
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and biological activity evaluation of benzoate compounds as local anesthetics
Huiying Zouab,
Guangying Chen*ab and
Shiyang Zhou *ab
*ab
aCollege of Chemistry and Chemical Engineering, Hainan Normal University, Haikou 571158, China. E-mail: chgying123@163.com; zhoushiyang520@126.com
bKey Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education, Hainan Normal University, Haikou 571158, China
First published on 26th February 2019
Abstract
Tetracaine and pramocaine were used as the lead compounds to design benzoate compounds. The combination principle was used to design the target molecule, and the target molecule was modified by bioisostere formation and modification with alkyl groups. In this research, a total of 16 compounds were designed and synthesized. In the process of synthesis, we selected a route with high total yields, mild conditions and simple operation. Three steps were used in the synthesis of the new target compounds, namely, alkylation, esterification and alkylation. The newly designed target compounds were evaluated via surface anesthesia, infiltration anesthesia, block anesthesia and acute toxicity tests. The results of biological activity experiments showed that compounds 4d, 4g, 4j, 4k, 4n, and 4o had a good local anesthetic effect, and the results of acute toxicity tests showed that the target compounds had low toxicity.
1. Introduction
An anesthetic is a drug that can act on the nervous system and inhibit it and is thus a drug that produces anesthesia.1,2 Local anesthetics act on nerve endings and nerve trunks and can reversibly block the conduction of nerve impulses. This results in a loss of local sensation without affecting consciousness, which is convenient for local surgery and treatment.3–5 Local anesthetics are commonly used in stomatology, ophthalmology, gynecology and minor surgical operations to temporarily relieve pain.6–9 Local anesthetics bind to specific parts of the sodium ion (Na+) channel on the nerve membrane and affect the membrane potential by reducing the passage of sodium ions through the sodium ion channel, and thereby block the conduction of nerve impulses. Local anesthetics can reduce the excitability of the membrane and have no effect on the resting potential.10–14The first local anesthetic that was used in clinical practice comprised active ingredients derived from coca leaves from South America. As early as the 16th century, Peruvians were known to chew coca leaves to relieve pain. In the 1860s Niemann extracted crystals of an alkaloid from coca leaves and named it as cocaine.15–17 In 1868 Maiz was the first to suggest that cocaine might be a local anesthetic, and in 1884 Koller (an Austrian ophthalmologist) was the first to use cocaine for clinical surgery, which led to the discovery of new anesthesia techniques and the rapid development of local anesthetic drugs.18–21 However, in the process of use, cocaine was found to cause addiction and some other toxic effects, such as allergic reactivity, tissue stimulation and instability in aqueous solutions. Hence, the clinical application of cocaine was limited. As a result, structural studies and modifications of cocaine have been carried out in the search for better local anesthetics. From studies of the structure of cocaine, it was gradually realized that the removal of the methyl carboxylate group in the structure of cocaine could eliminate addiction. The local anesthetic effect of ethyl-4-aminobenzenecarboxylate (benzocaine) was first confirmed in 1890, and procaine was successfully developed in 1904. The discovery of procaine made people realize the important role of the 4-aminobenzoate structure in local anesthetic drugs and started the study of the 4-aminobenzoate structure22–24 (tetracaine) and the development of local anesthetic drugs with amide25,26 (lidocaine), aminoketone27 (dyclonine) and aminoether28,29 (pramocaine) structures (Fig. 1).
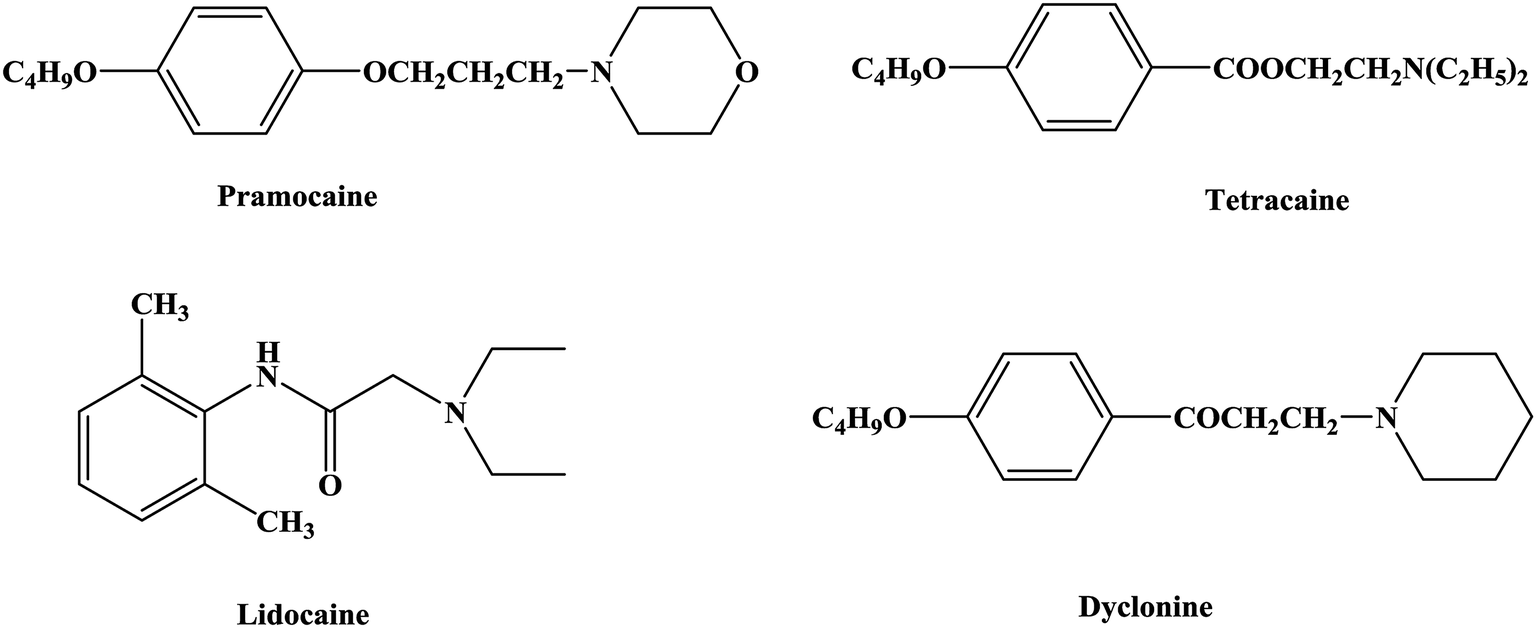 | ||
| Fig. 1 Chemical structures of local anesthetics. | ||
In this research, tetracaine and pramocaine were used as the lead compounds to design the target molecules. The lead compounds were integrated into an organic molecule by the combination principle. Then the integrated organic molecule was modified by bioisostere formation and modification with alkyl groups (Fig. 2). A series of novel target compounds with local anesthetic effects were designed. In terms of the structure–activity relationship (SAR) of the drugs, the designed compounds were composed of three basic skeleton structures, namely, a lipophilic section, a link section and a hydrophilic section (Fig. 3). According to the SAR, the target compounds should have a local anesthetic effect. Structural modification of the integrated molecules was performed in order to ensure that the onset time was short, the duration of action was long and the toxic effect was slight. The newly designed benzoate compounds, with 4-aminobenzoic acid (1a) and 4-hydroxybenzoic acid (1b) as starting materials, were synthesized by the three steps of alkylation, esterification and alkylation to give the target compounds (Scheme 1). The synthetic route was characterized by simple operation, high total yields, and mild reaction conditions.
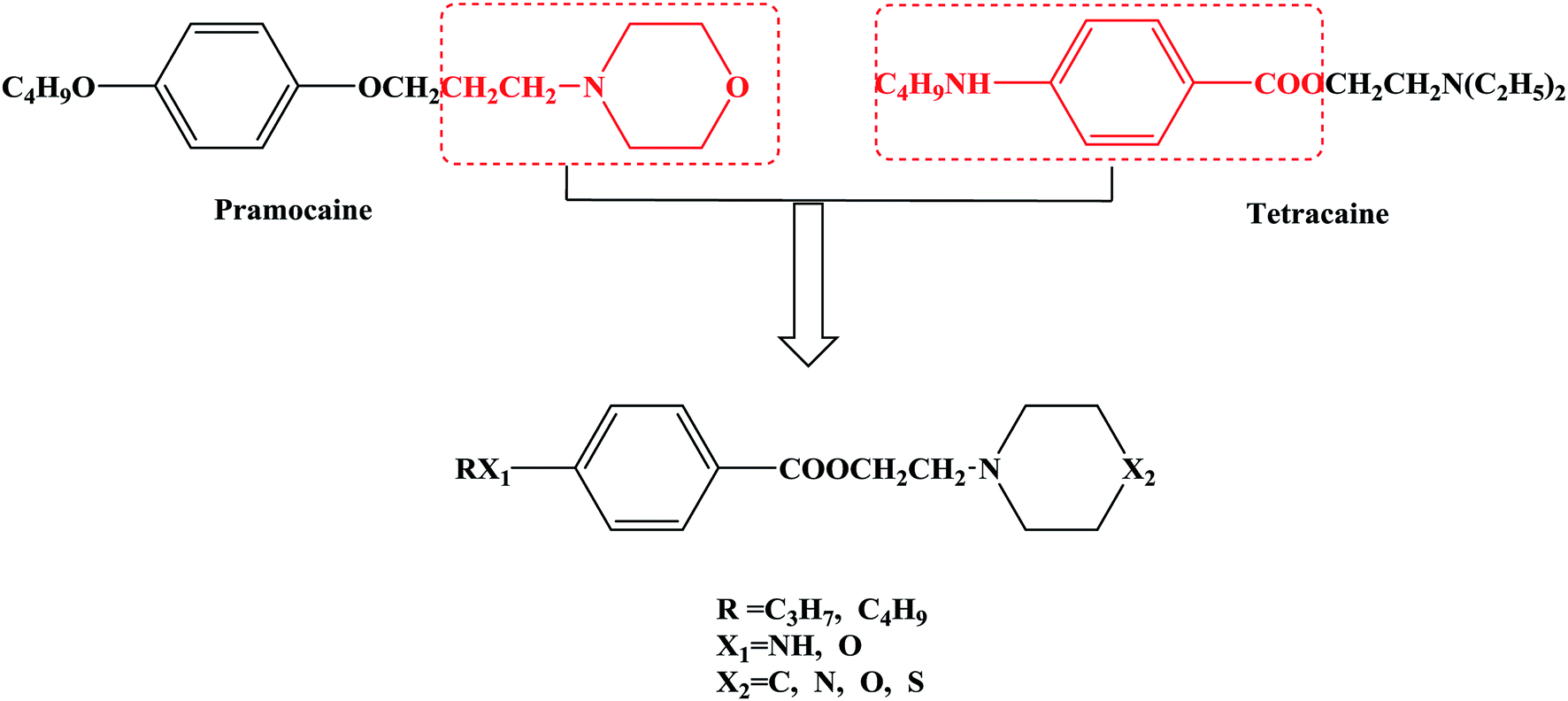 | ||
| Fig. 2 Design of target compounds. | ||
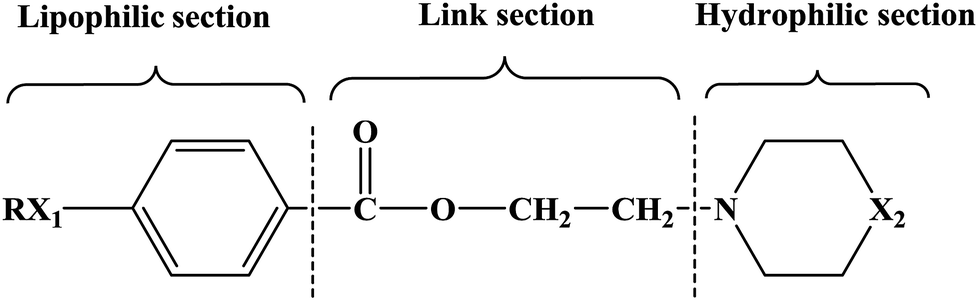 | ||
| Fig. 3 Basic skeleton structures of target compounds. | ||
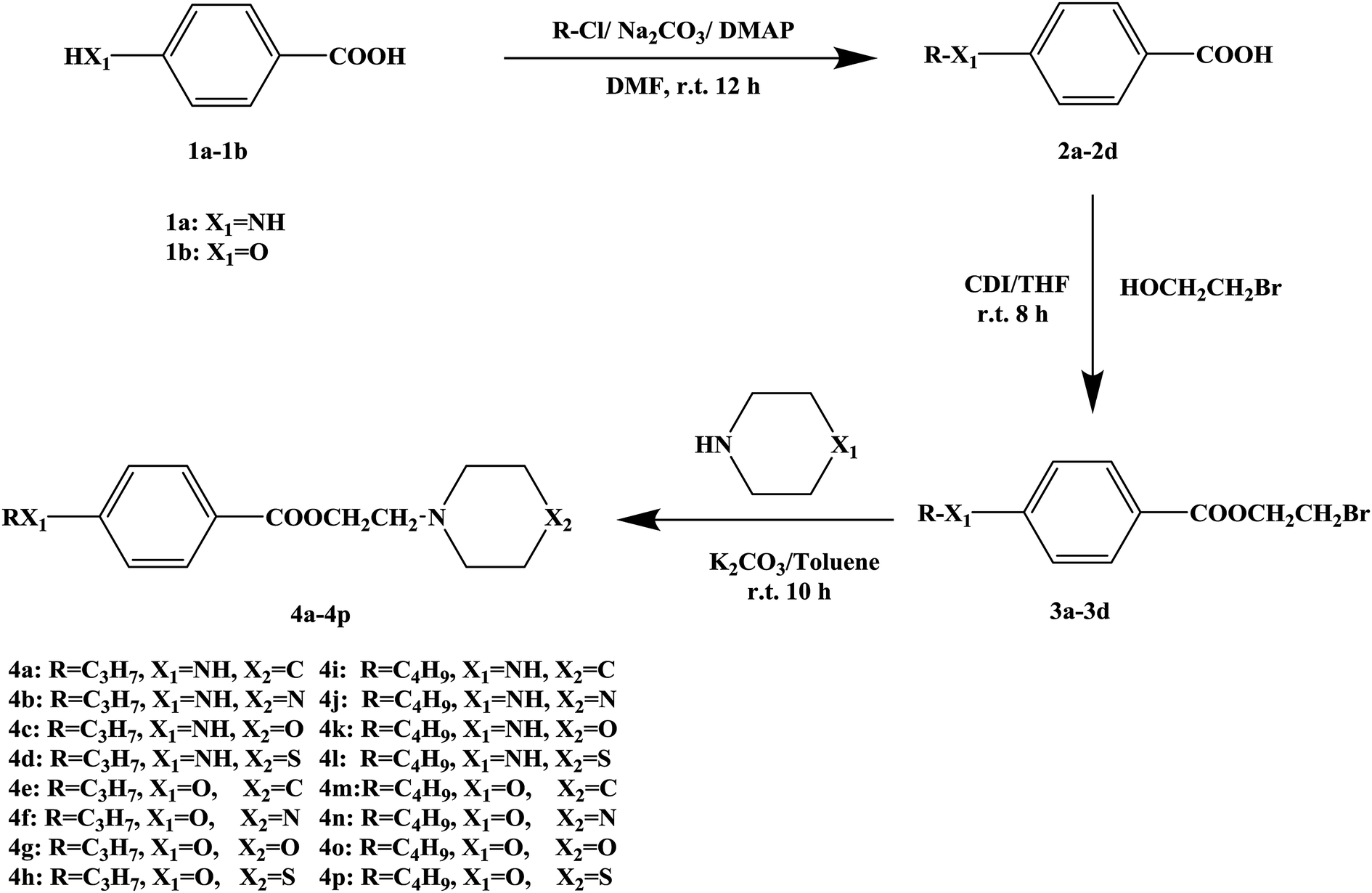 | ||
| Scheme 1 Synthetic route to target compounds. | ||
2. Materials and methods
2.1. Chemistry section
Chemistry materials and general methods. The reagents (chemicals) were purchased and used without further purification. Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker AMX-400 spectrometer (IS as TMS). Mass spectroscopy was performed with an Agilent 6460 system. HPLC analysis of all final biologically tested compounds was carried out using an Agilent 1260 series HPLC system. The purity was determined by reversed-phase HPLC and was ≥99% for all biologically tested compounds.2-(Piperidin-1-yl)ethyl-4-(propylamino)benzoate (4a). White crystals, yield 82.1%, mp 141–143 °C; 1H NMR (300 MHz, DMSO) δ: 0.90 (3H, t, J = 7.1 Hz, –CH3), 1.38 (2H, dtt, J = 10.8, 3.3, 2.4 Hz, –CH2–), 1.50 (4H, ddd, J = 10.8, 3.3, 2.4 Hz, –CH2–), 1.57 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 2.42 (4H, ddd, J = 12.5, 2.9, 2.6 Hz, –CH2–), 3.15 (2H, t, J = 7.1 Hz, –CH2–), 3.36 (2H, t, J = 7.4 Hz, –CH2–), 4.29 (2H, t, J = 7.1 Hz, –CH2–), 6.79 (2H, ddd, J = 8.6, 1.2, 0.5 Hz, Ph-H), 7.69 (2H, ddd, J = 8.6, 1.7, 0.5 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 11.4, 23.2, 24.6, 25.9, 47.1, 54.6, 56.7, 63.1, 112.1, 118.4, 130.6, 151.8, 165.9; HR-ESI-MS m/z: calcd for C17H26N2O2 {[M + H]+} 290.4073, found 290.1998; anal. calcd for C17H26N2O2: C, 70.31; H, 9.02; N, 9.65; O, 11.02; found: C, 70.33; H, 9.01; N, 9.64; O, 11.02%.
2-(Piperazin-1-yl)ethyl-4-(propylamino)benzoate (4b). White crystals, yield 86.3%, mp 180–182 °C; 1H NMR (300 MHz, DMSO) δ: 0.90 (3H, t, J = 7.1 Hz, –CH3), 1.59 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 2.33 (4H, ddd, J = 14.1, 10.2, 2.5 Hz, –CH2–), 2.66 (4H, ddd, J = 10.2, 9.4, 2.5 Hz, –CH2–), 3.16 (4H, t, J = 7.1 Hz, –CH2–), 3.34 (2H, t, J = 7.4 Hz, –CH2–), 4.31 (2H, t, J = 7.1 Hz, –CH2–), 6.77 (2H, ddd, J = 8.6, 1.2, 0.5 Hz, Ph-H), 7.69 (2H, ddd, J = 8.6, 1.7, 0.5 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 11.4, 23.2, 46.3, 47.3, 54.3, 57.4, 63.3, 112.0, 118.4, 130.6, 151.8, 165.8; HR-ESI-MS m/z: calcd for C16H25N3O2 {[M + H]+} 291.3954, found 291.1945; anal. calcd for C16H25N3O2: C, 65.95; H, 8.65; N, 14.42; O, 10.98; found: C, 65.96; H, 8.64; N, 14.43; O, 10.99%.
2-Morpholinoethyl-4-(propylamino)benzoate (4c). White crystals, yield 90.4%, mp 140–142 °C; 1H NMR (300 MHz, DMSO) δ: 0.90 (3H, t, J = 7.1 Hz, –CH3), 1.57 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 2.50 (4H, ddd, J = 9.4, 3.1, 2.5 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.34 (2H, t, J = 7.4 Hz, –CH2–), 3.56 (4H, ddd, J = 11.8, 10.2, 2.5 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 6.77 (2H, ddd, J = 8.6, 1.2, 0.5 Hz, Ph-H), 7.67 (2H, ddd, J = 8.6, 1.7, 0.5 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 11.4, 23.2, 47.3, 54.4, 55.7, 63.3, 66.7, 112.0, 118.4, 130.6, 151.8, 165.8; HR-ESI-MS m/z: calcd for C16H24N2O3 {[M + H]+} 292.3793, found 292.1789; anal. calcd for C16H24N2O3: C, 65.73; H, 8.27; N, 9.58; O, 16.42; found: C, 65.74; H, 8.26; N, 9.59; O, 16.41%.
2-Thiomorpholinoethyl-4-(propylamino)benzoate (4d). White crystals, yield 85.3%, mp 172–174 °C; 1H NMR (300 MHz, DMSO) δ: 0.90 (3H, t, J = 7.1 Hz, –CH3), 1.57 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 2.55 (4H, ddd, J = 10.9, 10.2, 2.3 Hz, –CH2–), 2.74 (4H, ddd, J = 8.7, 3.4, 2.3 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.34 (2H, t, J = 7.4 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 6.77 (2H, ddd, J = 8.6, 1.2, 0.5 Hz, Ph-H), 7.67 (2H, ddd, J = 8.6, 1.7, 0.5 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 11.4, 23.2, 28.4, 47.3, 53.7, 60.4, 63.3, 112.0, 118.4, 130.6, 151.8, 165.8; HR-ESI-MS m/z: calcd for C16H24N2O2S {[M + H]+} 308.4403, found 308.1554; anal. calcd for C16H24N2O2S: C, 62.31; H, 7.84; N, 9.08; O, 10.37; S, 10.39; found: C, 62.33; H, 7.83; N, 9.07; O, 10.38; S, 10.38%.
2-(Piperidin-1-yl)ethyl-4-propoxybenzoate (4e). White crystals, yield 80.5%, mp 108–110 °C; 1H NMR (300 MHz, DMSO) δ: 0.98 (3H, t, J = 7.5 Hz, –CH3), 1.36 (2H, dtt, J = 10.8, 3.3, 2.4 Hz, –CH2–), 1.48 (4H, ddd, J = 10.8, 3.3, 2.6 Hz, –CH2–), 1.74 (2H, t, J = 7.5 Hz, –CH2–), 2.43 (4H, ddd, J = 12.5, 2.9, 2.6 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 4.02 (2H, t, J = 7.5 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 10.5, 22.5, 24.4, 25.8, 54.4, 56.7, 63.3, 69.4, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C17H25NO3 {[M + H]+} 291.3912, found 291.1836; anal. calcd for C17H25NO3: C, 70.07; H, 8.65; N, 4.81; O, 16.47; found: C, 70.06; H, 8.66; N, 4.82; O, 16.46%.
2-(Piperazin-1-yl)ethyl-4-propoxybenzoate (4f). White crystals, yield 84.2%, mp 170–172 °C; 1H NMR (300 MHz, DMSO) δ: 0.98 (3H, t, J = 7.5 Hz, –CH3), 1.74 (2H, h, J = 7.5 Hz), 2.35 (4H, ddd, J = 14.1, 10.2, 2.5 Hz, –CH2–), 2.64 (4H, ddd, J = 10.2, 9.4, 2.5 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 4.02 (2H, t, J = 7.5 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 10.5, 22.5, 46.3, 54.3, 57.4, 63.3, 69.4, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C16H24N2O3 {[M + H]+} 292.3793, found 292.1788; anal. calcd for C16H24N2O3: C, 65.73; H, 8.27; N, 9.58; O, 16.42; found: C, 65.74; H, 8.28; N, 9.57; O, 16.41%.
2-Morpholinoethyl-4-propoxybenzoate (4g). White crystals, yield 88.7%, mp 113–115 °C; 1H NMR (300 MHz, DMSO) δ: 0.98 (3H, t, J = 7.5 Hz, –CH3), 1.74 (2H, t, J = 7.5 Hz, –CH2–), 2.50 (4H, ddd, J = 9.4, 3.1, 2.5 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.56 (4H, ddd, J = 11.8, 10.2, 2.5 Hz, –CH2–), 4.02 (2H, t, J = 7.5 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 10.5, 22.5, 54.4, 55.7, 63.3, 66.7, 69.4, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C16H23NO4 {[M + H]+} 293.3631, found 293.1629; anal. calcd for C16H23NO4: C, 65.51; H, 7.90; N, 4.77; O, 21.81; found: C, 65.52; H, 7.91; N, 4.76; O, 21.80%.
2-Thiomorpholinoethyl-4-propoxybenzoate (4h). White crystals, yield 83.1%, mp 150–152 °C; 1H NMR (300 MHz, DMSO) δ: 0.98 (3H, t, J = 7.5 Hz, –CH3), 1.74 (2H, t, J = 7.5 Hz, –CH2–), 2.55 (4H, ddd, J = 10.9, 3.4, 2.2 Hz, –CH2–), 2.74 (4H, ddd, J = 10.2, 8.7, 2.2 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 4.02 (2H, t, J = 7.5 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 10.5, 22.5, 28.4, 53.7, 60.2, 63.3, 69.4, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C16H23NO3S {[M + H]+} 309.4243, found 309.1401; anal. calcd for C16H23NO3S: C, 62.11; H, 7.49; N, 4.53; O, 15.51; S, 10.36; found: C, 62.13; H, 7.48; N, 4.52; O, 15.52; S, 10.35%.
2-(Piperidin-1-yl)ethyl-4-(butylamino)benzoate (4i). White crystals, yield 76.3%, mp 148–150 °C; 1H NMR (300 MHz, DMSO) δ: 0.88 (3H, t, J = 7.1 Hz, –CH3), 1.33 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.36 (4H, dtt, J = 10.8, 3.3, 2.4 Hz, –CH2–), 1.46 (2H, t, J = 7.4 Hz, –CH2–), 1.48 (4H, ddd, J = 10.8, 3.3, 2.6 Hz), 2.43 (4H, ddd, J = 12.5, 2.9, 2.6 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.30 (2H, t, J = 7.4 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 6.77 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.67 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 13.7, 20.1, 24.4, 25.8, 26.0, 43.5, 54.4, 56.7, 63.3, 112.0, 118.4, 130.6, 151.8, 165.8; HR-ESI-MS m/z: calcd for C18H28N2O2 {[M + H]+} 304.4341, found 304.2153; anal. calcd for C18H28N2O2: C, 71.02; H, 9.27; N, 9.20; O, 10.51; found: C, 71.01; H, 9.28; N, 9.21; O, 10.50%.
2-(Piperazin-1-yl)ethyl-4-(butylamino)benzoate (4j). White crystals, yield 80.4%, mp 193–195 °C; 1H NMR (300 MHz, DMSO) δ: 0.88 (3H, t, J = 7.1 Hz, –CH3), 1.33 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.46 (2H, t, J = 7.4 Hz, –CH2–), 2.35 (4H, ddd, J = 14.1, 10.2, 2.5 Hz, –CH2–), 2.64 (4H, ddd, J = 10.2, 9.4, 2.5 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.30 (2H, t, J = 7.4 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 6.77 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.67 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 13.7, 20.1, 26.0, 43.5, 46.3, 54.3, 57.4, 63.3, 112.0, 118.4, 130.6, 151.8, 165.8; HR-ESI-MS m/z: calcd for C17H27N3O2 {[M + H]+} 305.4223, found 305.2105; anal. calcd for C17H27N3O2: C, 66.85; H, 8.91; N, 13.76; O, 10.48; found: C, 66.86; H, 8.90; N, 13.75; O, 10.49%.
2-Morpholinoethyl-4-(butylamino)benzoate (4k). White crystals, yield 86.7%, mp 152–154 °C; 1H NMR (300 MHz, DMSO) δ: 0.88 (3H, t, J = 7.1 Hz, –CH3), 1.33 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.46 (2H, t, J = 7.4 Hz, –CH2–), 2.50 (4H, ddd, J = 9.4, 3.1, 2.5 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.30 (2H, t, J = 7.4 Hz, –CH2–), 3.56 (4H, ddd, J = 11.8, 3.1, 2.5 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 6.77 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.67 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 13.7, 20.1, 26.0, 43.5, 54.4, 55.7, 63.3, 66.7, 112.0, 118.4, 130.6, 151.8, 165.8; HR-ESI-MS m/z: calcd for C17H26N2O3 {[M + H]+} 306.4062, found 306.1944; anal. calcd for C17H26N2O3: C, 66.64; H, 8.55; N, 9.14; O, 15.66; found: C, 66.63; H, 8.54; N, 9.15; O, 15.67%.
2-Thiomorpholinoethyl-4-(butylamino)benzoate (4l). White crystals, yield 79.2%, mp 184–186 °C; 1H NMR (300 MHz, DMSO) δ: 0.88 (3H, t, J = 7.1 Hz, –CH3), 1.33 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.46 (2H, t, J = 7.4 Hz, –CH2–), 2.55 (4H, ddd, J = 10.9, 3.4, 2.2 Hz, –CH2–), 2.74 (4H, ddd, J = 8.7, 3.4, 2.3 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.30 (2H, t, J = 7.4 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 6.77 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.67 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 13.7, 20.1, 26.0, 28.4, 43.5, 53.7, 60.4, 63.3, 112.0, 118.4, 130.6, 151.8, 165.8; HR-ESI-MS m/z: calcd for C17H26N2O2S {[M + H]+} 322.4671, found 306.1944; anal. calcd for C17H26N2O2S: C, 63.32; H, 8.13; N, 8.69; O, 9.92; S, 9.94; found: C, 63.34; H, 8.12; N, 8.68; O, 9.93; S, 9.93%.
2-(Piperidin-1-yl)ethyl-4-butoxybenzoate (4m). White crystals, yield 78.2%, mp 115–117 °C; 1H NMR (300 MHz, DMSO) δ: 0.95 (3H, t, J = 7.1 Hz, –CH3), 1.37 (4H, dtt, J = 10.8, 3.3, 2.4 Hz, –CH2–), 1.46 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.49 (4H, ddd, J = 10.8, 10.2, 3.3, 2.6 Hz, –CH2–), 1.75 (2H, t, J = 7.4 Hz, –CH2–), 2.43 (4H, ddd, J = 12.5, 2.9, 2.6 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 4.00 (2H, t, J = 7.4 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 14.2, 19.0, 24.4, 25.8, 31.7, 54.4, 56.7, 63.3, 68.5, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C18H27NO3 {[M + H]+} 305.4183, found 305.1992; anal. calcd for C18H27NO3: C, 70.79; H, 8.91; N, 4.59; O, 15.72; found: C, 70.78; H, 8.92; N, 4.58; O, 15.73%.
2-(Piperazin-1-yl)ethyl-4-butoxybenzoate (4n). White crystals, yield 80.6%, mp 183–185 °C; 1H NMR (300 MHz, DMSO) δ: 0.95 (3H, t, J = 7.1 Hz, –CH3), 1.46 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.75 (2H, t, J = 7.4 Hz, –CH2), 2.35 (4H, ddd, J = 14.1, 10.2, 2.5 Hz, –CH2), 2.64 (ddd, J = 10.2, 9.4, 2.5 Hz, –CH2), 3.14 (2H, t, J = 7.1 Hz, –CH2), 4.00 (2H, t, J = 7.4 Hz, –CH2), 4.30 (2H, t, J = 7.1 Hz, –CH2), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 14.2, 19.0, 31.7, 46.3, 54.3, 57.4, 63.3, 68.5, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C17H26N2O3 {[M + H]+} 306.4065, found 306.1947; anal. calcd for C17H26N2O3: C, 66.64; H, 8.55; N, 9.14; O, 15.66; found: C, 66.65; H, 8.54; N, 9.13; O, 15.67%.
2-Morpholinoethyl-4-butoxybenzoate (4o). White crystals, yield 85.5%, mp 120–122 °C; 1H NMR (300 MHz, DMSO) δ: 0.95 (3H, t, J = 7.1 Hz, –CH3), 1.46 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.75 (2H, t, J = 7.4 Hz, –CH2–), 2.50 (4H, ddd, J = 9.4, 3.1, 2.5 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 3.56 (4H, ddd, J = 11.8, 3.1, 2.5 Hz, –CH2–), 4.00 (2H, t, J = 7.4 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 14.2, 19.0, 31.7, 54.4, 55.6, 63.3, 66.7, 68.5, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C17H25NO4 {[M + H]+} 307.3902, found 307.1787; anal. calcd for C17H25NO4: C, 66.43; H, 8.20; N, 4.56; O, 20.82; found: C, 66.41; H, 8.20; N, 4.57; O, 20.83%.
2-Thiomorpholinoethyl-4-butoxybenzoate (4p). White crystals, yield 80.1%, mp 162–164 °C; 1H NMR (300 MHz, DMSO) δ: 0.95 (3H, t, J = 7.1 Hz, –CH3), 1.46 (2H, tq, J = 7.4, 7.1 Hz, –CH2–), 1.74 (2H, t, J = 7.4 Hz, –CH2–), 2.55 (4H, ddd, J = 10.9, 3.4, 2.2 Hz, –CH2–), 2.74 (4H, ddd, J = 8.7, 3.4, 2.3 Hz, –CH2–), 3.14 (2H, t, J = 7.1 Hz, –CH2–), 4.00 (2H, t, J = 7.4 Hz, –CH2–), 4.30 (2H, t, J = 7.1 Hz, –CH2–), 7.06 (2H, ddd, J = 8.6, 1.2, 0.4 Hz, Ph-H), 7.95 (2H, ddd, J = 8.6, 1.4, 0.4 Hz, Ph-H); 13C NMR (75 MHz, DMSO) δ: 14.2, 19.0, 28.4, 31.7, 53.7, 60.4, 63.3, 68.5, 114.4, 121.6, 130.4, 163.6, 165.8; HR-ESI-MS m/z: calcd for C17H25NO3S {[M + H]+} 323.4513, found 323.1558; anal. calcd for C17H25NO3S: C, 63.13; H, 7.79; N, 4.33; O, 14.84; S, 9.91; found: C, 63.15; H, 7.78; N, 4.32; O, 14.85; S, 9.90%.
2.2. Biological section
Surface anesthesia. 190 healthy rabbits without eye diseases were taken and the eyelashes were cut off, and the rabbits were then randomly divided into an average of 19 groups. For each target compound, the rabbits were divided into one experimental group, two positive control groups and one blank control group. Each rabbit's lower eyelid was pulled into a cup shape with the thumb and forefinger, the middle finger was pressed against the nasolacrimal duct to prevent the liquid from flowing into the nasolacrimal duct, and a solution of the corresponding drugs (1% target compound and 1% positive control substance) was dropped, with two drops in each eye. The lower eyelid was gently rubbed to allow the drug solution to fully contact the cornea, and it was allowed to overflow after 2 min. Tetracaine and pramocaine were used as the positive controls and NS as the blank control. The effects of surface anesthesia were evaluated by a number of corneal stimulations. Corneal stimulation was used to gently touch the cornea at around 5 points in the upper, lower, middle, left and right parts. After stimulation had been performed 100 times, the blink reflex occurred less than 5 times, and the anesthetic effect was considered to be effective.
The corneal reflex was tested every 10 min for 1 hour before the completion of the infusion and every 20 min for 1 hour afterward, and the tests were continued until the blink reflex occurred more than 5 times. Blinks were compared between groups and in terms of how they changed over time.
Invasive anesthesia. 90 healthy mice were taken and randomly divided into 9 groups. For each target compound, the mice were divided into one experimental group, two positive control groups and one blank control group. The hair on the abdominal skin of the mice was shaved in an area with a diameter of about 2.5 cm. A circle with a diameter of 2 cm was drawn in this area with a marker, and a solution of the corresponding drugs (1% target compound and 1% positive control substance) was injected, of which 1 mL was injected subcutaneously to form a pichu. Tetracaine and pramocaine were used as the positive controls and NS as the blank control. A home-made test needle was used to test the response of the skin to the pinprick after the injection. In the beginning, it was tested every 15 min 10 times for each acupuncture and 1.5 hours after the injection, and the tests were performed every 0.5 hours until the mice had completely recovered to a state without anesthesia. The skin contraction reaction of the mice during acupuncture was positive (no pain inhibition). The changes in the pain inhibition rate over time were compared in each group.
Block anesthesia. A certain number of rats were taken and subjected to thermal irradiation, and the normal tail flick reaction time of the rats was routinely measured. Eighty rats with a tail length of 2–4 cm were randomly divided into 8 groups. The radiation intensity of the pain-inducing tail flick instrument was adjusted to 75, and then the rats were fixed and only the rats' tails were exposed. Five points were selected in each tail, and a solution of the corresponding drugs (1% target compound and 1% positive control substance) was injected, of which 0.2 mL was injected at each point (no injection into the blood vessels was allowed). Tetracaine and pramocaine were used as the positive controls and NS as the blank control. After the tail nerve block operation was completed, the length of tail flicks was measured every 2 min with the pain-inducing tail flick instrument, and the onset of analgesia and duration of analgesia were recorded.
Acute toxicity tests. An orally administered Korbor test was performed.
2.3. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P and pKa tests
P and pKa tests
Firstly, an oil phase (octanol) and a water phase were saturated with each other for 24 hours. Then an appropriate amount of the respective compound was added to a test tube, followed by 1 mL of the pre-saturated two-phase solution. The mouth of the test tube was closed and the test tube was oscillated for 72 hours to make the distribution reach equilibrium. After the distribution was balanced, centrifugal separation was carried out to measure the sample concentrations in the two respective phases, and then log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P was calculated. The compound was also prepared as a dilute solution with a certain concentration, and half of the equivalent sodium hydroxide solution was added. Then, after the solution had been shaken well, the pH of the solution was directly measured with a pH meter, and then the pKa was calculated.
P was calculated. The compound was also prepared as a dilute solution with a certain concentration, and half of the equivalent sodium hydroxide solution was added. Then, after the solution had been shaken well, the pH of the solution was directly measured with a pH meter, and then the pKa was calculated.
3. Results and discussion
3.1. Synthesis
At present, many types of local anesthetic drugs are used in clinical practice, such as tetracaine, lidocaine, dyclonine and pramocaine (Fig. 1). However, these local anesthetic drugs have many deficiencies; for example, tetracaine causes adverse reactions (toxicity), and an excessive dosage of drugs is likely to cause toxic reactions in the central nervous system (CNS) and cardiovascular system. In addition, pramocaine has an anesthetic effect of limited duration. In terms of their chemical structure, these local anesthetic drugs all contain a benzene ring, which is the parent structure (pharmacophore) of this kind of drug. In this research, the target compounds were designed with tetracaine and pramocaine as the lead compounds, and a new organic molecule was formed by the combination principle (Fig. 2). On this basis, the structure of the new organic molecule was modified, and a series of novel benzeneamide compounds were designed by the use of bioisostere formation and modification of the compound with alkyl groups (Fig. 2). The structure of the new organic molecule was determined, the substituent R on the benzene ring was modified, and two substituents, namely, –C3H7 and –C4H9, were selected in the designed process. For X1, N and O were selected as the connecting atoms. In addition, in the process of modification of the six-membered heterocyclic ring, for X2 C, N, O and S atoms were selected. From the perspective of the structure–activity relationship (SAR), the newly designed series of compounds had three basic skeleton structures, namely, a lipophilic section, a link section and a hydrophilic section (Fig. 3). The purpose of the modification of R and X1 was to design target compounds with rapid onset and long duration of anesthesia (lipophilic section), whereas the modification of X2 was performed to obtain compounds with low toxicity (hydrophilic section). In the process of synthesizing the target compounds, we selected a route with high total yields, mild conditions and simple operation. Three steps were used in the synthesis of the new target compounds, namely, alkylation, esterification and alkylation. The first alkylation step used 4-(propylamino)benzoic acid (2a), DMF as a solvent, anhydrous sodium carbonate as a deacidifying reagent and DMAP as a catalyst, and the reflux reaction was completed in 12 hours. The esterification used 2-bromoethyl-4-(propylamino)benzoate (3a), THF as a solvent, and CDI as a dehydrating agent, and the reflux reaction was completed in 8 hours. The second alkylation step used 2-(piperidin-1-yl)ethyl-4-(propylamino)benzoate (4a), toluene as a solvent, and anhydrous potassium carbonate (K2CO3) as a deacidifying reagent, and the reflux reaction was completed in 10 hours. The products were analyzed by 1H NMR, 13C NMR, MS, and elemental analysis. Moreover, some physical and chemical properties (logP and pKa) of the compounds were studied (Table 1). From Table 1, we can find that these target compounds had high lipid solubility (logP ranged from 1.57 to 3.73) and were easily absorbed.
| Compound | R | X1 | X2 | logP |
pKa |
|---|---|---|---|---|---|
| 4a | C3H7– | NH | C | 2.93 | 8.79 |
| 4b | C3H7– | NH | N | 1.57 | 7.90 |
| 4c | C3H7– | NH | O | 1.78 | 7.76 |
| 4d | C3H7– | NH | S | 2.54 | 8.00 |
| 4e | C3H7– | O | C | 3.29 | 8.77 |
| 4f | C3H7– | O | N | 1.96 | 7.86 |
| 4g | C3H7– | O | O | 2.18 | 7.73 |
| 4h | C3H7– | O | S | 2.90 | 7.92 |
| 4i | C4H9– | NH | C | 3.26 | 8.81 |
| 4j | C4H9– | NH | N | 1.98 | 7.93 |
| 4k | C4H9– | NH | O | 2.21 | 7.76 |
| 4l | C4H9– | NH | S | 2.94 | 7.98 |
| 4m | C4H9– | O | C | 3.73 | 8.76 |
| 4n | C4H9– | O | N | 2.36 | 7.85 |
| 4o | C4H9– | O | O | 2.59 | 7.75 |
| 4p | C4H9– | O | S | 3.31 | 7.95 |
3.2. Biological activity
Local anesthetics act on nerve endings and nerve trunks and play a role in local anesthesia. Local anesthetics are commonly used in stomatology, ophthalmology, gynecology and minor surgical operations. The mechanism of action of a local anesthetic is that the drug binds to the receptor of the sodium ion (Na+) channel on the nerve membrane, reduces the passage of Na+ through the ion channel and blocks the conduction of nerve impulses so as to achieve the purpose of local anesthesia. People expect that local anesthetics should have the characteristics of rapid onset, long duration and low toxicity. At present, the commonly used local anesthetics have some disadvantages, such as slow onset, short duration and high toxicity, and hence it is necessary to design new local anesthetics. The newly designed target compounds were evaluated via surface anesthesia, infiltration anesthesia, block anesthesia and acute toxicity tests to select target compounds with good biological activity. The mechanism of action of these compounds was studied. The results of the biological activity tests showed that these compounds had good local anesthetic effects (Tables 2–4). In particular, compounds 4d, 4g, 4j, 4k, 4n, and 4o had excellent effects in terms of surface anesthesia, infiltration anesthesia and block anesthesia. In the process of the local anesthesia experiment, it was found that the anesthetic effects of compounds 4k, 4n and 4o had the characteristics of rapid onset and long duration. In addition, the results of the acute toxicity test showed that the median lethal dose (LD50) of compounds 4k, 4n and 4o was higher and indicated that the target compounds had low toxicity. In the study of the mechanism of local anesthetic action, we found that the Na+ channels bound to by this kind of drug bind to the receptor, and thus the passage of Na+ was affected. The binding of these compounds to the receptor involves multiple bonds (Fig. 4), including van der Waals forces, dipole–dipole interactions, electrostatic attraction, and hydrophobic action.| Compound | Blink reflexes (n = 10) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 min | 20 min | 30 min | 40 min | 50 min | 60 min | 80 min | 100 min | 120 min | 140 min | |
| 4a | 7 ± 0.3 | 7 ± 0.2 | 6 ± 0.2 | 5 ± 0.2 | 4 ± 0.2 | 3 ± 0.1 | 6 ± 0.6 | 25 ± 2.3 | 44 ± 4.1 | 72 ± 6.3 |
| 4b | 7 ± 0.4 | 6 ± 0.6 | 5 ± 0.3 | 4 ± 0.3 | 3 ± 0.2 | 3 ± 0.3 | 5 ± 0.5 | 21 ± 2.1 | 40 ± 4.2 | 70 ± 6.2 |
| 4c | 6 ± 0.3 | 6 ± 0.7 | 4 ± 0.2 | 3 ± 0.2 | 3 ± 0.2 | 4 ± 0.5 | 6 ± 0.6 | 27 ± 2.5 | 49 ± 3.9 | 89 ± 7.3 |
| 4d | 4 ± 0.3 | 3 ± 0.1 | 3 ± 0.2 | 2 ± 0.1 | 2 ± 0.1 | 2 ± 0.1 | 4 ± 0.5 | 20 ± 1.9 | 39 ± 4.0 | 71 ± 7.3 |
| 4e | 6 ± 0.4 | 6 ± 0.6 | 5 ± 0.3 | 4 ± 0.7 | 4 ± 0.5 | 6 ± 0.8 | 8 ± 0.9 | 30 ± 3.2 | 53 ± 4.7 | 92 ± 8.2 |
| 4f | 7 ± 0.5 | 6 ± 0.5 | 5 ± 0.5 | 4 ± 0.8 | 4 ± 0.5 | 5 ± 0.4 | 9 ± 1.1 | 32 ± 3.1 | 57 ± 5.1 | 94 ± 8.3 |
| 4g | 4 ± 0.2 | 3 ± 0.2 | 2 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 2 ± 0.1 | 4 ± 0.7 | 21 ± 2.0 | 38 ± 2.6 | 82 ± 7.5 |
| 4h | 6 ± 0.3 | 6 ± 0.5 | 5 ± 0.6 | 4 ± 0.5 | 3 ± 0.2 | 3 ± 0.3 | 5 ± 0.7 | 23 ± 2.1 | 41 ± 4.0 | 83 ± 7.6 |
| 4i | 7 ± 0.5 | 6 ± 0.7 | 5 ± 0.6 | 4 ± 0.5 | 4 ± 0.5 | 3 ± 0.3 | 6 ± 0.8 | 26 ± 2.2 | 46 ± 4.3 | 86 ± 8.3 |
| 4j | 5 ± 0.3 | 4 ± 0.3 | 3 ± 0.2 | 3 ± 0.2 | 2 ± 0.1 | 2 ± 0.1 | 5 ± 0.6 | 24 ± 2.3 | 48 ± 4.1 | 90 ± 8.5 |
| 4k | 4 ± 0.3 | 3 ± 0.2 | 2 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 3 ± 0.2 | 14 ± 1.3 | 33 ± 2.8 | 69 ± 5.1 |
| 4l | 6 ± 0.6 | 5 ± 0.5 | 4 ± 0.3 | 3 ± 0.2 | 3 ± 0.5 | 4 ± 0.4 | 6 ± 0.7 | 28 ± 2.7 | 50 ± 4.7 | 93 ± 8.9 |
| 4m | 6 ± 0.5 | 5 ± 0.6 | 4 ± 0.3 | 3 ± 0.3 | 3 ± 0.3 | 4 ± 0.5 | 5 ± 0.6 | 21 ± 2.0 | 40 ± 4.1 | 86 ± 8.0 |
| 4n | 3 ± 0.2 | 2 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 1 ± 0.1 | 2 ± 0.1 | 2 ± 0.1 | 10 ± 1.2 | 38 ± 3.5 | 80 ± 7.3 |
| 4o | 4 ± 0.2 | 3 ± 0.2 | 2 ± 0.1 | 2 ± 0.1 | 1 ± 0.1 | 2 ± 0.1 | 2 ± 0.1 | 8 ± 0.9 | 24 ± 1.9 | 65 ± 5.3 |
| 4p | 8 ± 0.7 | 7 ± 0.9 | 6 ± 0.8 | 5 ± 0.7 | 4 ± 0.6 | 4 ± 0.5 | 7 ± 0.9 | 29 ± 3.0 | 50 ± 4.1 | 96 ± 9.5 |
| Tetracaine | 6 ± 0.4 | 5 ± 0.6 | 4 ± 0.4 | 3 ± 0.2 | 2 ± 0.1 | 2 ± 0.1 | 5 ± 0.6 | 18 ± 1.3 | 38 ± 3.0 | 79 ± 6.4 |
| Pramocaine | 6 ± 0.5 | 5 ± 0.8 | 3 ± 0.3 | 2 ± 0.1 | 2 ± 0.1 | 2 ± 0.1 | 6 ± 0.8 | 23 ± 2.1 | 44 ± 4.0 | 89 ± 7.8 |
| NS | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Compound | Pain inhibition (n = 10, %) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15 min | 30 min | 45 min | 60 min | 75 min | 90 min | 2 h | 2.5 h | 3 h | 3.5 h | 4 h | 4.5 h | 5 h | |
| 4d | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 93.2 ± 0.5 | 85.2 ± 1.3 | 76.5 ± 1,5 |
| 4g | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 98.9 ± 0.2 | 86.7 ± 1.2 | 73.8 ± 1.4 |
| 4j | 98.4 ± 0.3 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 91.1 ± 0.6 | 78.9 ± 2.1 | 56.4 ± 2.1 |
| 4k | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 99.7 ± 0.2 | 90.8 ± 0.9 |
| 4n | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 94.3 ± 0.5 |
| 4o | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 92.0 ± 0.6 |
| Tetracaine | 98.7 ± 0.2 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 94.1 ± 0.5 | 89.6 ± 0.7 | 70.3 ± 1.0 | 46.3 ± 3.1 | 32.1 ± 3.2 |
| Pramocaine | 99.1 ± 0.2 | 100 | 100 | 100 | 100 | 100 | 100 | 98.5 ± 0.4 | 90.3 ± 0.8 | 81.5 ± 0.6 | 63.9 ± 2.1 | 40.2 ± 3.0 | 25.5 ± 3.4 |
| NS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Compound | Onset of analgesia (min) | Duration of analgesia (min) | LD50 (mg kg−1) |
|---|---|---|---|
| 4d | 8.8 ± 0.4 | 230.7 ± 3.2 | 543.7 ± 2.3 |
| 4g | 8.3 ± 0.4 | 254.6 ± 3.6 | 854.6 ± 3.7 |
| 4j | 12.5 ± 0.7 | 221.8 ± 3.0 | 735.1 ± 3.2 |
| 4k | 7.2 ± 0.3 | 279.9 ± 3.2 | 907.2 ± 4.3 |
| 4n | 6.4 ± 0.2 | 293.5 ± 3.4 | 976.5 ± 4.5 |
| 4o | 6.6 ± 0.2 | 286.4 ± 3.3 | 928.9 ± 4.9 |
| Tetracaine | 12.3 ± 0.9 | 172.3 ± 2.6 | 435.4 ± 3.3 |
| Pramocaine | 10.4 ± 0.8 | 166.5 ± 2.3 | 472.1 ± 3.6 |
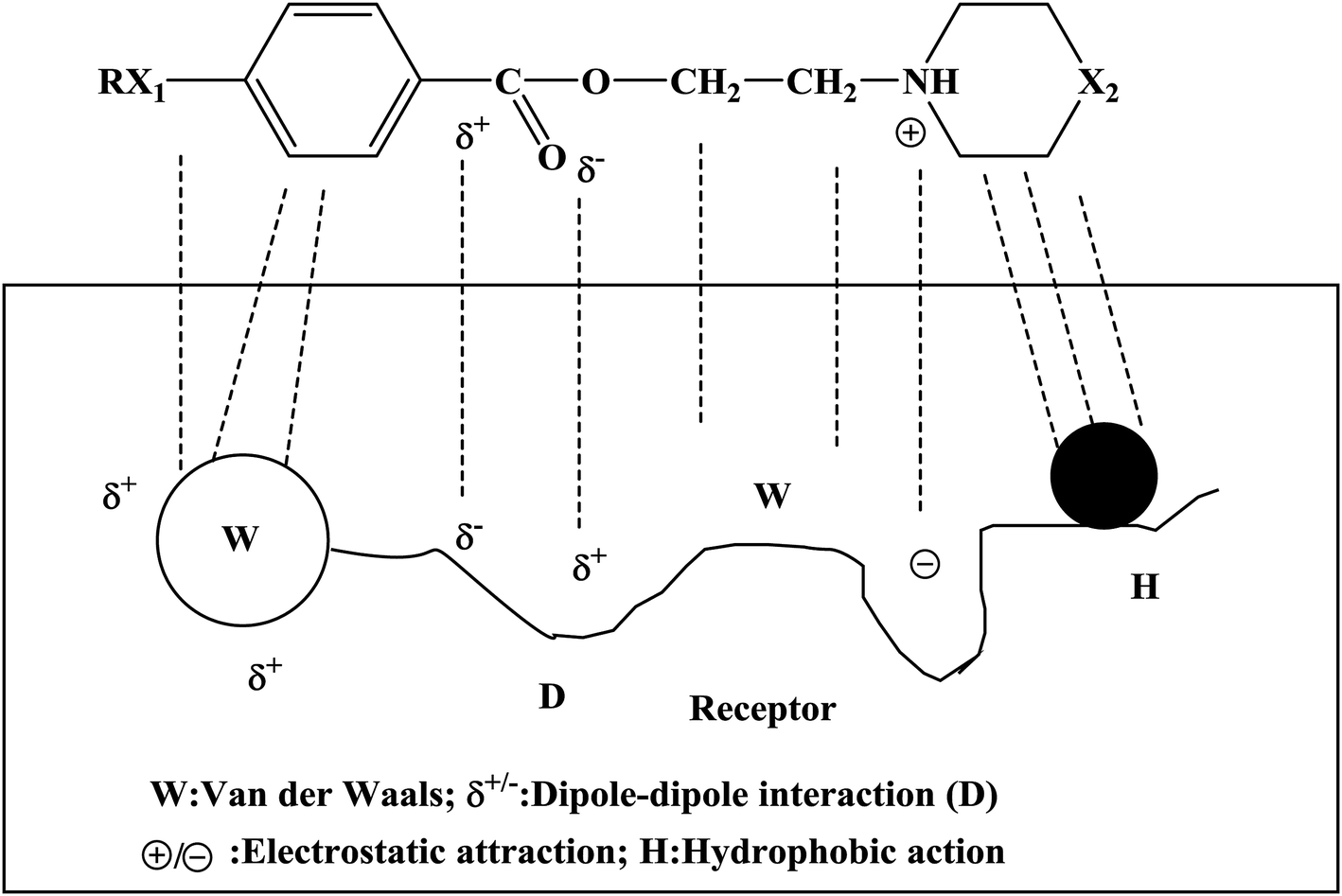 | ||
| Fig. 4 Binding of the target compounds to the receptor. | ||
4. Conclusion
Tetracaine and pramocaine were used as the lead compounds, and a total of 16 target compounds were designed and synthesized. The combination principle was used to design the target molecule, which was modified by bioisostere formation and modification with alkyl groups to design benzoate compounds. We selected three steps for the synthesis of the target compounds via a route with high total yields, mild conditions and simple operation.The biological activity was evaluated by surface anesthesia, infiltration anesthesia, block anesthesia and acute toxicity tests, and the experimental results showed that compounds 4d, 4g, 4j, 4k, 4n, and 4o had a good local anesthetic effect. The results of acute toxicity tests showed that the target compounds had low toxicity.
Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The Project was sponsored by the Innovation Team Project of Universities in Chongqing (No. CXTDX201601018). The Project was also supported by the Chongqing Scientific and Technological Innovation Special Project of Social Undertakings and People’s Livelihood Guarantee (No. cstc2015shmszx80060), Chongqing University Students' Training Project of Innovation and Undertaking (201510637085), Doctoral Program of Chongqing Normal University (No. 12XLB006), Outstanding Achievements Transformation Project in Chongqing Normal University (No. 15XZH08), National Natural Science Foundation (No. 216602012, No. 41866005), and Postgraduate Research and Innovation Project of Hainan Normal University (Hsyx2018-8). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Chongqing Institute of Chinese Materia Medica, and the experiments were approved by the Animal Ethics Committee of Chongqing Institute of Chinese Materia Medica.References
- A. Brown, R. Weiss and C. Greenberg, et al., Interscalene block for shoulder arthroscopy: comparison with general anesthesia, Arthroscopy, 1993, 9(3), 295–300 CrossRef CAS
.
- G. Weinberg, T. VadeBoncouer and G. Ramaraju, et al., Pretreatment or resuscitation with a lipid infusion shifts the dose-response to bupivacaine-induced asystole in rats, Anesthesiology, 1998, 88(4), 1071–1075 CrossRef CAS PubMed
- G. Weinberg, R. Ripper and D. Feinstein, et al., Lipid emulsion infusion rescues dogs from bupivacaine-induced cardiac toxicity, Reg. Anesth. Pain Med., 2003, 28(3), 198–199 CrossRef CAS PubMed
- J. Picard and T. Meek, Lipid emulsion to treat overdose of local anaesthetic: the gift of the glob, Anaesthesia, 2006, 61(2), 107–109 CrossRef PubMed
- M. Rosenblatt, M. Abel and G. Fischer, et al., Successful use of a 20% lipid emulsion to resuscitate a patient after a presumed bupivacaine-related cardiac arrest, Anesthesiology, 2006, 105(1), 217–225 CrossRef PubMed
- R. Litz, M. Popp and S. Stehr, et al., Successful resuscitation of a patient with ropivacaine-induced asystole after axillary plexus block using lipid infusion, Anaesthesia, 2006, 61(8), 800–801 CrossRef CAS PubMed
- G. Cave and M. Harvey, Intravenous lipid emulsion as antidote beyond local anesthetic toxicity: a systematic review, Acad. Emerg. Med., 2009, 16(9), 815–824 CrossRef PubMed
- A. Sirianni, K. Osterhoudt and D. Calello, et al., Use of lipid emulsion in the resuscitation of a patient with prolonged cardiovascular collapse after overdose of bupropion and lamotrigine, Acad. Emerg. Med., 2008, 51(4), 412–415 Search PubMed
- M. Harvey and G. Cave, Intralipid outperforms sodium bicarbonate in a rabbit model of clomipramine toxicity, Acad. Emerg. Med., 2007, 49(2), 178–185 Search PubMed
- M. Harvey, G. Cave and K. Hoggett, et al., Correlation of plasma and peritoneal diasylate clomipramine concentration with hemodynamic recovery after intralipid infusion in rabbits, Acad. Emerg. Med., 2009, 16(2), 151–156 CrossRef PubMed
- D. Crandell and G. Weinberg, Moxidectin toxicosis in a puppy successfully treated with intravenous lipids, J. Vet. Emerg. Crit. Care, 2009, 19(2), 181–186 CrossRef PubMed
- S. Kampe, M. Warm and S. Kasper, et al., Concept for postoperative analgesia after pedicled TRAM flaps: Continuous wound instillation with 0.2% ropivacaine via multilumen catheters. A report of two cases, Br. J. Plast. Surg., 2003, 56(5), 478–483 CrossRef CAS PubMed
- L. Nielsen, P. Lumholt and L. Halmich, Local anaesthesia with vasoconstrictor is safe to use in areas with end-arteries in fingers, toes, noses and ears, Ugeskr. Laeg., 2014, 176(44), 44 Search PubMed
- S. Györke and V. Lukyanenko, Dual effects of tetracaine on spontaneous sodium release in rat ventricular myocytes, J. Physiol., 1997, 500(2), 297–309 CrossRef
- S. Derry, P. Wiffen and R. Moore, et al., Topical lidocaine for neuropathic pain in adults, Cochrane Database Syst. Rev., 2014, 7, CD010958 Search PubMed
- A. Martí, D. Simancas and V. Anand, et al., Prophylactic lidocaine for myocardial infarction, Cochrane Database Syst. Rev., 2015, 8, CD008553 Search PubMed
- L. Slaughter, A. Patel and J. Slaughter, Pharmacological treatment of neonatal seizures: a systematic review, J. Child Neurol., 2013, 28(3), 351–364 CrossRef PubMed
- R. Morabito, A. Marino and S. Dossena, et al., Nematocyst discharge in Pelagia noctiluca (Cnidaria, Scyphozoa) oral arms can be affected by lidocaine, ethanol, ammonia and acetic acid, Toxicon, 2014, 83, 52–58 CrossRef CAS PubMed
- M. Segal, G. Rogers and H. Needleman, et al., Hypokalemic sensory overstimulation, J. Child Neurol., 2007, 22(12), 1408–1410 CrossRef PubMed
- D. Jackson, A. Chen and C. Bennett, Identifying true lidocaine allergy, J. Am. Dent. Assoc., 1994, 125(10), 1362–1366 CrossRef CAS PubMed
- J. Picard, S. Ward and R. Zumpe, et al., Guidelines and the adoption of 'lipid rescue' therapy for local anaesthetic toxicity, Anaesthesia, 2009, 64(2), 122–125 CrossRef CAS PubMed
- A. Gulihar, S. Robati and H. Twaij, et al., Articular cartilage and local anaesthetic: A systematic review of the current literature, J. Orthop., 2015, 12, S200–S210 CrossRef PubMed
- W. A. Carterall, Molecular mechanisms of gating and drug block of sodium channels, Sodium Channels and Neuronal Hyperexcitability, Novartis Foundation Symposia, 2001, vol. 241, pp. 206–225 Search PubMed
- S. Sheu and W. Lederer, Lidocaine's negative inotropic and antiarrhythmic actions. Dependence on shortening of action potential duration and reduction of intracellular sodium activity, Circ. Res., 1985, 57(4), 578–590 CrossRef CAS PubMed
- K. Collinsworth, S. Kalman and D. Harrison, The clinical pharmacology of lidocaine as an antiarrhythymic drug, Circulation, 1974, 50(6), 1217–1230 CrossRef CAS PubMed
- A. Pupka, J. Sikora and J. Mauricz, et al., The usage of synthol in the body building, Polim. Med., 2009, 39(1), 63–65 Search PubMed
- N. Bernardo, M. Siqueira and M. DePaiva, et al., Caffeine and other adulterants in seizures of street cocaine in Brazil, Int. J. Drug Policy, 2003, 14(4), 331–334 CrossRef
- A. Gargiulo, G. Burns and C. Huck, Dyclonine hydrochloride-a topical agent for managing pain, Ill. Dent. J., 1992, 61(4), 303–304 CAS
- J. Schmidt, L. Blockus and R. Richards, The Pharmacology of Pramoxine Hydrochloride: A New Topical Local Anesthetic, Curr. Res. Anesth. Analg., 1953, 32(61), 418–425 CAS
| This journal is © The Royal Society of Chemistry 2019 |