
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic molecular junctions between graphene sheets: a molecular dynamics screening for enhanced thermal conductance†
Alessandro Di Pierro a,
Maria Mar Bernala,
Diego Martineza,
Bohayra Mortazavib,
Guido Saraccoa and
Alberto Fina*a
a,
Maria Mar Bernala,
Diego Martineza,
Bohayra Mortazavib,
Guido Saraccoa and
Alberto Fina*a
aDipartimento di Scienza Applicata e Tecnologia, Politecnico di Torino, Alessandria Campus, Viale Teresa Michel 5, 15121 Alessandria, Italy. E-mail: alberto.fina@polito.it; Tel: +39 0131 229 316
bInstitute of Structural Mechanics, Bauhaus-Universität Weimar, Marienstraße 15, D-99423 Weimar, Germany
First published on 17th May 2019
Abstract
The proper design and synthesis of molecular junctions for the purpose of establishing percolative networks of conductive nanoparticles represent an opportunity to develop more efficient thermally-conductive nanocomposites, with several potential applications in heat management. In this work, theoretical classical molecular dynamics simulations were conducted to design and evaluate thermal conductance of various molecules serving as thermal bridges between graphene nanosheets. A wide range of molecular junctions was studied, with a focus on the chemical structures that are viable to synthesize at laboratory scale. Thermal conductances were correlated with the length and mechanical stiffness of the chemical junctions. The simulated tensile deformation of the molecular junction revealed that the mechanical response is very sensitive to small differences in the chemical structure. The analysis of the vibrational density of states provided insights into the interfacial vibrational properties. A knowledge-driven design of the molecular junction structures is proposed, aiming at controlling interfacial thermal transport in nanomaterials. This approach may allow for the design of more efficient heat management in nanodevices, including flexible heat spreaders, bulk heat exchangers and heat storage devices.
1 Introduction
Thermal management is central in several fast-growing technologies, from semiconductors industry,1 to avionics2 and rechargeable lithium-ion batteries.3 Inappropriate thermal management can result in overheating and consequent shortening of service life4 and may even lead to hazardous conditions such as fire.5 Aluminum and copper heat exchangers are common add-on solutions for high-drain integrated circuits, but they increase the number of components, system weight and assembly time, and therefore cost. Thermally-conductive chip packages made out of polymeric composites may represent a considerable improvement, but satisfactory materials are still lacking, due to insufficient thermal conductivity. In fact, research efforts are currently focused on the development of polymer composites and nanocomposites for efficient heat transfer.6–8 Carbon nanotubes (CNT) and a variety of 2D materials as graphene-related materials (GRM),9 boron nitride,10 fosforene,11 silicone12 and recently borophene,13 attracted great research interest due to the outstanding properties evaluated through in silico approaches. In particular, thermal properties of graphene were studied in details and strictly correlated to its phonon transport properties.14–16 Indeed, graphene was recognized as an excellent candidate for applications in thermally conductive materials. However, thermal transport in (nano)composites is strongly limited by the thermal resistances occurring between conductive particles in direct contact, as well as between particles and the surrounding matrix.8,17 The thermal resistance is caused by phonon scattering in the presence of structural defects and discontinuity,18 as well as acoustic mismatch19 phenomena between the two components of the interface. In particular, the inefficiency of phonon transfer within a network of conductive particles in contact has been related to the “soft” interface, which does not allow transfer of efficient vibrational modes of phonons.7,20 A possible approach to decrease this contact resistance is by grafting molecular junctions21 between nanoparticles, to increase the contact stiffness and enhance phonon transfer.22,23 However, controlled chemical functionalization of graphene remains challenging and presents drawbacks, as covalent bonding on graphene induces rehybridization,24 acting as lattice defects25 and altering the in-plane phonon modes,26 ultimately affecting the thermal properties of graphene.27,28 On the other hand, non-covalent functionalization based on secondary interactions29,30 is known to preserve the hybridization of graphene but is expected to deliver rather limited reductions in contact resistance.Non-Equilibrium Molecular Dynamics (NEMD) is a well-established numerical method to investigate thermal conductivity and interfacial thermal properties.31 The Interfacial Thermal Conductance (ITC), defined in this paper as the conductance between solid particles and the polymer matrix, reflects the physico-chemical properties of the interface;6,20 in fact, chemical compatibilization through organic chain grafting onto the solid particles was typically found to be effective to improve thermal transport.32–34 Wang et al.33 simulated covalently alkyl functionalized graphene platelets inside a polyethylene matrix, showing that an higher amount of linkers, and longer protruding linkers in matrix increases ITC. The Vibrational Density of States (VDOS) highlighted that functionalization induced a shift in frequencies where phonon match occurs more between polymer and fillers. A similar layout was reported by Wang et al.,34 via covalent functionalization of graphene with a variety of small dangling molecules: in this case, the VDOS spectra of butyl functionalized graphene found in the best phonon match with paraffin polymer matrix, resulting in a remarkable ITC increase. Later, Shen and coworkers35 investigated the ITC in epoxy resin/graphene nanocomposites, as a function of the chemical functionalization of graphene, and showed that the highest ITC reduction occurs with triethylenetetramine moieties, because of their ability to penetrate in the epoxy resin and form covalent bonds. On the other hand, the thermal conductance associated with filler–filler contacts, in this work referred to as Thermal Boundary Conductance (TBC), was also studied numerically by various authors, specifically between carbon nanotubes and graphene platelets surface-functionalized with small bridging groups such as oxygen or methylene bridges,36–39 short alkyl chains40 or benzene.41 TBC of alkyl molecular junctions covalently bound between two graphene flakes was investigated in details by Li and coworkers40 via Density Functional Theory (DFT), showing a correlation between TBC and the stretching imposed to the molecular junction. Recently, we also reported NEMD simulations of edge-to-edge alkyl junctions between graphene nanoribbons,42 where covalent bonding was compared to van der Waals forces between interpenetrating molecules.
While the above mentioned numerical simulations focused on small chemical moieties or alkyl chains as bridging agents between nanoparticles, the synthesis of these types of junctions between carbon nanoparticles is experimentally very challenging. Therefore, more viable chemical structures need to be addressed to allow for practical applications. Li et al.30 addressed for the first time aromatic molecules as thermal junctions in π-stacked graphene sheets, and compared it with a para-disubstituted benzene covalently bound junction. Han et al.43 reported aminosilane functionalization between a silicon substrate and GRM for heat management in electronic hotspots achieving lower operating temperatures than unfunctionalized samples. Furthermore, we recently reported the experimental and computational study of graphene nanoplatelets edge-functionalized with 1,5-bis(4-aminophenyloxy)pentane or 4-aminophenol to produce molecular junctions based on covalent bonding or as secondary interactions, respectively.44
Building on previous work, the present contribution addresses the TBC calculation of various aromatic molecules as possible candidates for the use in thermal molecular junctions, focusing on chemically viable systems that are synthesizable at laboratory scale. Different edge-grafted molecules, which act as thermal bridges between graphene nanosheets, are evaluated with respect to TBC via full atomistic NEMD. The contribution of vibrational modes, molecule stiffness, chain chemistry and segments lengths were systematically analyzed, providing a computational screening for molecules to be utilized experimentally.
2 Molecular dynamics modeling
NEMD calculations were carried out with Graphics Processing Unit (GPU) accelerated45 Large-scale Atomistic Molecular Massively Parallel Simulator (LAMMPS) code. The COMPASS force field developed by Sun46 was chosen for the introduction of the atomic interactions in this work. COMPASS is a widely used force field in the molecular dynamics simulation of organic materials, including GRM polymer nanocomposite.35,47 Although the Tersoff potential is more frequently used to calculate graphene thermal conductivity, COMPASS was also used by Zhang et al.48 to calculate a value of 550 W m−1 K−1 for thermal conductivity in graphene finite slab. COMPASS includes a much larger chemical species database than Tersoff and AIREBO, allowing simulating organic chemical structures as well as non-bond interactions in the condensed phase. It also contains anharmonic bonding terms, providing a more accurate representation of thermal transport processes,49 which appear relevant in molecular junctions.50 In this work, non-bond interactions were defined by the van der Waals contribution from the COMPASS built-in Lennard Jones 9-6 function and the Coulomb electrostatic term, by the use of atomic partial charges,46 with a 10 Å cut-off. Velocity Verlet algorithm recalculated positions and velocities every 0.25 fs while the initial partial charges assignment and equilibration was set through Qeq algorithm.51 Periodic Boundary Conditions (PBC) were set in x,y,z. However, being a supercell system, the simulation domain was padded to fit the model and empty space was set to more than double of the cut-off distance in order to avoid PBC interaction.The model was composed of about 4100 atoms, divided in two graphene nanoribbons (approximately 100 Å by 50 Å) grafted through the armchair edge by six parallel molecular junctions. Because the model size is shorter than the phonon mean free path in graphene, no inner flake phonon scattering flake is expected.16 This edge-to-edge layout is depicted in Fig. 1A, with six 1,5-bis(p-phenylenoxy)pentane junctions as an example. All aromatic molecules ends were bonded in para (p-) with the graphene nanoribbons (Fig. 1B′) except for the fully aromatic acene-based thermal bridges (Fig. 1B′′). The molecular junction length refers to the distance “d” between platelets edges (Fig. 1B). All the distances reported herein were determined through VMD52 tools from time-averaged measurements of the six junctions. Furthermore, the C–C distance was found 1.39 Å, in fair agreement with literature values (1.40 Å) for other force fields commonly adopted in thermal investigations with graphene.53
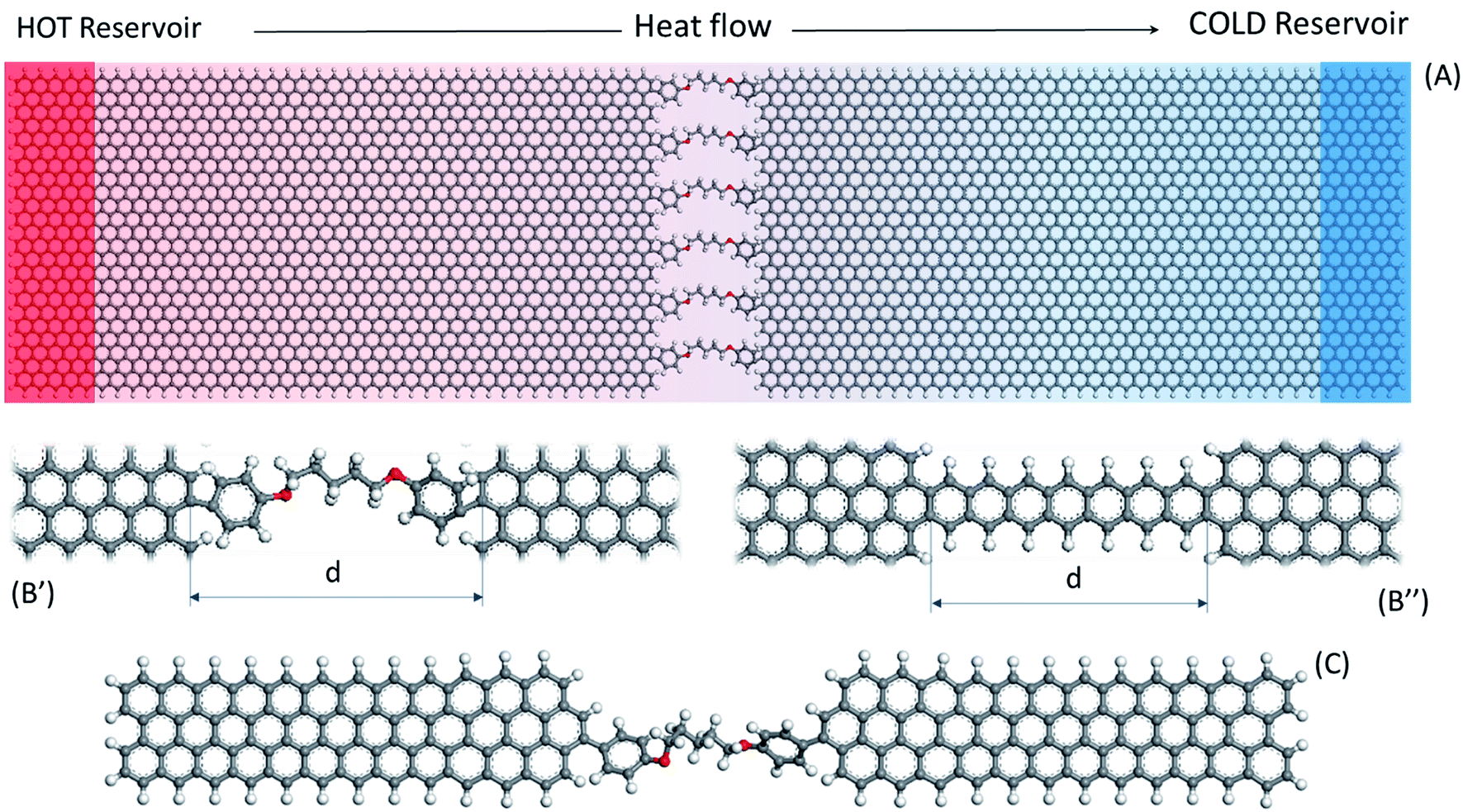 | ||
| Fig. 1 (A) Planar model representation and NEMD layout, (B′) junctions detail of 1,5-bis(p-phenylenoxy)pentane, C5OP, and (B′′) heptacene, with dimensioning (“d”) for platelets distance. (C) The model for tensile testing with C5OP, as example molecule. | ||
The simulation procedure followed a well-established scheme: the whole system was initially relaxed through an equilibration period of 125 ps in NVT (constant number of atoms, volume and temperature) canonical ensemble at 300 K, within a temperature range in which the quantum effect was previously reported to be negligible.54 At the end of the thermal equilibration, followed 250 ps of thermo-stated preheating with thermostats. In this stage, Nosé–Hoover thermostats, were set to 310 K and 290 K and applied to the model ends, about 8 Å along the x coordinate. Here, all the atoms in the system, except those in thermo-stated regions, run in NVE (constant number of atoms, volume and energy) and a thermal gradient is gradually established inside the model. With the system in steady state, for 4.8 ns, data collection27,55 of energy and temperature was performed. The thermal flow through thermostats was obtained from the slope the energy versus time plot,56 while the group temperatures were computed from the averaging the instantaneous local kinetic temperature27,55 as reported in eqn (1):
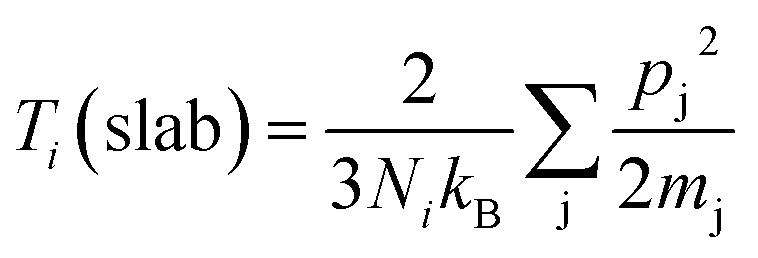 | (1) |
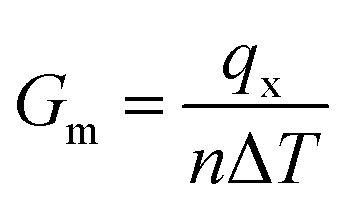 | (2) |
To investigate the phonons in molecules and graphene,26 vibrational spectra has been calculated for the isolated molecules as well as for a hydrogen-terminated graphene nanoribbon. The VDOS were calculated through discrete Fourier transform of the x, y and z velocity autocorrelation function in eqn (3):
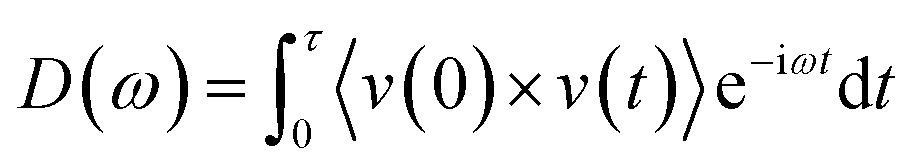 | (3) |
The elastic modulus was calculated through linear fitting up to 10% of deformation in elastic regime. Stress–strain plots are reported in Fig. S4.† The result of this test should be considered only in relative terms between the molecules and not as absolute Young modulus. Uniaxial strain simulations were conducted for six replicas along z at 0.1 K, similarly to metal nanowires,57,58 to minimize the velocity fluctuation noise in stress tensors calculation. For this study, a different specimen made out of two small graphene ribbons (about 80 × 10 Å2) in single junction has been designed for every molecule (as example, C5OP in shown in Fig. 1C). The simulation box PBC were set as fixed in x and periodic in y and z, adopting a 50 Å padding room as already adopted in VDOS calculation. An initial relaxation step in NVT was set bringing the temperature from 5 K to 0.1 K in 5 ps. Then, the left slab of the specimen was fixed and a longitudinal uniaxial velocity of 0.5 Å ps−1 was applied to the right-end graphene ribbon generating a constant pulling force overall the system. The simulation run in NVE for 20 ps or until the molecule broke, whichever occurred first.
3 Results and discussion
Sixteen molecular junctions were considered in this study, as summarized in Table 1. Aliphatic/aromatic molecules represent the largest subset, and include a variety of diphenyloxyalkanes (A–G, in Table 1): diphenoxymethane (C1OP); 1,2-diphenoxyethane (C2OP); 1,3-diphenoxypropane (C3OP); 1,4-diphenoxybutane (C4OP); 1,5-diphenoxypentane (C5OP); 1,6-diphenoxyhexane (C6OP); 1,7-diphenoxyheptane (C7OP). 1,3-Dibenzylbenzene (PCP) and 1,7-diphenylheptane (C7P) were also addressed as ether-free counterparts. Diphenoxybenzene (POP, H in Table 1) exploited a central aromatic moiety instead of the aliphatic one. Fully aromatic junctions include, biphenyl (BP), phenanthrene (PH) and pyrene (PY) (K–M in Table 1). It is worth mentioning that all of these junctions may in principle be obtained between graphene sheets via aryldiazonium chemistry, as demonstrated experimentally for C5OP.44 For sake of comparison, junctions based on acenes containing 3 (anthracene, ACN), 5 (pentacene, PCN) or 7 (heptacene, HCN) benzene rings, were also simulated (N–P in Table 1). While their use as thermal junction remains theoretical owing to the difficulties in the synthesis of such molecular junctions, six-member carbon rings between graphene nanoribbons were previously addressed as a model system for graphitized polyimide/graphene composites.59 It is worth noting that acene junctions preserves the conjugation of sp2 carbon across the contact of the two graphene sheets; in fact acene-bridged graphene sheets could even be considered as a single graphene with rectangular holes in its structure, that was previously addressed by Yarifard et al.60| # | Long name | Short name | Gm [pW K−1] | E [GPa] | d [Å] | Structure |
|---|---|---|---|---|---|---|
| A | Diphenoxymethane | C1OP | 208 ± 53 | 145 | 12.5 | 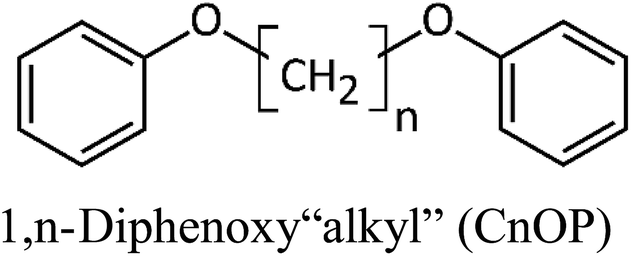 |
| B | 1,2-Diphenoxyethane | C2OP | 182 ± 5 | 80 | 13.8 | |
| C | 1,3-Diphenoxypropane | C3OP | 161 ± 20 | 94 | 14.9 | |
| D | 1,4-Diphenoxybutane | C4OP | 148 ± 23 | 72 | 16.3 | |
| E | 1,5-Diphenoxypentane | C5OP | 139 ± 7 | 93 | 17.2 | |
| F | 1,6-Diphenoxyhexane | C6OP | 130 ± 9 | 53 | 18.8 | |
| G | 1,7-Diphenoxyheptane | C7OP | 123 ± 6 | 70 | 20.0 | |
| H | 1,3-Diphenoxybenzene | POP | 260 ± 16 | 152 | 16.6 |  |
| I | 1,3-Dibenzylbenzene | PCP | 221 ± 14 | 85 | 16.3 | 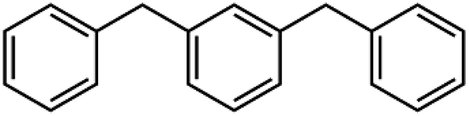 |
| J | 1,7-Diphenylheptane | C7P | 139 ± 33 | 65 | 17.5 | 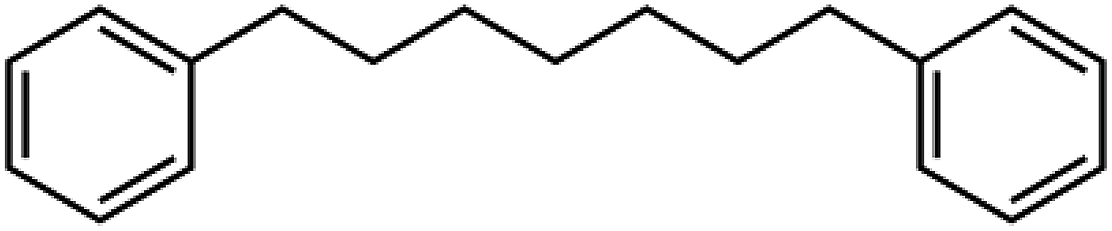 |
| K | Biphenyl | BP | 472 ± 26 | 141 | 9.7 |  |
| L | Phenanthrene | PH | 608 ± 25 | 165 | 9.8 | 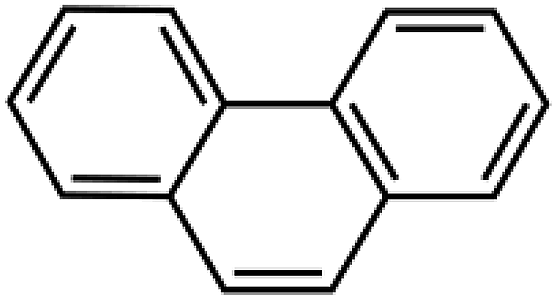 |
| M | Pyrene | PY | 648 ± 14 | 179 | 9.7 | 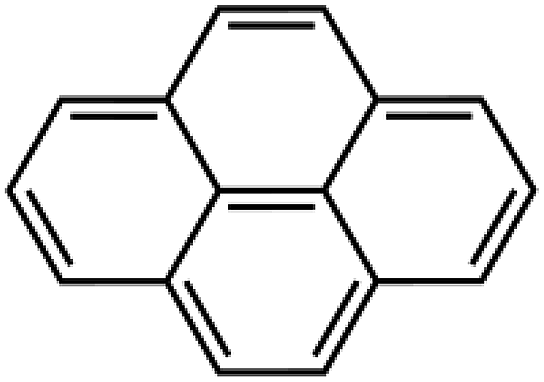 |
| N | Anthracene | ACN | 1121 ± 35 | 279 | 7.4 | 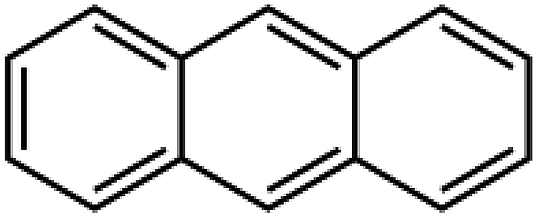 |
| O | Pentacene | PCN | 1076 ± 44 | 285 | 12.3 | 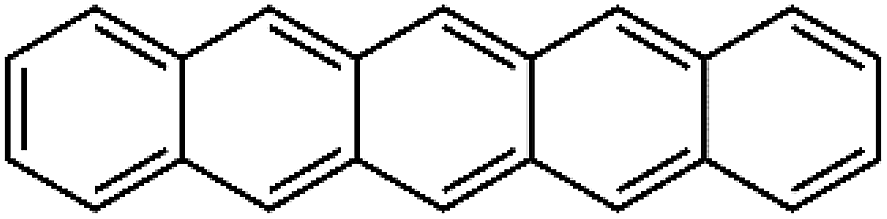 |
| P | Heptacene | HCN | 1007 ± 40 | 268 | 17.1 | 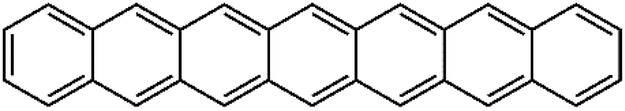 |
As a first case study, C5OP was analyzed in a range of parameters, including Thermostat Temperature Difference (TTD), molecular conformation and junction topology. The TTD effect was evaluated keeping the simulation equilibrium temperature centered at 300 K and increasing the TTD from 20 K to 25 K and 30 K, leading to Gm values of 139 ± 7, 131 ± 6 and 135 ± 4 pW K−1, respectively, which are considered constant within the experimental uncertainty. When shortening the alkyl chain in aliphatic/aromatic (CnOP) junctions to C4OP and C3OP, an enhancement of Gm to 148 ± 23 and 161 ± 20 pW K−1 was obtained, respectively. Further shortening to C2OP and C1OP exhibited further increased values up to 182 ± 5 and 208 ± 53 pW K−1, thus confirming the increasing conductance with shortening junction length. On the other hand, as expected, longer component of the alkyl portion reported lower Gm: 130 ± 9 pW K−1 in C6OP and 123 ± 6 pW K−1 in C7OP. It is also worth noting that the calculated thermal conductances of those molecules were in the same order of magnitude of similar length40,42 alkyl ones.
To investigate the CnOP molecular junctions stiffness as a function of the chain length, a set of tensile simulations have been performed. Resulting elastic moduli (E) were found in the range between 145 and 53 GPa for C1OP and C6OP, respectively, with a decreasing trend over values with increasing alkyl chain length (Table 1). As matter of comparison, elastic modulus of 487 GPa was calculated for pristine graphene slab. Despite this value is significantly lower, compared to 980 GPa previously reported,27, (which may be related to the different force field as well as to the small model size), a comparison of the obtained modulus remains possible within this set of results. Interestingly, the tensile modulus trend vs. CnOP junction length was found to be consistent with the decrease in thermal conductance (Fig. 2A), despite an odd–even effect is observed in calculated elastic modulus. To gain further insight about the role of chain length, VDOS were calculated for the different junctions (Fig. 2B). Graphene exhibits a broad region of vibrational states up to 5 THz, a sharp peak at 45.2 THz (called G peak from Raman analysis, usually at 47 THz15) and broad peak around 17 THz. For molecular junctions, several additional bands are observable in the regions 0 to 25 THz and 42 to 46 THz. However, very limited match is observed between VDOS spectra for pristine graphene and in the presence of CnOP molecular junctions. The almost linear decay in thermal conductance reported in Fig. 2, combined with the phonon spectra mismatch, is indicative of a diffusive regime with scattering at the interface strongly limiting heat transfer.
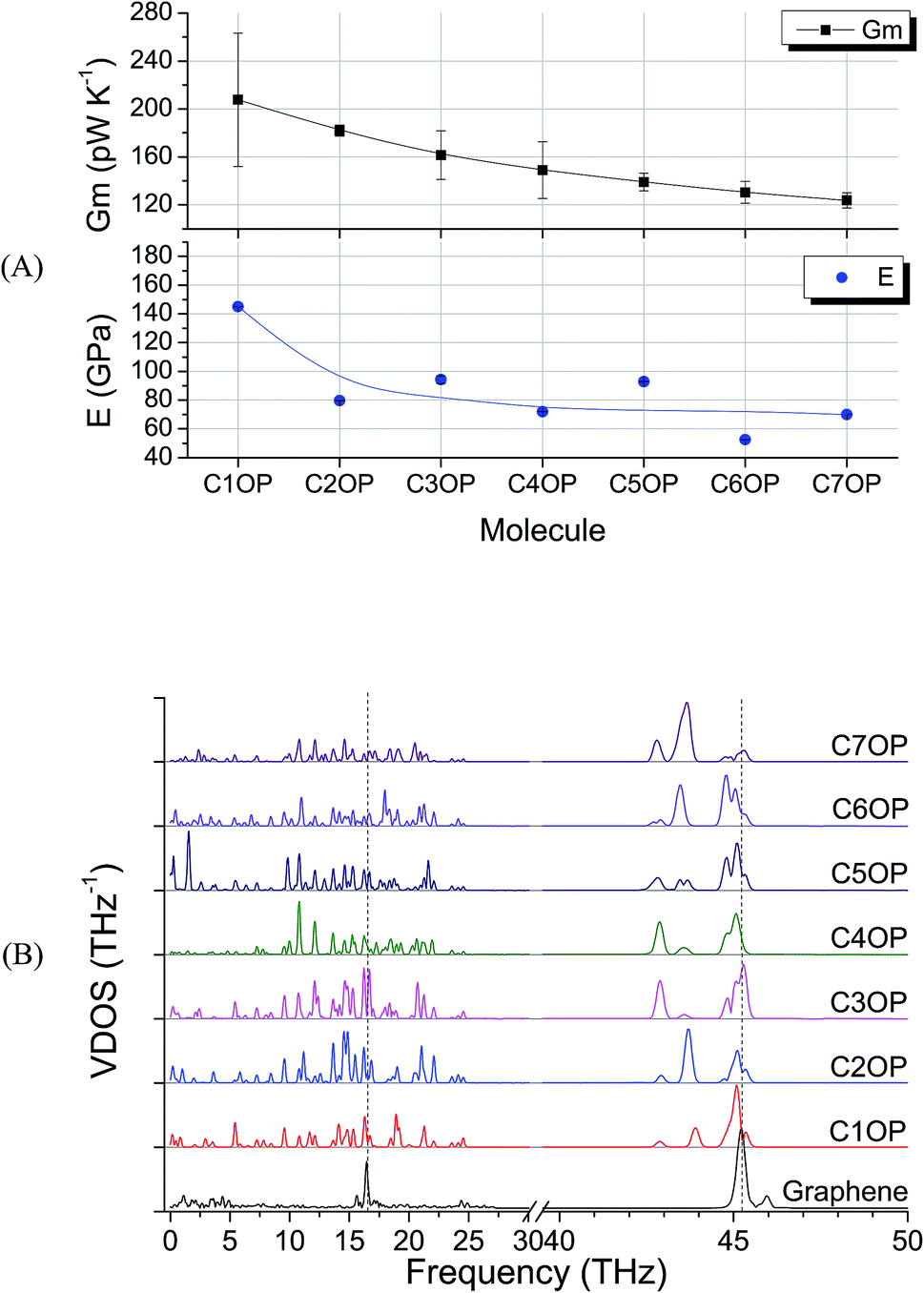 | ||
| Fig. 2 (A) Gm and elastic modulus E for aliphatic–aromatic molecular junctions, the line guides the eye through values reported in Table 1. Error bars in E are below 1% and barely visible. (B) VDOS for the same aliphatic–aromatic molecular junctions, scale in 40–50 THz is magnified by a factor of three. The dashed lines highlight the most intense graphene peaks at 16.5 THz and 45.2 THz (G-peak). | ||
The role of ether bond was studied comparing junctions with similar length, namely C5OP and C7P (E, J in Table 1). Despite the stiffness of C7P (65 GPa) was found approx. 30% lower than C5OP, the 139 ± 33 pW K−1 conductance for C7P is equivalent to the value for C5OP. This finding suggests that the presence of the soft alkyl component in the center of the molecular junction limits the overall heat transfer and ether substitution in a relatively long alkyl chain has a negligible effect. The VDOS for C7P is reported in Fig. 3, which shows the main contribution from alkyl moieties as broad peaks in the 42–44 THz region, in fair agreement with the spectrum for C5OP. The role of flexible groups (ether vs. methylene bridges) was further investigated in more rigid aromatic structure. Junctions based on diphenoxybenzene (POP, d = 16.6 Å, H in Table 1) and dibenzylbenzene (PCP, d = 16.3 Å, I in Table 1) were compared in both thermal conductance and stiffness. The thermal conductances for these systems were found about 260 ± 16 pW K−1 for POP and 221 ± 14 for PCP, suggesting ether bridges are more effective that methylene in phonon transfer. Furthermore, these values were compared to a junction of comparable length, C4OP, (148 ± 23 pW K−1, d = 16.3 Å, D in Table 1), which clearly evidenced the advantage of aromatic structures in terms of thermal conductance. The analysis of VDOS spectra (Fig. 3) provided further support for the differences in heat transfer. Indeed, signal in the region of 43–44 THz in VDOS spectra progressively reduced from C7P to PCP, while no significant counts in this band remains for POP. Furthermore, a stronger peak rises around 16.5 THz in PCP and POP, while the G peak for POP junctions appears to best overlap with the main vibration of pristine graphene.
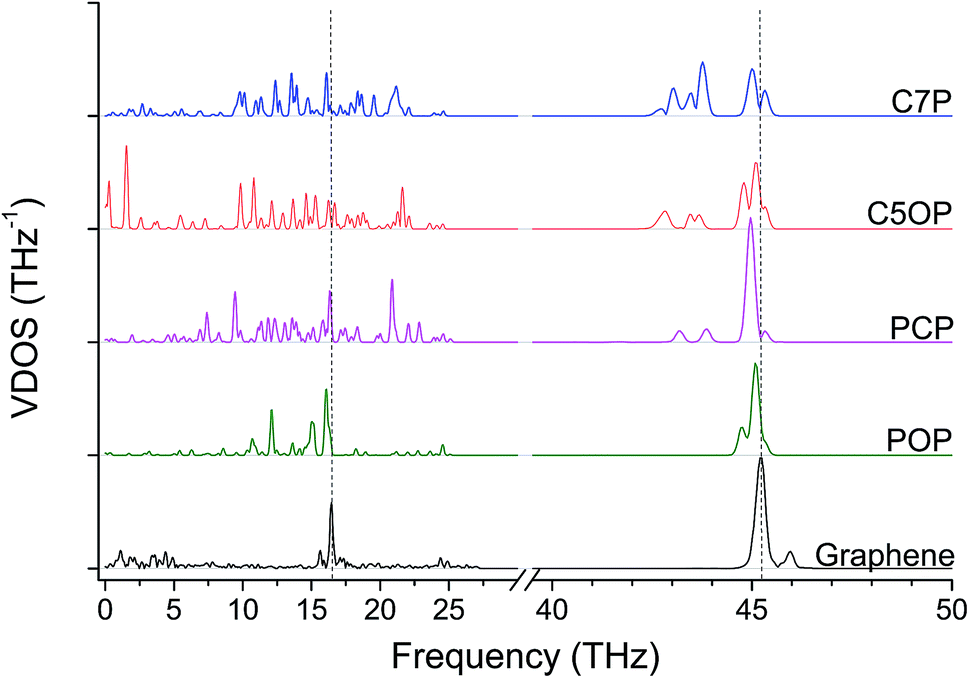 | ||
| Fig. 3 VDOS for molecular junctions with/without ether bridge, the scale in 40–50 THz is magnified by a factor of three for clarity. Dashed lines highlights the most intense graphene peaks at 16.5 THz and 45.2 THz (G-peak). | ||
Tensile simulations highlighted a wide gap between elastic moduli for POP (152 GPa) and PCP (85 GPa). This difference suggested that the deformability of flexible moieties is indeed crucial in controlling the overall junction stiffness, also when rigid aromatic rings are present in the molecular structure. Indeed, carbon–oxygen bond parameters in COMPASS evidence for significantly higher stiffness compared to C–C (2nd, 3rd and 4th power coefficients for c4o-o2e are remarkably higher than c4-c4,46 details in ESI, Table S1 and Fig. S4†), directly affecting the vibrational states. Based on this landmark, polyaromatic hydrocarbons were also studied as potentially effective molecular junctions. In fact, polyaromatic molecules exhibits highly delocalized electronic structures, and limited conformational freedom of the molecule, becoming progressively more similar to graphene itself when increasing the number of condensed aromatic rings. Indeed, VDOS analysis of selected polyaromatic junctions (Fig. 4) confirmed the general excellent overlapping of the two most intense signal for polyaromatic junction with the 16.5 THz and 45.2 THz peaks for graphene. Some differences are clearly observable between the aromatic junctions and graphene, the molecular junctions own more vibrational modes in the region 5–20 THz. A thermal conductance of 472 ± 26 pW K−1 was calculated for biphenyl junction (BP, K in Table 1) and further enhancement was obtained for higher aromatic condensation, as 608 ± 25 pW K−1 was calculated for phenanthrene (PH, L in Table 1) and 648 ± 14 pW K−1 for pyrene (PY, M in Table 1). The tensile deformation of aromatic junctions reflects a higher stiffness than the alkyl based ones, with calculated elastic moduli of 141 GPa for biphenyl, 165 GPa for phenanthrene and 179 GPa for pyrene.
 | ||
| Fig. 4 VDOS for aromatic molecules, scale in 40–50 THz is magnified by a factor of three. Dashed lines highlights the most intense graphene peaks at 16.5 THz and 45.2 THz (G-peak). | ||
Based on the results obtained for aliphatic and aromatic junctions described so far, the combination of length and stiffness of the bridging chain appear to control the overall efficiency of the thermal transport through the interface. In fact, short and stiff junctions lead to the highest values of thermal conductance, as depicted in Fig. 5.
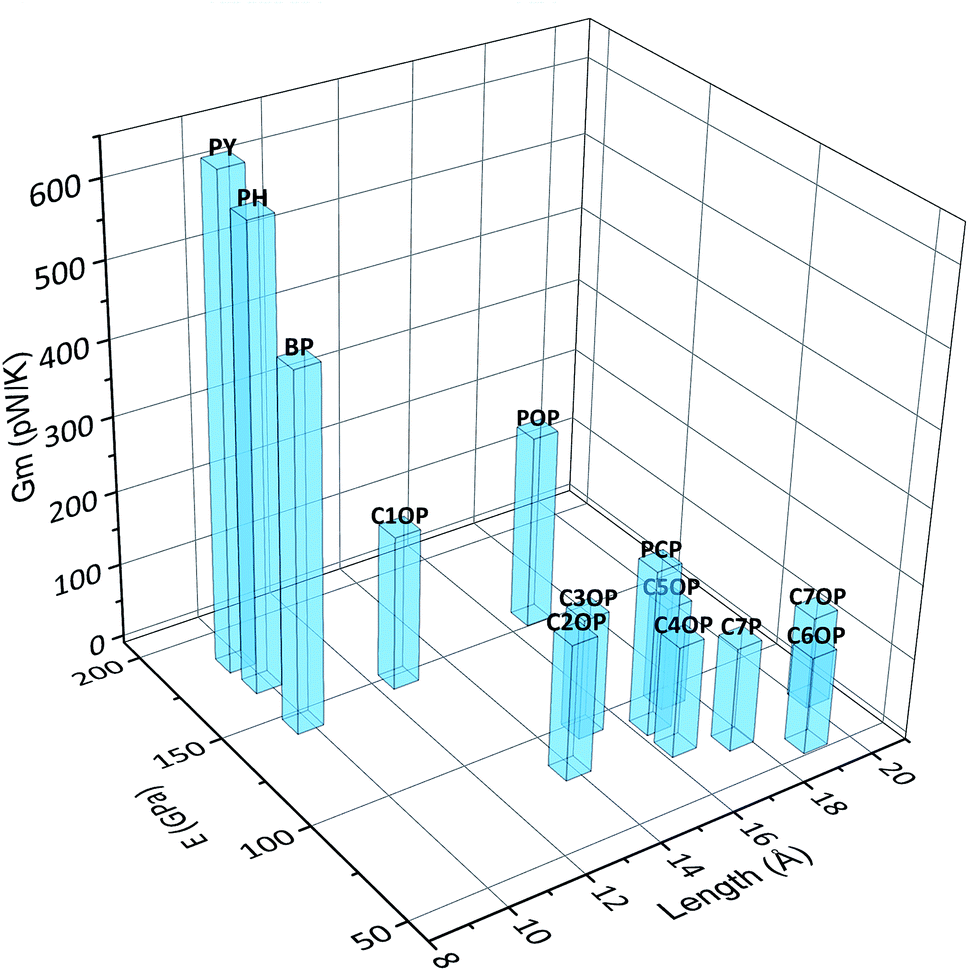 | ||
| Fig. 5 Calculated thermal conductance Gm of thermal junctions as a function of length and stiffness. | ||
Finally, acene-based junctions made of 3, 5 and 7 aromatic rings (anthracene, ACN, N in Table 1; pentacene, PCN, O in Table 1; heptacene, HCN, P in Table 1) were addressed. Despite the exploitation of acenes in molecular junctions is extremely challenging, these junctions were addressed here as a kind of theoretical “upper bound” for the thermal conductance for thermal bridges between graphene sheets, as it preserves the conjugation of sp2 carbon across the contact of the two graphene sheets. The tensile testing revealed almost constant elastic modulus values, namely 279 GPa for ACN, 285 GPa for PCN and 268 GPa for HCN. On the other hand, Gm values of 1121 ± 35, 1076 ± 44 and 1007 ± 40 pW K−1 were obtained for ACN, PCN and HCN, respectively. These values appear to be only slightly affected by the acene length, which more than doubles, rising from 7.4 Å for ACN to 17.1 Å for HCN. These results suggest the phonon transfer on these junctions to be ballistic, in contrast with the diffusive regime observed in alkyl junctions. As expected, the VDOS (Fig. 6) for polyaromatic junctions closely matches the main bands for pristine graphene, similarly to other aromatic junctions (Fig. 4).
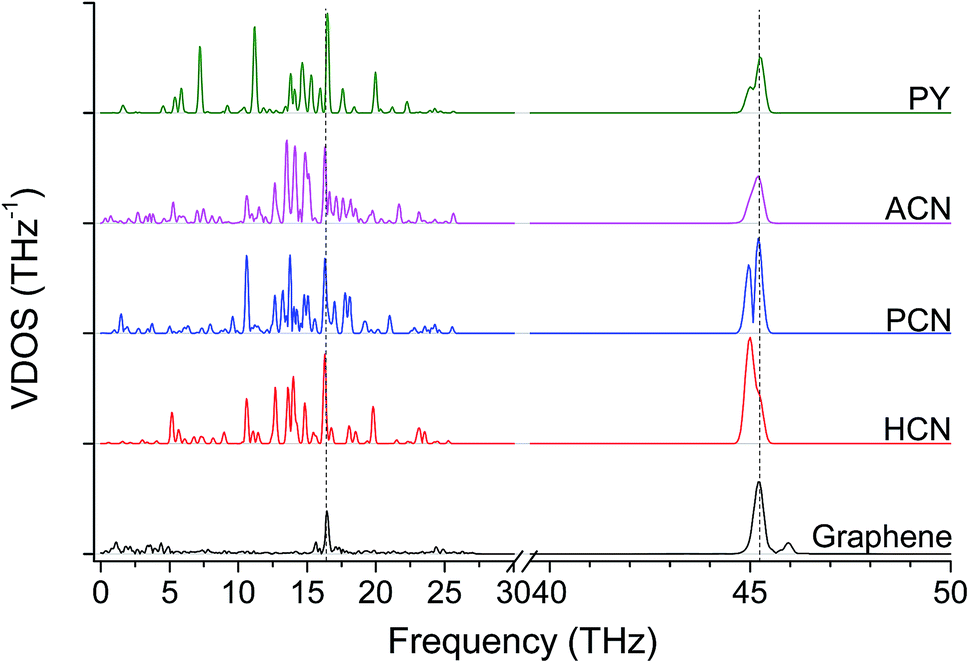 | ||
| Fig. 6 VDOS in acenes, scale in 40–50 THz is magnified by a factor of three. Dashed lines highlights the most intense graphene peaks at 16.5 THz and 45.2 THz (G-peak). Pyrene is added as a matter of comparison. | ||
4 Conclusions
The thermal conductance of molecular junctions between graphene nanoribbons were investigated by means of non-equilibrium molecular dynamics simulations. Particular focus was dedicated to experimentally synthesizable junctions for the purpose of improving the thermal percolation within networks of graphene nanoflakes. Thermal boundary conductance was found to strongly correlate with molecular junction length and stiffness. Calculated thermal conductance was found to be lowest for aliphatic/aromatic junctions characterized by the long and flexible alkyl chain and highest for the short and rigid polyaromatic bridging molecules (e.g. phenanthrene and pyrene). Chemical structures that are able to change structural conformations due to the presence of free rotating single bonds, appeared to directly limit the thermal transport capability. Differences were also observed between junctions containing methylene and ether groups, related to the different features of their bonds to carbon.In terms of phonon density of states, the overlapping of molecular junction VDOS with the spectra of graphene was found to correlate with the thermal conductance of the junctions. While VDOS for aliphatic/aromatic junctions exhibited limited overlapping with the main vibration bands of graphene at both 16.5 THz and 45.2 THz, aromatic and polyaromatic structures displayed vibrational spectra similar to graphene.
Finally, acene-based junctions, which preserve the conjugation of sp2 carbon across the contact of two graphene sheets, were included in this study for sake of comparison. Their vibrational density of state was found comparable to graphene, resulting in values of thermal conductance that exceed 1000 pW K−1. We found that thermal conductance varies little with acene length, suggesting ballistic phonon transfer through the interface. While acene junctions synthesis is currently very challenging, the simulated conductance values obtained here should be considered as a theoretical upper limit to the thermal performance of any experimentally viable junction.
The screening of the different covalent junction structures addressed in this paper provides a guideline for the development of thermally efficient interfaces to be exploited in graphene-based thermal management materials. Furthermore, the molecular thermal conductance values calculated here can be used in upper-scale continuum models to predict effective thermal conductivity in graphene-based laminates or polymer nanocomposites. Proposed methodology can be also extended for the engineering and design of thermally percolative networks for the enhancement of heat transfer in other nanostructured systems.
Authors' contributions
A. Fina conceived the experiments, interpreted results and led the project, A. Di Pierro carried out MD simulations, post processing and interpreted results, Diego Martinez provided VDOS spectra interpretation. B. Mortazavi supported NEMD calculation and discussion of the results. Maria Mar Bernal supported chemical interpretation and G. Saracco participated to the discussion of results. Manuscript was mainly written by A. Di Pierro and A. Fina.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme grant agreement 639495—INTHERM—ERC-2014-STG. B. M. acknowledges the financial support by ERC for COMBAT project (Grant number 615132).References
- S. Ghosh, I. Calizo, D. Teweldebrhan, E. P. Pokatilov, D. L. Nika, A. A. Balandin, W. Bao, F. Miao and C. N. Lau, Extremely high thermal conductivity of graphene: Prospects for thermal management applications in nanoelectronic circuits, Appl. Phys. Lett., 2008, 92(15), 151911 CrossRef.
- S. V. Garimella, A. S. Fleischer, J. Y. Murthy, A. Keshavarzi and R. Prasher, Thermal Challenges in Next-Generation Electronic Systems, IEEE Trans. Compon. Packag. Technol., 2008, 31(4), 801–815 Search PubMed.
- J. M. Tarascon and M. Armand, Issues and challenges facing rechargeable lithium batteries, Nature, 2001, 414(6861), 359–367 CrossRef CAS PubMed.
- N. Vichare, P. Rodgers, V. Eveloy and M. G. Pecht, In situ temperature measurement of a notebook computer - A case study in health and usage monitoring of electronics, IEEE Trans. Device Mater. Reliab., 2004, 4(4), 658–663 CrossRef.
- A. Hammami, N. Raymond and M. Armand, Lithium-ion batteries: runaway risk of forming toxic compounds, Nature, 2003, 424(6949), 635–636 CrossRef CAS PubMed.
- Z. D. Han and A. Fina, Thermal conductivity of carbon nanotubes and their polymer nanocomposites: A review, Prog. Polym. Sci., 2011, 36(7), 914–944 CrossRef CAS.
- N. Burger, A. Laachachi, M. Ferriol, M. Lutz, V. Toniazzo and D. Ruch, Review of thermal conductivity in composites: Mechanisms, parameters and theory, Prog. Polym. Sci., 2016, 61, 1–28 CrossRef CAS.
- H. Y. Chen, V. V. Ginzburg, J. Yang, Y. F. Yang, W. Liu, Y. Huang, L. B. Du and B. Chen, Thermal conductivity of polymer-based composites: Fundamentals and applications, Prog. Polym. Sci., 2016, 59, 41–85 CrossRef CAS.
- E. Chiavazzo and P. Asinari, Reconstruction and modeling of 3D percolation networks of carbon fillers in a polymer matrix, Int. J. Therm. Sci., 2010, 49(12), 2272–2281 CrossRef CAS.
- B. Mortazavi, H. Yang, F. Mohebbi, G. Cuniberti and T. Rabczuk, Graphene or h-BN paraffin composite structures for the thermal management of Li-ion batteries: A multiscale investigation, Appl. Energy, 2017, 202, 323–334 CrossRef CAS.
- Y. Y. Zhang, Q. X. Pei, J. W. Jiang, N. Wei and Y. W. Zhang, Thermal conductivities of single- and multi-layer phosphorene: a molecular dynamics study, Nanoscale, 2016, 8(1), 483–491 RSC.
- B. Mortazavi, A. Dianat, G. Cuniberti and T. Rabczuk, Application of silicene, germanene and stanene for Na or Li ion storage: A theoretical investigation, Electrochim. Acta, 2016, 213, 865–870 CrossRef CAS.
- B. Mortazavi, A. Dianat, O. Rahaman, G. Cuniberti and T. Rabczuk, Borophene as an anode material for Ca, Mg, Na or Li ion storage: A first-principle study, J. Power Sources, 2016, 329, 456–461 CrossRef CAS.
- A. A. Balandin, Thermal properties of graphene and nanostructured carbon materials, Nat. Mater., 2011, 10(8), 569–581 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Superior thermal conductivity of single-layer graphene, Nano Lett., 2008, 8(3), 902–907 CrossRef CAS PubMed.
- G. Fugallo, A. Cepellotti, L. Paulatto, M. Lazzeri, N. Marzari and F. Mauri, Thermal conductivity of graphene and graphite: collective excitations and mean free paths, Nano Lett., 2014, 14(11), 6109–6114 CrossRef CAS PubMed.
- Y. Su, J. J. Li and G. J. Weng, Theory of thermal conductivity of graphene-polymer nanocomposites with interfacial Kapitza resistance and graphene-graphene contact resistance, Carbon, 2018, 137, 222–233 CrossRef CAS.
- A. Maiti, G. D. Mahan and S. T. Pantelides, Dynamical simulations of nonequilibrium processes – Heat flow and the Kapitza resistance across grain boundaries, Solid State Commun., 1997, 102(7), 517–521 CrossRef CAS.
- E. T. Swartz and R. O. Pohl, Thermal-Resistance at Interfaces, Appl. Phys. Lett., 1987, 51(26), 2200–2202 CrossRef CAS.
- X. Xu, J. Chen, J. Zhou and B. Li, Thermal Conductivity of Polymers and Their Nanocomposites, Adv. Mater., 2018, 30(17), e1705544 CrossRef PubMed.
- F. Sun, T. Zhang, M. M. Jobbins, Z. Guo, X. Zhang, Z. Zheng, D. Tang, S. Ptasinska and T. Luo, Molecular bridge enables anomalous enhancement in thermal transport across hard-soft material interfaces, Adv. Mater., 2014, 26(35), 6093–6099 CrossRef CAS PubMed.
- J. C. Klockner, J. C. Cuevas and F. Pauly, Transmission eigenchannels for coherent phonon transport, Phys. Rev. B, 2018, 97(15), 155432 CrossRef.
- D. Martinez Gutierrez, A. Di Pierro, A. Pecchia, L. M. Sandonas, R. Gutierrez, M. Bernal, B. Mortazavi, G. Cuniberti, G. Saracco and A. Fina, Thermal bridging of graphene nanosheets via covalent molecular junctions: a non-equilibrium Green's functions–density functional tight-binding study, Nano Res., 2019, 12(4), 791–799 CrossRef CAS.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Functionalization of graphene: covalent and non-covalent approaches, derivatives and applications, Chem. Rev., 2012, 112(11), 6156–6214 CrossRef CAS PubMed.
- R. Gulotty, M. Castellino, P. Jagdale, A. Tagliaferro and A. A. Balandin, Effects of functionalization on thermal properties of single-wall and multi-wall carbon nanotube-polymer nanocomposites, ACS Nano, 2013, 7(6), 5114–5121 CrossRef CAS PubMed.
- D. L. Nika and A. A. Balandin, Phonons and thermal transport in graphene and graphene-based materials, Rep. Prog. Phys., 2017, 80(3), 036502 CrossRef PubMed.
- B. Mortazavi and S. Ahzi, Thermal conductivity and tensile response of defective graphene: A molecular dynamics study, Carbon, 2013, 63, 460–470 CrossRef CAS.
- Y. Y. Zhang, Q. X. Pei, X. Q. He and Y. W. Mai, A molecular dynamics simulation study on thermal conductivity of functionalized bilayer graphene sheet, Chem. Phys. Lett., 2015, 622, 104–108 CrossRef CAS.
- T. F. Luo and J. R. Lloyd, Enhancement of Thermal Energy Transport Across Graphene/Graphite and Polymer Interfaces: A Molecular Dynamics Study, Adv. Funct. Mater., 2012, 22(12), 2495–2502 CrossRef CAS.
- Q. Li, M. Strange, I. Duchemin, D. Donadio and G. C. Solomon, A Strategy to Suppress Phonon Transport in Molecular Junctions Using pi-Stacked Systems, J. Phys. Chem. C, 2017, 121(13), 7175–7182 CrossRef CAS.
- F. Müller-Plathe, A simple nonequilibrium molecular dynamics method for calculating the thermal conductivity, J. Chem. Phys., 1997, 106(14), 6082–6085 CrossRef.
- Y. Gao and F. Muller-Plathe, Increasing the Thermal Conductivity of Graphene-Polyamide-6,6 Nanocomposites by Surface-Grafted Polymer Chains: Calculation with Molecular Dynamics and Effective-Medium Approximation, J. Phys. Chem. B, 2016, 120(7), 1336–1346 CrossRef CAS PubMed.
- M. Wang, N. Hu, L. Zhou and C. Yan, Enhanced interfacial thermal transport across graphene–polymer interfaces by grafting polymer chains, Carbon, 2015, 85, 414–421 CrossRef CAS.
- Y. Wang, H. F. Zhan, Y. Xiang, C. Yang, C. M. Wang and Y. Y. Zhang, Effect of Covalent Functionalization on Thermal Transport across Graphene-Polymer Interfaces, J. Phys. Chem. C, 2015, 119(22), 12731–12738 CrossRef CAS.
- X. Shen, Z. Y. Wang, Y. Wu, X. Liu and J. K. Kim, Effect of functionalization on thermal conductivities of graphene/epoxy composites, Carbon, 2016, 108, 412–422 CrossRef CAS.
- M. B. Bigdeli and M. Fasano, Thermal transmittance in graphene based networks for polymer matrix composites, Int. J. Therm. Sci., 2017, 117, 98–105 CrossRef CAS.
- V. Varshney, J. Lee, A. K. Roy and B. L. Farmer, Modeling of interface thermal conductance in longitudinally connected carbon nanotube junctions, J. Appl. Phys., 2011, 109(8), 084913 CrossRef.
- W. Chen, J. C. Zhang and Y. A. Yue, Molecular dynamics study on thermal transport at carbon nanotube interface junctions: Effects of mechanical force and chemical functionalization, Int. J. Heat Mass Transfer, 2016, 103, 1058–1064 CrossRef CAS.
- M. Fasano, M. B. Bigdeli, M. R. V. Sereshk, E. Chiavazzo and P. Asinari, Thermal transmittance of carbon nanotube networks: Guidelines for novel thermal storage systems and polymeric material of thermal interest, Renewable Sustainable Energy Rev., 2015, 41, 1028–1036 CrossRef CAS.
- Q. Li, I. Duchemin, S. Y. Xiong, G. C. Solomon and D. Donadio, Mechanical Tuning of Thermal Transport in a Molecular Junction, J. Phys. Chem. C, 2015, 119(43), 24636–24642 CrossRef CAS.
- J. T. Lu and J. S. Wang, Quantum phonon transport of molecular junctions amide-linked with carbon nanotubes: A first-principles study, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78(23), 235436 CrossRef.
- A. Di Pierro, G. Saracco and A. Fina, Molecular junctions for thermal transport between graphene nanoribbons: Covalent bonding vs. interdigitated chains, Comput. Mater. Sci., 2018, 142, 255–260 CrossRef CAS.
- H. Han, Y. Zhang, N. Wang, M. K. Samani, Y. Ni, Z. Y. Mijbil, M. Edwards, S. Xiong, K. Saaskilahti, M. Murugesan, Y. Fu, L. Ye, H. Sadeghi, S. Bailey, Y. A. Kosevich, C. J. Lambert, J. Liu and S. Volz, Functionalization mediates heat transport in graphene nanoflakes, Nat. Commun., 2016, 7, 11281 CrossRef CAS PubMed.
- M. M. Bernal, A. Di Pierro, C. Novara, F. Giorgis, B. Mortazavi, G. Saracco and A. Fina, Edge-Grafted Molecular Junctions between Graphene Nanoplatelets: Applied Chemistry to Enhance Heat Transfer in Nanomaterials, Adv. Funct. Mater., 2018, 28(18), 1706954 CrossRef.
- W. M. Brown, P. Wang, S. J. Plimpton and A. N. Tharrington, Implementing molecular dynamics on hybrid high performance computers – short range forces, Comput. Phys. Commun., 2011, 182(4), 898–911 CrossRef CAS.
- H. Sun, COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase Applications Overview with Details on Alkane and Benzene Compounds, J. Phys. Chem. B, 1998, 102(38), 7338–7364 CrossRef CAS.
- X. Shen, X. Lin, N. Yousefi, J. Jia and J.-K. Kim, Wrinkling in graphene sheets and graphene oxide papers, Carbon, 2014, 66, 84–92 CrossRef CAS.
- J. Zhang, X. He, L. Yang, G. Wu, J. Sha, C. Hou, C. Yin, A. Pan, Z. Li and Y. Liu, Effect of tensile strain on thermal conductivity in monolayer graphene nanoribbons: a molecular dynamics study, Sensors, 2013, 13(7), 9388–9395 CrossRef CAS PubMed.
- X. F. Wu and T. F. Luo, The importance of anharmonicity in thermal transport across solid-solid interfaces, J. Appl. Phys., 2014, 115(1), 014901 CrossRef.
- N. Mingo, Anharmonic phonon flow through molecular-sized junctions, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74(12), 125402 CrossRef.
- A. K. Rappe and W. A. Goddard, Charge Equilibration for Molecular-Dynamics Simulations, J. Phys. Chem., 1991, 95(8), 3358–3363 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed , 27–28.
- C. H. Diao, Y. Dong and J. Lin, Reactive force field simulation on thermal conductivities of carbon nanotubes and graphene, Int. J. Heat Mass Transfer, 2017, 112, 903–912 CrossRef CAS.
- A. Khan, I. Navid, M. Noshin, H. Uddin, F. Hossain and S. Subrina, Equilibrium Molecular Dynamics (MD) Simulation Study of Thermal Conductivity of Graphene Nanoribbon: A Comparative Study on MD Potentials, Electronics, 2015, 4(4), 1109–1124 CrossRef CAS.
- B. Mortazavi and T. Rabczuk, Multiscale modeling of heat conduction in graphene laminates, Carbon, 2015, 85, 1–7 CrossRef CAS.
- A. Cao, Molecular dynamics simulation study on heat transport in monolayer graphene sheet with various geometries, J. Appl. Phys., 2012, 111(8), 083528 CrossRef.
- H. F. Zhan and Y. T. Gu, Theoretical and numerical investigation of bending properties of Cu nanowires, Comput. Mater. Sci., 2012, 55, 73–80 CrossRef CAS.
- H. A. Wu, Molecular dynamics study on mechanics of metal nanowire, Mech. Res. Commun., 2006, 33(1), 9–16 CrossRef.
- J.-c. Xu, S.-c. Dai, H.-l. Li and J.-h. Yang, Molecular dynamics simulation of the thermal conductivity of graphitized graphene/polyimide films, New Res. Carbon Mater., 2018, 33(3), 213–220 CrossRef.
- M. Yarifard, J. Davoodi and H. Rafii-Tabar, Computation of the thermal resistance in graphene sheets with a rectangular hole, Comput. Mater. Sci., 2017, 126, 29–34 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Plots of NEMD energy, temperatures and stress–strain with tensile simulation details and force field parameters description. See DOI: 10.1039/c9ra00894b |
| This journal is © The Royal Society of Chemistry 2019 |