
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructured poly(L-lactic acid)–poly(ethylene glycol)–poly(L-lactic acid) triblock copolymers and their CO2/O2 permselectivity†
Yun Xueyana,
Li Xiaofanga,
Pan Pengju b and
Dong Tungalag*a
b and
Dong Tungalag*a
aCollege of Food Science and Engineering, Inner Mongolia Agricultural University, 306 Zhaowuda Road, Hohhot, Inner Mongolia 010018, China. E-mail: dongtlg@163.com
bCollege of Chemical and Biological Engineering, Zhejiang University, 38 Zheda Road, Hangzhou 310027, China
First published on 23rd April 2019
Abstract
Biodegradable poly(L-lactic acid)–poly(ethylene glycol)–poly(L-lactic acid) (PLLA–PEG–PLLA) copolymers were synthesized by ring-opening polymerization of L-lactide using dihydroxy PEG as the initiator. The effects of different PEG segments in the copolymers on the mechanical and permeative properties were investigated. It was determined that certain additions of PEG result in composition-dependent microphase separation structures with both PLLA and PEG blocks in the amorphous state. Amorphous PEGs with high CO2 affinity form gas passages that provide excellent CO2/O2 permselectivity in such a nanostructure morphology. The gas permeability and permselectivity depend on the molecular weight and content of the PEG and are influenced by the temperature. Copolymers that have a higher molecular weight and content of PEG present better CO2 permeability at higher temperatures but provide better CO2/O2 permselectivity at lower temperatures. In addition, the hydrophilic PEG segments improve the water vapor permeability of PLLA. Such biodegradable copolymers have great potential for use as fresh product packaging.
Introduction
Fruits and vegetables are perishable products that continue to metabolize actively after harvest. During storage and transportation of fresh produce, control of respiration plays a significant role in prolonging the shelf life. By decreasing the amount of available O2 and increasing the CO2 gas concentration surrounding the product, the respiration rate and metabolism are correspondingly decreased.1 In addition, a high relative humidity in the atmosphere surrounding a fresh product diminishes dehydration and preserves freshness, but excessive humidity in the package may stimulate the development of microbial pathogens.2Modified atmosphere packaging (MAP) was first reported in 1927 and refers to the technique of sealing actively respiring produce in polymeric film packages to modify the O2 and CO2 levels within the package.3 This technique is considered to be one of most effective and convenient ways to decrease the respiration rate and extend the shelf life of fresh produce.4 MAP is a dynamic process, and the internal atmosphere depends on the natural interplay between the respiration of product and the permeation of gases through the plastic films in sealed packagings.5 Therefore, the CO2, O2 and H2O permeabilities of films are very important for providing optimal storage conditions for fresh produce.6 For most fruits and vegetables, a suitable MAP packaging film must have a relatively high CO2, O2 and H2O permeability and be much more permeable to CO2 than to O2.3 However, most plastics have relatively poor CO2/O2 permselectivity and H2O permeability and are still not optimal for fruit and vegetable packagings.
Recently, increasing use of traditional nondegradable plastics has caused serious environmental problems; consequently, biodegradable polymers are under growing consideration as materials for food packaging. Poly(L-lactic acid) (PLLA) is a natural polymer that is produced from 100% renewable resources, such as corn and sugar beets.7 It is highly transparent, nontoxic, biodegradable and biocompatible, and offers great promise in a wide range of commodity applications.8 Compared with most petrochemical-based polymers, such as poly(ethylene), poly(propylene) and even poly(ethylene terephthalate), the oxygen and moisture permeabilities of PLLA are much higher.9 Therefore, PLLA may be a useful biodegradable packaging material for fruits and vegetables. However, the CO2/O2 selectivity of PLLA films cannot satisfy the demand of MAP packagings.10,11 In addition, its applications are also greatly limited by its low glass transition temperature, poor toughness and ductility.12
Poly(ethylene glycol) (PEG) is a polyether compound that is soluble in water and is generally considered biologically inert and safe. PEGs are commercially available over a wide range of molecular weights from 300 to tens of thousands. PEGs with different molecular weights have different physical properties that are dependent on the chain length and find use in different applications. They have been used in numerous applications, including as surfactants, in foods, cosmetics, pharmaceutics and biomedicines, as dispersing agents, as solvents, in ointments and suppository bases, as tablet excipients and as laxatives.13–16 PEG is a flexible polymer and the polar ether oxygens in PEG increase the solubility and selectivity of CO2.17–22 However, it has extremely high crystallinity, and neat PEG is very difficult to process into gas separation membranes. A series of copolymers and blends with highly branched, cross linked networks and repeating units of ethylene oxide segments can be designed to produce membranes with ideal selectivities.23–27 These polymers can be used for the separation of CO2, CH4, N2 and H2, which is one of the most important membrane gas separation processes of industrial applications such as natural gas, landfill or tail gas deacidification. However, few studies have reported on the use of PEG to improve the CO2/O2 permselectivity of PLLA for applications in food packaging.
In this study, soft PEG segments were employed to improve the permeability and toughness of PLLA. To avoid the migration of PEG from the plastic which would result in lower physical properties of packaging film and contamination of food, high molecular weight PLLA–PEG–PLLA (PLGL) triblock copolymers with various PEG content and different PEG molecular weights were synthesized by copolymerization. The effects of PEG content and molecular weights in copolymers on the crystallization behavior, microstructure, mechanical properties, gas permeability and selectivity were investigated.
Experimental
Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 20000 g mol−1 were obtained from Sigma-Aldrich, USA. Stannous octoate (Sn(Oct)2), chloroform and methanol were purchased from Sinopharm Chemical Reagent Co., Ltd, China.
000 and 20000 g mol−1 were obtained from Sigma-Aldrich, USA. Stannous octoate (Sn(Oct)2), chloroform and methanol were purchased from Sinopharm Chemical Reagent Co., Ltd, China.PLGL triblock copolymers were prepared by ring-opening polymerization of L-lactide in the presence of PEG. Sn(Oct)2 was used as the catalyst. As an example, to synthesize the PLGL (PLLA75000–PEG20000–PLLA75000) triblock copolymer, PEG (2.67 g, 0.134 mmoL, Mn = 20000) was added to a 3 neck flask and vacuum dried at 100 °C for 4 h. L-Lactide (20 g, 0.25 mmoL) and Sn(Oct)2 (0.06 g, 0.148 mmoL) were added to the reaction mixture and stirred at 120 °C for 24 h under argon gas. The product was dissolved in chloroform and precipitated with excess cold methanol and the white precipitation was air dried in a fume hood then vacuum dried at room temperature for 72 h.
For simplicity, the samples were labeled as PLGLxGy, and PLLA75000–PEG20000–PLLA75000 was labeled as PLGL75G20. The numbers after PLGLG denote the theoretical molecular weights/1000 of the PLLA and PEG blocks, respectively. The molecular weights and the polydispersity values were determined by gel permeation chromatography (GPC). The copolymer composition was determined by 1H NMR signals at 5.09 (–COCH(CH3)O–), 1.6 (–CH3) and 3.6 (–OCH2CH2–). The molecular weight of the PLLA block in the copolymer was determined from the copolymer composition on the basis of PEG's molecular weight. The molecular weights and compositions of copolymers together with the neat PLLA used for the experiments are listed in Table 1.
| Sample | EG/LA (wt/wt) | PLLA–PEG–PLLAa (cycle unit number) | EG/LAa (wt/wt) | PEGa wt/% | Molecular weight a | Mnb | Pdb |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR based on ethylene glycol (EG) unit (4H, 3.6 ppm) and lactic acid (LA) unit (1H, 5.09 ppm) of the PLGL copolymers.b Determined by GPC. | |||||||
| PLLA | — | — | — | — | — | 92069 |
2.12 |
| PLGL75G06 | 1/25.0 | (LA)1045–(EG)136–(LA)1045 | 1/25.1 | 3.8 | 156415 |
119214 |
1.75 |
| PLGL55G06 | 1/18.3 | (LA)775–(EG)136–(LA)775 | 1/18.6 | 5.1 | 117534 |
99976 |
1.82 |
| PLGL35G06 | 1/11.7 | (LA)510–(EG)136–(LA)510 | 1/12.2 | 7.6 | 73440 |
75743 |
1.64 |
| PLGL75G12 | 1/12.5 | (LA)1042–(EG)273–(LA)1042 | 1/12.5 | 7.4 | 162022 |
108395 |
1.44 |
| PLGL55G12 | 1/9.2 | (LA)769–(EG)273–(LA)769 | 1/9.2 | 9.7 | 122749 |
95403 |
1.41 |
| PLGL35G12 | 1/5.8 | (LA)501–(EG)273–(LA)501 | 1/6.0 | 14.2 | 84262 |
75344 |
1.68 |
| PLGL75G20 | 1/7.5 | (LA)1064–(EG)454–(LA)1064 | 1/7.7 | 11.5 | 173164 |
101924 |
2.09 |
| PLGL55G20 | 1/5.5 | (LA)800–(EG)454–(LA)800 | 1/5.8 | 14.8 | 135200 |
96882 |
1.37 |
| PLGL35G20 | 1/3.5 | (LA)509–(EG)454–(LA)509 | 1/3.7 | 21.4 | 93309 |
77632 |
1.34 |
| PLGL25G20 | 1/2.5 | (LA)391–(EG)454–(LA)391 | 1/2.8 | 26.2 | 76291 |
60927 |
1.51 |
Characterization of PLGL copolymers and films
To simulate humidity conditions during the storage of fresh produce, the oxygen permeability was determined at different humidity levels using a constant pressure gas permeability tester (Model 8001 Illinois Instruments, Inc., USA). Each test was repeated four times. The oxygen permeabilities (OP) and carbon dioxide permeabilities (CP) were calculated as previously described.28
Results and discussion
Molecular characterization of PLGL copolymers
PLGL triblock copolymers with different PLLA chain lengths were prepared by ring opening polymerization of L-lactide in the presence of Sn(Oct)2 as a catalyst. PEGs with different molecular weights were used as the middle block.The total molecular weight and polydispersity (Pd) of the polymers were determined by GPC and the results are shown in Table 1. The relative mass ratio (determined by molar ratios) of ethylene glycol/L-lactic acid (EG/LA) in copolymers as determined by 1H NMR (Fig. S1†) were lower than the fitting ratio, which may be due to an incomplete reaction of monomers. Since different EG/LA feeding ratio, PEG content varied from 3.8 to 26.2 wt%. All copolymers exhibited relatively high molecular weights, greater than 7.3 × 104, indicating good film forming properties. In addition, it can also be seen that the molecular weight decreased from 1.7 × 105 to 7.3 × 104 as the PEG in the copolymers increased. The number average molecular weights of copolymers that were determined by GPC were lower than those determined by 1H NMR. This may be because the systems contain some PLLA homopolymer, which can lead to higher molecular weights in NMR analysis. However, all copolymers have a relatively low Pd value, indicating a narrow molecular weight distribution. Taken together, both 1H NMR and GPC results demonstrated that high molecular weight PLGL copolymers with different PLLA chain lengths and PEG content were successfully synthesized in this work.
WAXD analysis
The crystalline structure was analyzed by wide angle X-ray diffraction and the results are shown in Fig. 1. Fig. 1(A) shows the WAXD patterns of pure PLLA and PLGL55Gy copolymers. It can be seen that the pure PLLA sample exhibits a typical amorphous broad band.29 For PLGL55Gy copolymers, the length of PLLA chains in the copolymers is certain, and it can be seen from Fig. 1(A) that no obvious diffraction peaks were observed for PLGL55G06 and PLGL55G12, which revealed amorphous structures. However, as the molecular weight of PEG increased to 20000 in the copolymer, a small diffraction peak at approximately 16.9° appeared in the WAXD pattern of PLGL55G20, which is attributed to the crystallization of PLLA block.30
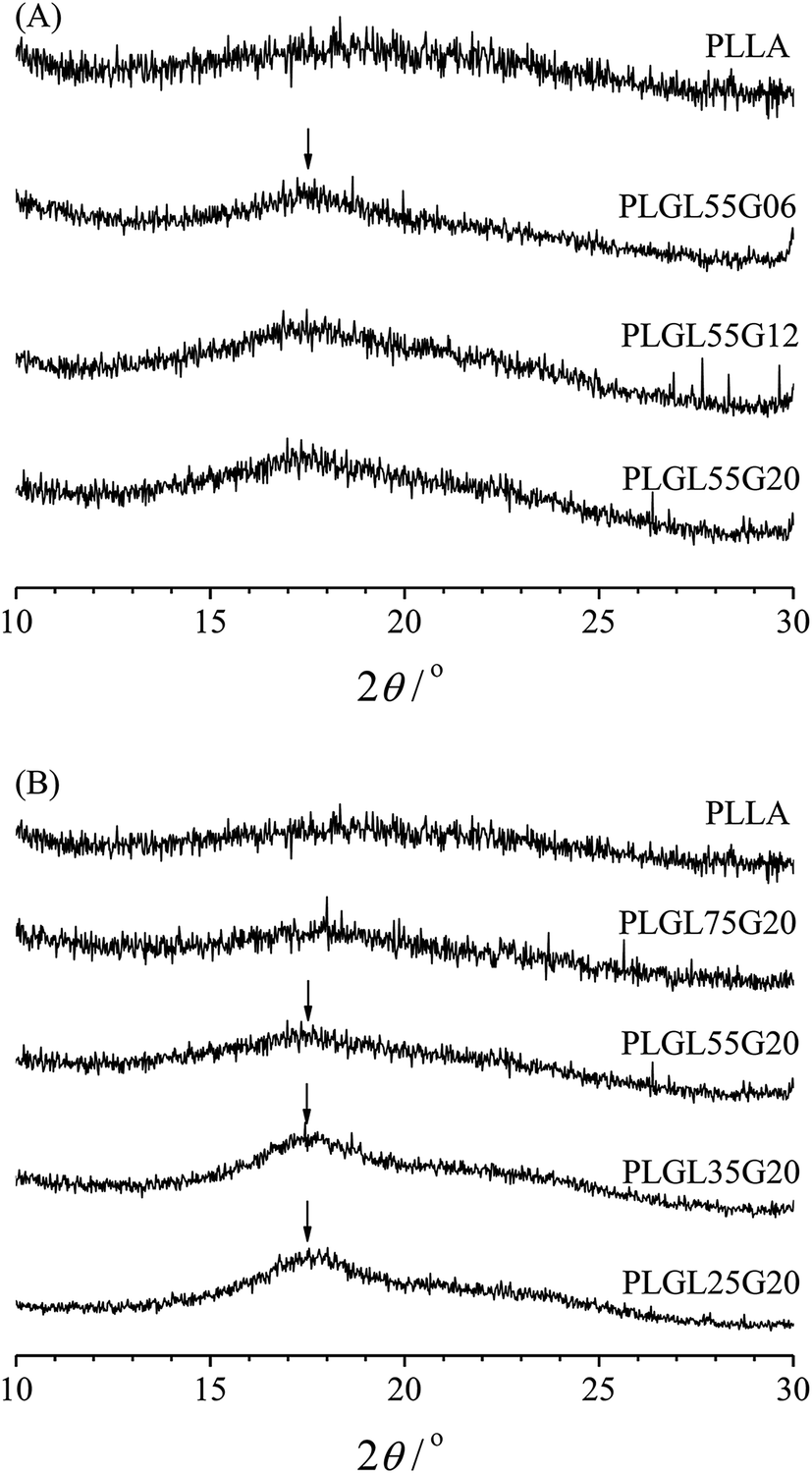 | ||
| Fig. 1 The WAXD pattern of PLGL55Gy and PLGLxG20 copolymers. | ||
Fig. 1(B) presents the WAXD patterns of pure PLLA and PLGLxG20 copolymers. As shown in Fig. 1(B), there are no obvious diffraction peaks that can be attributed to crystalline PLLA block in PLGL75G20. With decreasing PLLA chain lengths, the relative content of PEG increased in the copolymers, and a broad diffraction peak appeared at 17.6° in the WAXD patterns of PLGL55G20, indicating the presence of a small amount of PLLA crystallites. The intensity of this peak slowly increased from PLGL55G20 to PLGL25G20, suggesting that the incorporation of additional high molecular weight PEG improved the crystallization of PLLA in the copolymers during film formation. However, most of the PLLA was in an amorphous state. In addition, it should be noted that no clear crystal diffraction peaks for PEG were observed in any of the samples, indicating that the PEG was essentially in an amorphous state in the copolymers (Fig. 1 and S2†).
ATR-FTIR analysis
Fig. 2 shows ATR-FTIR spectra in the 1850–1700 cm−1 and 3200–2700 cm−1 regions of PLLA, PEG and PLGL55Gy copolymers. As seen in Fig. 2, for pure PLLA, the bands at 1750 cm−1 and at 2995 and 2944 cm−1 are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CH3 symmetric stretching modes of PLLA.31 The CH2 symmetric stretching in PEG appeared at approximately 2886 cm−1, which partly overlapped with IR absorption peaks for CH3 in PLLA, since the changes in the FTIR spectra in those regions are relatively small.
O and CH3 symmetric stretching modes of PLLA.31 The CH2 symmetric stretching in PEG appeared at approximately 2886 cm−1, which partly overlapped with IR absorption peaks for CH3 in PLLA, since the changes in the FTIR spectra in those regions are relatively small.
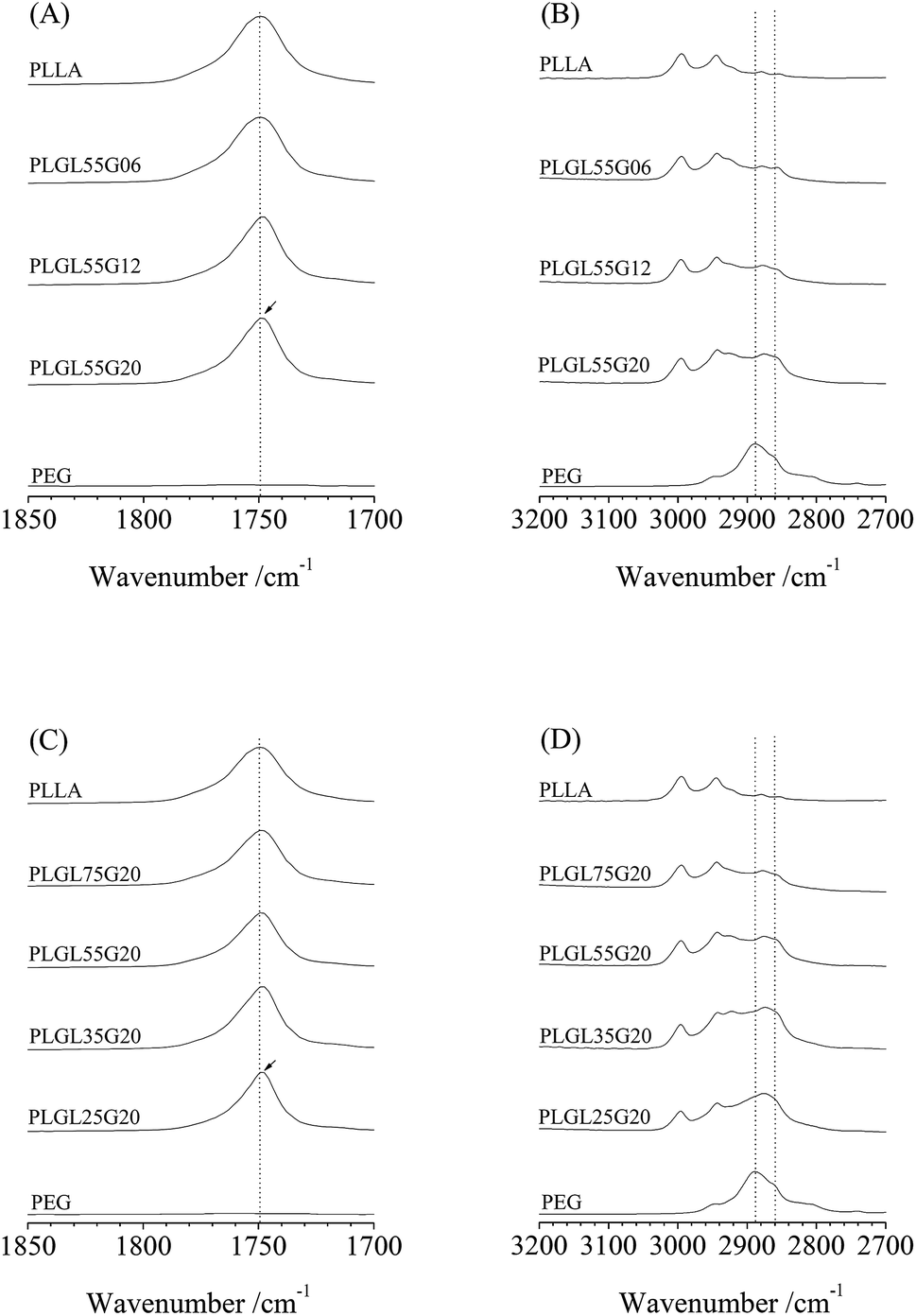 | ||
| Fig. 2 The FTIR spectra of PLLA, PEG and PLGLxGy copolymers. | ||
As seen in Fig. 2(A), the characteristic absorption bands for PLLA at 1750 cm−1 in PLGL55G06 and PLGL55G12 are almost identical. As the PEG molecular weight increased to 20000, the PEG content in PLGL55G20 increased to 14.8%. Furthermore, a red shift phenomenon was observed as the band at 1750 cm−1 shifted to 1748.5 cm−1. This may be due in part to PLLA segments with an ordered arrangement or to crystallization in PLGL55G20.
Fig. 2(B) shows that the spectrum of pure PEG contains two peaks. The first peak was at 2886 cm−1, which was attributed to the stretching vibrations of CH2 groups in crystalline PEG. The second peak was a small shoulder at 2860 cm−1 and was due to vibrations of CH2 in amorphous regions of PEG.32 For the copolymers, on one hand, the stretching band assigned to crystalline PEG at 2886 cm−1 was almost eliminated in PLGL55G20. On the other hand, the intensity of the band for amorphous PEG at 2860 cm−1 increased, implying that most of the PEG blocks exist in an amorphous state in all of the copolymers. Therefore, the spectra for PLGL55Gy (as well as the spectra for PLGLxG6 and PLGLxG12 in Fig. S3 and S4†) indicate that the PLLA and PEG blocks are in an amorphous state in those copolymers. The above results agree with those from the WAXD analysis and demonstrate that PLLA and PEG blocks maintain an amorphous state in all of the copolymers. Only a small amount of PLLA crystal was present in PLGL55G20, indicating that the presence of large amounts of high molecular weight PEG can promote the crystallization of the PLLA block. This result agrees with the results in Tables S1, S2 and Fig. S6–S9 in the ESI.†
For PLGLxG20 copolymers, as shown in Fig. 2(C) and (D), the PEG content increased with decreasing PLLA lengths in the copolymers. In addition, the intensity of the characterized absorption bands showed a similar trend with PLGL55Gy copolymers. That is, the intensity of bands assigned to amorphous PEG increased, and a redshift also occurred for the band of PLLA with increasing PEG content in the copolymers. Fig. 2(C) and (D) show that most of the PLLA and PEG were also in an amorphous state in the PLGLxG20 copolymers. This result agrees well with the DSC result. No evidence for crystallization of PEG was observed in the DSC curves (Fig. S5†). The crystallinity of neat PLLA was approximately 3.3%, and the crystallization of PLLA was only slightly influenced by the presence of large amounts of high molecular weight PEG. PLLA in PLGL copolymers produces a relatively low level of crystallinity of approximately 1.5–11.7% (as shown in Table S1 and Fig. S6†).
AFM analysis
During the cooling scans for all of the copolymers from 200 to −50 °C, no crystallization peaks for PEG were observed, except for PLGLxG20 copolymers (Table S2, Fig. S7†). In the DSC curve for PLGL75G20, a crystallization peak for PEG appeared at −14.7 °C, and the crystal enthalpy was only 1 J g−1. With increasing PEG content, the crystal enthalpy of PEG gradually increased to 32.8 J g−1 in PLGL25G20. This result indicated that additional high molecular weight PEG was easier to gather and form a microphase segregation structure.To further observe the microstructure of the copolymers, atomic force microscopy (AFM) images were obtained. Fig. 3 shows two dimensional AFM surface images of copolymer films containing various compositions that were obtained at room temperature.
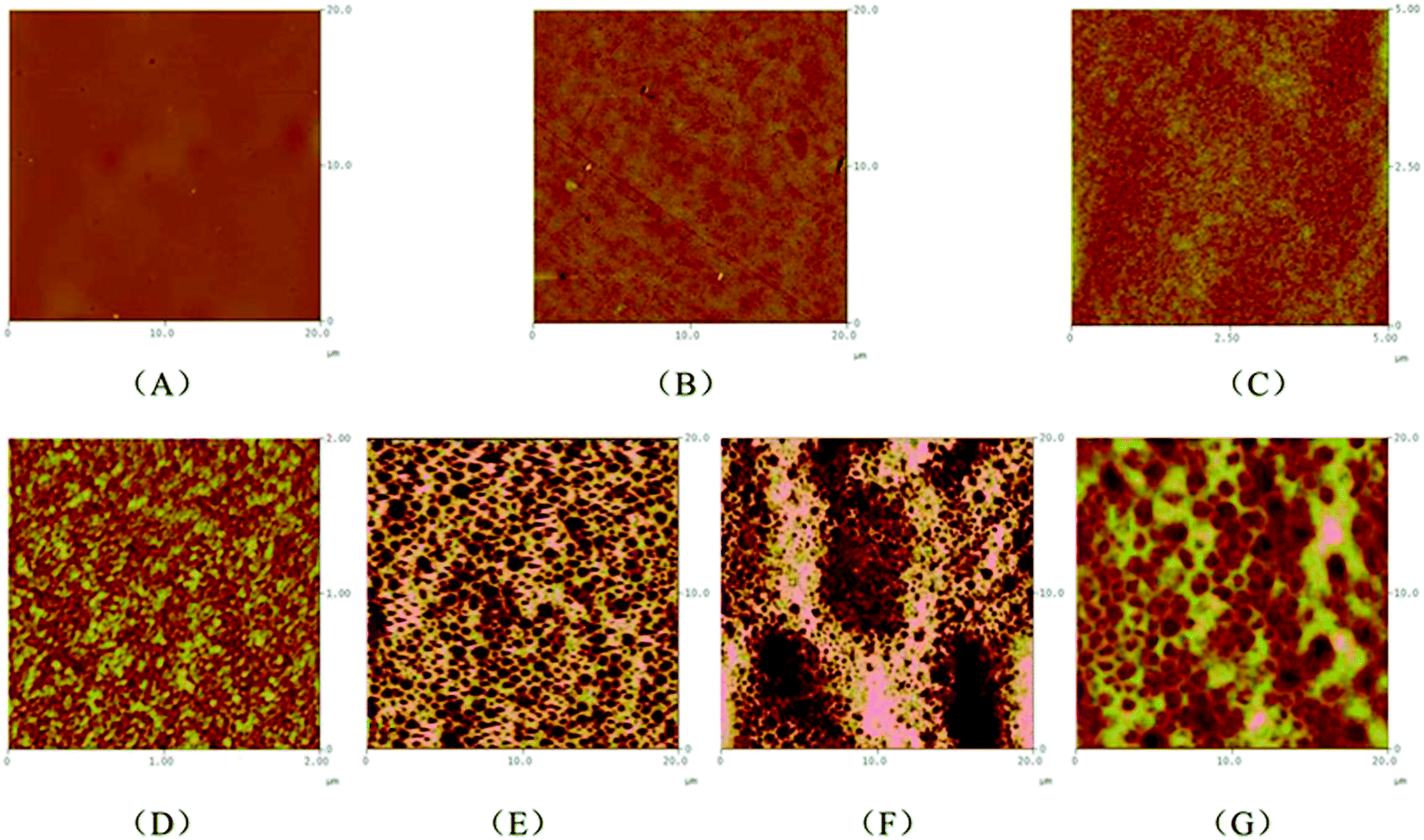 | ||
| Fig. 3 The two dimensional AFM surface images of PLLA (A), PLGL35G06 (B), PLGL35G12 (C), PLGL75G20 (D), PLGL55G20 (E), PLGL35G20 (F) and PLGL25G20 (G) films. | ||
As shown in Fig. 3(A), pure PLLA exhibited a homogeneous and continuous phase structure. For PLGL35Gy copolymers, Fig. 3(B), (C) and (F) show that an obvious microphase separation appeared with the addition of PEG. The microphase separation size of PLGL35G12 was approximately 10–20 nm, and the size of PLGL25G20 was approximately 400 nm. That is to say the degree of phase separation increased with the molecular weight of PEG. For PLGLxG20 copolymers, different degrees of phase separation can be seen from Fig. 3(D)–(G). It is noteworthy that AFM images show the presence of island structures on the surfaces of PLGL75G20 and PLGL55G20 films. The microphase separation size of PLGL75G20 was approximately 20 nm. With increasing PEG content, wormlike structures are observed on the surface of PLGL35G20 and PLGL25G20 films in the AFM images. The microphase separation size of PLGL25G20 increased to approximately 900 nm. We speculate that the dark phase was mainly amorphous PEG blocks, and the bright yellow phase was mainly amorphous PLLA blocks.
Considering both the AFM and DSC results, the physical state may be speculated to be as shown in Fig. 4. For a series of PLGLxG06 and PLGLxG12 copolymers, PLLA and PEG chains can intertwine to form a homogeneous “blend”. The two phases present good compatibility since the molecular weight and content of PEG are both low. Thus, the thermal behavior of PEG cannot be easily determined from DSC curves. However, by further increasing the molecular weight and content of PEG, phase separation occurred in PLGLxG20 copolymers. Few PEG chains intertwined with PLLA. Most of the PEG was in an amorphous state and was arranged alternately and orderly with PLLA chains.
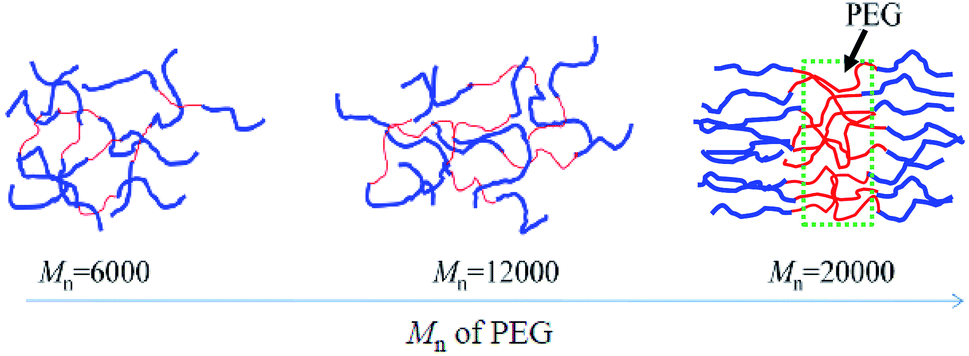 | ||
| Fig. 4 Schematic diagram of PLGLxGy block copolymers. | ||
Gas permeability analysis
For fresh food packaging, plastic films with excellent carbon dioxide (CO2) and oxygen (O2) permeability and permselectivity can quickly adjust gas proportions in sealed packaging to a low O2 and high CO2 atmosphere. This can reduce respiration by the product and prolong the shelf-life of fresh produce.Fig. 5–7 (and Tables S3 and S4†) show the CO2 and O2 permeability and permselectivity of PLLA and PLGLxGy copolymers at different temperatures. As shown in Fig. 5(A), the CO2 permeability coefficient (CP) of pure PLLA was approximately 1.99 × 10−8 cm3 m m−2 h−1 Pa−1 at 5 °C, and the CP value of PLLA slowly increased with increasing temperature to 40 °C. For all PLGLxG06 copolymers, the CP values differ minimally from neat PLLA at 5 °C. As the temperature increased from 5 to 40 °C, the CP values of the PLGLxG06 copolymers gradually increased, and became larger than the CP of PLLA at a high temperature. In addition, it should be noted that the greater the PEG content, the faster the CP value increased with increasing temperature. PLGL35G06 has the largest CP value (approximately 9.73 × 10−8 cm3 m m−2 h−1 Pa−1) of all PLGLxG06 copolymers at 40 °C.
 | ||
| Fig. 5 The CO2 and O2 permeability of PLLA and PLGLxG06 copolymer films at different temperatures. | ||
Fig. 5(B) shows the O2 permeability coefficient (OP) of PLLA and PLGLxG06 copolymers; the OP of all the films also showed an increasing tendency with increasing temperature. However, it should be noted that the OP increased slower than the CP values under the same conditions. Pure PLLA had an OP value of approximately 5.66 × 10−9 cm3 m m−2 h−1 Pa−1 at 5 °C, which slowly increased to 1.03 × 10−8 cm3 m m−2 h−1 Pa−1 at 25 °C. By further increasing the temperature, the increase of the OP of PLLA gradually slowed, and the OP of PLLA was approximately 1.74 × 10−8 cm3 m m−2 h−1 Pa−1 at 40 °C. With insertion of PEG blocks, the OP of PLGLxG06 copolymers decreased slightly, and all of the copolymers maintained slightly lower OP values than PLLA. The OP values for PLLA and PLGLxG06 copolymers showed minimal difference below 25 °C. At temperatures greater than 25 °C, PLGL75G06 and PLGL55G06 maintained OP values close to those for PLLA. However, the OP for PLGL35G06 increased faster than for pure PLLA above 35 °C, and PLGL35G06 has a slightly higher OP than pure PLLA. That may be because the PEG block has no special ability to dissolve and diffuse O2 molecules. Compared with PLLA, PEG alone presents a slight barrier to O2. At low temperatures, the presence of a small amount of PEG decreased the free volume between the molecular chains. Thus, gas permeation decreased slightly and led to slightly smaller OP values for the copolymers. At a temperature close to the melting point, the impact of flexible PEG blocks on the mobility of copolymers became large. Thus, the OP for PLGL35G06 increased faster and became even higher than for PLLA above 35 °C.
Fig. 5(C) shows the permselectivity ratio of CO2 to O2 (PC/O) for PLLA and PLGLxG06 copolymers. The PC/O value for pure PLLA was approximately 3.51 at 5 °C, and the PC/O decreased slowly to 2.79 at 40 °C. Therefore, pure PLLA has poor permselectivity for CO2 and O2 and cannot meet the demand of MAP packaging of fresh products with strong respiration.33 For PLGLxG06 copolymers, the PC/O values were approximately 3.48–6.08 and are more suitable for fruit and vegetable packaging.
As determined with pure PLLA, the PC/O of copolymers also exhibited a decreasing tendency, and decreased even more rapidly with increasing temperature. However, the PC/O increased with an increase of the PEG content in copolymers at the same temperature (also shown in Fig. S9(C)†). PLGL35G06 has the highest PC/O value, approximately 6.08 at 5 °C. There are two reasons for this phenomenon: on one hand, ethylene oxide (EO) groups in PEG blocks have a strong affinity for CO2. With decreasing lengths of PLLA at the ends, the relative content of PEG in copolymers increased. Thus, the EO group content was increased, which further improved the solubility and diffusion selectivity of CO2 of the copolymers at a certain temperature.34 On the other hand, although the gas permeability coefficient was improved, the solubility of CO2 in the films decreased with increasing temperatures. Generally, the increase of the gas permeability coefficient occurred faster than the decrease of the solubility. However, increased temperatures will finally cause a decrease of both solubility selectivity and diffusion selectivity for most polymer films,35,36 and the PLGL35G06 with the highest PEG content presented the best permselectivity at lower temperatures.
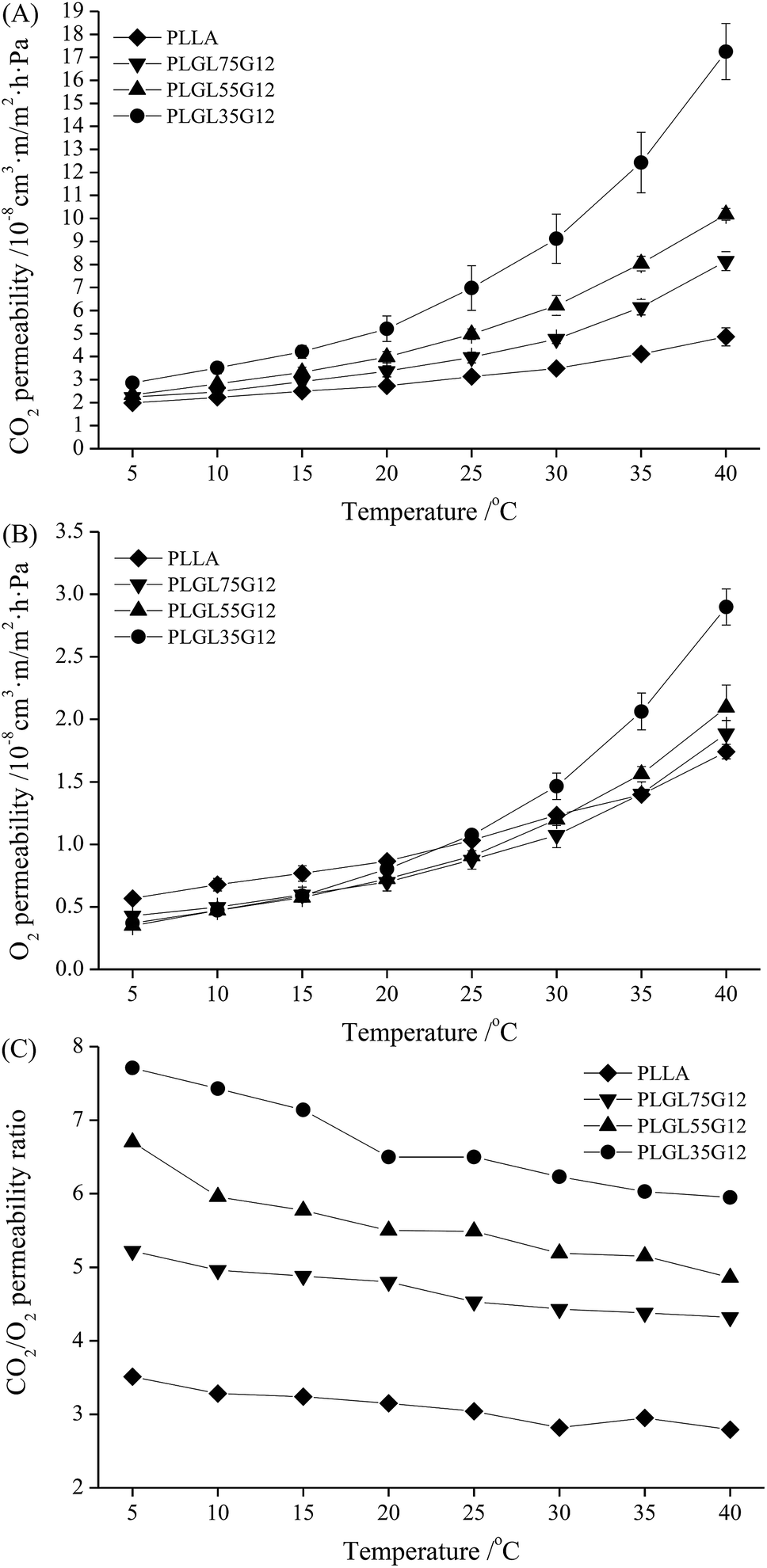 | ||
| Fig. 6 The CO2 and O2 permeability of PLLA and PLGLxG12 copolymer films at different temperatures. | ||
000, the CO2 permeability of the PLGLxG20 copolymers became much more different than the PLGLxG06 and PLGLxG12 copolymers under the same conditions. In particular, when the temperature was greater than 20 °C, the CO2 permeability of PLGLxG20 copolymers increased rapidly, and the difference became larger. However, the O2 permeability of PLGL75G20, PLGL55G20 and PLGL35G20 showed minimal difference below 25 °C. The O2 permeabilities of copolymers with higher PEG content increased more rapidly above 25 °C, and these three copolymers began to show obvious differences. PLGL25G20, which has the highest PEG content, presented a much higher O2 permeability than the other PLGLxG20 copolymers, and the OP reached 6.1 × 10−8 cm3 m m−2 d−1 Pa−1 at 40 °C.
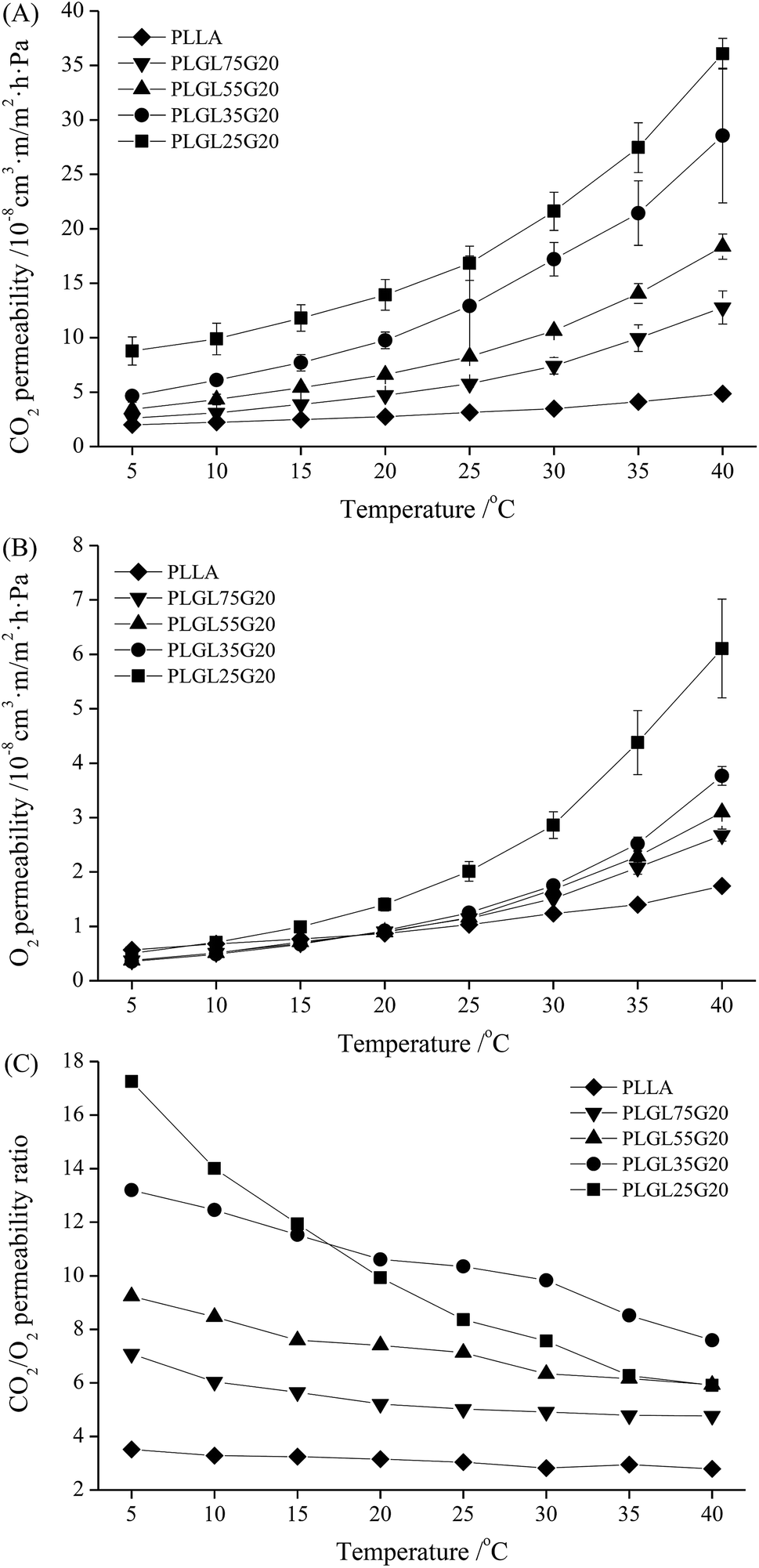 | ||
| Fig. 7 The CO2 and O2 permeability of PLLA and PLGLxG20 copolymer films at different temperatures. | ||
Fig. 7(C) shows the permselectivity for PLGLxG20. It can be seen that those copolymers exhibit a major difference in CO2/O2 selectivity. The PC/O values for PLGL75G20, PLGL55G20 and PLGL35G20 were approximately 4.77–13.20, and they decreased slowly with increasing temperature. It should be noted that PLGL25G20, which has the highest PEG content of all the PLGLxG20 copolymers, has a relatively high PC/O value of approximately 17.3 at 5 °C. Nevertheless, the PC/O was sharply decreased and was similar to the PC/O of PLGL55G20 at 40 °C. There are a number of explanations for this behavior: one is that the PLGLxG20 copolymer contained approximately 11.5–26.2% soft PEG blocks. The strong affinity for CO2 of PEG plays a leading role in the permselectivity of films at low temperature, and the copolymers with greater PEG content have a higher solubility and selectivity for CO2. However, as the temperature rises closer to the melting point of PEG, the mobility of copolymers with more PEG blocks was greatly increased. The soft PEG and hard PLLA segments in the molecular chains were more likely to aggregate. The increase of flexile PEG concentrations finally changed the phase balance in the PLGLxG20 copolymer system, which became unstable and various phase separations occurred in those copolymers. Both the permeability of CO2 and O2 of films were improved and eventually lead to a more rapid decrease of the PC/O values for PLGL25G20. In addition, the rigidity of PLGLxG20 membranes will decrease at high temperatures, allowing gases to pass through more easily. The affinity for CO2 of PEG was limited and was no longer a predominant factor for influencing the gas selectivity of copolymer membranes.37
Oxygen permeability of polymers at different relative humidity
A partial pressure difference test method cannot simulate a humid environment for fruit and vegetable storage, and a constant pressure test method cannot simulate a low-temperature environment. In the present study, an isostatic permeometer was used to determine the O2 permeability at different levels of humidity at 23 °C. Fig. 8 shows the variation of the CP values of PLLA and PLGLxG20 copolymers with a change of humidity.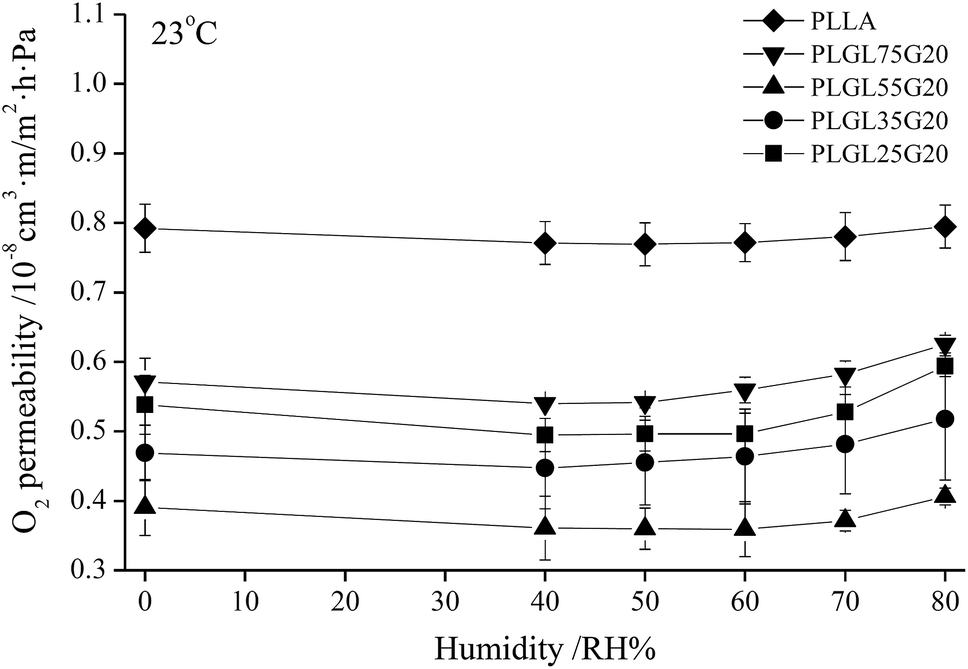 | ||
| Fig. 8 The variation of O2 permeability of PLLA and PLGLxG20 triblock copolymer films with different humidity. | ||
It can be seen that different test methods have certain effects on the CP values. However, since the OP represents the basic physical property of a polymer, the OP values obtained from both methods have no significant difference. As shown in Fig. 8, PLLA has the highest OP value. However, the OP was quite stable with a changing humidity, indicating that PLLA was insensitive to humidity. After copolymerizing with PEG, the OP values of PLGLxG20 copolymers were lower than for PLLA. Furthermore, the OP values decreased slightly at first, and then slowly increased with increasing humidity. It should be noted that the OP changed minimally with humidity below RH60%. By further increasing the humidity to above RH60%, the OP exhibited an obvious increasing trend with increasing relative humidity. Overall, PLLA and PLGLxG20 copolymers could maintain a stable O2 permeability under different levels of humidity. That is, those copolymers provide a fresh produce stable humidity environment because of their stable permeability.
Water vapor permeability
A suitable humidity environment is extremely important for food preservation. On one hand, fresh produce requires that the film provides a water vapor barrier to prevent a massive loss of water. On the other hand, excessive accumulation of water vapor results in condensation. The relative humidity in a sealed package greatly depends on the water vapor permeability of the packaging film. The water vapor transmission rate (WVTR) and water vapor permeability (WVP) are used to characterize the permeability of films. Table 2 and Fig. 9 present the water vapor permeability for PLLA and PLGLxGy copolymers.| Sample | Thickness/μm | Water vapor transmit rate/g m−2 d−1 | Water vapor permeability/10−7 g m m−2 h−1 Pa−1 |
|---|---|---|---|
| a The testing condition is 23 °C, RH 65%. | |||
| PLLA | 39.5 ± 1.1 | 396 ± 57 | 3.72 ± 0.47 |
| PLGL75G06 | 50.3 ± 4.2 | 336 ± 9 | 3.48 ± 0.15 |
| PLGL55G06 | 46.9 ± 1.9 | 349 ± 61 | 3.30 ± 0.19 |
| PLGL35G06 | 62.7 ± 0.4 | 297 ± 79 | 4.23 ± 0.27 |
| PLGL75G12 | 35.3 ± 1.1 | 433 ± 49 | 4.02 ± 0.72 |
| PLGL55G12 | 45.3 ± 3.3 | 375 ± 83 | 4.20 ± 0.55 |
| PLGL35G12 | 48.1 ± 1.6 | 482 ± 80 | 5.87 ± 0.49 |
| PLGL75G20 | 53.2 ± 1.1 | 485 ± 139 | 4.99 ± 0.70 |
| PLGL55G20 | 37.5 ± 2.2 | 634 ± 146 | 6.12 ± 0.71 |
| PLGL35G20 | 49.4 ± 4.1 | 974 ± 77 | 11.89 ± 0.47 |
| PLGL25G20 | 42.1 ± 2.1 | 1827 ± 36 | 17.51 ± 0.48 |
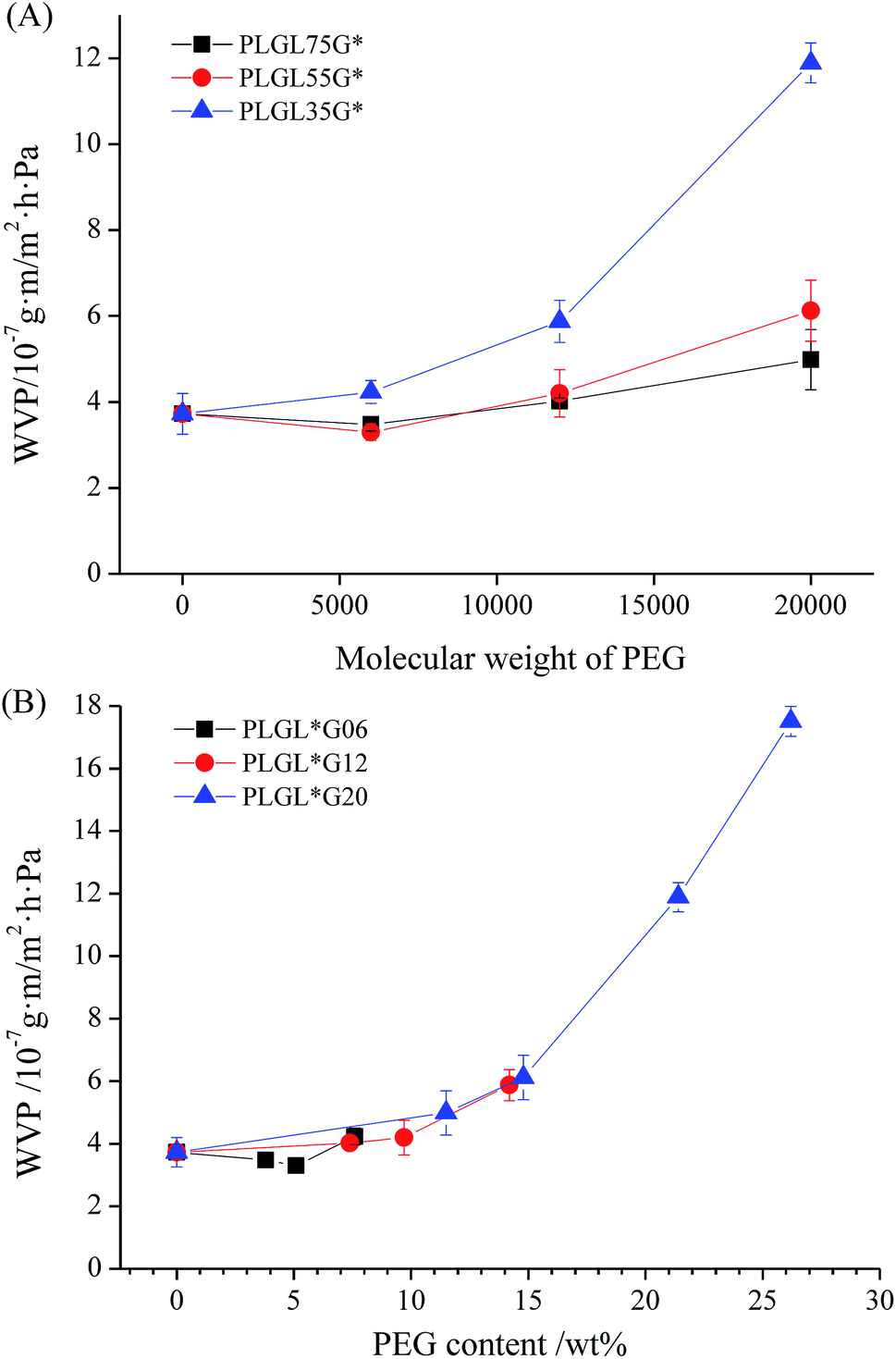 | ||
| Fig. 9 The variation of WVP of PLLA and PLGLxGy triblock copolymer films with different molecular weight of PEG. | ||
Fig. 9(A) summarizes the variation of the WVP value with the PEG content and molecular weight. It can be seen that the WVP values show a dependence on both the content and the molecular weight of the PEG. For PLGL35Gy and PLGL55Gy copolymers with certain PLLA lengths, the WVP values decreased slightly at first, and then increased with increasing PEG molecular weights. In the case of PLGL75Gy copolymers, the WVP values showed an obvious increasing trend with increasing PEG molecular weight from the beginning. For copolymers with certain PEG molecular weights, the shorter the PLLA length, the higher was the WVP value. Furthermore, copolymers with higher molecular weight PEG exhibited a greater increase of the permeability. As shown in Table 2, PLGL25G20 has the highest WVTR of approximately 1827 g m−2 d−1 due to the high molecular weight PEG and the highest PEG content of approximately 26.2%. The WVP of the most widely used polyethylene (PE) film is approximately 5.9 g m−2 d−1.38 When PE is used for packaging of fresh fruits and vegetables, condensation can occur easily in the package and cause local corruption, and perforation is the most commonly used method to avoid condensation. However, perforation causes the film to lose its control of the gas proportions in sealed packaging. For PLGLxGy copolymers, the existence of a sufficient number of hydrophilic PEG blocks can adjust the water vapor permeability of films and may be able to prevent the condensation in fresh product packagings.
It can be seen from Fig. 9(B) and Table 2 that the WVTR of neat PLLA was approximately 396 g m−2 d−1, and the WVP was 3.72 × 10−7 g m m−2 h−1 Pa−1. For PLGLxGy copolymers, compared with neat PLLA, the water permeability of PLGL75G06 and PLGL55G06 were slightly decreased. The WVP of PLGL55G06 was 3.30 × 10−7 g m m−2 h−1 Pa−1. However, the WVP of PLGL35G06 suddenly increased to 4.23 × 10−7 g m m−2 h−1 Pa−1. That is, the PLGL35G06 exhibited better water vapor permeability than neat PLLA because, despite the fact that PEG has good water vapor hydrophilicity, the molecular weight of the PEG was 6000 and the content of PEG in PLGL75G06 and PLGL55G06 was less than 6%. As shown in Fig. 3 and 4, low molecular weight PEG had good PLLA compatibility and formed a relatively homogeneous polymer. The H2O permeability channels in amorphous PLLA were filled with PEG chains and the hydrophilicity of the PEG was relatively weak. Thus PLGL75G06 and PLGL55G06 possess lower water vapor permeabilities.
In the case of PLGLxG12 copolymers, the WVP of PLGL75G12 was higher than for pure PLLA and was approximately 4.02 × 10−7 g m m−2 h−1 Pa−1. As the PLLA chains decreased in PLGL35G12, the PEG content increased, and the WVP increased to 5.87 × 10−7 g m m−2 h−1 Pa−1. As the molecular weight of the PEG increased to 20000, the WVP value for PLGL75G20 increased to 4.99 × 10−7 g m m−2 h−1 Pa−1. The WVP for PLGL35G20 increased further to 1.19 × 10−6 g m m−2 h−1 Pa−1 with increasing PEG content. The WVP of PLGL25G20 reached 1.75 × 10−6 g m m−2 h−1 Pa−1.
This is because as the content of PEG increased further in the copolymers, the hydrophilic PEG chains aggregated easily. Gas permeability channels can be formed by the PEG. H2O molecules were easily adsorbed onto the surface of the membrane and diffused into the membrane, and the DSC and WAXD results indicated that most of the PLLA chains were in an amorphous state. The presence of large amounts of high molecular weight PEG results in microphase separation. In consideration of the above factors, the water vapor permeability of PLGLxG12 and PLGLxG20 copolymers increased with increasing PEG content.
Conclusions
The effects of inserting different molecular weights and percentages of PEG blocks on the microstructure and permeation properties for CO2, O2 and H2O of triblock copolymers were determined. It was found that certain additions of PEG could results in composition-dependent microphase separation structures with both PLLA and PEG blocks in an amorphous state. PEG blocks with high CO2 affinity in such microphase separation structures provide additional permeability channels, which allow CO2 to permeate more readily. The CO2 permeability and CO2/O2 selectivity of copolymers depend on both the molecular weight and the weight of the PEG blocks. Copolymers with higher molecular weight PEGs provide better CO2 permeability at higher temperatures, but a better CO2/O2 perm-selectivity at lower temperatures. The highest CO2/O2 permselective ratio was obtained at approximately 17.3 at 5 °C. Hydrophilic PEG also improved the water vapor permeability of copolymers. Such biodegradable triblock copolymers have great potential for use as fresh product packaging membranes due to their unusually high CO2/O2 permselectivity and water vapor permeability.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21564012), Double First-class Disciplines Innovation Team Building and Talent Cultivation Project of Inner Mongolia Agriculture University (2018) and The Scrolling Support of Prairie Talents Project (2017).References
- R. K. Prange and J. M. Delong, Controlled-atmosphere related disorders of fruits and vegetables, Stewart Postharvest Review, 2006, 2, 1–10 Search PubMed.
- S. Ben-Yehoshua, S. P. Burg and R. Young, Resistance of citrus fruit to mass transport of water vapor and other gases, Plant Physiol., 1985, 79, 1048–1053 CrossRef CAS PubMed.
- S. Mangaraj, T. K. Goswami and P. V. Mahajan, Applications of Plastic Films for Modified Atmosphere Packaging of Fruits and Vegetables: A Review, Food Eng. Rev., 2009, 1, 133–158 CrossRef CAS.
- R. E. Hardenburg, Effect of in-package environment on keeping quality of fruits and vegetables, HortScience, 1971, 6, 194–202 Search PubMed.
- P. V. Mahajan, F. A. R. Oliveira, J. C. Montanez and J. Frias, Development of user-friendly software for design of modified atmosphere packaging for fresh and fresh-cut produce, Innovative Food Sci. Emerging Technol., 2007, 8, 84–92 CrossRef.
- X. Yun, Y. Wang, M. Li, Y. Jin and T. Dong, Application of permselective poly(ε-caprolactone) film for equilibrium-modified atmosphere packaging of strawberry in cold storage, J. Food Process. Preserv., 2017, 41, e13247 CrossRef.
- F. C. Pavia, V. L. Carrubba and V. Brucato, Polymeric scaffolds based on blends of poly-L-lactic acid (PLLA) with poly-D-L-lactic acid (PLA) prepared via thermally induced phase separation (TIPS): demixing conditions and morphology, Polym. Bull., 2013, 70, 563–578 CrossRef.
- J. R. Dorgan, H. Lehermeier and M. Mang, Thermal and Rheological Properties of Commercial-Grade Poly(Lactic Acid)s, J. Polym. Environ., 2000, 8, 1–9 CrossRef.
- M. Jamshidian, E. A. Tehrany, F. Cleymand, L. Stéphane, T. Falher and D. Stéphane, Effects of synthetic phenolic antioxidants on physical, structural, mechanical and barrier properties of poly lactic acid film, Carbohydr. Polym., 2012, 87, 1763–1773 CrossRef CAS.
- T. Dong, S. Song, M. Liang, Y. Wang, X. Qi, Y. Zhang and Y. Jin, Gas Permeability and Permselectivity of Poly(L-Lactic Acid)/SiOx Film and Its Application in Equilibrium-Modified Atmosphere Packaging for Chilled Meat: Modified PLLA Film For pork EMA, J. Food Sci., 2016, 82, 97–107 CrossRef PubMed.
- H. J. Lehermeier, J. R. Dorgan and J. D. Way, Gas permeation properties of poly(lactic acid), J. Membr. Sci., 2001, 190, 243–251 CrossRef CAS.
- T. Dong, Z. Yu, J. Wu, Z. Zhao, X. Yun, Y. Wang, Y. Jin and J. Yang, Thermal and barrier properties of stretched and annealed polylactide films, Polym. Sci., Ser. A, 2015, 57, 738–746 CrossRef CAS.
- L. Singh, M. F. Siddiqui, A. Ahmad, A. R. Mohd Hasbi, M. Sakinah and Z. A. Wahid, Application of polyethylene glycol immobilized Clostridium sp. LS2 for continuous hydrogen production from palm oil mill effluent in upflow anaerobic sludge blanket reactor, Biochem. Eng. J., 2013, 70, 158–165 CrossRef CAS.
- S. K. Spear, J. G. Huddleston and R. D. Rogers, Polyethylene glycol and solutions of polyethylene glycol as green reaction media, Green Chem., 2005, 7, 64–82 RSC.
- G. P. Tang, J. M. Zeng, S. J. Gao, Y. X. Ma, L. Shi, Y. Li, H. P. Too and S. Wang, Polyethylene glycol modified polyethylenimine for improved CNS gene transfer: effects of PEGylation extent, Biomaterials, 2003, 24, 2351–2362 CrossRef CAS PubMed.
- V. S. Murali, Rapid detection of polyethylene glycol sonolysis upon functionalization of carbon nanomaterials, Exp. Biol. Med., 2015, 240, 1147–1151 CrossRef CAS PubMed.
- K. I. Okamoto, M. Fuji, S. Okamyo, H. Suzuki, K. Tanaka and H. Kita, Gas permeation properties of poly(ether imide) segmented copolymers, Macromolecules, 1995, 28, 6950–6956 CrossRef CAS.
- H. P. Schlanger and L. Paterson, Gas permeabilities of cellulose nitrate/poly(ethylene glycol) blend membranes, J. Appl. Polym. Sci., 2010, 27, 2387–2393 Search PubMed.
- Y. Yin, L. Yang, M. Yoshino, M. Yoshino, J. Fang, K. Tanaka, H. Kita and K. Okamoto, Synthesis and Gas Permeation Properties of Star-like Poly(ethylene oxide)s Using Hyperbranched Polyimide as Central Core, Polym. J., 2004, 36, 294–302 CrossRef CAS.
- C. Charmette, J. Sanchez, P. Gramain and A. Rudatsikira, Gas transport properties of poly(ethylene oxide-co-epichlorohydrin) membranes, J. Membr. Sci., 2004, 230, 161–169 CrossRef CAS.
- P. Cláudio, J. R. Ribeiro and B. D. Freeman, Solubility and partial molar volume of carbon dioxide and ethane in crosslinked poly(ethylene oxide) copolymer, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 456–468 CrossRef.
- H. Lin and B. D. Freeman, Gas solubility, diffusivity and permeability in poly(ethylene oxide), J. Membr. Sci., 2004, 239, 105–117 CrossRef CAS.
- L. Kwisnek, J. Goetz, K. P. Meyers, S. R. Heinz, J. S. Wiggins, S. Nazarenko, L. Kwisnek, J. Goetz and K. P. Meyers, PEG Containing Thiol-Ene Network Membranes for CO2 Separation: Effect of Cross-Linking on Thermal, Mechanical, and Gas Transport Properties, Macromolecules, 2014, 47, 3243–3253 CrossRef CAS.
- N. P. Patel and R. J. Spontak, Gas-Transport and Thermal Properties of a Microphase-Ordered Poly(styrene-b-ethylene oxide-b-styrene) Triblock Copolymer and Its Blends with Poly(ethylene glycol), Macromolecules, 2004, 37, 2829–2838 CrossRef CAS.
- M. Sadeghi, M. P. Chenar, M. Rahimian, S. Moradi and A. H. S. Dehaghani, Gas permeation properties of polyvinylchloride/polyethyleneglycol blend membranes, J. Appl. Polym. Sci., 2008, 110, 1093–1098 CrossRef CAS.
- M. A. Semsarzadeh and B. Ghalei, Characterization and gas permeability of polyurethane and polyvinyl acetate blend membranes with polyethylene oxide-polypropylene oxide block copolymer, J. Membr. Sci., 2012, s401–402, 97–108 CrossRef.
- Q. Yuan, H. Zhao, Y. Cao, X. Ding and M. Zhou, Poly(n,n-dimethylaminoethyl methacrylate)-poly(ethylene oxide) copolymer membranes for selective separation of CO2, J. Membr. Sci., 2008, 310, 365–373 CrossRef.
- T. Dong, X. Yun, M. Li, W. Sun, Y. Duan and Y. Jin, Biodegradable high oxygen barrier membrane for chilled meat packaging, J. Appl. Polym. Sci., 2015, 132(16) DOI:10.1002/app.41871.
- W. Channuan, J. Siripitayananon, R. Molloy, M. Sriyai, F. J. Davis and G. R. Mitchell, The structure of crystallisable copolymers of L-lactide, ε-caprolactone and glycolide, Polymer, 2005, 46, 6411–6428 CrossRef CAS.
- J. Zhang, K. Tashiro, H. Tsuji and A. J. Domb, Disorder-to-Order Phase Transition and Multiple Melting Behavior of Poly(l-lactide) Investigated by Simultaneous Measurements of WAXD and DSC, Macromolecules, 2008, 41, 1352–1357 CrossRef CAS.
- J. Li, Y. He and Y. Inoue, Thermal and mechanical properties of biodegradable blends of poly(L-lactic acid) and lignin, Polym. Int., 2003, 52, 949–955 CrossRef CAS.
- X. Yun, X. Li, W. Sun, Y. Jin and T. Dong, Fast Crystallization and Toughening of Poly(L-lactic acid) by Incorporating with Poly(ethylene glycol) as a Middle Block Chain, Polym. Sci., Ser. A, 2018, 60, 141–155 CrossRef CAS.
- Sandhya, Modified atmosphere packaging of fresh produce: Current status and future needs, LWT–Food Sci. Technol., 2010, 43, 381–392 CrossRef CAS.
- J. Xia, S. Liu and T. S. Chung, Effect of end groups and grafting on the CO2 separation performance of poly(ethylene glycol) based membranes, Macromolecules, 2011, 44, 7727–7736 CrossRef CAS.
- S. R. Reijerkerk, A. Arun, R. J. Gaymans, K. Nijmeijer and M. Wessling, Tuning of mass transport properties of multi-block copolymers for CO2 capture applications, J. Membr. Sci., 2010, 359, 54–63 CrossRef CAS.
- C. Ma and W. J. Koros, High-performance ester-crosslinked hollow fiber membranes for natural gas separations, J. Membr. Sci., 2013, 428, 251–259 CrossRef CAS.
- M. M. Talakesh, M. Sadeghi, M. P. Chenar and A. Khosravi, Gas separation properties of poly(ethylene glycol)/poly(tetramethylene glycol) based polyurethane membranes, J. Membr. Sci., 2012, s415–416, 469–477 CrossRef.
- A. Grumezescu and M. Nutraceuticals, Nanotechnol. Agri-Food Ind., 2016, 7, p147–174 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectroscopy, differential scanning calorimetry, Fourier transform infrared spectrum, X-ray scattering, CO2 and O2 permeability, water vapor permeability, Scanning Electron Microscopy (SEM) (of all copolymers except as mentioned in the main text). See DOI: 10.1039/c9ra00656g |
| This journal is © The Royal Society of Chemistry 2019 |