
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective approach to rationally design tetrahydro-β-carboline–isatin conjugates as potential SERMs against breast cancer†
Bharvi Sharmaa,
Amandeep Singha,
Liang Gub,
Sourav Taru Sahab,
Ashona Singh-Pillayc,
Nosipho Celec,
Parvesh Singhc,
Mandeep Kaur*b and
Vipan Kumar *a
*a
aDepartment of Chemistry, Guru Nanak Dev University, Amritsar-143005, India. E-mail: vipan_org@yahoo.com
bSchool of Molecular and Cell Biology, University of the Witwatersrand, Private Bag 3, Wits-2050, Johannesburg, South Africa. E-mail: mandeep.kaur@wits.ac.za
cSchool of Chemistry and Physics, University of KwaZulu Natal, P/Bag X54001, Westville, Durban 4000, South Africa
First published on 28th March 2019
Abstract
A series of tetrahydro-β-carboline–isatin conjugates, with varying substituents as well as stereochemistry at C-1 and C-5 position of tetrahydro-β-carboline (THβC) and isatin ring, were prepared and assayed for anti-proliferative efficacy on Estrogen Responsive ER(+) (MCF-7) and ER(−ve) MDA-MB-231 cell-lines. The synthesized scaffolds displayed selective anti-proliferative efficacy against MCF-7 cell-line with the most active conjugate 8b exhibiting an IC50 value of 37.42 μM, comparable to that of peganumine A, a tetrahydro-β-carboline analogue, isolated from Peganum harmala. The synthesized compound 8b was also more potent than the standard drug tamoxifen (IC50 = 50 μM against MCF-7). The observed activities were further corroborated via docking studies in ER-α (PDB ID: 3ERT).
Introduction
Breast cancer (BC) is the most prevalent neoplasm amongst women and considered as the second largest cause of death globally after lung cancer. In 2018, an estimated 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 735350 new cases of BC were diagnosed and over 609640 deaths were expected to occur in United States.1 The occurrence of breast cancer in India is continually increasing over the last few decades and has now ranked number one amongst Indian females with an age adjusted rate as high as 25.8 and mortality of 12.7 per 100000 women in 2017.2 Naturally occurring tetrahydro-β-carboline (THβC) derivatives such as vinblastine,3 reserpine,4 manzamine5 and callophycin A;6 are indole-based alkaloids known for their diverse pharmacological potential. Peganumine A, an octacyclic THβC analogue, extracted from the seeds of Peganum harmala, exhibited significant anti-proliferative activity against MCF-7 with an IC50 value of 38.5 μM.7 N-(Biphenyl-2-yl)tryptoline (BPT) is another example of a saturated tricyclic framework exhibiting better anti-proliferative potential via inhibition of mitosis and blocking the G0/G1 phase of the cell cycle and has low cytotoxicity (Fig. 1).8 Structurally modified tadalafil derivatives displayed anti-proliferative activity against MDA-MB-231 cell-lines with IC50 values in the μM range.9 The anti-proliferative potential of THβCs along with their low cytotoxicity, attributed to the lack of DNA intercalating properties owing to its non-planar structure, has recently attracted the attention of the scientific fraternity for the development of selective anti-proliferatives.8
735350 new cases of BC were diagnosed and over 609640 deaths were expected to occur in United States.1 The occurrence of breast cancer in India is continually increasing over the last few decades and has now ranked number one amongst Indian females with an age adjusted rate as high as 25.8 and mortality of 12.7 per 100000 women in 2017.2 Naturally occurring tetrahydro-β-carboline (THβC) derivatives such as vinblastine,3 reserpine,4 manzamine5 and callophycin A;6 are indole-based alkaloids known for their diverse pharmacological potential. Peganumine A, an octacyclic THβC analogue, extracted from the seeds of Peganum harmala, exhibited significant anti-proliferative activity against MCF-7 with an IC50 value of 38.5 μM.7 N-(Biphenyl-2-yl)tryptoline (BPT) is another example of a saturated tricyclic framework exhibiting better anti-proliferative potential via inhibition of mitosis and blocking the G0/G1 phase of the cell cycle and has low cytotoxicity (Fig. 1).8 Structurally modified tadalafil derivatives displayed anti-proliferative activity against MDA-MB-231 cell-lines with IC50 values in the μM range.9 The anti-proliferative potential of THβCs along with their low cytotoxicity, attributed to the lack of DNA intercalating properties owing to its non-planar structure, has recently attracted the attention of the scientific fraternity for the development of selective anti-proliferatives.8
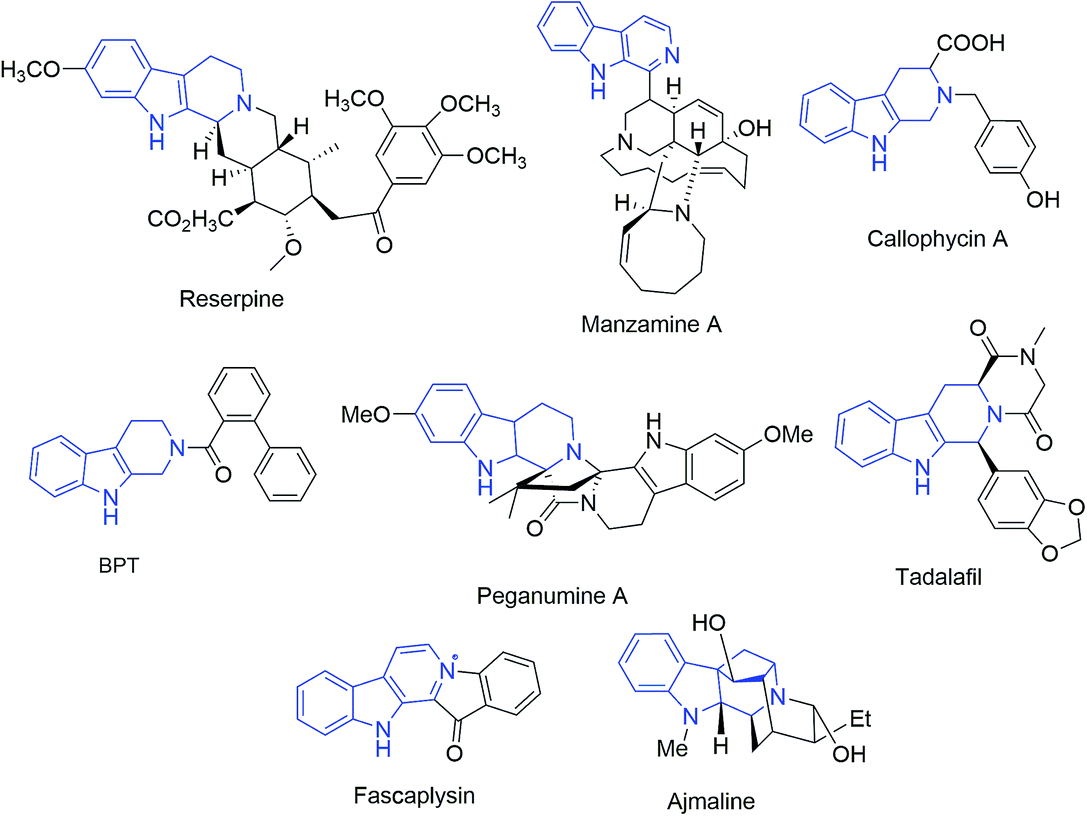 | ||
| Fig. 1 Natural and synthetic structural motifs with a tetrahydro-β-carboline core. | ||
Isatin is another indole-based moiety with proven anti-proliferative potential via inhibition of tyrosine kinases like VEGFR-1, VEGFR-2 and cyclic dependent kinases (CDKs).10 Sutent®, an isatin analogue (5-fluoro-3-alkenyl-2 oxindole) has been approved by the FDA for the treatment of gastrointestinal stromal tumors and advanced renal cell carcinoma.11 Although market is saturated with first line drugs and potential SERMS sanctioned by US Food and Drug Administration (FDA) viz. tamoxifen (TAM), raloxifene having significant anti-BC activities, yet they are also associated with adverse side effects such as endometrial cancer, insomnia, dizziness.12 Recent reports from our lab have shown in vitro anti-proliferative activities of ospemifene–isatins against breast cancer cell lines. The inclusion of isatin ring among these conjugates enhanced the anti-proliferative activities in general with the most potent conjugate exhibiting an IC50 of 1.56 μM against MCF-7, being ∼30 folds potent than tamoxifen.13 The methodology was further extended towards evaluating the anti-proliferative efficacy of 1H-1,2,3-triazole linked nitro-imidazole–isatin and isatin–ferrocenyl chalcone conjugates.14,15
In continuation to our recent reports on the synthesis of molecular conjugates with biological relevance and their evaluation,16–18 the present manuscript encompasses the design, synthesis and anti-proliferative evaluation of 1H-1,2,3-triazole linked THβC–isatin conjugates, as depicted in Fig. 2, against MCF-7 and MDA-MB-231 cell-lines using MTT assay. The use of L-tryptophan in the present series of compounds is not only owed to its natural availability but also because of the promising anti-proliferative potential of L-tryptophan derived THβC against MDA-MB-231 cell-line. The nature of substituents as well as the stereochemistry at C-1 of THβC and C-5 of isatin ring was varied so as to study their effect on the anti-proliferative activities.
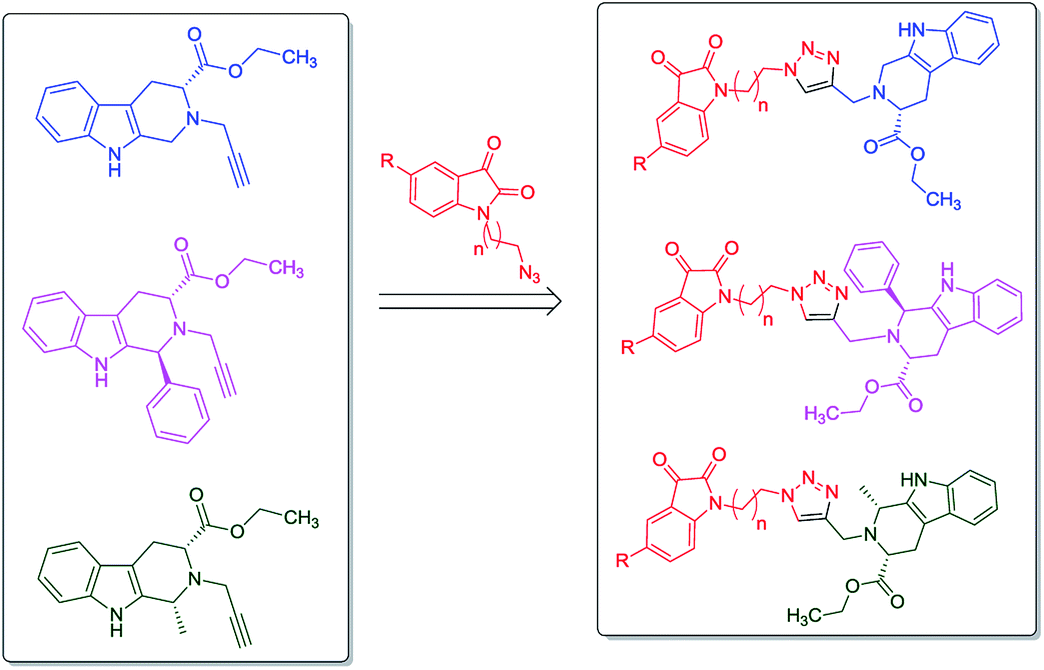 | ||
| Fig. 2 Designed tetrahydro-β-carboline–isatin conjugates. | ||
Results and discussion
Chemistry
The synthetic methodology involved the enantioselective Pictet–Spengler cyclization of L-tryptophan with various aldehydes to afford the corresponding tetrahydro-β-carbolines with varying stereochemistry at C-1 position.19,20 Depending upon the reactivity of different aldehydes, different reaction conditions were employed for the synthesis of corresponding THβC derived carboxylic acids. The preparation of 2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic acid 2a involved the base-promoted Pictet–Spengler reaction of L-tryptophan with 30% aqueous solution of formalin at room temperature. In the case of aliphatic aldehydes viz. acetaldehyde, a kinetically controlled Pictet–Spengler reaction of L-tryptophan with 40% solution of acetaldehyde yielded cis-isomer(1S,3S)-1-methyl-2,3,4,9-tetrahydro-1H-pyrido-[3,4-β]indole-3-carboxylic acid 2b as the major product.With a less reactive aromatic aldehyde viz. benzaldehyde, the reaction upon heating in acetic acid followed a thermodynamically controlled pathway to afford a diastereomeric mixture which was used in subsequent steps without purification. The treatment of THβC derived acids 2a–c with thionyl chloride in dry ethanol afforded the corresponding esters 3a–c. Compound 3c having the phenyl substituent at C-1 was successfully purified using column chromatography and only the trans isomer was used in subsequent steps because of its prevalence in naturally occurring alkaloids viz. ajmaline,21 sarpagine22 as well as the non-natural analogue tadalafil. Base-promoted N-propargylation of 3a–c in dry acetonitrile resulted in the formation of the corresponding ‘click’ precursors 4a–c (Scheme 1). The second precursor viz. N-alkylazidoisatins 7 was obtained via base-promoted alkylation of C-5 substituted isatin 5 with dibromoalkane to afford 6 with successive treatment with sodium azide (Scheme 2).
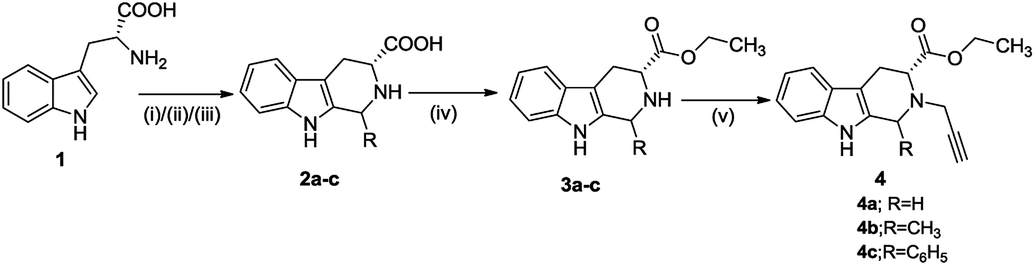 | ||
| Scheme 1 Synthesis of N-propargylated tetrahydro-β-carboline ethyl ester. (i) 30% formaldehyde, NaOH, H2O, 8 h, reflux, or (ii) 40% acetaldehyde, H2SO4,, 6–8 h, rt, or (iii) benzaldehyde, acetic acid, 2 h, reflux (iv) SOCl2, EtOH, 6 h, reflux (v) propargyl bromide, acetonitrile, rt, 6–8 h. (vi) CuSO4·5H2O, sodium ascorbate, EtOH, rt, 8 h. | ||
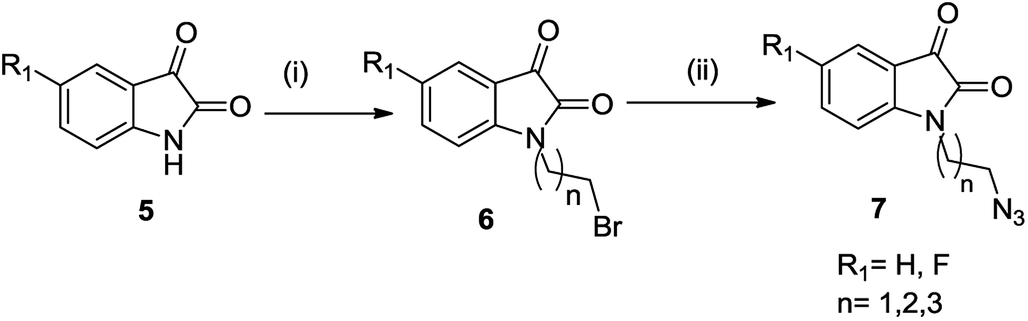 | ||
| Scheme 2 Synthesis of substituted N-alkyl-azido-isatin (i) NaH, dibromoalkane, anhy. DMF, 60 °C, 6–8 h (ii) NaN3, DMF, 60 °C, 2 h. | ||
Cu-promoted azide–alkyne cycloaddition reactions of 4a–c with 7a–f gave the desired THβC–isatin conjugates 8a–r in good yields as shown in Scheme 3. The compounds thus synthesized were purified using ethylacetate:hexane (80:20) on alumina as a stationary phase.
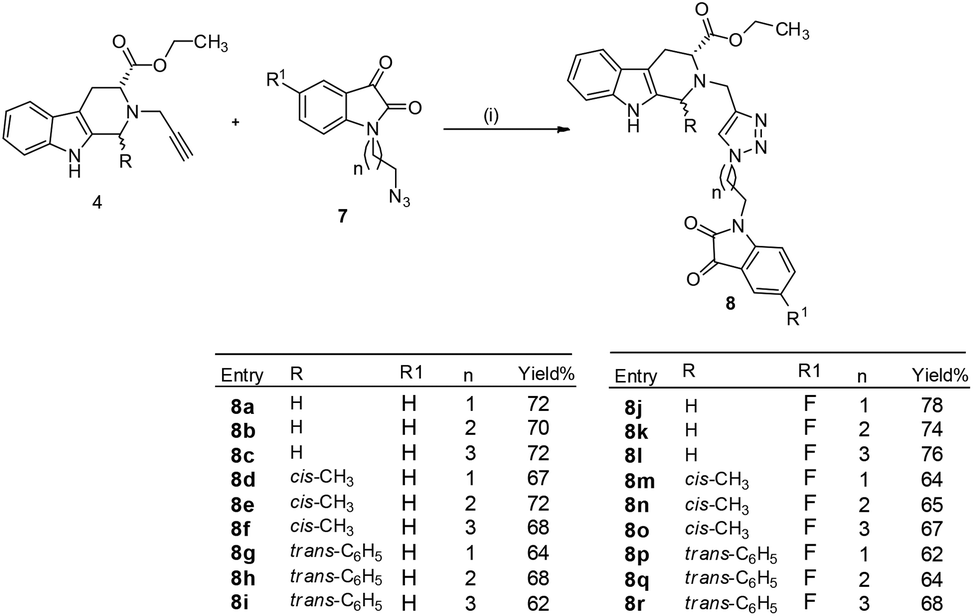 | ||
| Scheme 3 Synthesis of 1H-1,2,3-triazole-linked tetrahydro-β-carboline–isatin conjugates 8a–r (i) CuSO4, sodium ascorbate, EtOH, 6–8 h, rt. | ||
The structure to the compounds was assigned on the basis of analytical evidences and spectral data. For example, the most active compound 2-{1-[3-(2,3-dioxo-2,3-dihydro-indol-1-yl)-propyl]-1H-[1,2,3]triazol-4-ylmethyl}-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic acid ethyl ester (8b) displayed molecular ion peak m/z at [M + 1]+ 513.2206 in its High Resolution Mass Spectrum (HRMS). The prominent features in its 1H NMR include the appearance of triplet at δ 1.23 corresponding to methyl protons, multiplet corresponding to methylene (H-4) of THβC ring at δ 3.09–3.21 and singlet because of triazole-H at δ 7.67. Further, 13C NMR confirmed the assigned structure by the appearance of characteristic absorptions at δ 172.7 representing the carbonyl carbon of THβC along with δ 158.5 and 183.0 corresponding to isatin ring carbonyls.
Anti-proliferative activities of synthesized conjugates
The newly synthesized conjugates were screened for their anti-proliferative activities against human breast cancer cell-lines viz. MCF-7 and MDA-MB-231 and the results were compared with reference drugs, plumbagin23 and peganumine A.7 Detailed results of these studies, in terms of % age growth inhibition of MCF-7 and MDA-MB-231 cells, have been depicted in Fig. 3. The IC50 value i.e. the necessary concentration for inhibiting 50% of cell viability by the synthesized compounds, has been enlisted in Table 1.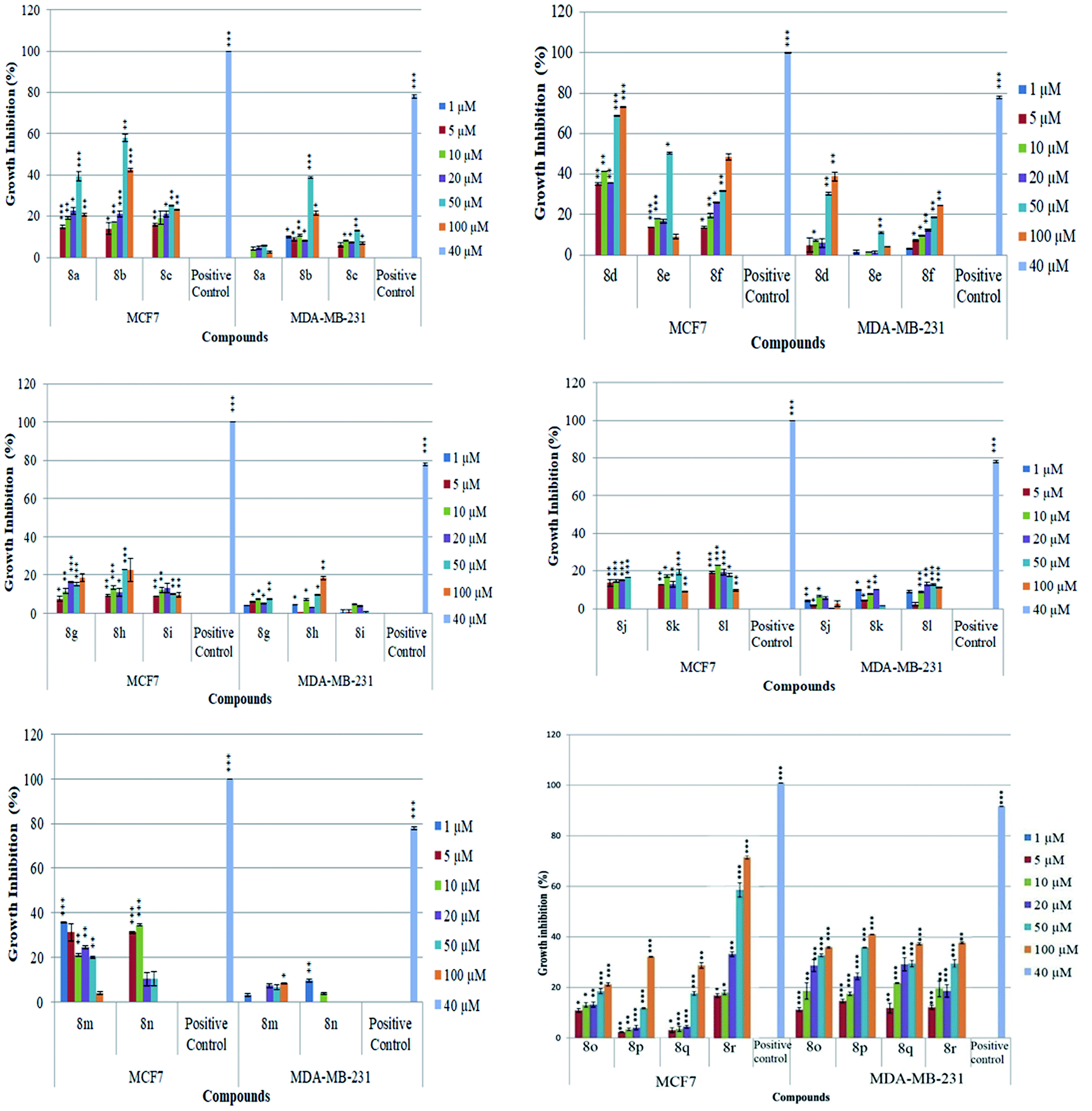 | ||
| Fig. 3 Representative graph of percentage growth inhibition on MCF7 and MDA-MB-231 cells at selected concentrations of test compounds 8a–r. 40 μM plumbagin was used as a positive control. Data shows mean ± standard deviation S.D. (n = 3), where *p < 0.05, **p < 0.01 and ***p < 0.001 significance compared to untreated control. | ||
| Entry | Compound | MCF-7 (μM) | MDA-MB231 (μM) | Entry | Compound | MCF-7 (μM) | MDA-MB231 (μM) |
|---|---|---|---|---|---|---|---|
| 8a | 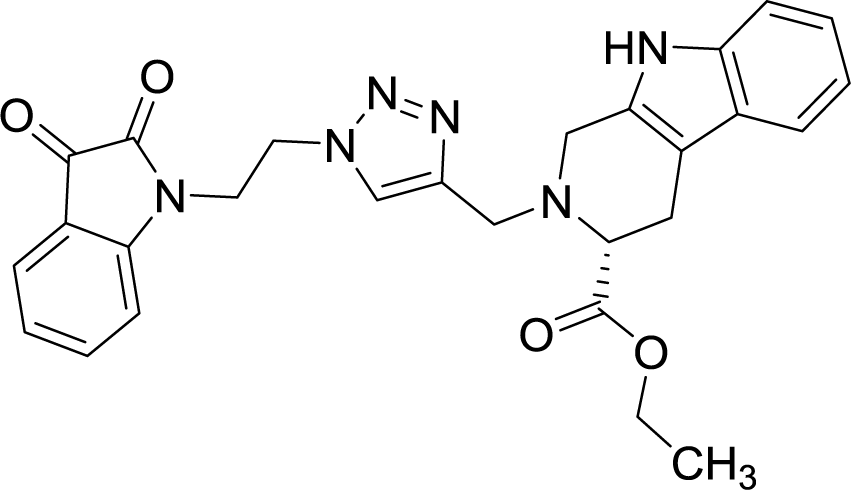 |
>100 | >100 | 8k | 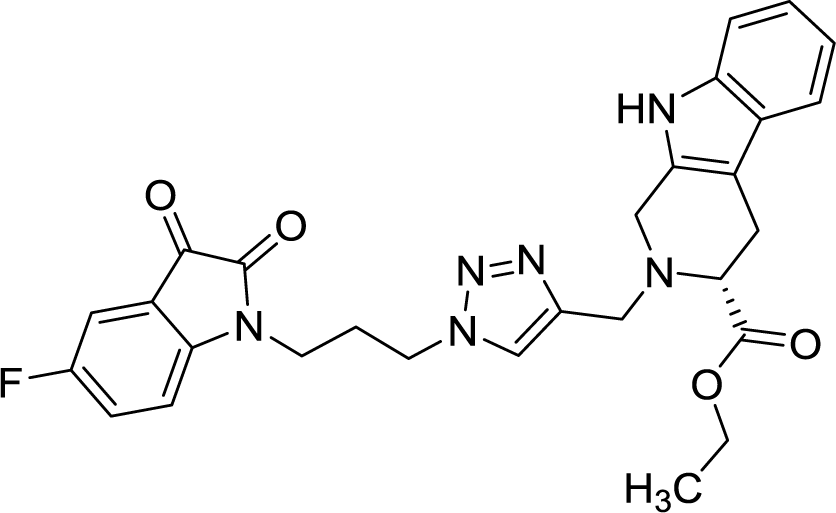 |
>100 | >100 |
| 8b | 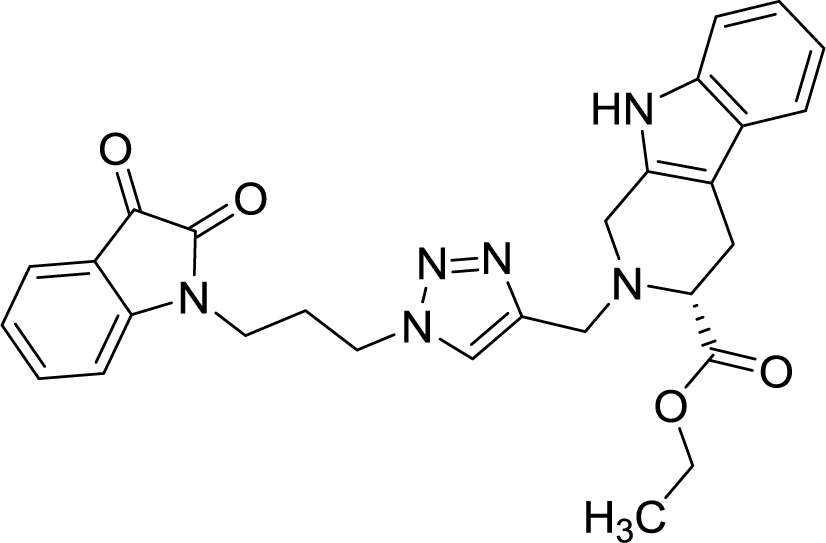 |
37.42 | >100 | 8l | 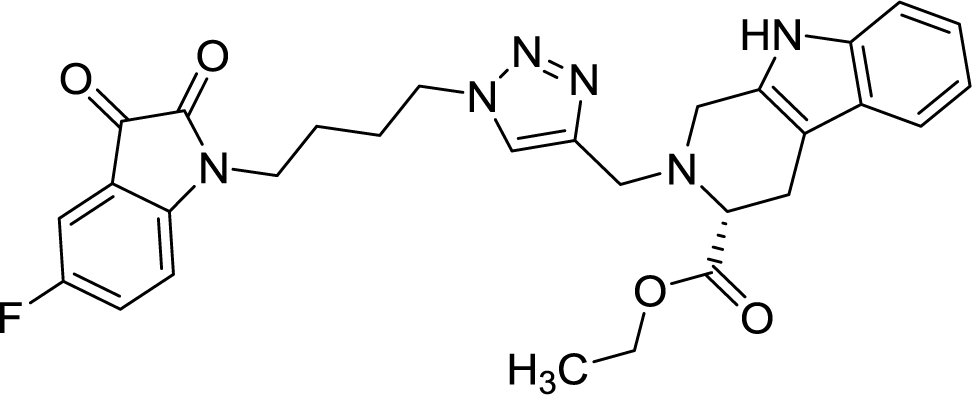 |
>100 | >100 |
| 8c |  |
>100 | >100 | 8m |  |
>100 | >100 |
| 8d | 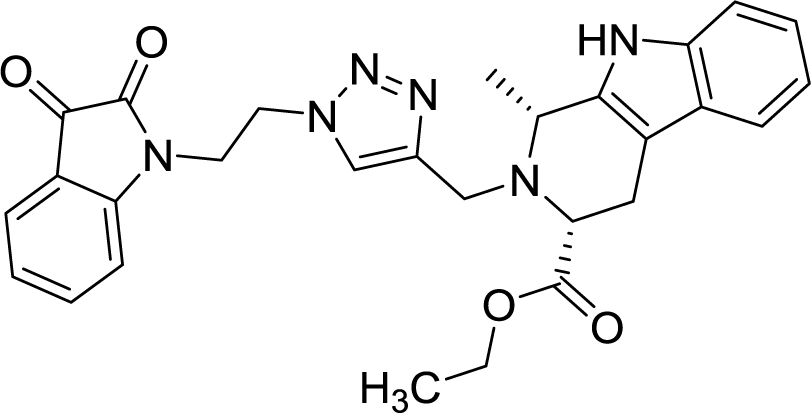 |
45.3 | >100 | 8n | 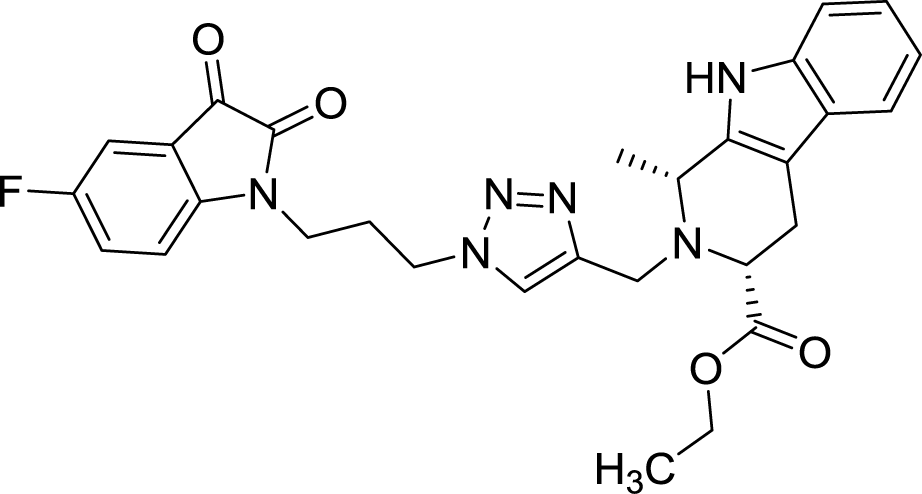 |
>100 | >100 |
| 8e | 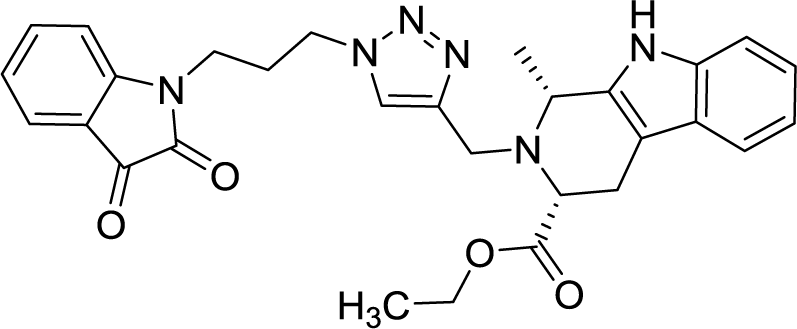 |
50 | >100 | 8o | 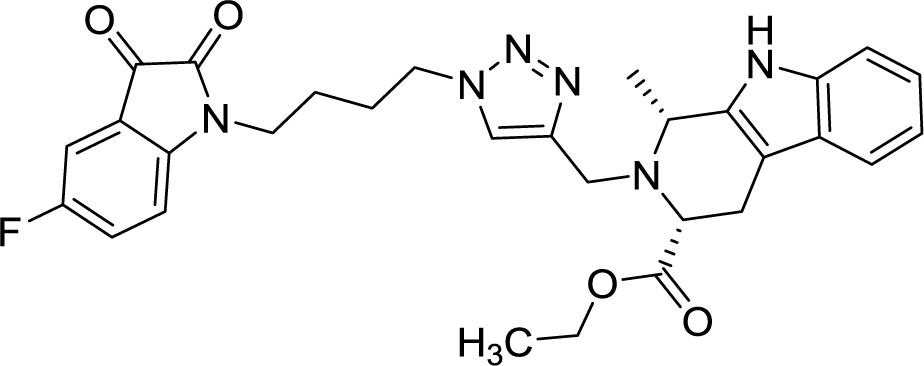 |
>100 | >100 |
| 8f | 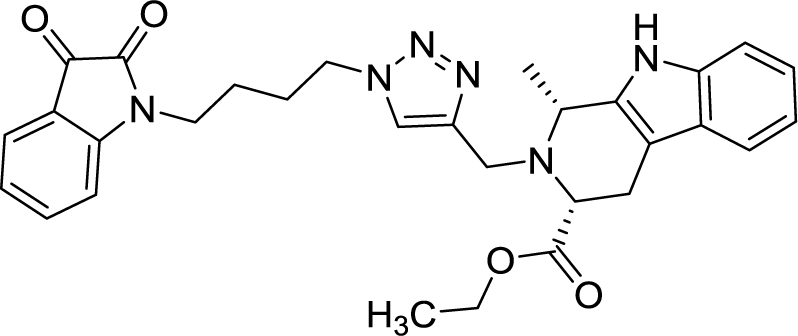 |
>100 | >100 | 8p | 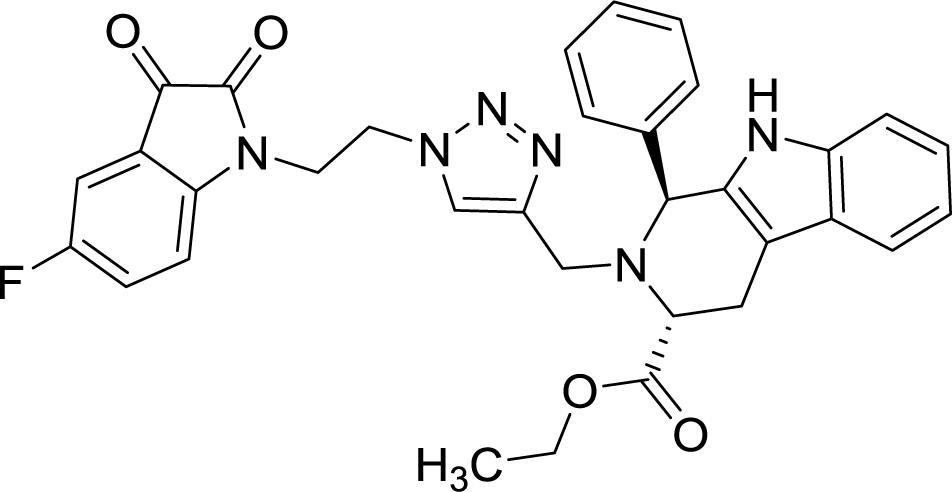 |
>100 | >100 |
| 8g | 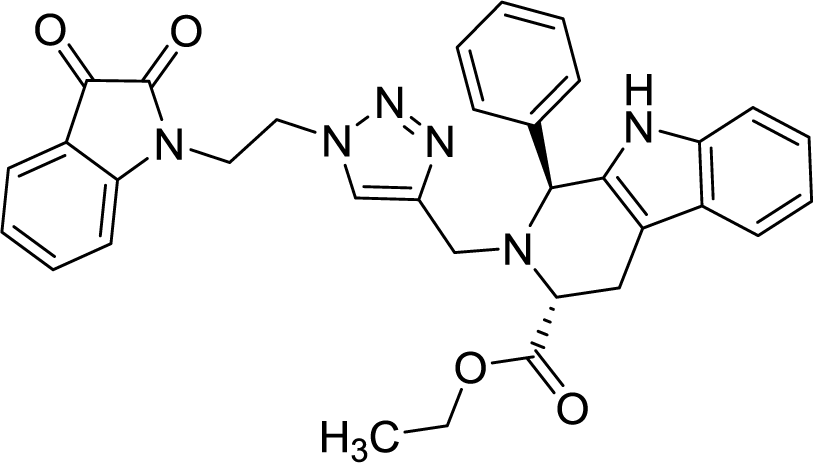 |
>100 | >100 | 8q | 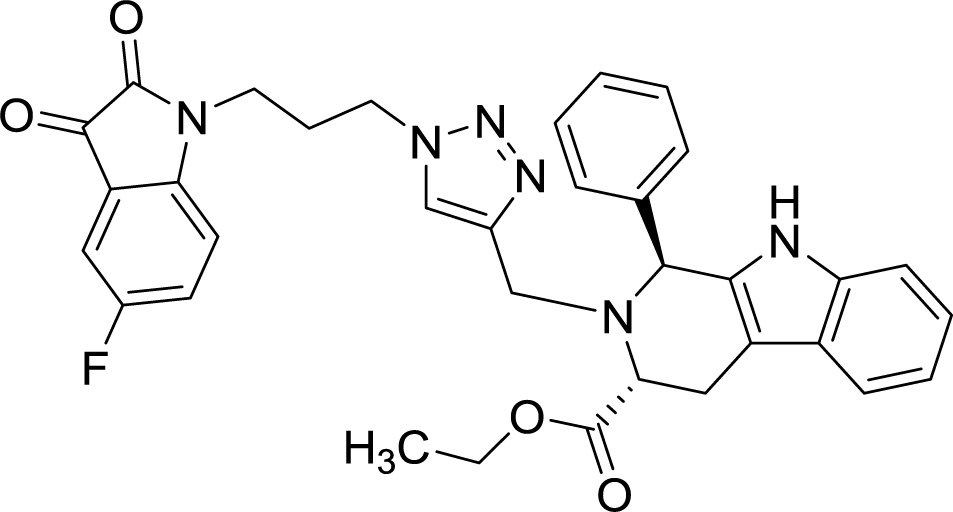 |
>100 | >100 |
| 8h | 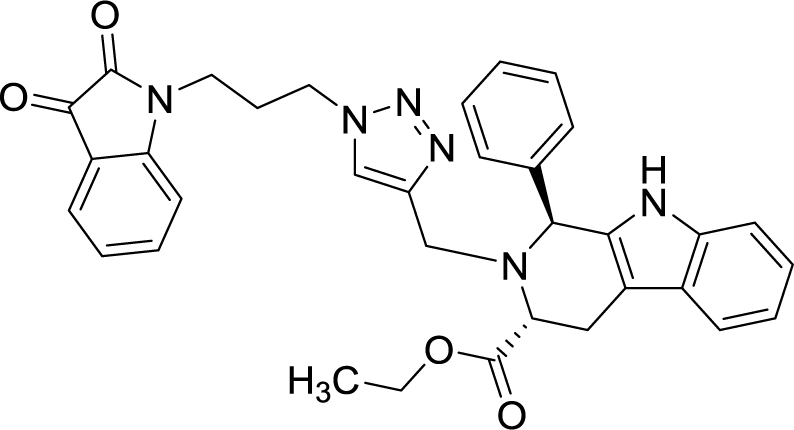 |
>100 | >100 | 8r | 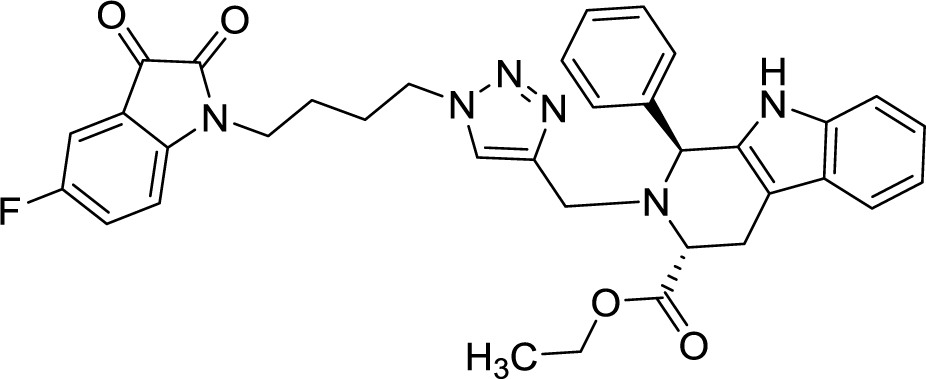 |
42.11 | >100 |
| 8i | 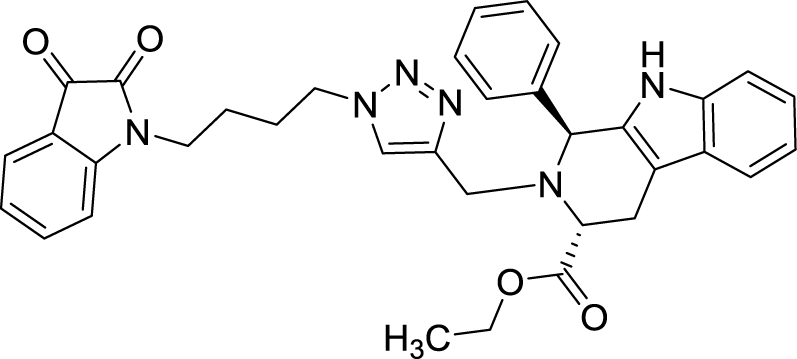 |
>100 | >100 | Plumbagin | 3.5 | 4.4 | |
| 8j | 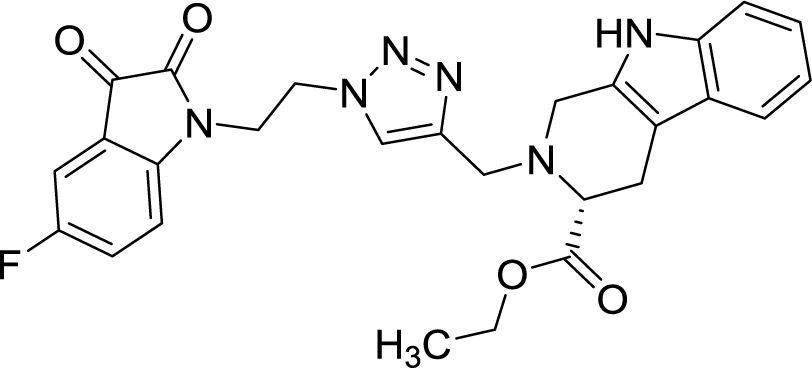 |
>100 | >100 | Peganumine A7 | 38.5 | — | |
| Tamoxifen | 50 | 75 |
As evident from Table 1, all the synthesized conjugates proved to be inactive against MDA-MB-231 (ER−) cell-line and were selective towards estrogen responsive MCF-7 cell-line. Structure activity relationship analysis showed the dependence of anti-proliferative activity on the nature of substituent at C-1 and C-5 position of THβC and isatin ring respectively while the alkyl chain length did not seem to play any substantial role. Among un-substituted (R = H) isatin based conjugates 8a–8i, the presence of either hydrogen or methyl substituents at C-1 position of THβC resulted in the loss of activities as evident from 8g–8i. The induction of fluoro-substituent at C-5 position of isatin ring proved to be detrimental for anti-proliferative activities as most of the compounds were inactive except 8r, having a phenyl substituent at C-1, exhibiting an IC50 value of 42.11 μM. Interestingly, several compounds (8k–n) showed a negative correlation, where an increase in drug concentration induced a decrease in growth inhibition, possibly suggesting that these compounds are supporting cancer cell proliferation. However, it could also suggest that at high concentrations, these compounds might have precipitated out of culture media and thus lost their effectiveness.
Molecular docking studies
In order to identify novel binding cores, molecular docking of the active ligands in the binding site of estrogen receptor-α (ER-α) was performed to determine the crucial binding interactions responsible for the inhibitory effect observed.24 The native ligand in the crystal structure of ER-α (PDB ID: 3ERT) is afimoxifene which inhibited the receptor by binding to the predominantly hydrophobic pocket. The binding interaction profile of the ligand–protein complex largely comprised of hydrophobic interactions with residues Leu41, Ala45, Trp78, Leu82, Leu86, Phe99, Leu220 and Leu231. These interactions were accompanied by two distinct hydrogen bonds with Glu48 and Arg89, two water bridges established between residues Thr42 and Leu86 as well as a single salt bridge with amino acid Asp46.Compound 8b shared a similar hydrophobic interaction network to that of the native ligand. The binding landscape of compound 8b comprised a significant hydrophobic contribution between residues, Leu41, Leu44, Ala45, Leu49, Trp78, Leu82, Leu220 and Leu231 with the phenyl fused heterocyclic rings and the hydrophobic linker (bond distance = 3.93/3.77, 3.99, 3.81, 3.78, 3.62/3.87, 3.95, 3.95 and 3.84 Å, respectively). A single salt bridge exists between the positively charged tert-amine and Asp46 at a distance of 5.11 Å. Despite the binding profile of compound 8b being very simple, it was identified as the most potent inhibitor (Fig. 4).
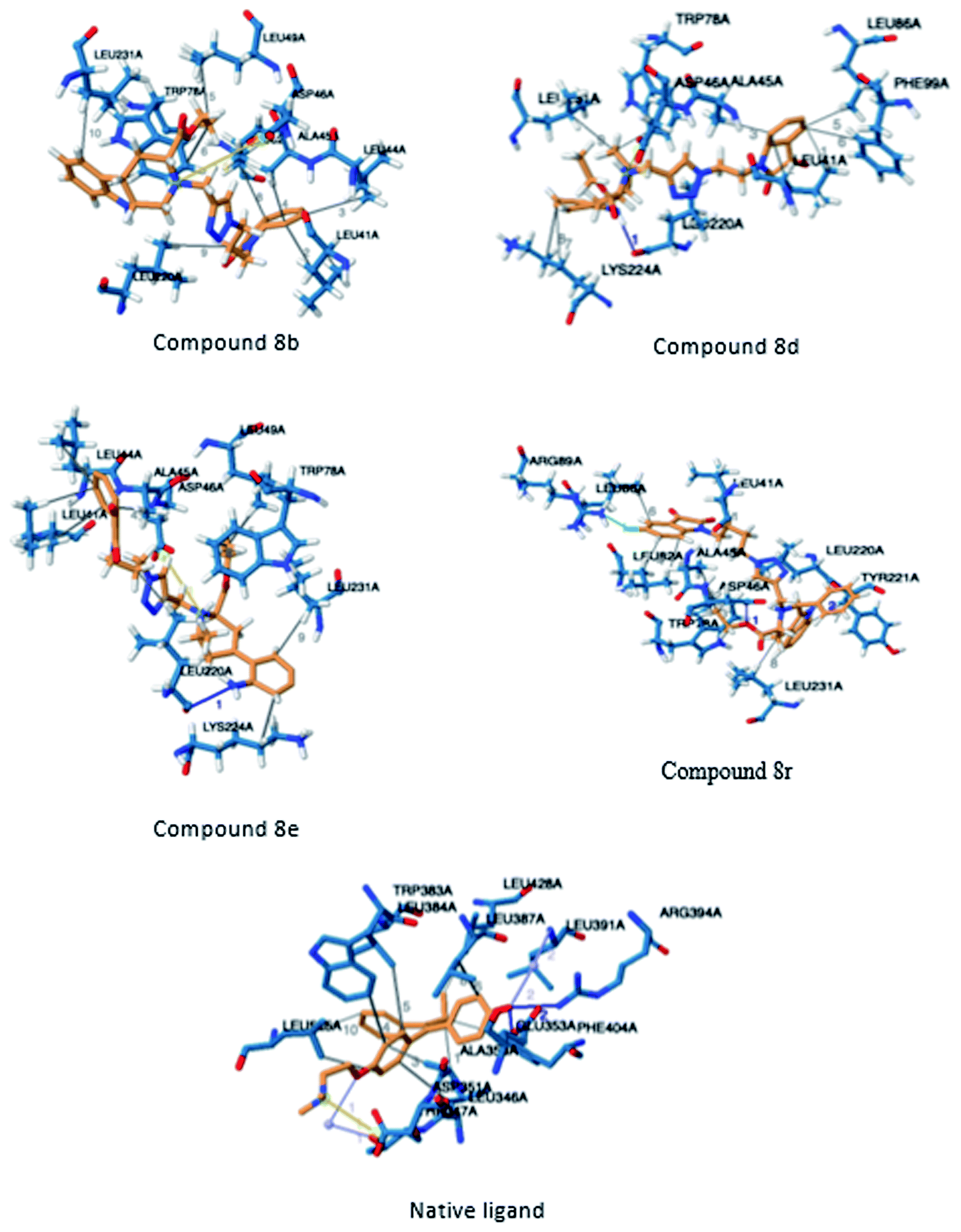 | ||
| Fig. 4 Three-dimensional illustration of docked ligands interacting in the binding site of the estrogen receptor (PDB: 3ERT). | ||
Compounds 8d, 8e and 8r demonstrated promising activity against the MCF-7 cell-line in comparison to peganumine A. Compounds 8d and 8e have a near identical binding interaction profile. The binding of these compounds to the active site of ER-α comprised an extensive network of van der Waals interactions between residues Leu41, Leu44, Ala45, Leu49, Trp78, Leu86, Phe99, Lys224 and Leu231 and the aromatic moieties of the ligands (bond distance = 4.00/3.90//3.92/3.78, 3.97, 3.68//3.76, 3.83, 3.83//3.54, 3.87, 3.83, 3.96/3.95//3.39 and 3.85//3.82 Å; respectively). The protein–ligand interaction was further supported by a hydrogen bond between the oxygen of the hydrogen acceptor Leu220 and the triazole nitrogens (bond distance = 3.00//3.54 Å; bond angle = 136.41°//105.19°) as well as a salt bridge between the anion Asp46 and the tertamine (bond distance = 4.27//5.06 Å).
The inhibitory activity of compound 8r was largely driven by the existence of the hydrophobic network between residues Leu41, Ala45, Trp78, Leu82, Leu86, Tyr221 and Leu231 at a bond distance of 3.70, 3.75, 3.64, 3.80/3.87, 3.52, 3.99 and 3.69 Å, respectively. Also, the ligand interaction included two hydrogen bonds between the donor residue Asp46 and the carbonyl oxygen of the ligand ethoxy group as well as the acceptor residue Leu220 and the aromatic nitrogen (bond distance = 3.82 and 2.93 Å; bond angle = 100.07° and 166.19°; respectively). The fluorine moiety of compound 8r also formed a distinct electrostatic interaction with residue Arg89, (bond distance = 3.18 Å, donor angle = 141.31°, acceptor angle = 109.50°). The thermodynamic and conformational stability of the complexes 8b, 8d, 8e and 8r in the binding site has been well depicted by high binding affinities with energy value of −9.6, −8.8, −9.9 and −10.5 kcal mol−1 respectively (Table 2). The parameters describing the physicochemical properties viz. logP and topological polar surface area (TPSA) of the target compounds have been well demonstrated in Table 3 (see ESI†). The synthesized conjugates 8b, 8d, 8e and 8f (TPSA/Å = 110.01, 110.01, 110.01 and 110.10 respectively) possess good potential to permeate cell membrane. Further, the moderate positive value of partition coefficient logP clearly defines the lipophilic nature of the compounds 8b, 8d, 8e and ability to permeate cell membrane effectively and efficiently.
| Ligand identifier | Docking score/(kcal mol−1) | RMSD i.b. | RMSD u.b. |
|---|---|---|---|
| 8a | −9.7 | 0.000 | 0.000 |
| 8b | −9.6 | 0.000 | 0.000 |
| 8c | −9.7 | 0.000 | 0.000 |
| 8d | −8.8 | 1.960 | 2.689 |
| 8e | −9.9 | 0.000 | 0.000 |
| 8f | −8.7 | 1.978 | 2.557 |
| 8g | −9.3 | 2.080 | 2.582 |
| 8h | −9.6 | 2.577 | 4.014 |
| 8i | −10.4 | 0.000 | 0.000 |
| 8j | −10.2 | 0.000 | 0.000 |
| 8k | −10.2 | 0.000 | 0.000 |
| 8l | −9.3 | 1.193 | 1.427 |
| 8m | −9.5 | 2.061 | 2.912 |
| 8n | −9.6 | 1.265 | 1.506 |
| 8o | −8.9 | 2.170 | 2.873 |
| 8p | −9.1 | 4.238 | 6.490 |
| 8q | −10.9 | 0.000 | 0.000 |
| 8r | −10.5 | 0.000 | 0.000 |
| Native ligand | −7.9 | 0.000 | 0.000 |
| Plumbagin | −6.2 | 1.357 | 2.189 |
The molecular docking of the discussed conjugates has highlighted critical pharmacophoric features of the potential drug candidates which directly affect their inhibitory activity against MCF-7 cell-line. It is evident that the synthesized tetrahydro-β-carboline–isatin conjugates are promising anti-proliferative agents. With further derivatization, the pharmacological efficacy of the potential drug candidates can be improved to reveal potent inhibitors. The binding pattern of the active compound and potential anti-proliferative agents i.e. compounds 8b, 8d, 8e and 8r, respectively; emphasises the need for the active ligand to comprise hydrophobic pharmacophores. Effective inhibitory activity can be achieved from the presence of hydrogen bonds created between the protein and ligand, specifically with the conjugate being the hydrogen bond acceptor and protein the donor. Further to this, the interaction of the isatin conjugate in the binding pocket should be facilitated with the presence of ionised non-covalent bonds. The structural incentive by introducing a halogen at position C-5 position of the isatin appears not critical as is observed in compound 8r.
Conclusion
A series of tetrahydro-β-carboline–isatin conjugates having varied substituents at C-5 position of isatin as well as at C-1 position of THβC ring were prepared via azide alkyne cycloaddition reactions. The compound, 8b with an optimum combination of un-substituted THβC and isatin ring along with a propyl-chain as spacer, proved to be most potent among the synthesized scaffolds exhibiting an IC50 value of 37.42 μM, comparable to that of peganumine A. The anti-proliferative evaluation studies of synthesized conjugates revealed them to be selective against estrogen responsive MCF-7 cell line suggestive of the fact that they can serve as molecular templates in the expansion of cancer chemotherapies competent of targeting ER-dependent breast cancers. Docking results further support that the most potent compound displays great binding affinity with ER(+) owing to occupation of binding sites via various hydrophobic, H-bonding and cation-π interactions with ER(+) active site.Experimental section
General note
All the reactions were carried out using standard techniques. The column chromatography was carried out using silica gel (60–120 mesh) with ethyl acetate:hexane as eluent. Melting points were recorded in capillaries and are uncorrected. The spectra of 1H NMR and 13C NMR spectra were recorded on JEOL 400, BRUKER 500 and 125 MHz spectrometers and obtained as CDCl3 solutions using tetramethylsilane (TMS) as an internal standard. Chemical shifts were reported in parts per million (ppm) and coupling constants J indicated in hertz. Mass spectral data was gathered on BrukermicrOTOF QII equipment using ESI as the source.
Preparation of 2-prop-2-ynyl-2,3,4,9-tetrahydro-β-carboline-3-carboxylic acid ethyl ester 4
To a solution of secondary amine (3 mmol) and K2CO3 (3.6 mmol) in 15 mL acetone, propargyl bromide (3.6 mmol) was added and reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and concentrated under reduced pressure to yield crude product which was purified via column chromatography using (20:80) ethylacetate:hexane as eluent. 4a pale yellow solid; 92%; 4b pale yellow solid 90%; 4c light brown solid 84%.
Procedure for the synthesis of conjugates (8a–r)
To the stirred solution of N-alkylazido-isatin 7 (1 mmol) and 2-prop-2-ynyl-2,3,4,9-tetrahydro-β-carboline-3-carboxylic acid ethyl ester 4 in a mixture of ethanol:water (85:15), was added CuSO4·5H2O (0.055 mmol) and sodium ascorbate (0.143 mmol). The reaction mixture was allowed to stir at rt for 7–8 h. Upon completion of the reaction as monitored through TLC, the resulting mixture was extracted with ethyl acetate (2 × 30 mL) and water (2 × 25 mL). The organic layers were combined, dried over anhydrous Na2SO4 and concentrated under reduced pressure to yield the desired conjugates (8a–r); which were purified via chromatography using alumina as stationary phase and ethyl-acetate:hexane (80:20) as eluent mixture.
Cell culturing
MCF-7 cells were cultured in DMEM media with 10% FBS and 1% penicillin–streptomycin and MDA-MB-231 cells were cultured in DMEM supplemented with 5% FBS and 1% penicillin–streptomycin, both incubated at 37 °C and 5% carbon dioxide.22MTT assay
Cells were seeded in 96 well plates at a density of 5000 cells per well in triplicate in media. After 24 hours (h), the test compounds, diluted in complete Dulbecco's media Eagle's medium (DMEM) were added to each well. Cells were treated with a range of different concentrations of test compounds (1 μM, 5 μM, 10 μM, 20 μM, 50 μM, 100 μM) for 24 h at 37 °C and 5% carbon dioxide. Subsequently, sterile 5 μL of 5 mg mL−1 MTT (Sigma-Aldrich) dissolved in PBS was added to each well and incubated with cells for 2 h. Solubilization solution (10% SDS, 10 mM HCl) of equal volume to the wells was then added to each well, which was incubated with cells for 16 h at 37 °C. The optical density of each well was read at 570 nm using a microtiter plate reader (Thermo Fisher Scientific Multiskan GO Microplate Reader, SkanIt™ software).Statistical analysis
The statistical analysis was carried out using Excel® and IC50 values were determined using Graph pad Prism5 software (Hearne Scientific Software). The experiments were performed in duplicate and the statistical significance was calculated using student's t-test. A p-value of less than 0.05 was used to estimate the significance of the observations. A Z-factor was calculated for each 96-well plate and assays having Z-factor above >0.5 were included in the statistical analysis.25Molecular docking
The X-ray crystal structure of the estrogen receptor-α (PDB ID: 3ERT) was obtained from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). Using the UCSF Chimera package the ligand and receptor complexes were prepared for docking.26 Chem3D Ultra was used to construct a three-dimensional structure of the native ligand and most active isatin conjugates. The 3D structure of the compounds was further optimised using the force field, MMFF94s in the Avogadro package (V 1.2.0). AutoDock Tools (V 1.5.6) was used to establish the grid box parameters. All non-polar hydrogens were merged, Gasteiger partial atomic charges added, and rotatable bonds assigned. The grid box of the complex had a grid spacing of 0.375 Å. The parameter of the grid points are given in Table 4 (ESI†). Autodock Vina was used to perform the docking of each ligand into the IGF-1 receptor in triplicate.27 The docking systems were validated by redocking the ligand of the protein crystal structures into the binding site. The Protein–Ligand Interaction Profiler (PLIP) was used to analyse the non-polar interactions between the protein and ligand in complex.28Conflicts of interest
The authors declare no conflict of interest.Abbreviations
| BC | Breast cancer |
| THβC | Tetrahydro-β-carboline |
| SERMs | Selective estrogen receptor modulators |
| ER | Estrogen receptor |
| IC50 | 50% Inhibitory concentration |
| SAR | Structure–activity relationship |
Acknowledgements
Financial assistance from Council of Scientific and Industrial Research, New Delhi, India, under CSIR-JRF fellowship CSIR Ref No. 09/254(0257)/2016-EMR-I gratefully acknowledged. The authors also would like to thank the National Research Foundation of South Africa for their support in facilitating our research (UID: 99563). We would also like to thank the Centre for High Performance Computing based in Cape Town for access to computational resources. MCF7 (The European Collection of Authenticated Cell Cultures (ECACC), UK). MDA-MB-231 (The American Type Culture Collection (ATCC), USA).Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- S. Malvia, S. A. Bagadi, U. S. Dubey and S. Saxena, Asia-Pac. J. Clin. Oncol., 2017, 13, 289–295 CrossRef PubMed.
- N. Neuss, M. Gorman and W. Hargrove, J. Am. Chem. Soc., 1964, 86, 1440–1442 CrossRef CAS.
- L. Garrett, O. Carrier Jr and B. H. Douglas, Eur. J. Pharmacol., 1967, 2, 236–238 CrossRef CAS.
- P. Jakubec, A. Hawkins, W. Felzmann and D. J. Dixon, J. Am. Chem. Soc., 2012, 134, 17482–17485 CrossRef CAS.
- P. B. Simon, J. L. Ovenden, C. H. Nielson, R. H. Liptrot, D. M. Willis, A. D. Tapiolas, C. A. Wright and C. A. Motti, Phytochem. Lett., 2011, 4, 69–71 CrossRef.
- K. B. Wang, Y. T. Di, Y. Bao, C. M. Yuan, G. Chen, D. H. Li, J. Bai, H. P. He, X. J. Hao, Y. H. Pei, Y. K. Jing, Z. L. Li and H. M. Hua, Org. Lett., 2014, 16, 4028–4031 CrossRef CAS PubMed.
- (a) S. Mahale, S. B. Bharate, S. Manda, P. Joshi, P. R. Jenkins, R. A. Vishwakarma and B. Chaudhuri, Cell Death Dis., 2015, 6, 1743 CrossRef PubMed; (b) S. Kumar, A. Singh, K. Kumar and V. Kumar, Eur. J. Med. Chem., 2017, 142, 48 CrossRef CAS PubMed.
- H. A. Mohamed, N. M. R. Girgis, R. Wilcken, M. R. Bauer, H. N. Tinsley, B. D. Gary, G. A. Piazza, F. M. Boeckler and A. H. Abadi, J. Med. Chem., 2011, 54, 495–509 CrossRef CAS PubMed.
- C. L. Tourneau, E. Raymond and S. Faivre, Ther. Clin. Risk Manage., 2017, 2, 341 Search PubMed.
- R. J. Motzer, M. D. Michaelson, B. G. Redman, G. R. Hudes, G. Wilding, R. A. Figlin, M. S. Ginsberg, S. T. Kim, C. M. Baum, S. E. De Primo, J. Z. Li, C. L. Bello, C. P. Theuer, D. J. George and B. I. Rini, J. Clin. Oncol., 2006, 24, 16 CrossRef CAS PubMed.
- C. D. Teaux, V. Leblanc, G. B. Langer, S. Parent, E. Asselin and G. B. Rube, Steroids, 2008, 73, 1077–1089 CrossRef PubMed.
- S. Kumar, L. Gu, G. Palma, M. Kaur, A. S. Pillay, P. Singh and V. Kumar, New J. Chem., 2018, 42, 3703 RSC.
- S. Kumar, L. Gu, G. Palma, M. Kaur, A. S. Pillay, P. Singh and V. Kumar, ACS Omega, 2018, 3, 12106–12113 CrossRef CAS PubMed.
- A. Singh, S. T. Saha, S. Perumal, M. Kaur and V. Kumar, ACS Omega, 2018, 3, 5808–5813 CrossRef CAS PubMed.
- S. Kumar, T. bains, A. S. W. Kim, C. Tam, J. Kim, L. W. Cheng, K. M. Land, A. Debnath and V. Kumar, Front. Cell. Infect. Microbiol., 2018, 8, 380 CrossRef PubMed.
- Shalini, A. Viljoen, L. Kremer and V. Kumar, Bioorg. Med. Chem. Lett., 2018, 28, 1309–1312 CrossRef CAS PubMed.
- A. Rani, A. Singh, J. Gut, P. J. Rosenthal and V. Kumar, Eur. J. Med. Chem., 2018, 143, 150–156 CrossRef CAS PubMed.
- H. Song, Y. Liu, L. Wang and Q. Wang, J. Agric. Food Chem., 2014, 5, 1010–1018 CrossRef PubMed.
- M. L. V. Linn and J. M. Cook, J. Org. Chem., 2010, 75, 3587–3599 CrossRef PubMed.
- X. Fu and J. M. Cook, J. Org. Chem., 1993, 58, 661–672 CrossRef CAS.
- T. Wang and J. M. Cook, Org. Lett., 2000, 2, 2057–2059 CrossRef CAS PubMed.
- S. Sagar, L. Esau, B. Moosa, N. M. Khashab, V. B. Bajic and M. Kaur, Anti-Cancer Agents Med. Chem., 2014, 14, 170–180 CrossRef CAS PubMed.
- C. D. Savi, R. H. Bradbury, A. A. Rabow, R. A. Norman, C. D. Almeida, D. M. Andrews, P. Ballard, D. Buttar, R. J. Callis, G. S. Currie, J. O. Curwen, C. D. Davies, C. S. Donald, L. J. L. Feron, H. Gingell, S. C. Glossop, B. R. Hayter, S. Hussain, G. Karoutchi, S. G. Lamont, P. MacFaul, T. A. Moss, S. E. Pearson, M. Tonge, G. E. Walker, H. M. Weir and Z. Wilson, J. Med. Chem., 2015, 58, 8128–8140 CrossRef.
- J. H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, Nucleic Acids Res., 2015, 43, 443–447 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectral data of the synthesized compounds along with scanned 1H and 13C NMR spectra of representative compounds viz. 8b, 8c, 8f, 8g, 8h, 8i, 8j and Table 4. See DOI: 10.1039/c9ra00744j |
| This journal is © The Royal Society of Chemistry 2019 |