
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyaluronic acid-mediated multifunctional iron oxide-based MRI nanoprobes for dynamic monitoring of pancreatic cancer†
Yu Luo‡
b,
Yuehua Li‡c,
Jing Li‡c,
Caixia Fud,
Xiangrong Yu*e and
Li Wu*acf
aInstitute of Biomedical Manufacturing and Life Quality Engineering, School of Mechanical Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, P. R. China. E-mail: fdwl525@163.com
bSchool of Chemical Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, P. R. China
cInstitute of Diagnostic and Interventional Radiology, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai, China
dMR Application Development, Siemens Shenzhen Magnetic Resonance Ltd, Shenzhen, China
eDepartment of Radiology, Zhuhai Hospital of Jinan University, Zhuhai People's Hospital, 79 Kangning Road, Zhuhai, Guangdong 519000, P. R. China
fDepartment of Pediatrics, University of Washington, Seattle, USA
First published on 4th April 2019
Abstract
Regarded as the most promising technology for an early and precise detection of pancreatic cancer, a sensitive nanoprobe has been developed to enhance the accumulation of contrast agent at the tumor site. Hyaluronic acid (HA)-mediated multifunctional Fe3O4 nanoparticles (NPs) were used to target pancreatic cancer cells because on their cytomembrane they overexpress CD44, a receptor protein that has a high affinity to HA. The formation of HA-mediated multifunctional Fe3O4 nanoparticles began with the synthesis of polyethyleneimine (PEI·NH2) stabled Fe3O4 NPs by slight reduction. Subsequently, the formed Fe3O4@PEI·NH2 NPs were modified with fluorescein isothiocyanate (FITC), polyethylene glycol (mPEG-COOH) and HA in succession to form the multifunctional Fe3O4 NPs denoted as HA-Fe3O4 NPs. HA served as a targeting molecule to identify the surface antibody of CD44. As nanoparticles with a diameter of ca. 11.9 nm, the HA-Fe3O4 NPs exhibited very high r2 relaxivity of 321.4 mM−1 s−1 and this has proved that HA-Fe3O4 NPs could be efficient T2-weighted magnetic resonance imaging (MRI) contrast agents. In a CCK8 cell proliferation assay of HA-Fe3O4 NPs, there was no toxic response for Fe concentrations up to 100 μg mL−1. Flow cytometry, confocal microscopy observation, and cell MRI results show that the HA-targeted groups had significantly higher cellular uptake than the nontargeted groups. This demonstrates that the HA-Fe3O4 NPs are uptaken by pancreatic cells via an HA-mediated targeting pathway. The HA-mediated active targeting strategy could be applied to other biomedical projects.
Introduction
Pancreatic cancer is a relatively common cancer with a 5 year survival rate of less than 8%.1 The optimal treatment prolonging the patients lives with pancreatic cancer is radical surgery. However, metastasis has frequently begun during diagnosis, and the resection rate is less than 20%. For the survival of the patient, earlier detection and diagnosis of pancreatic cancer is particularly important.Magnetic resonance imaging (MRI) is considered an effective method for the diagnosis of pancreatic cancer because of its high resolution and noninvasive imaging mode.2 In one examination, with the use of safer intravenous contrast agents, MRI can perform noninvasive assessments of the pancreatic parenchyma, vascular network, and neighboring soft tissues. MR contrast agents are divided into two major categories: T1 and T2 MR contrast agents based on gadolinium and superparamagnetic iron oxide nanoparticles (Fe3O4 NPs).3–7 Currently, gadolinium-based contrast agents (GBCAS) are used in clinical applications of MRI.8,9 However, severe complications associated with GBCAS are increasing as they are being linked with nephrogenic systemic fibrosis (NSF).10 Fe3O4 NPs provide several advantages compared to traditional T1 contrast agents by having a higher magnetic signal intensity, longer contrast enhancement, and in particular, lower cytotoxicity.11 However, there are two main limitations to using Fe3O4 NPs as MR contrast agents. Firstly, they were engulfed by macrophage and gathered in the reticuloendothelial system (RES).12 The passive targeting of cancer is due to the differences between the tumor and normal tissue with respect to their number of blood vessels, the wider gap in the vascular wall and poor structural integrity in solid tumor tissue, resulting in, enhanced permeability and retention (EPR) effect of tumor blood vessel.13,14 The EPR effect difference is minor between pancreatic cancer tissues and normal pancreatic tissue. Also, the tumor can show hyperintensity or isointensity in the contrast-enhanced MRI. In addition, during the early stage of lesions, tumors are smaller and more difficult to detect. Nanoprobes that are modified with specific ligand molecules to target delivery to the tumor site are essential for highly sensitive MRI of pancreatic tumors.15
Recent studies have shown that CD44, a protein that has an important part in the aggressive pancreatic cancer, is overexpressed in pancreatic cancer cell lines.13,16–19 CD44 is a receptor with a high affinity for HA and contributes to cellular processes such as cell survival, differentiation, proliferation, adhesion, migration, and chemo-resistance.20–23 Studies have shown that HA has been an effective ligand for targeting therapy for pancreatic cancer.24,25 However, there are few reports using CD44 as a target for diagnosis of pancreatic cancer with MR.
In this study, multifunctional HA modified Fe3O4 NPs were developed by covalently modifying synthesized Fe3O4-PEI NPs with HA, fluorescein isothiocyanate (FITC), and mPEG via a PEI-mediated conjugation chemistry strategy. The final product was denoted as HA-Fe3O4 NPs (Scheme 1). Fe3O4 NPs modified with multifunctional HA were applied for MRI of MIAPaca-2 cells, a CD44 overexpressing cancer cell line, and orthotopic transplantation model. The in vivo MRI was performed to monitor the growth of the tumor. This study provides the experimental basis for increased accuracy and observation of the development of pancreatic tumors via the targeting MRI-guidance strategy and could have enormous potential application in earlier detection of pancreatic cancer.
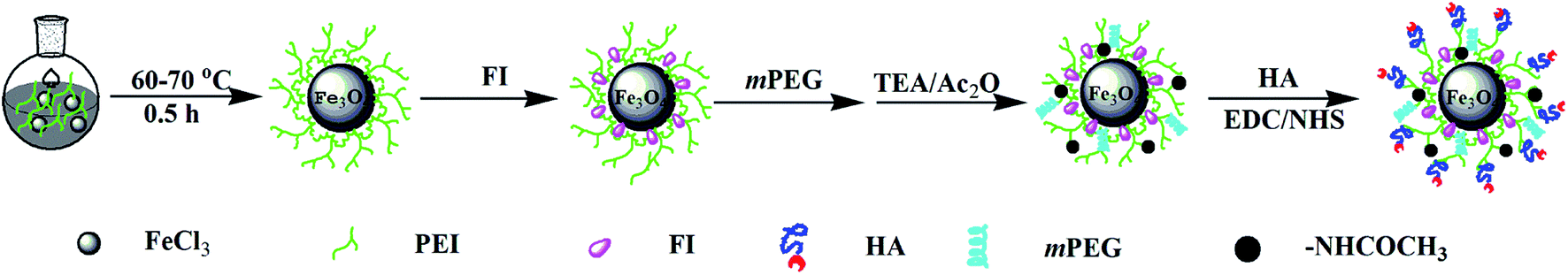 | ||
| Scheme 1 Synthesis Routes of the HA-Fe3O4 NPs. | ||
Experimental
Preparation of Fe3O4-PEI·NH2
The method of Fe3O4-PEI NPs synthesis is as follows.26 Argon gas was pumped into FeCl3·6H2O solution (2.6 g/40 mL) to remove oxygen for 30 minutes under intense stirring before sodium sulfite solution (0.4 g/20 mL) added with the same time. The reaction mixture was transferred to a 500 mL three-necked flask and a PEI·NH2 solution (1.0 g/10 mL) was then added to the flask. After mixing, heated the flask up to 60–70 °C for 30 min, cooled it to room temperature and continued stirring for about 2 h. The mixture was separated and purified by an external magnetic field, and the same steps were repeated three to five times with water. Finally, the obtained Fe3O4 were redissolved in water (25 mL) and stored at 4 °C for further use.Synthesis of FITC labeled Fe3O4 NPs
The Fe3O4 nanoparticles (120 mg) prepared in the last step were dispersed in 20 mL. FITC (4.2 mg, 1 mL in DMSO) solution was added to the suspension of Fe3O4 nanoparticles to avoid light and stirred vigorously at ambient temperature for one day. After that, the FITC labeled Fe3O4 NPs were fabricated.Synthesis of HA-Fe3O4 NPs
The polyethylene glycol modification method was adopted to improve the colloid stability and biocompatibility of Fe3O4 NPs. Firstly, dissolved Fe3O4 NPs (60 mg) into 5 mL DMSO, activated the carboxyl group of mPEG-COOH with EDC (1.7 mg) and NHS (1.5 mg) for 3 h. The solution was added to FITC-labeled Fe3O4 suspension by dropwise manner, the reaction lasted for 3 days and avoid light. The reaction products were divided into two equal parts. And one part of the solution was added triethylamine (187.5 μL) and acetic anhydride (200 μL) respectively to form the non-HA materials (denoted as non-HA targeted control group materials: nHA-Fe3O4 NP). Another part of the solution was respectively added with triethylamine (75 μL) and acetic anhydride (80 μL) to obtain the partially acetylated iron oxide nanoparticles. 116 mg of hyaluronic acid was weighed and dissolved in 5 mL water. Then, adopted the similar carboxyl activation method of mPEG-COOH to active the –COOH of hyaluronic acid (EDC 1.3 mg for 30 min and then added NHS 1.1 mg for 3 h). After that, the solution was added to the partially acetylated iron oxide nanoparticles and continued reaction 3 days to form the multifunctional iron oxide nanoparticles modified by hyaluronic acid. The formed multifunctional iron oxide particles (denoted as HA-Fe3O4 NPs) were then collected after removing excess reactants and by-products via separation and purification. Finally, the NPs was released into water (5 mL) and stored at 4 °C.Cytotoxicity testing and cell morphology observation
MIAPa-Ca-2 cells were cultivated by the similar manner of our previous report.10 After being treated with nHA-Fe3O4 NPs or HA-Fe3O4 NPs, the morphology, and cell viability were observed under microscopy (Leica DMIL LED inverted phase contrast microscope) and measured by the CCK-8 assay.MR imaging in vivo
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shanghai Jiao Tong University and experiments were approved by the Animal Ethics Committee of Shanghai Jiao Tong University. The management of animals and the establishment of in situ pancreatic cancer tumors model are similar to our previous work.27 Scanning was performed at 7, 14 and 21 days after the operation on a 3.0 tesla MR (MAGNETOM Verio, Siemens) with a mouse coil.Results and discussion
Preparation of HA-Fe3O4 NPs
The morphology and size of the synthesized nHA and HA-Fe3O4 NPs were determined by using TEM (Fig. 1). They were clearly seen that there was a uniform size distribution for particles with an approximate circular morphology. The nHA and HA-Fe3O4 NPs were measured to have a mean diameter of 12.2 ± 1.7 nm (Fig. 1a) and 11.9 ± 1.9 nm (Fig. 1b) respectively. Table S1 (ESI†) showed the hydrodynamic sizes and zeta potentials of the Fe3O4-PEI, nHA and HA-Fe3O4 NPs. With respect to zeta potentials, Fe3O4-PEI, due to the abundance of amino groups, had a zeta potential of +32.1 mV while the zeta potentials of the nHA-Fe3O4 and HA-Fe3O4 NPs NP decrease to +12.2 and −16.6 mV. The hydrodynamic size of Fe3O4-PEI, nHA-Fe3O4, and HA-Fe3O4 NPs are 240.9 nm, 289.3 nm, and 306.1 nm, respectively. The size difference between the various types of NPs showed that the surface modification was successful. The colloid stability of nHA-Fe3O4 and HA-Fe3O4 NPs were one of the core premises for the further biologic application. It is found that after dispersing in water for 7 days, the hydrodynamic sizes of nHA-Fe3O4 and HA-Fe3O4 NPs changed slightly, but without visible aggregations or precipitations (Fig. S1 and S2, ESI†) indicating, that the prepared NPs were colloidal stable. The characteristic absorption peak at 520 nm of FITC was observed in the HA-Fe3O4 NPs (Fig. S3, ESI†) thus, the UV-Vis results indicated that FITC had been successfully conjugated with HA-Fe3O4 NPs.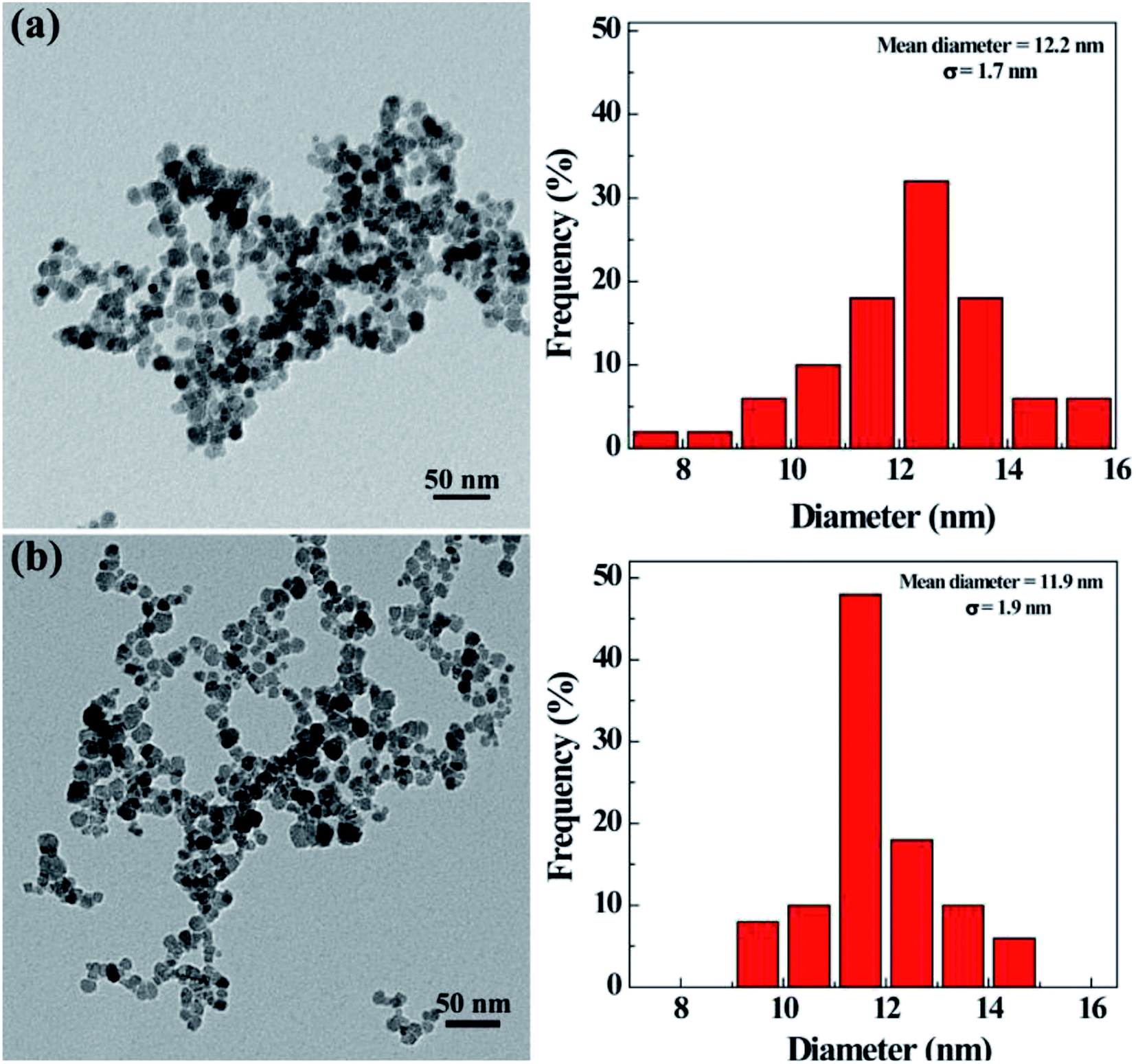 | ||
| Fig. 1 TEM image of nHA-Fe3O4 NPs (a) and HA-Fe3O4 NPs (b), respectively. | ||
The percentage of HA in the Fe3O4 NPs was analyzed quantitatively by using TGA (Fig. 2). The weight of PEI and HA in the Fe3O4 NPs was measured to be 9.8% and 5.7% respectively, further indicating there was a successful surface modification of FITC-mPEG and HA. The curve of room temperature magnetic performance confirms the nHA-Fe3O4 or HA-Fe3O4 NPs presents obvious superparamagnetism, and the saturation magnetization of nHA-Fe3O4 or HA-Fe3O4 NPs is calculated to be 90.5 emu g−1 and 101.3 emu g−1, respectively (Fig. S4, ESI†).
 | ||
| Fig. 2 Thermogravimetric analysis of Fe3O4-PEI NPs (black curve), nHA-Fe3O4 NPs (red curve), and HA-Fe3O4 NPs (blue curve), respectively. | ||
T2 relaxivity measurements
The relaxation times of nHA-Fe3O4 and HA-Fe3O4 NPs were quantified on a 0.5 T NMI20 Analysing and Imaging System to examine the feasibility of these NPs as a T2-weighted MR contrast agent (Shanghai NIUMAG Corporation, Shanghai, China). Fig. 3 contains the T2-weighted MR images that showed decreasing MRI signal intensity with increasing Fe concentrations in both the nHA-Fe3O4 and HA-Fe3O4 NPs (Fig. 3a). The r2 of the nHA and HA-Fe3O4 NPs were counted. As shown in Fig. 3b, the r2 of nHA-Fe3O4 and HA-Fe3O4 NPs were counted to be 204.1 and 321.4 mM−1 s−1. The high r2 of nHA-Fe3O4 and HA-Fe3O4 NPs indicated that these NPs can be used as potential T2 weighted contrast agents for MRI. | ||
| Fig. 3 T2 MR image and r2 relaxivity of the nHA-Fe3O4 NP or HA-Fe3O4 NPs as a function of Fe concentration. | ||
Cell morphology and cytotoxicity assay observation
The cytotoxicity of nHA-Fe3O4 and HA-Fe3O4 NPs were tested before MR scanning. After the MIAPaCa-2 cells were cultivated for 24 hours with nHA-Fe3O4 and HA-Fe3O4 NPs at particle concentrations of 10, 20, 60, 80 and 100 μg mL−1, the cell viability was assessed by the CCK8 assay (Fig. 4). When compared with a PBS control, the viability of MIAPaCa-2 cells had no significant influence after incubation with either nHA-Fe3O4 or HA-Fe3O4 NPs within the tested concentration range (10–100 μg mL−1). The results indicated that both particles were no cytotoxicity at the given concentration.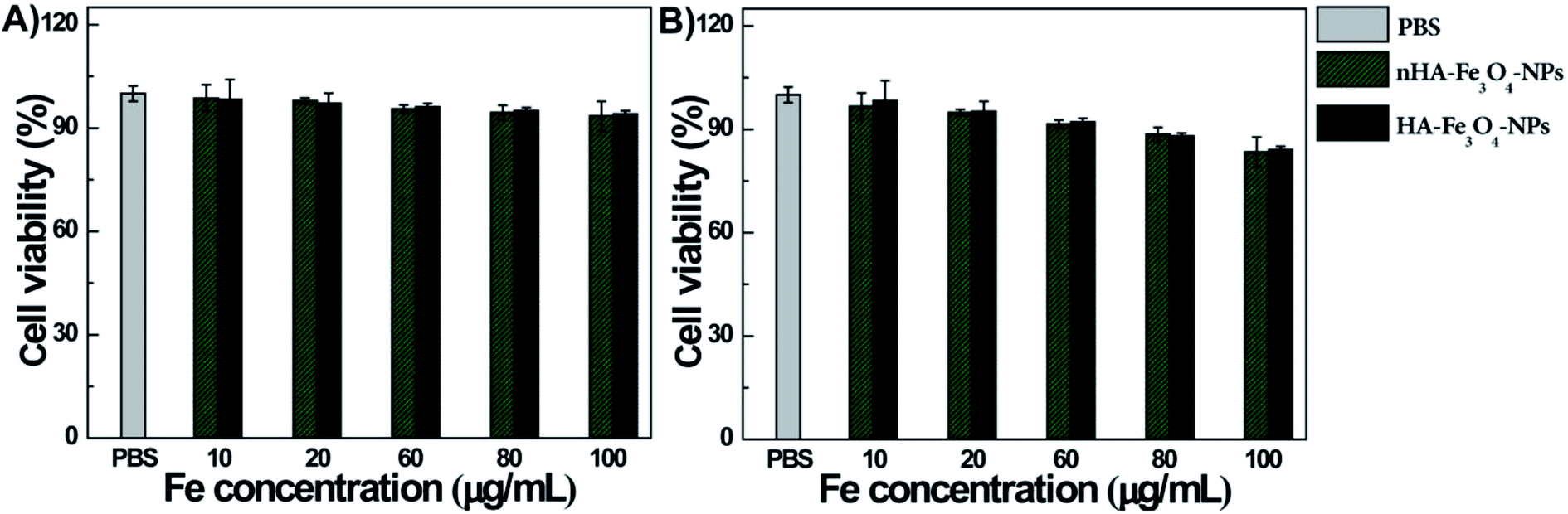 | ||
| Fig. 4 The viability of MIAPaCa-2 cells after treated with PBS, nHA-Fe3O4 NP and HA-Fe3O4 NPs at the different Fe concentrations for 24 h (A) or 48 h (B) at 37 °C by the CCK-8 assay. | ||
The cytocompatibility of the nHA-Fe3O4 or HA-Fe3O4 NPs were further explored by observed the morphology of MIAPaCa-2 cells treated with the NPs (Fig. S5, ESI†). The MIAPaCa-2 cells did not show any morphological changes at the studied Fe concentrations when compared with the PBS control. Both the morphological and CCK8 data was consistent further validating that both nHA-Fe3O4 and HA-Fe3O4 NPs had a good cytocompatibility.
Cellular uptake
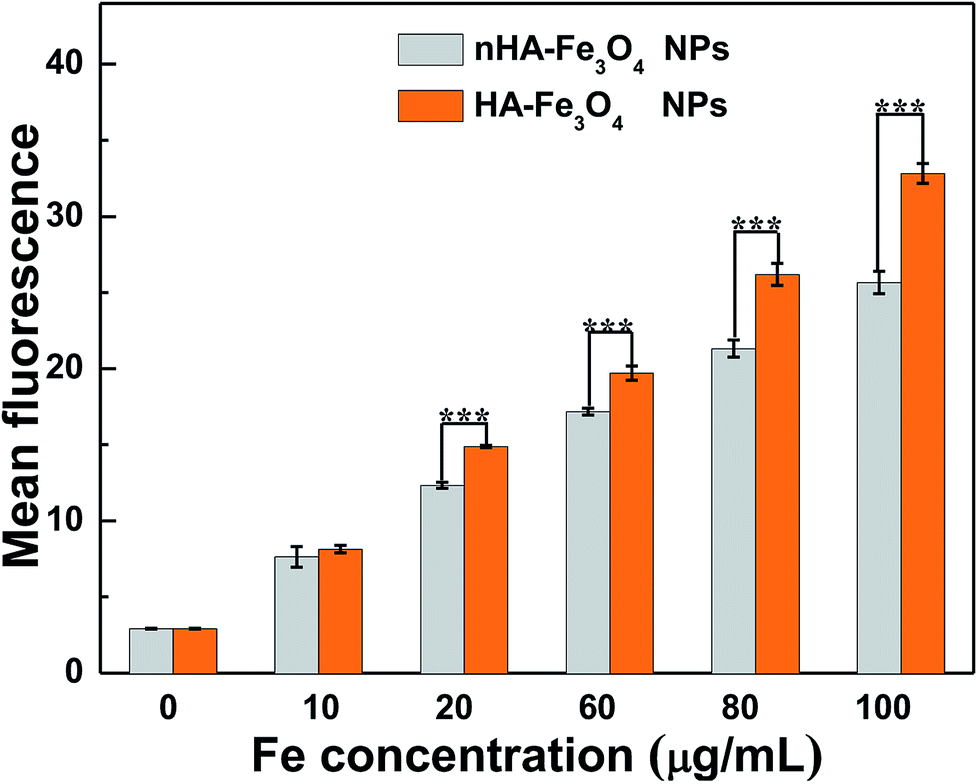
The conjugation of FI moiety and Fe3O4 NPs was necessary to observe the cellular uptake of the nHA and HA-Fe3O4 NPs by confocal microscopy and flow cytometry assay. The mean fluorescence intensity between the two NPs treatments was compared (Fig. 5 and S6, ESI†). MIAPaCa-2 cells incubated with HA-Fe3O4 NPs showed the fluorescence intensity higher than that with nHA-Fe3O4 NPs as the Fe concentration was increased. The ligand–receptor interaction between CD44 and HA ligands should be related to the improved cellular uptake of the HA-Fe3O4 NPs by MIAPaCa-2 cells.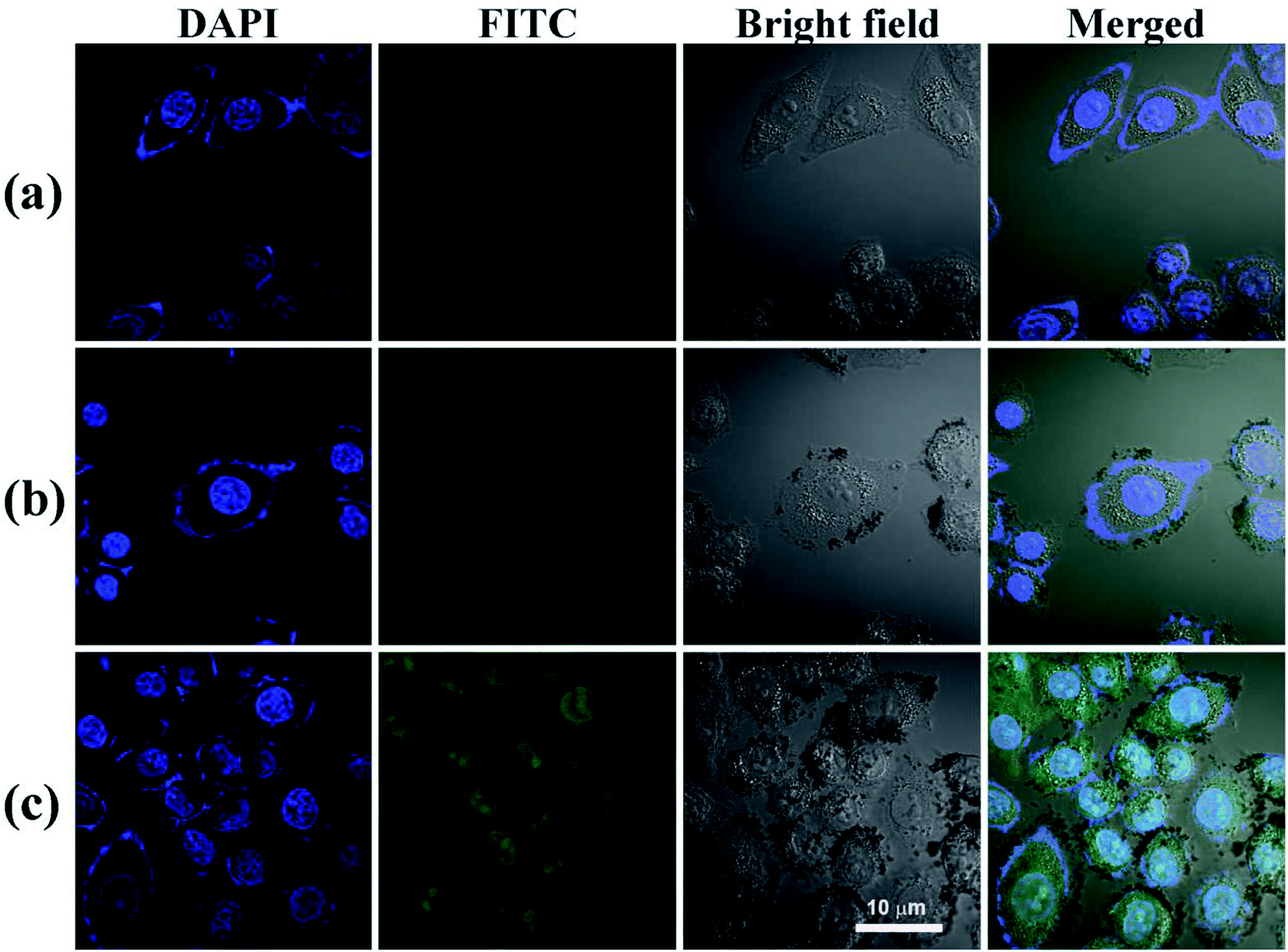 | ||
| Fig. 5 The ability of MIAPaCa-2 cells to uptake PBS (a), nHA-Fe3O4 NP (b) and HA-Fe3O4 NPs (c) ([Fe] 50 μg mL−1) 4 hours after treatment, MIAPaCa-2 cells treated with PBS were used as control, scale bar = 10 μm. | ||
Nuclei appeared blue when MIAPaCa-2 cells were added with PBS (Fig. 5a). When MIAPaCa-2 cells treated with either nHA or HA-Fe3O4 NPs at a Fe concentration of 60 μg mL−1, they displayed intense green fluorescent signals that originated from FI on the cell surfaces and in the cytosol (Fig. 5b and c). However, the cells treated with HA-Fe3O4 NPs had an increased number of cells displaying a green fluorescence. This indicated that the HA-Fe3O4 NPs were uptaken by MIAPaCa-2 cells by active targeting pathway.
Prussian blue staining was applied to qualitatively assess the specific cellular uptake of the HA-Fe3O4 NPs by MIAPaCa-2 (Fig. S7, ESI†). The MIAPaCa-2 cells that were treated with the HA-Fe3O4 NPs displayed significantly deep blue staining than cells treated with the nHA-Fe3O4 NPs with the same Fe concentrations. Blue staining was not displayed in control cells added with PBS. The flow cytometry assay (Fig. 6), confocal microscopy observation results, and the Prussian blue staining results demonstrated the function of HA-mediated targeting.
 | ||
| Fig. 6 Flow cytometric analysis of the MIAPaCa-2 cells treated with the nHA-Fe3O4 NP or HA-Fe3O4 NPs for 4 h at the different Fe concentration. | ||
In vitro MRI of cancer cells
The nHA and HA-Fe3O4 NPs were used as probes for MR scanning of MIAPaCa-2 cells. Before MR scanning, MIAPaCa-2 cells overexpressing CD44 receptor were incubated with nHA-Fe3O4 or HA-Fe3O4 NPs at a range of Fe concentrations from 5 to 80 μg mL−1 at 37 °C and 5% CO2 for 4 hours. T2 WI of the cells were collected (Fig. 7). | ||
| Fig. 7 In vitro T2 MR images of MIAPaCa-2 cells treated with the nHA-Fe3O4 NP or HA-Fe3O4 NPs for 4 h. | ||
It was showed in cells treated with both nHA-Fe3O4 and HA-Fe3O4 NPs that the MR signal intensity decreased with Fe concentration increasing. This further demonstrates that by attaching the HA ligand, the NPs could be directly targeting to cells.
MRI of tumor in mice
The nHA-Fe3O4 NPs or HA-Fe3O4 NPs were injected intravenously into mice (0.3 mL in PBS, 0.6 mg Fe per mouse) for MR scanning at 7, 14 and 21 days after tumor seeding (Fig. 8). After the HA-Fe3O4 NPs were injected, the MR signal of tumor gradually decreased (Fig. 8b, d and 8f). The highest contrast enhancement was induced by the particles at 90 min post-injection while the MR signal started to recover at 180 min. Compare to HA-Fe3O4 NPs, the tumor MR signal injected with nHA-Fe3O4 NPs did not decrease during the post-injection (Fig. 8a, c and e). It was well known that the decreased MR signal of tumor after injection with nHA-Fe3O4 NPs was associated with the EPR effect. Results showed that the HA-directed targeting role enabled specific delivery of the particles to the tumor, thus, in conjunction with the passive EPR effect, there was specific MRI of the tumor. The MR signal intensity of tumors injected with both nHA-Fe3O4 and HA-Fe3O4 NPs were recovered to some extent at 180 min post-injection. Likely, the NPs have undergone the metabolism process decreased distribution in the tumor. At 7, 14 and 21 days after tumor seeding, there was a gradual decrease in MR signal intensity with the tumor.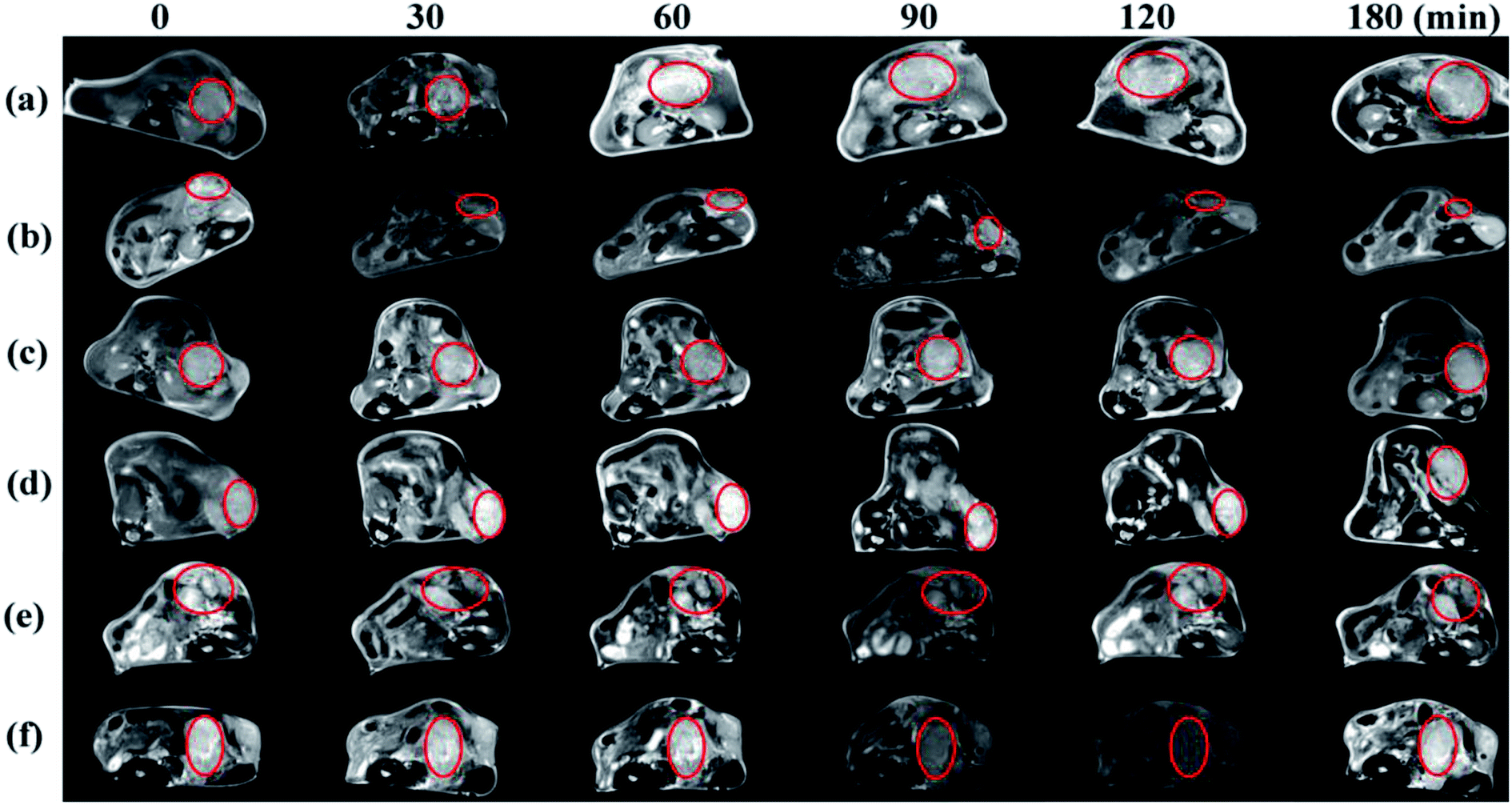 | ||
| Fig. 8 In vivo transverse T2 MR images of tumors after intravenous injection of the nHA-Fe3O4 NP ((a) 7 days; (c) 14 days; (e) 21 days) and HA-Fe3O4 NPs ((b) 7 days; (d) 14 days; (f) 21 days) ([Fe] 1 mg mL−1, in 200 μL saline) at different time points post i.v.-injection. | ||
21 days after tumor seeding, MR scanning was performed, then, the mice were sacrificed and the tumors were removed for HE staining (Fig. S8 and S9, ESI†). Microscopically, the tumor cells were spindle-shaped and had large nuclei with visible nucleoli. The nuclear fission of the tumor cells was inconspicuous at 7 and 14 days after tumor seeding. The tumor cells invade the surrounding acinar tissue and destroy the acinar forming a single acinar like “island” at 21 days after tumor seeding. The brown refractive particles were seen in tumor interstitial tissue during this period. The quantity of particles in HA-Fe3O4 NPs group is significantly higher than that nHA-Fe3O4 NPs group. Given the different time points, the results may suggest that HA-Fe3O4 NPs can early detect a pancreatic tumor as contrast agent, especially compared to nHA-Fe3O4 NPs.
In vivo biodistribution and acute toxicity assessment
ICP-AES was used to analyze the Fe concentration in main organs to further observe the biodistribution behavior of the nHA and HA-Fe3O4 NPs (Fig. 9). 24 hours after the injection of nHA-Fe3O4 or HA-Fe3O4 NPs, the Fe concentration in tumor tissue and tested organs increased more than that of the control mice. Fe was uptaken mainly by the liver and spleen. However, a small quantity remains in the hearts, lungs, kidneys, and tumors. The Fe gather in the liver and spleen were closely related to the clearance effect of RES. The biological acute toxicity assessment was performed by the routine blood-biochemical blood testing (Fig. S10, ESI†). The nHA-Fe3O4 or HA-Fe3O4 NPs saline solution was intravenously injected into mice at a dosage (40 mg kg−1). Afterward, mice were sacrificed 10 days post-injection (n = 10). Healthy mice with age and weight matched were the control group. The liver and kidney function indexes including alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (AKP), blood urea nitrogen (BUN) and creatinine (CREA) appeared normal. The blood indexes had no significant differences between the treatment groups and the control group. | ||
| Fig. 9 In vivo biodistribution of hearts, livers, spleens, lungs, kidneys, and tumors 24 h post intravenous injection of the nHA-Fe3O4 NP and HA-Fe3O4 NPs s (600 μg Fe, in 0.3 mL PBS). | ||
Conclusions
In this study, we developed an HA-Fe3O4 NPs for targeted MRI of pancreatic cancer. These Fe3O4 NPs were proved to be stable, water soluble, and cytocompatible in the given concentration set. The results of cellular uptake analysis indicated that the HA-Fe3O4 NPs were specifically uptaken by MIAPaCa-2 cells which overexpress the CD44 receptor. In summary, as effective nanoprobes for MRI of pancreatic cancer cells and an orthotopic pancreatic cancer model through the HA-mediated active targeting pathway, the developed HA-Fe3O4 NPs will play a great role as MR contrast agent for early detection of pancreatic cancer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Medical and Technology Intercrossing Research Foundation of Shanghai Jiaotong University (YG2014QN07) and the National Natural Science Foundation of China (Grant No. 51602334). The authors would like to thank Thomas Derezes for making significant edits to the manuscript and Pathologist Fuqiang Wang for interpretation to the tumor HE staining.Notes and references
- P. L. S. Uson Junior, D. Callegaro-Filho, D. D. G. Bugano, F. Moura and F. C. Maluf, J. Gastrointest. Cancer, 2018, 49, 481–486 CrossRef PubMed
.
- C. Multari, M. Miola, S. Ferraris, D. Movia, K. Z. Rozman, N. Kostevsek, A. Follenzi, E. Verne and A. Prina-Mello, Int. J. Appl. Ceram. Technol., 2018, 15, 947–960 CrossRef CAS
- J. Li, L. Zheng, H. Cai, W. Sun, M. Shen, G. Zhang and X. Shi, Biomaterials, 2013, 34, 8382–8392 CrossRef CAS PubMed
- S. Wen, K. Li, H. Cai, Q. Chen, M. Shen, Y. Huang, C. Peng, W. Hou, M. Zhu and G. Zhang, Biomaterials, 2013, 34, 1570–1580 CrossRef CAS PubMed
- C. Guo, L. Sun, H. Cai, Z. Duan, S. Zhang, Q. Gong, K. Luo and Z. Gu, J. Gastrointest. Cancer, 2017, 9, 23508–23519 CAS
- Q. Luo, X. Xiao, X. Dai, Z. Duan, D. Pan, H. Zhu, X. Li, L. Sun, K. Luo and Q. Gong, J. Gastrointest. Cancer, 2018, 10, 1575–1588 CAS
- C. Hao, X. Wang, Z. Hu, S. Ling, D. Pan, Q. Gong, Z. Gu and K. Luo, Appl. Mater. Today, 2018, 11, 207–218 CrossRef
- Y. Luo, J. Yang, J. Li, Z. Yu, G. Zhang, X. Shi and M. Shen, Colloids Surf., B, 2015, 136, 506–513 CrossRef CAS PubMed
- Y. Luo, L. Zhao, X. Li, J. Yang, L. Guo, G. Zhang, M. Shen, J. Zhao and X. Shi, J. Mater. Chem. B, 2016, 4, 7220–7225 RSC
- Y. Luo, J. Yang, Y. Yan, J. Li, M. Shen, G. Zhang, S. Mignani and X. Shi, Nanoscale, 2015, 7, 14538–14546 RSC
- C. Zhang, Y. Yan, Q. Zou, J. Chen and C. Li, Asia-Pac. J. Clin. Onco., 2016, 12, 13–21 CrossRef PubMed
- J. Li, Y. He, W. Sun, Y. Luo, H. Cai, Y. Pan, M. Shen, J. Xia and X. Shi, Biomaterials, 2014, 35, 3666–3677 CrossRef CAS PubMed
- B. B. S. Cerqueira, A. Lasham, A. N. Shelling and R. Al-Kassas, Eur. J. Pharm. Biopharm., 2015, 97, 140–151 CrossRef CAS PubMed
- S. Sharifi, H. Seyednejad, S. Laurent, F. Atyabi, A. A. Saei and M. Mahmoudi, Contrast Media Mol. Imaging, 2015, 10, 329–355 CrossRef CAS PubMed
- J. Xie, K. Chen, H. Y. Lee, C. Xu, A. R. Hsu, S. Peng, X. Chen and S. Sun, J. Am. Chem. Soc., 2008, 130, 7542–7543 CrossRef CAS PubMed
- S. Zhao, C. Chen, K. Chang, A. Karnad, J. Jagirdar, A. P. Kumar and J. W. Freeman, Clin. Cancer Res., 2016, 22, 5592–5604 CrossRef CAS PubMed
- J. Wei, Y. Zhang, K. T. Kane, M. A. Collins, D. M. Simeone, M. P. D. Magliano and K. T. Nguyen, Mol. Cancer Res., 2015, 13, 9–15 CrossRef PubMed
- N. J. Wood, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 427–428 Search PubMed
- M. Kumazoe, M. Takai, J. Bae, S. Hiroi, Y. Huang, K. Takamatsu, Y. Won, M. Yamashita, S. Hidaka and S. Yamashita, Oncogene, 2016, 36, 2643 CrossRef PubMed
- K. H. Bae, J. J. Yoon and T. G. Park, Biotechnol. Prog., 2006, 22, 297–302 CrossRef CAS PubMed
- G. Jiang, K. Park, J. Kim, K. S. Kim, E. J. Oh, H. Kang, S. E. Han, Y. K. Oh, T. G. Park and H. S. Kwang, Biopolymers, 2010, 89, 635–642 CrossRef PubMed
- M. Kamat, K. Elboubbou, D. C. Zhu, T. Lansdell, X. Lu, W. Li and X. Huang, Bioconjugate Chem., 2015, 21, 2128–2135 CrossRef PubMed
- S. Kiuchi, S. Ikeshita, Y. Miyatake and M. Kasahara, Exp. Mol. Pathol., 2015, 98, 41–46 CrossRef CAS PubMed
- P. Kesharwani, S. Banerjee, S. Padhye, F. H. Sarkar and A. K. Iyer, Biomacromolecules, 2015, 16, 3042–3053 CrossRef CAS PubMed
- X. P. Li, X. W. Zhang, L. Z. Zheng and W. J. Guo, Int. J. Clin. Exp. Pathol., 2015, 8, 6724–6731 CAS
- Y. Hu, J. Li, J. Yang, P. Wei, Y. Luo, L. Ding, W. Sun, G. Zhang, X. Shi and M. Shen, Biomater. Sci., 2015, 3, 721–732 RSC
- L. Wu, P. Lv, H. Zhang, C. Fu, X. Yao, C. Wang, M. Zeng, Y. Li and X. Wang, Magn. Reson. Imaging, 2015, 33, 737–760 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Part of experimental details and additional experimental results. See DOI: 10.1039/c9ra00730j |
| ‡ Yu Luo, Yuehua Li, and Jing Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |