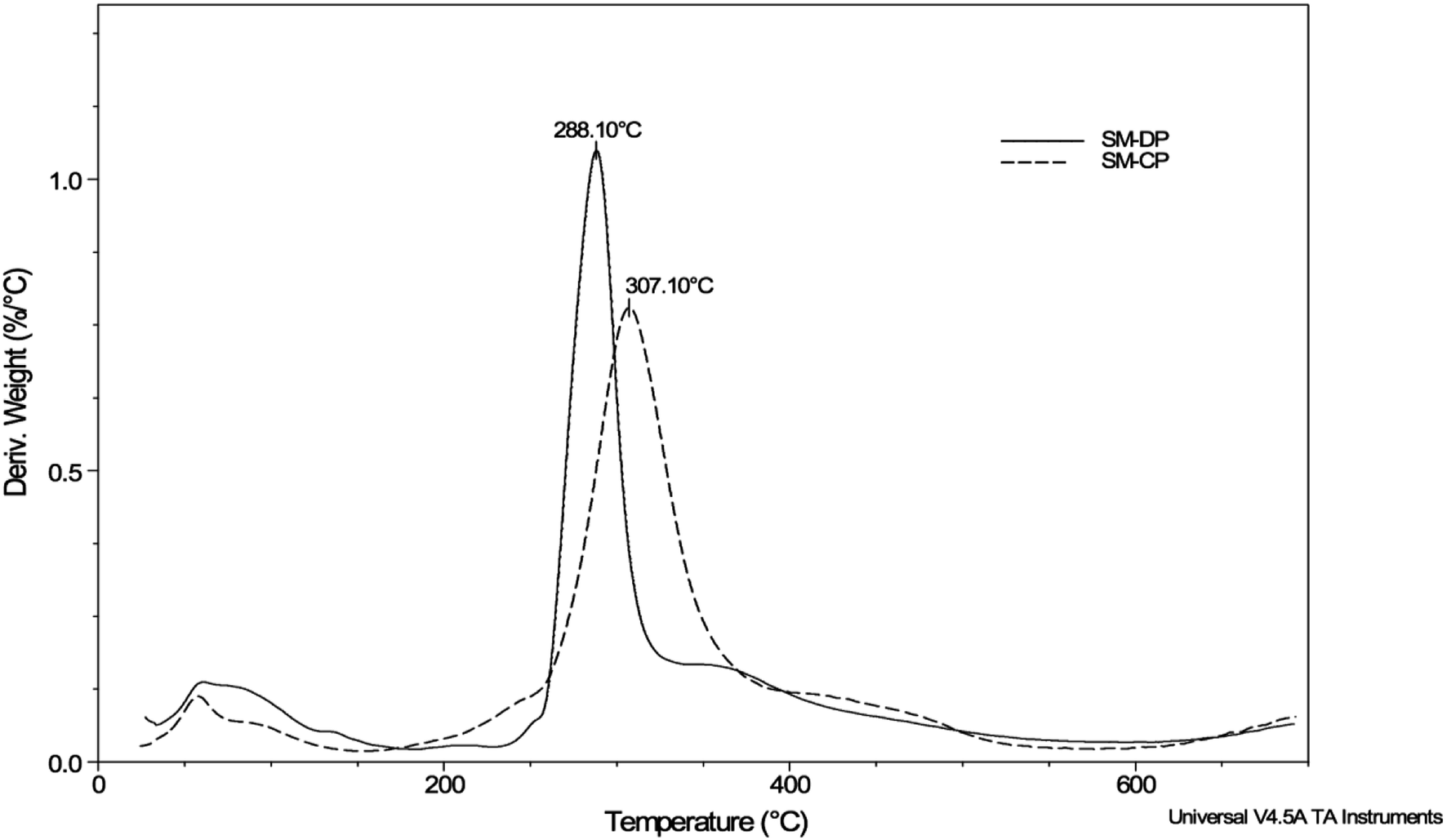 | ||
| Fig. 4 First derivative of the thermogravimetric curve of SM-CP and SM-DP. | ||
3.6. In vitro antioxidant activities of SM-CP and SM-DP
 | ||
| Fig. 5 Antioxidant properties of SM-CP and SM-DP as a function of their concentrations: (a) DPPH radical-scavenging activity, (b) metal chelating activity, (c) reducing power essay, (d) total antioxidant capacity and (e) β-carotene bleaching test, and (f) gel electrophoresis pattern of the plasmid pGapZαA® incubated with Fenton's reagent in the presence and absence of SM-CP and SM-DP; lane 1: untreated control (native DNA), lane 2: DNA incubated with Fenton's reagent, lanes 3 and 4: DNA + Fenton's reagent + SM-DP or SM-CP (1 mg ml−1), respectively. | ||
Hence, it may be suggested that the potential antioxidant activity, especially that of SM-CP – regardless of whether it acts as a proton donor or as a free radical scavenger – is mainly related to its sulfation degree rather than its MW.
3.7. Antibacterial activity evaluation
The antibacterial activity of SM-CP and SM-DP was evaluated against Gram-positive (S. aureus, M. luteus and L. monocytogenes) and Gram-negative (S. enterica, E. coli and P. aeruginosa) bacteria at the concentration of 50 mg ml−1 (Table 3). The antibacterial activity of both the polysaccharide extracts varied as a function of the tested strain. Indeed, the two extracts exhibited significant antibacterial activity against all bacterial tested strains except for M. luteus and P. aeruginosa. Moreover, the best bacterial growth inhibition was detected for SM-CP against S. aureus and L. monocytogenes with a clear diameter zone of 10.75 ± 0.35 mm and 10.25 ± 0.35 mm, respectively. These results are of great importance particularly in the case of S. aureus, which is well-known by its resistance to a number of antibiotics and for its production capacity of several types of enterotoxins that cause many types of enteritis and septicaemia.50 However, the obtained inhibition growth values are still lower than those of gentamicin used as the reference.| Bacteria strains | SM-CP | SM-DP | Gentamicin (30 μg) | |
|---|---|---|---|---|
| a nd: not detected. Diameter values are expressed in mm and given as means ± SD. a,b Different letters in the same line indicate significant differences (P ≤ 0.05). | ||||
| Gram+ | S. aureus | 10.75 ± 0.35a | 6.0 ± 0.2b | 37 |
| M. luteus | nd | nd | 18 | |
| L. monocytogenes | 10.25 ± 0.35a | 8.5±0b | 22 | |
| Gram− | S. enterica | 7.25 ± 0.35a | 8.75 ± 0.35a | 15 |
| E. coli | 7.5 ± 0.5a | 8.5 ± 0.5a | 29 | |
| P. aeruginosa | nd | nd | 14 | |
Differences in the observed antibacterial activities of the two extracts could be related to their chemical structures and their sulfate groups, as mentioned by.51 These authors reported, in fact, that highest polysaccharide sulfate content is associated with the greatest antibacterial activity. These results reveal that the polysaccharides extracted from S. marginatus can be an adequate agent to inhibit the growth of microorganisms that strike the quality and the safety of industrial food products.
3.8. In vitro anticoagulant activities of the SM-CP and SM-DP
The blood coagulation mechanisms maintain the stability of the vascular system by activating a series of clotting factors, resulting in the production of thrombin and the formation of fibrin clots. Coagulation can happen either by activation of the blood components in the vascular system (endogenous coagulation pathway or common pathway) or by exposure of the tissue factor at the site of a lesion, specifically at the vessel wall (exogenous coagulation pathway).52In vitro anticoagulant activities of the SM-CP and SM-DP were assessed by measuring the activated partial thromboplastin time (aPTT), the prothrombin time (PT) and the fibrinogen level (FL) of normal human plasma in the presence of polysaccharide extracts, and the results are shown in Fig. 6. The aPTT assay is the most studied assay that evaluates the endogenous coagulation pathway, leading to the identification of the deficiency of coagulation factors, which are XII, XI, IX, VIII, X, V, II (prothrombin), and I (fibrinogen). PT mainly reflects the performance of exogenous coagulation pathway factors such as factors I (fibrinogen), II (prothrombin), V, VII and X. FL reveals the last step of coagulation cascade in which the soluble fibrinogen is transformed into insoluble fibrin threads (common pathway).53
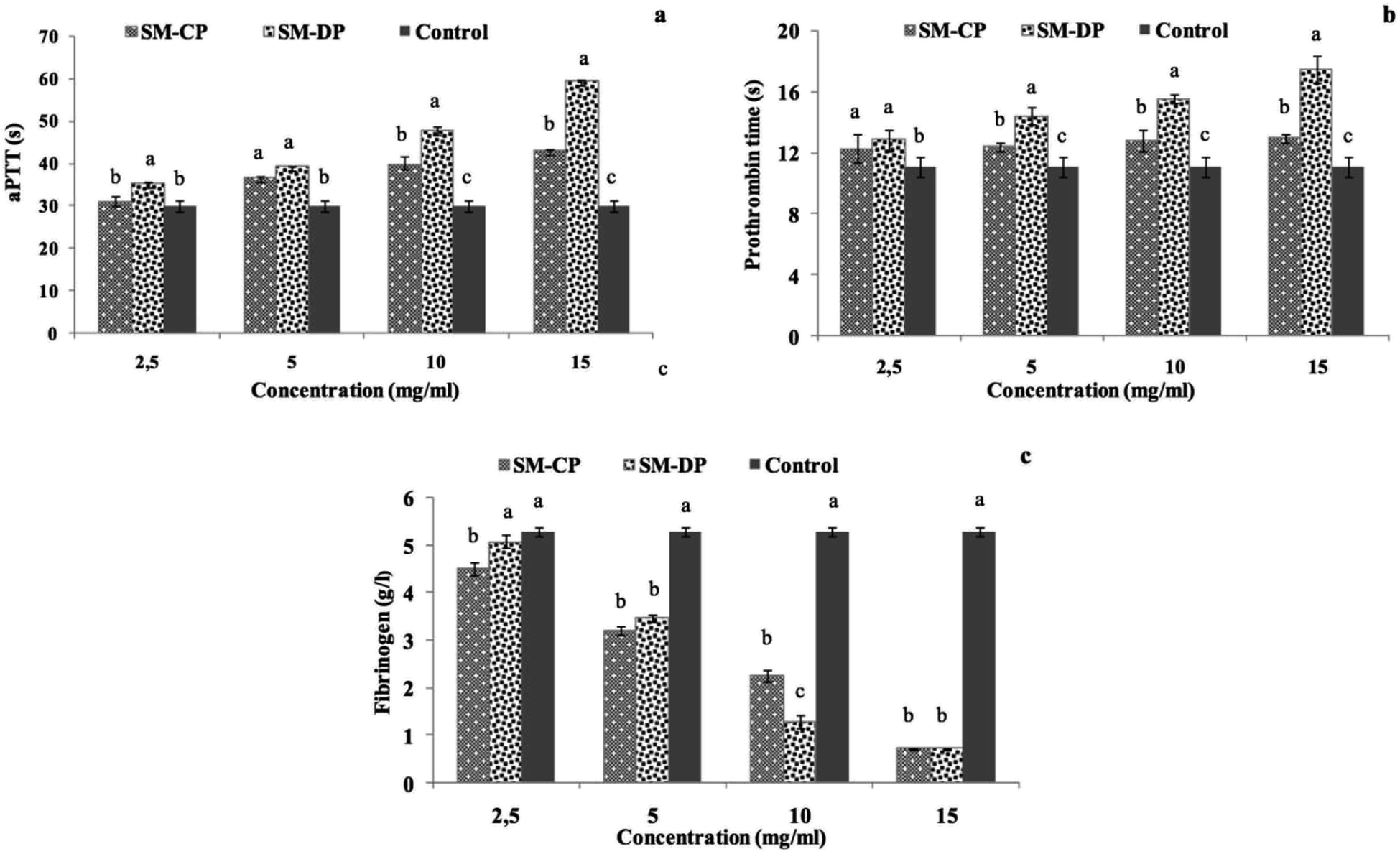 | ||
| Fig. 6 Anticoagulant properties of SM-CP and SM-DP as a function of their concentrations; (a) activated partial thromboplastin time aPTT, (b) prothrombin time PT and (c) fibrinogen level. Different letters within different samples indicate significant differences at p ≤ 0.05. | ||
Results represented in Fig. 6 prove that the anticoagulant activity of both polysaccharides samples depends on their concentration. In fact, SM-CP and SM-DP reached their maximum activity at the concentration of 15 mg ml−1 for all tests. The aPTT results showed that the two polysaccharides displayed strong aPTT prolonging activities. At low concentrations (2.5 and 5 mg ml−1), both samples exhibited similar aPTT prolongation, which remained greater than that of the control sample (30.03 s). Moreover, with the increasing sample concentration, this activity was higher in the SM-DP. In fact, the aPTT prolonging activity of SM-DP at 15 mg ml−1 was estimated at 59.4 s, whereas it was at 43 s in the SM-CP at the same concentration (Fig. 6a). For the PT, the data indicated that the highest SM-CP and SM-DP effect was obtained at the highest concentration tested (Fig. 6b). The prolonged PT suggest the inhibition of the exogenous coagulation pathway, which is more potent for SM-DP (17.45 s) as compared to that for SM-CP (12.93 s). These findings were in accordance with those found by Jung et al.54 who proved, through the PT test, that sulfated polysaccharides purified from a brown algae Ecklonia cava enhanced the inhibition of the coagulation factors II, VII and X and blocked the extrinsic and the common coagulation pathways.
Moreover, the FL, exploring the common thrombotic route, indicated that both polysaccharide extracts displayed markedly strong activity as compared to the control (Fig. 6c). The fibrinogen level is equal to 5.29 g l−1 for the control against 2.26 and 1.28 g l−1 for SM-CP and SM-DP at a concentration of 10 mg ml−1, respectively. The highest fibrinogen level reduction (0.7 g l−1) was obtained at a concentration of 15 mg ml−1 for both samples. Lang et al.55 reported that an increase in the fibrinogen level indicated the formation of a firm clot. Therefore, the decrease in the FL for SM-CP and SM-DP suggested the potentiality of SM-CP and SM-DP as novel anticoagulant agents.
The obtained results indicate that SM-CP and SM-DP acted mainly via the intrinsic pathway of the coagulation system. Differences detected in the anticoagulant activity values of SM-CP and SM-DP suggested that SM-DP was able to bind to the antithrombine III better than SM-CP; this was due to chemical modification induced during the deproteinization treatment.53 Briefly, due to the formation of an antithrombin-polysaccharide complex, the polysaccharide interacts with FIIa via electrostatic interactions between the negative charges of the sulfate groups and carboxyl groups of the polysaccharide and the positive charges of FIIa;56 the resulting complex induces the halt of the FIIa activity.
In this context, Yang et al.57 reported that the polysaccharides extracted from the sea cucumber Holothuria coluber displayed potential anticoagulant activities due to their high degree of sulfation. In addition, Liang et al.58 proved that the anticoagulant activities of polysaccharides would increase as a function of their sulfation degree and molecular mass. In fact, depending on their structures, average molar weights, and sulfate contents, polysaccharides display different anticoagulant properties. The structural interactions between polysaccharides with coagulation cofactors and their target proteases are also affected by the charge density and the sulfate content in polysaccharides.59,60
3.9. Fractionation of the S. marginatus polysaccharide by anion exchange chromatography
To fractionate the polysaccharides extracted from the flesh of S. marginatus, anion exchange chromatography (DEAE-c) was applied to the crude polysaccharide fraction, showing the best biological activity. The total sugars as well as the uronic acid contents were determined in each sub-fraction, as depicted in Fig. 7.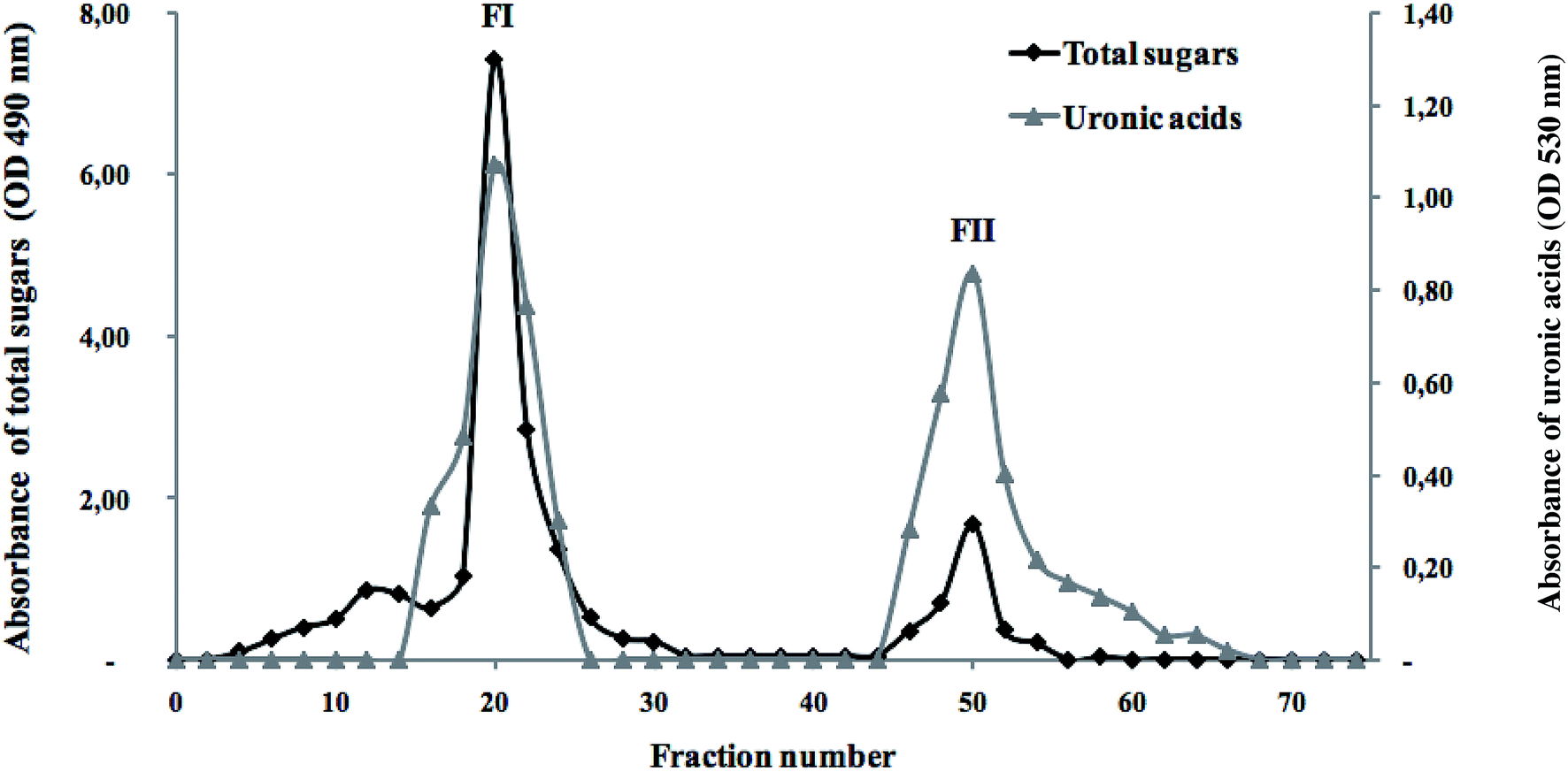 | ||
| Fig. 7 Elution profile of the SM-CP fractionated by anion-exchange chromatography (DEAE-cellulose) in terms of total sugars and uronic acids. | ||
The elution profile showed an excellent superposition between the total sugars and the uronic acids. Moreover, two peaks were eluted using a linear gradient of NaCl solution (1 M). Note that an increase in the NaCl concentration (3 M) did not affect the elution profile. These results revealed the presence of differently charged sugars in SM-CP, and the last peak (FII) represented the negatively charged polysaccharides. These peaks were pooled separately, desalted through UF and freeze-dried to test their antioxidant and anticoagulant activities.
3.10. Antioxidant and anticoagulant activities of the DEAE-c polysaccharide fractions
The antioxidant activities of the obtained fractions FII and FIII were evaluated in terms of DPPH radical-scavenging and ferrous chelating activities at a concentration of 5 mg ml−1. The results illustrated in Fig. 8a show that the best antioxidant activities are obtained in FII, the last eluted fraction. In fact, FII is found to be the most effective fraction in scavenging the DPPH free radicals and chelating the ferrous ions with 34% and 29% values, respectively.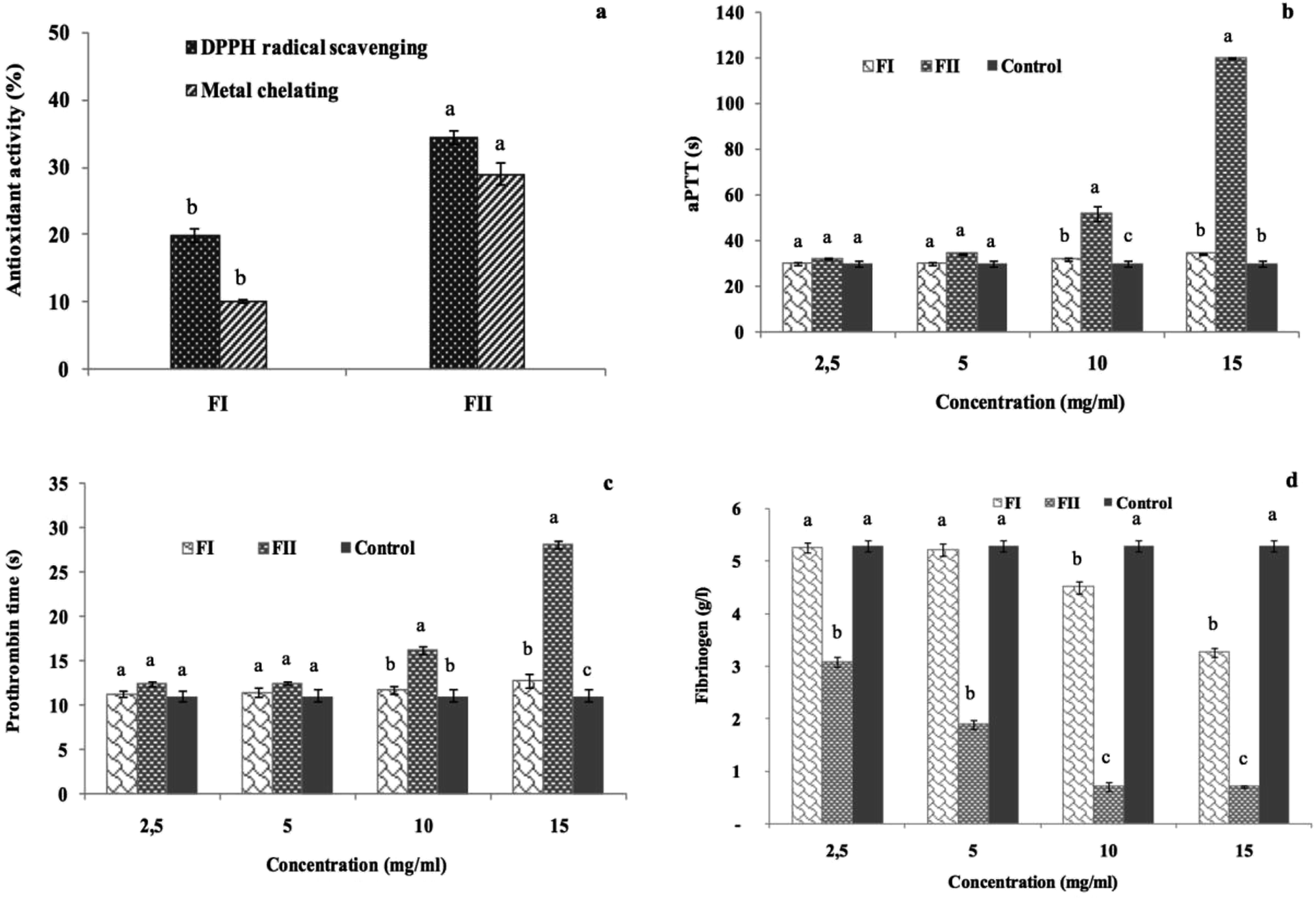 | ||
| Fig. 8 Antioxidant and anticoagulant activities of the FI and FII fractions; (a) DPPH radical-scavenging activity and metal chelating activity, (b) activated partial thromboplastin time (s), (c) prothrombin time (s), and (d) fibrinogen level (g l−1). Different letters within different fractions indicate significant differences at p ≤ 0.05. | ||
While the FI sub-fraction showed weak anticoagulant effect, FII prolonged significantly the aPTT and the PT clotting times as compared to the control, and the values reached 120 s and 28.15 s, respectively, against 30.03 s and 11.10 s for the control (p ≤ 0.05). However, the values obtained with the FI are still similar to those of the control (Fig. 8b and c). The effect of the FII addition reduced the fibrinogen level to 0.7 g l−1 vs. a fibrinogen concentration of 5.29 g l−1 in the absence of the sample (Fig. 8d).
These results prove the strong anticoagulant effect of the negatively charged sub-fraction (FII) due to its interaction with the intrinsic and the extrinsic pathway cofactors; in this context, Jung et al. (2007) (ref. 54) have reported that the sulfated polysaccharides purified from a brown alga, Ecklonia cava, enhance the inhibition of the coagulation factors II, VII and X and then block the extrinsic coagulation pathways, as observed through PT clotting-time evaluation. Recently, Mansour et al.61 reported through the aPTT test that the purified sulfated polysaccharide extracted from Holothuria polii exhibited a great anticoagulant effect under the intrinsic coagulation pathway. As the time required for clot formation in the plasma sample is inversely proportional to the concentration of fibrinogen, the lowest quantity of fibrinogen indicates highest anticoagulant effect. Subsequently, FIII showing the lowest FL demonstrated the best anticoagulant activity.
The overall data suggested that S. marginatus polysaccharides and mainly FII could be considered as a biologically promising anticoagulant agent. Hence, note that the anticoagulant activity of FII could be recovered through the inhibition of intrinsic, extrinsic and common coagulation pathways.
4 Conclusions
In the present study, we investigated the biological characteristics of polysaccharides extracted from the bivalve species S. marginatus via ethanol precipitation followed by anion exchange fractionation. The results revealed interesting antioxidant and anticoagulant activities of both the extracted polysaccharides and demonstrated, particularly, that the most negatively charged DEAE-c fraction (FII) showed the highest anticoagulant effects. In the antibacterial test, the best bacterial inhibitions were detected for SM-CP against S. aureus and L. monocytogenes. The overall results emphasize the potential biomedical effects of SP extracted from S. marginatus. However, further cellular experiments are needed to better understand the mechanism of action of polysaccharides before their concrete use in the production of functional foods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Ministry of Higher Education and Scientific Research, Tunisia.References
- S. B. Natarajan, Y.-S. Kim, J.-W. Hwang and P.-J. Park, RSC Adv., 2016, 6, 26163–26177 RSC.
- P. Laurienzo, Mar. Drugs, 2010, 8, 2435–2465 CrossRef CAS PubMed.
- S. S. Bandyopadhyay, M. H. Navid, T. Ghosh, P. Schnitzler and B. Ray, Phytochemistry, 2011, 72, 276–283 CrossRef CAS PubMed.
- R. Saravanan, K. K. Ebenezar, S. Rajasekaran and R. Thamaraiselvan, J. Biol. Sci., 2013, 13, 283 CrossRef CAS.
- M. Berri, C. Slugocki, M. Olivier, E. Helloin, I. Jacques, H. Salmon, H. Demais, M. Le Goff and P. N. Collen, J. Appl. Phycol., 2016, 28, 2999–3008 CrossRef.
- P. Seedevi, M. Moovendhan, S. Vairamani and A. Shanmugam, Int. J. Biol. Macromol., 2016, 85, 117–125 CrossRef CAS PubMed.
- O. Abdelhedi, R. Nasri, N. Souissi, M. Nasri and M. Jridi, Carbohydr. Polym., 2016, 152, 605–614 CrossRef CAS PubMed.
- Y.-Y. Wang, W.-Y. Qiu, Z.-B. Wang, H.-L. Ma and J.-K. Yan, RSC Adv., 2017, 7, 11067–11075 RSC.
- C. Ai, M. Duan, N. Ma, X. Sun, J. Yang, C. Wen, Y. Sun, N. Zhao and S. Song, J. Funct. Foods, 2018, 47, 211–219 CrossRef CAS.
- Z. Formanek, J. Kerry, F. Higgins, D. Buckley, P. Morrissey and J. Farkas, Meat Sci., 2001, 58, 337–341 CrossRef CAS PubMed.
- D. Liu, Y. Cao, X. Zhang, C. Peng, X. Tian, C. Yan, Y. Liu, M. Liu and Y. Han, Biochim. Biophys. Acta, Mol. Basis Dis., 2018, 1864, 2901–2912 CrossRef CAS PubMed.
- N. Ayache, L. Hmida, J. F. Cardoso, Z. Haouas, F. D. Costa and M. S. Romdhane, J. Shellfish Res., 2016, 35, 389–397 CrossRef.
- F. da Costa and D. Martínez-Patiño, Aquaculture, 2009, 288, 57–64 CrossRef.
- D. Ballabio, V. Consonni and F. Costa, Chemom. Intell. Lab. Syst., 2012, 111, 28–33 CrossRef CAS.
- G. Huang, S. Shu, T. Cai, Y. Liu and F. Xiao, Int. J. Food Sci. Nutr., 2012, 63, 739–741 CrossRef CAS PubMed.
- AOAC, Official methods of analysis, Association of Official Analytical Chemists, Washington, DC, 15th edn, 1990 Search PubMed.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- M. DuBois, K. A. Gilles, J. K. Hamilton, P. t. Rebers and F. Smith, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- T. Bitter and H. M. Muir, Anal. Biochem., 1962, 4, 330–334 CrossRef CAS PubMed.
- C.-F. Ji, Y.-B. Ji and D.-Y. Meng, Exp. Ther. Med., 2013, 6, 1259–1264 CrossRef CAS PubMed.
- P. Bersuder, M. Hole and G. Smith, J. Am. Oil Chem. Soc., 1998, 75, 181–187 CrossRef CAS.
- A. Yıldırım, A. Mavi and A. A. Kara, J. Agric. Food Chem., 2001, 49, 4083–4089 CrossRef.
- I. I. Koleva, T. A. van Beek, J. P. Linssen, A. d. Groot and L. N. Evstatieva, Phytochem. Anal., 2002, 13, 8–17 CrossRef CAS PubMed.
- E. A. Decker and B. Welch, J. Agric. Food Chem., 1990, 38, 674–677 CrossRef CAS.
- P. Prieto, M. Pineda and M. Aguilar, Anal. Biochem., 1999, 269, 337–341 CrossRef CAS PubMed.
- D. Vanden Berge and A. Vlietinck, Screening Methods for Antimicrobial and Antiviral Agents from Higher Plants, Methods in Plant Biochemistry, Academic Press, London, England, 1991 Search PubMed.
- M. Y. Celik, S. T. Çulha, M. Çulha, H. Yildiz, S. Acarli, I. Celik and P. Celik, Indian J. Geo-Mar. Sci., 2014, 43, 601–606 Search PubMed.
- V. Brock and M. Wolowicz, Oceanol. Acta, 1994, 17, 97–103 Search PubMed.
- N. Sayari, R. Balti, M. B. Mansour, I. B. Amor, I. Graiet, J. Gargouri and A. Bougatef, Biomed. Pharmacother., 2016, 80, 322–330 CrossRef CAS PubMed.
- C. Costachel, B. Coddeville, J.-P. Latgé and T. Fontaine, J. Biol. Chem., 2005, 280, 39835–39842 CrossRef CAS PubMed.
- C. G. De Kruif, F. Weinbreck and R. de Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340–349 CrossRef CAS.
- B.-w. Zhu, D.-m. Li, D.-y. Zhou, S. Han, J.-f. Yang, T. Li, W.-x. Ye and G. H. Greeley, Food Chem., 2011, 125, 1273–1278 CrossRef CAS.
- M. B. Mansour, M. Dhahri, M. Hassine, N. Ajzenberg, L. Venisse, V. Ollivier, F. Chaubet, M. Jandrot-Perrus and R. M. Maaroufi, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2010, 156, 206–215 CrossRef PubMed.
- I. S. Fernando, K. A. Sanjeewa, K. W. Samarakoon, W. W. Lee, H.-S. Kim, E.-A. Kim, U. Gunasekara, D. Abeytunga, C. Nanayakkara and E. de Silva, Algae, 2017, 32, 75–86 CrossRef CAS.
- M. Xin, L. Ren, Y. Sun, H.-h. Li, H.-S. Guan, X.-X. He and C.-X. Li, Eur. J. Med. Chem., 2016, 114, 33–40 CrossRef CAS PubMed.
- Q. Li, C. Li, C. Yang, C. Liu, G. Yu and H. Guan, Int. J. Biol. Macromol., 2013, 62, 281–286 CrossRef CAS PubMed.
- A. Alves, S. G. Caridade, J. F. Mano, R. A. Sousa and R. L. Reis, Carbohydr. Res., 2010, 345, 2194–2200 CrossRef CAS PubMed.
- W. Garnjanagoonchorn, L. Wongekalak and A. Engkagul, Chem. Eng. Process.: Process Intens., 2007, 46, 465–471 CrossRef CAS.
- V. Vicente-García, E. Ríos-Leal, G. Calderón-Domínguez, R. O. Cañizares-Villanueva and R. Olvera-Ramírez, Biotechnol. Bioeng., 2004, 85, 306–310 CrossRef PubMed.
- Y. Guo, S. Cong, J. Zhao, Y. Dong, T. Li, B. Zhu, S. Song and C. Wen, Carbohydr. Polym., 2018, 188, 54–59 CrossRef CAS PubMed.
- M. Jridi, M. Mezhoudi, O. Abdelhedi, S. Boughriba, W. Elfalleh, N. Souissi, R. Nasri and M. Nasri, Carbohydr. Polym., 2018, 194, 319–327 CrossRef CAS PubMed.
- Y. Zhang, X. Lu, Z. Fu, Z. Wang and J. Zhang, Food Chem., 2011, 127, 1084–1090 CrossRef CAS PubMed.
- A. B. Hsouna, M. Trigui, G. Culioli, Y. Blache and S. Jaoua, Food Chem., 2011, 125, 193–200 CrossRef.
- H. Wang, X. Guo, X. Hu, T. Li, X. Fu and R. H. Liu, Food Chem., 2017, 217, 773–781 CrossRef CAS PubMed.
- B. W. Souza, M. A. Cerqueira, A. I. Bourbon, A. C. Pinheiro, J. T. Martins, J. A. Teixeira, M. A. Coimbra and A. A. Vicente, Food Hydrocolloids, 2012, 27, 287–292 CrossRef CAS.
- S. J. Stohs and D. Bagchi, Free Radical Biol. Med., 1995, 18, 321–336 CrossRef CAS PubMed.
- X. Yang, X. Gao, F. Han and R. Tan, Biochim. Biophys. Acta, Gen. Subj., 2005, 1725, 120–127 CrossRef CAS PubMed.
- L. Costa, G. Fidelis, S. Cordeiro, R. Oliveira, D. Sabry, R. Câmara, L. Nobre, M. Costa, J. Almeida-Lima and E. Farias, Biomed. Pharmacother., 2010, 64, 21–28 CrossRef CAS PubMed.
- Y. Chen, H. Zhang, Y. Wang, S. Nie, C. Li and M. Xie, Food Chem., 2015, 186, 231–238 CrossRef CAS PubMed.
- M. Hajji, O. Masmoudi, N. Souissi, Y. Triki, S. Kammoun and M. Nasri, Food Chem., 2010, 121, 724–731 CrossRef CAS.
- O. Berteau and B. Mulloy, Glycobiology, 2003, 13, 29R–40R CrossRef CAS PubMed.
- R. Woodruff, Y. Xu, J. Layzer, W. Wu, M. Ogletree and B. Sullenger, J. Thromb. Haemostasis, 2013, 11, 1364–1373 CrossRef CAS PubMed.
- F. Sun, Y. Liu, D. Wang, Z. Wang, H. Mu, F. Wang, K. Ding and J. Duan, Carbohydr. Polym., 2017, 184, 191–198 CrossRef PubMed.
- W.-K. Jung, Y. Athukorala, Y.-J. Lee, S. H. Cha, C.-H. Lee, T. Vasanthan, K.-S. Choi, S.-H. Yoo, S.-K. Kim and Y.-J. Jeon, J. Appl. Phycol., 2007, 19, 425–430 CrossRef CAS.
- T. Lang, K. Johanning, H. Metzler, S. Piepenbrock, C. Solomon, N. Rahe-Meyer and K. A. Tanaka, Anesth. Analg., 2009, 108, 751–758 CrossRef PubMed.
- H. H. Hwang and D. Y. Lee, Macromol. Res., 2016, 24, 767–772 CrossRef CAS.
- W. Yang, Y. Cai, R. Yin, L. Lin, Z. Li, M. Wu and J. Zhao, Int. J. Biol. Macromol., 2018, 115, 1055–1062 CrossRef CAS PubMed.
- L. Liang, L. Ao, T. Ma, Y. Ni, X. Liao, X. Hu and Y. Song, Int. J. Biol. Macromol., 2018, 106, 447–455 CrossRef CAS PubMed.
- F. R. Melo, M. S. Pereira, D. Foguel and P. A. Mourão, J. Biol. Chem., 2004, 279, 20824–20835 CrossRef CAS PubMed.
- S. Sudharsan, N. Subhapradha, P. Seedevi, V. Shanmugam, P. Madeswaran, A. Shanmugam and A. Srinivasan, Int. J. Biol. Macromol., 2015, 81, 1031–1038 CrossRef CAS PubMed.
- M. B. Mansour, R. Balti, V. Ollivier, H. B. Jannet, F. Chaubet and R. M. Maaroufi, Carbohydr. Polym., 2017, 174, 760–771 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |