
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA magnetically recyclable superparamagnetic silica supported Pt nanocatalyst through a multi-carboxyl linker: synthesis, characterization, and applications in alkene hydrosilylation†
Laiming Li,
Youxin Li *,
Jincong Yan,
Hang Cao,
Dongyun Shao and
James J. Bao
*,
Jincong Yan,
Hang Cao,
Dongyun Shao and
James J. Bao
Tianjin Key Laboratory for Modern Drug Delivery and High-Efficiency, Collaborative Innovation Center of Chemical Science and Engineering, School of Pharmaceutical Science and Technology, Tianjin University, Room 412-8, Building No. 24, 92 Weijin Road, Nankai District, Tianjin 300037, China. E-mail: lyx@tju.edu.cn; Fax: +86-22-2789-2820; Tel: +86-22-2789-2820
First published on 24th April 2019
Abstract
To simplify separation procedures, improve the reusability and decrease the loss of Pt, two Pt catalysts anchored on superparamagnetic silica (Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt) were prepared for the first time. The stable magnetic properties made them easily recyclable using a magnet rather than filtration, decantation or centrifugation. After 12 catalytic runs for both 30–50 nm Pt catalysts, the yield of 1-heptylmethyldichlorosilane was still up to 90%. The average loss of Pt in each reaction was only 0.87% for Fe3O4@SiO2-EDTA@Pt and 0.66% for Fe3O4@SiO2-DTPA@Pt owing to the strong interaction between Pt and carboxyl. The unprecedented activity and selectivity of the two Pt nanoparticle catalysts were observed in the hydrosilylation of alkenes. The turnover number in the reaction between 1-hexene and methyldichlorosilane using 5 × 10−8 mol of the Pt approached 662![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 733 for Fe3O4@SiO2-EDTA@Pt and 579947 for Fe3O4@SiO2-DTPA@Pt over 12 h. The corresponding hydrosilylation products in excellent yields were obtained when we employed a broad range of alkenes as substrates, including 5 isomerous hexenes and 14 important industry raw materials. Fe3O4@SiO2-DTPA@Pt showed a better activity. They have potential for catalyzing more reactions and replacing the current homogeneous Pt catalysts in industry.
733 for Fe3O4@SiO2-EDTA@Pt and 579947 for Fe3O4@SiO2-DTPA@Pt over 12 h. The corresponding hydrosilylation products in excellent yields were obtained when we employed a broad range of alkenes as substrates, including 5 isomerous hexenes and 14 important industry raw materials. Fe3O4@SiO2-DTPA@Pt showed a better activity. They have potential for catalyzing more reactions and replacing the current homogeneous Pt catalysts in industry.
1. Introduction
Magnetic materials such as magnetite (Fe3O4) and maghemite (γ-Fe2O3) are widely utilized in various fields, such as high-density data storage, chemical sensors, catalysts, drug delivery, biological assays, and electro photographic developers.1–5 Compared with traditional techniques such as filtration, decantation or centrifugation, magnetic materials could be easily separated from solution and re-dispersed through an external magnetic field,6 which overcomes time- and solvent-consuming procedures.6 Magnetite Fe3O4, especially nano-Fe3O4 possessing super paramagnetic behavior at a nanoparticle size with high surface area, is attractive for practical applications.7–10 An ongoing application of magnetic materials is to immobilize metal catalysts because efficient separation, recovery and recycling of the catalyst may enhance their lifetime and minimize their consumption to result in significant economical and environmental benefits.11–14Among these transition metal catalysts, such as copper (Cu), platinum (Pt), palladium (Pd), rhodium (Rh), nickel (Ni) or ruthenium (Ru),15–20 Pt is one of the most widely used owing to its unparalleled catalytic activity and mild reaction conditions in various fields, such as hydrosilylation, aldehyde oxidation, sulfuric acid decomposition. Although homogeneous Pt catalysts such as Speier's catalyst and the Karstedt's catalyst suffer from a number of drawbacks, like side reactions and low selectivity and reusability, there isn't efficient substitution in industry. The immobilization of platinum catalyst is a wise choice. Numerous researches were being reported on how to immobilize Pt on magnetic materials.21–26
Magnetic silica is used as an ideal inorganic support because of its merits, like magnetic property, easy modification, high biocompatibility, solvent compatibility, adsorption capacity, acid–base properties, and chemical and thermal stability when compared to other structures.27–29 A number of multi-carboxyl based affinity ligands, such as ethylenediaminetetraacetic acid (EDTA) and diethylenetriaminepentaacetic acid (DTPA), are well-known chelators for their strong metal-complexing property.30 Magnetic silica particles modified by EDTA or DTPA were widely used in industries including cleaning, the nuclear industry, pharmaceuticals, and the manufacturing of textile leather, rubber, and paper, etc.31–35 The immobilized Pt catalysts on silica gel through EDTA and DTPA were developed by our group.36,37 EDTA and DTPA showed a strong interaction with platinum, but the kinds of heterogeneous catalysts still need to reuse through centrifugation and 31% Pt was loss after 13 recycles.
Here, we first time try to immobilize Pt catalysts through carboxyl ligands like EDTA or DTPA grafted onto the surface of magnetite silica (Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt) to simplify separation procedure, improve the reusability and decrease the loss of Pt. In addition, the two catalysts are applied first time in 4 isomerization–hydrosilylation and hydrosilylation of 14 compounds, which are important industry raw materials. These make the current two catalysts more valuable industry.
2. Experimental section
2.1. Materials and reagents
All chemicals were used as received without further purification. Iron chloride hexahydrate (FeCl3·6H2O, >99%) and ferrous chloride tetrahydrate (FeCl2·4H2O, >99%) were from Yuanli Chemical. γ-Aminopropyltrimethoxysilane (APTMS) was from Qufu Chenguang Chemical. Tetraethylorthosilicate (TEOS) was from Tianjin Kemiou Chemical Reagent. Ethylenediaminetetraacetic acid (EDTA), diethylenetriaminepentaacetic acid (DTPA), sodium silicate, ammonia solution (25%), anhydrous pyridine, acetic anhydride, diethyl ether, hydrochloric acid (HCl), toluene, acetone, ethanol, methanol, n-decane, i-propanol, n-butanol and n-hexanol were from Jiangtian Chemical Technology. Methyldichlorosilane was from Kaihua Synthetic. 1-Hexene, trans-2-hexene, cis-2-hexene, trans-3-hexene and cis-3-hexene were form Tokyo Chemical Industry Co., Ltd. N,N-Dimethylacrylamide, 1,2-epoxy-4-vinylcyclohexane, methyl methacrylate, butyl acrylate, ethane-1,2-diyl bis(2-methylacrylate), crotonic acid ethyl ester and trans-2-hexenyl acetate were from Bid Pharmatech Ltd. 1,2-Epoxy-7-octene, allyl glycidyl ether, 5-hexen-2-one, allylchloride and trans-2-hexen-1-yl phenylacetate were from Nanjin Shengbicheng Chemical Technology Co., Ltd. Cyclohexene and norbornylene were from Shanghai Macklin Biochemical Co., Ltd. H2PtCl6·6H2O was from Tianjin Fengchuan Chemical Reagent Technologies. 1-Hexyl-methyldichlorosilane was from CNW Technologies. Deionized water was from Tianjin Yongyuan Distilled Water Manufacturing Center.2.2. Materials synthesis
The magnetite Fe3O4@SiO2 nanoparticles were prepared by two sol–gel approach. First, the surface of Fe3O4 nanoparticles was covered by silica layer (SiO2 encapsulated Fe3O4 particles, Fe3O4@SiO2). Briefly, sodium silicate was dissolved in deionized water in a glass beaker equipped with a mechanical stirrer and the pH was adjusted to 12–13 by sodium hydroxide. Fe3O4 nanoparticles were added to the sodium silicate solution. The mixture was treated ultrasonically for 30 min. Then, the temperature of the mixture was increased to 85 °C and nitrogen gas was used to prevent the intrusion of oxygen. 2 M hydrochloric acid was added dropwise to adjust pH value to 6.0. Then, the precipitates were washed several times with deionized water by magnetic decantation. The Fe3O4@SiO2 was coated again to ensure that the desired amount of silica on all magnetite Fe3O4.44,45 For this, Fe3O4@SiO2 was re-dispersed in 100 mL methanol–water (1:1, v/v) mixture and followed by the addition of 1 mL ammonium hydroxide (25% aqueous solution). Then, 1 mL of TEOS was dissolved in 10 mL methanol and added dropwise to the mixture by gently stirring for 1 h. The obtained nanoparticles (Fe3O4@SiO2) were collected by magnet and washed several times with distilled water and ethanol. Finally, the nanoparticles were dried at room temperature under vacuum and brown yellow solid was observed.
:1, v/v). After the addition of 25 g of EDTA dianhydride or 30 g DTPA dianhydride, the mixture was kept at 80 °C for 24 h to allow the amino groups on the silica gel to react with the carboxyl groups.48,49 Finally the yellowish-brown product was separated by magnet and washed with 4 M ammonia water solution to remove unreacted EDTA or DTPA.50 Then products were washed again with ethanol/acetic acid (1:1, v/v), water and methanol sequentially.51 The multi-carboxyl modified magnetic silica gels (Fe3O4@SiO2-EDTA or Fe3O4@SiO2-DTPA) were dried in an oven at 90 °C for 8 h.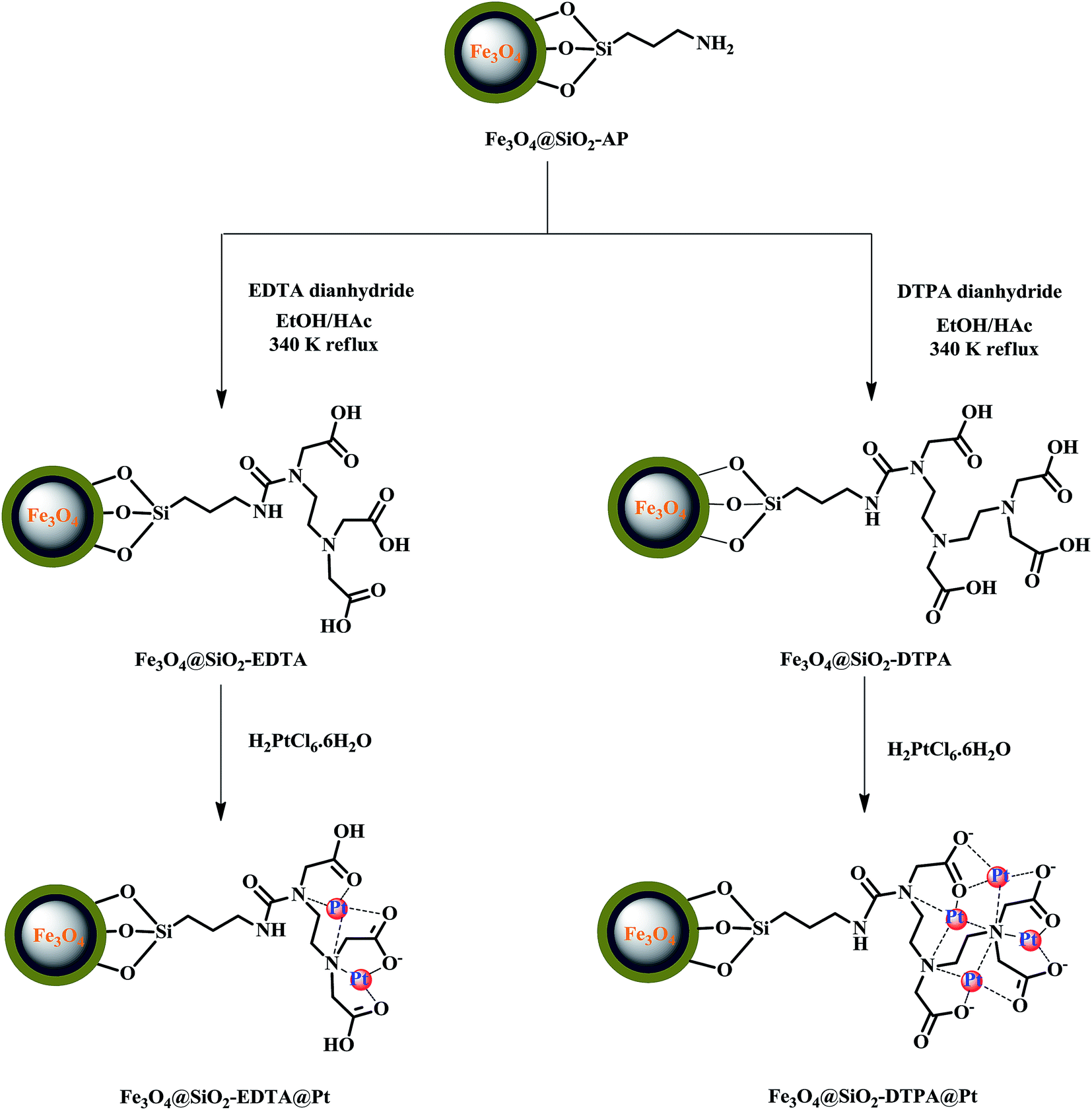 | ||
| Fig. 1 Preparation of Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt catalysts. | ||
2.3. Characterization of the catalysts
The crystal lattice structure of the magnetic intermediates (Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-AP), and products (Fe3O4@SiO2-EDTA@Pt, Fe3O4@SiO2-DTPA@Pt) were identified by XRD, in the 2θ range of 10–90° at a scan speed of 4° min−1, using a Rigaku D/MAX-2500 diffractometer with Cu-Kα radiation (λ = 0.1540 nm). Particle sizes of the catalysts were studied using a JEOL JEM-2100F apparatus with an accelerating voltage of 200 kV. TEM of Fe3O4@SiO2-EDTA@Pt, Fe3O4@SiO2-DTPA@Pt were prepared by placing a drop containing the nanoparticles in a coated carbon grid. The morphology of the catalysts was investigated using a Hitachi S2800 SEM. The EDS analysis, which was simultaneously performed during the SEM examinations, was conducted to confirm the element distribution on the surface of adsorbents. FT-IR spectra were taken in KBr pressed pellets on a TENSOR 27 Spectrometer. EA was used to measure the nitrogen content in the materials on an Elementar Vario Micro cube. The thermal stabilities were measured by TGA employing a NETZSCH STA 409 TG-DTA under a dynamic nitrogen atmosphere at a heating rate of 10 °C min−1. The magnetization and hysteresis loops were measured at room temperature by a LDJ9600 VSM. The BET measurement was carried out on a Quantachrome NOVA 1000 system at liquid-N2 temperature.2.4. Catalytic alkene hydrosilylation
In a general procedure, hydrosilylation was carried out in a 50 mL round-bottomed tube equipped with a homemade reflux condenser to the upper of which a heating system was attached. An appropriate amount of alkene and platinum magnetic complex were stirred at a specific reaction temperature for 30 min before 15 mmol methyldichlorosilane was added. The structure and yield of hydrosilylation products were determined based on calibration curves of standards. The catalyst was collected by simple magnetic separation and reused in subsequent runs without any treatment. All hydrosilylation products were characterized by comparing their spectra and physical parameters with authentic samples. The reaction product was quantified by gas chromatography (GC) and the structures of products were identified by nuclear magnetic resonance (NMR). GC equipped with a flame ionization detector (FID) (Wuhan Trustworthy Technology, China) and a long capillary column (30 m × 0.25 mm × 0.25 μm) coated by 5% phenyl and 95% methyl polysiloxane. Injector and detector temperature was 260 °C, carrier gas was nitrogen, injection volume at 0.8 μL, temperature programme: the column was kept at 60 °C for 3 min, then heated to 260 °C at a rate of 10 °C min−1. 1H NMR spectra were recorded on a Bruker AC-P400 (400 MHz) spectrometer in CDCl3 as solvent.3. Results and discussion
3.1. Synthesis
In the synthetic procedures described earlier, magnetite Fe3O4 nanoparticles were prepared by the co-precipitation method from ferrous and ferric ion solutions at a mol ratio of 1:2. The magnetic nanoparticles have extensive hydroxyl groups on its surface when contacted with aqueous phase.52 Silicic acid as silica precursors encapsulated the Fe3O4 nanoparticles by two sol–gel approaches. First silicate acid layer was formed gradually from sodium silicate solution by dropwise addition of hydrochloric acid. The second silicate acid layer was formed by dropwise addition of TEOS solution. However, the TEOS is non-polar in nature and it is necessary to use organic reagent like methanol as a mutual solvent to make TEOS and water miscible. Subsequently, the silanol groups (Si–OH) on the surface are transformed into siloxane bonds (Si–O–Si) via the condensation reaction and the silica coating is formed.53 Notably, the introduction of a dense silica coating on the magnetic nanoparticles is necessary for this study. It protects the magnetic Fe3O4 from oxidation and provides the surface charges needed to prevent aggregation in the subsequent condensation reactions and practical applications. The silanol groups change the surface properties making it easier and more effective to perform surface modification with organic silane molecules.
Before being modified by EDTA or DTPA group, the magnetic silica nanoparticles were functionalized with APTMS, which acted as an auto-catalyst. Those amino groups on the magnetic silica surface enable the magnetic silica materials to be functionalized with EDTA and DTPA by grafting the corresponding anhydrides on those amino groups. The EDTA or DTPA groups, in turn, serve as a new matrix to immobilize metallic Pt. The chemical structures of the resulting product were depicted in Fig. 1. The multi-carboxyl functionalized magnetic silica materials were found to be a yellow powder with an excellent shelf life. These magnetite nanoparticles functionalized with EDTA or DTPA will be referred to as Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt.
3.2. Characterization
The data from AAS analysis indicated the amounts of Pt in Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt were 173.80 and 192.77 μmol g−1, respectively. As a comparison, Pt was also immobilized on Fe3O4@SiO2-AP and Fe3O4@SiO2, which had only 33.40 and 0.10 μmol g−1 of Pt, respectively. It means the interaction between Pt and carboxyl is stronger than amino group, which is benefit to the immobilization and stability of Pt.Morphology of the two novel catalysts was characterized by SEM, EDS and TEM (Fig. 2). Results showed that the dried power of Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt nanoparticles had an irregular surface with sharp edge and slight agglomeration, which could be dispersed well in solution. The presence of Si, O and Fe signals in the EDS pattern confirmed that Fe3O4 was successfully mixed with silica. The higher intensity of Si peak compared with that of Fe peak showed that the Fe3O4 nanoparticles were coated by SiO2. The collected process by a magnet avoids the SiO2 nanoparticles without Fe3O4. The EDS analysis results also confirmed the elemental composition of the nanoparticles and the existence of Pt in the prepared nanoparticle catalysts. Through second robust sol–gel approach, the high magnification TEM images revealed that the Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt particles obtained were uniform with a diameter of about 30–50 nm. Each magnetic particle was composed of dark magnetic Fe3O4 nanoparticles of about 10–15 nm diameter. Even further, it clearly showed that those dark magnetic particles were coated with a light-gray SiO2 layer with a thickness of around 15 nm.
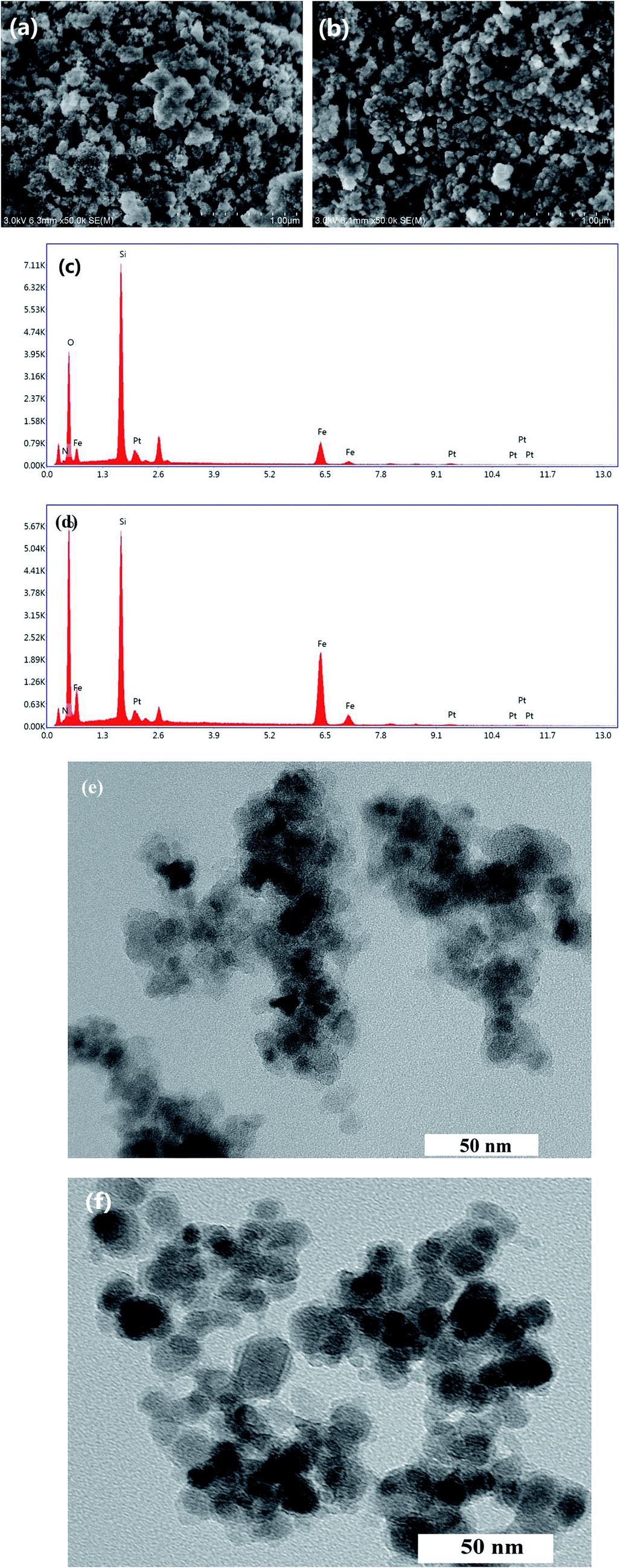 | ||
| Fig. 2 SEM micrographs of (a) Fe3O4@SiO2-EDTA@Pt and (b) Fe3O4@SiO2-DTPA@Pt, EDS of (c) Fe3O4@SiO2-EDTA@Pt and (d) Fe3O4@SiO2-DTPA@Pt, TEM micrographs of (e) Fe3O4@SiO2-EDTA@Pt and (f) Fe3O4@SiO2-DTPA@Pt. | ||
The XRD patterns of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt nanoparticles were shown in Fig. 3. It showed typical X-ray diffraction patterns of Fe3O4.54 The characteristic diffraction peaks appeared in Fe3O4 at 30.1°, 35.5°, 43.3°, 54.3°, 57.3° and 62.6° could be assigned to the planes of Fe3O4 (JCPDS No. 19629), respectively. These characteristic diffraction peaks were observed in Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt nanoparticles which indicated that Fe3O4 was existent in all synthesized samples and the whole preparation processes did not result in the phase change of Fe3O4. The obvious characteristic peak of Pt wasn't observed in Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt, which may indicate that the amount of Pt was low or the Pt particles were highly dispersed on the surface of support.55
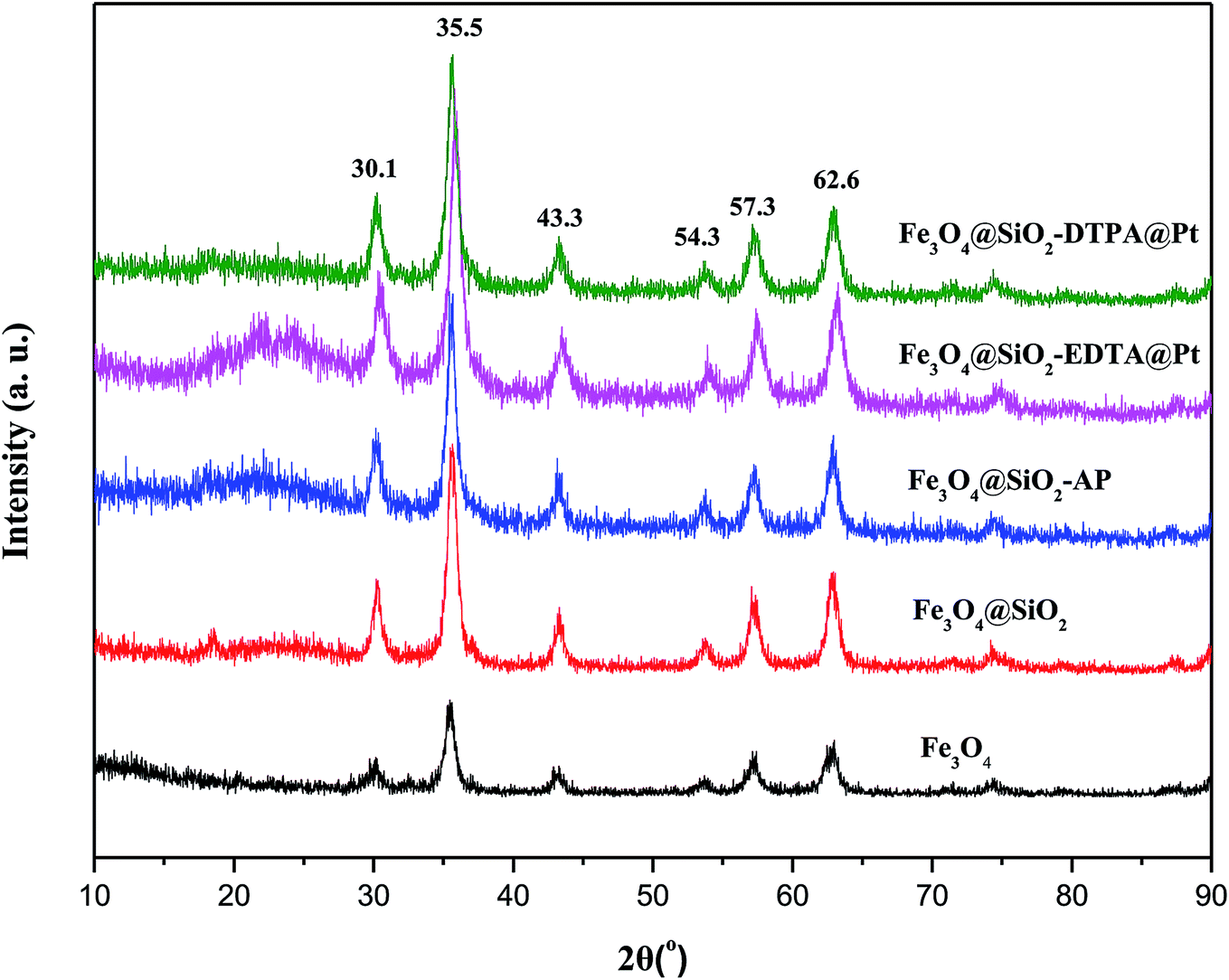 | ||
| Fig. 3 XRD patterns of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt nanoparticles. | ||
In order to confirm properties of the surface of composite nanoparticles, the molecular structure of the different composites was characterized using FT-IR spectra (Fig. 4.). The typical absorption peak for Fe3O4 at 635 and 576 cm−1 was attributed to the stretching vibration of Fe–O bond. The typical peaks of Fe3O4 were observed in all of spectra, which further confirmed Fe3O4 nanoparticles existed in all of intermediates and products. Whereas the gradual decrease of peak signals indicated the Fe3O4 nanoparticles were at core of Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA, Fe3O4@SiO2-EDTA@Pt, Fe3O4@SiO2-DTPA and Fe3O4@SiO2-DTPA@Pt. For Fe3O4@SiO2, a new board absorption peak at 1092 cm−1 resulted from Si–O–Si antisymmetric stretching vibration of silica layer, which verified the successful coating of silica layers onto Fe3O4.56,57 The peak around 1627 cm−1 was attributed to the absorbed water on the silica shell or the silanol group of the silica.58 The characteristic absorption band at 3422 cm−1 was from the stretching vibrations of hydroxyl groups. These peaks from Fe3O4@SiO2 were also observed in Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA, Fe3O4@SiO2-DTPA, Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt. After the functionalization of Fe3O4@SiO2 with APTMS, the new absorption peaks at 2925 and 2858 cm−1 represented the symmetric vibrations of aliphatic C–H stretching of the methylene group for silane coupling agent, which indicated that amino group was successfully grafted onto the surface of Fe3O4@SiO2. After grafting multi-carboxyl groups, the strong characteristic absorption band appeared at 1733 and 1404 cm−1 could be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibration of the –COO– group, which indicated the formation of carboxylic groups (EDTA and DTPA) on the surface of the Fe3O4@SiO2-AP.59 As a result of the presence of large quantities of metallic Pt on the surface of the nanoparticles, the decrease in vibration of CO and C–O suggests that the carboxyl group on the surface of the functional magnetic silica have coordinated with platinum, signifying the immobilization of platinum complex on the Fe3O4@SiO2-EDTA and Fe3O4@SiO2-DTPA surface.
O and C–O stretching vibration of the –COO– group, which indicated the formation of carboxylic groups (EDTA and DTPA) on the surface of the Fe3O4@SiO2-AP.59 As a result of the presence of large quantities of metallic Pt on the surface of the nanoparticles, the decrease in vibration of CO and C–O suggests that the carboxyl group on the surface of the functional magnetic silica have coordinated with platinum, signifying the immobilization of platinum complex on the Fe3O4@SiO2-EDTA and Fe3O4@SiO2-DTPA surface.
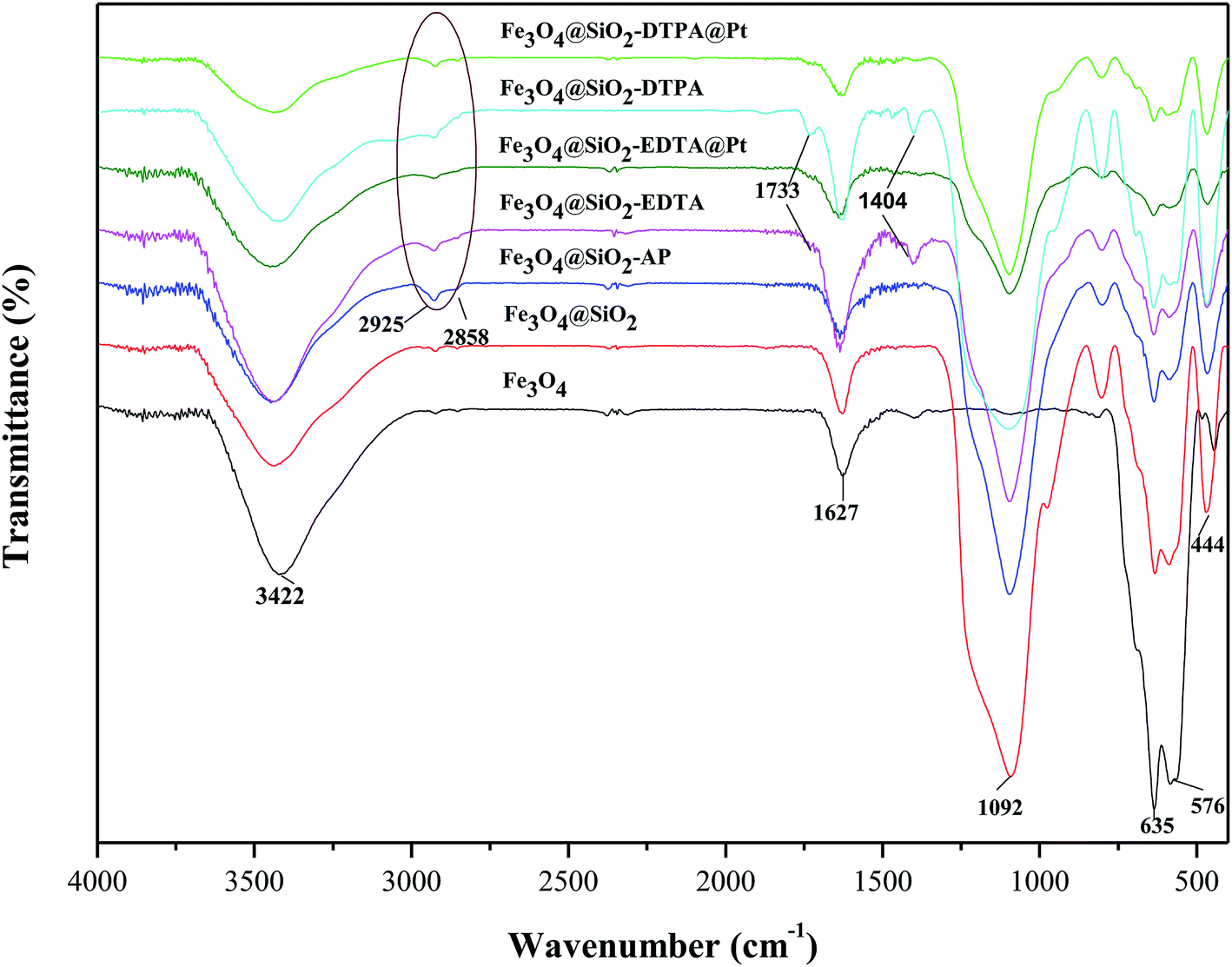 | ||
| Fig. 4 FT-IR spectra of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA, Fe3O4@SiO2-EDTA@Pt, Fe3O4@SiO2-DTPA, and Fe3O4@SiO2-DTPA@Pt nanoparticles. | ||
XPS was used to further characterize the above catalysts (Fig. 5.). XPS results showed distinct Fe, O, N, C, Si and Pt peaks from the two novel nanoparticles (Fig. 5a). The detail XPS data for Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA@Pt, Fe3O4@SiO2-DTPA@Pt and H2PtCl6·6H2O were listed in Table 1. It revealed that the binding energies of Si 2p and O 1s in Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt were similar to those of Fe3O4@SiO2-AP. The banding energy of Cl 2p in Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt was similar to that of H2PtCl6·6H2O. It was noteworthy that the banding energy of Pt 4f7/2 in Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt was 3–5 eV more than that in H2PtCl6·6H2O. It could be summarized that electron donation from carboxyl group to Pt to form the coordination bond between carboxyl and Pt in Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt, which was also consistent with the FT-IR results. The Pt 4f XPS spectra contain two peaks corresponding to Pt 4f7/2 and 4f5/2 states from the spin-orbital splitting and each peak was de-convoluted with Pt(0), Pt(II) and Pt(IV).60–62 In Fe3O4@SiO2-EDTA@Pt, the percentage of Pt4+, Pt2+ and Pt0 peaks were 28.9, 46.5 and 24.6%, respectively. In Fe3O4@SiO2-DTPA@Pt, the ratio of Pt2+ (50.3%) and Pt0 (26.4%) were higher than that of Fe3O4@SiO2-EDTA@Pt, whereas the Pt4+ decreased to 23.3%. Results indicated the Pt2+ and Pt0 were predominant species that may be corresponding to good catalytic activity and might be produced on the catalyst surface through passivation process during the sample preparation.
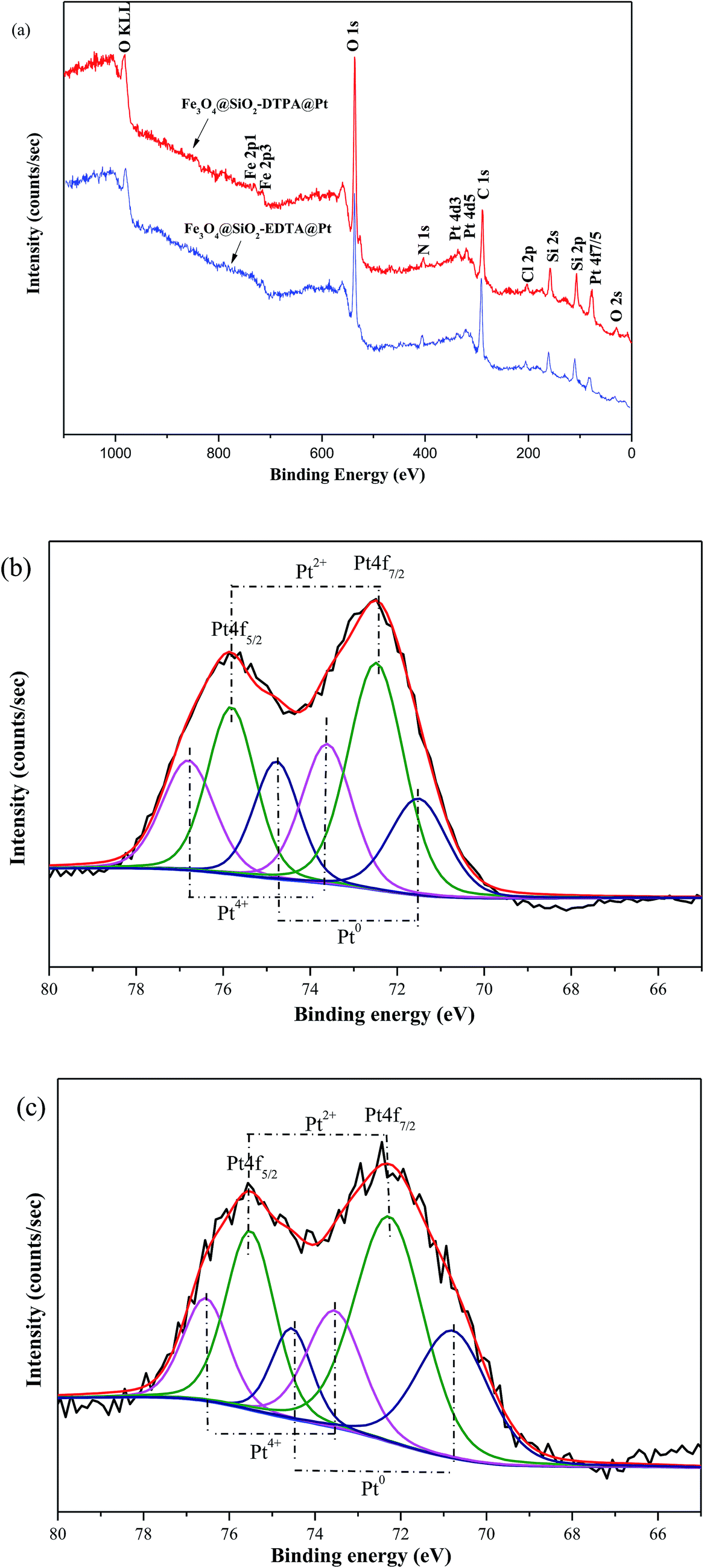 | ||
| Fig. 5 (a) XPS spectra of Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt. (b) High-resolution XPS spectra of Pt 4f for Fe3O4@SiO2-EDTA@Pt. (c) High-resolution XPS spectra of Pt 4f for Fe3O4@SiO2-DTPA@Pt. | ||
| Samples | Pt 4f7/2 | Si 2p | N 1s | O 1s | Cl 2p |
|---|---|---|---|---|---|
| a The banding energies are referred to C 1s (284.6 eV). | |||||
| Fe3O4@SiO2-EDTA@Pt | 78.40 | 107.21 | 405.59 | 536.88 | 201.29 |
| Fe3O4@SiO2-DTPA@Pt | 76.80 | 106.40 | 403.23 | 536.86 | 200.40 |
| Fe3O4@SiO2-AP | — | 108.78 | 407.24 | 537.60 | — |
| H2PtCl6·6H2O | 73.10 | — | — | — | 199.50 |
BET analysis showed another important characteristic of the Pt catalysts. Fig. 6a shows the nitrogen adsorption isotherms and pore-size distribution of Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt catalysts. Both of two samples showed type III isotherms with type H3 hysteresis loops which is related to the slit-like pores because of aggregation of particles.63 When we dispersed these nanoparticles in aqueous solution and observed using microscope, these nanoparticles were mono-disperse. It means the aggregation is temporary in BET analysis. The BET data including surface area, average pore size and total pore volume derived from the isotherms were calculated. The BET specific surface area, the pore size and the total pore volume of Fe3O4@SiO2-EDTA@Pt were 75.68 m2 g−1, 17.50 nm and 0.685 cm3 g−1, respectively. They were corresponding to 36.65 m2 g−1, 29.86 nm and 0.366 cm3 g−1 for Fe3O4@SiO2-DTPA@Pt.
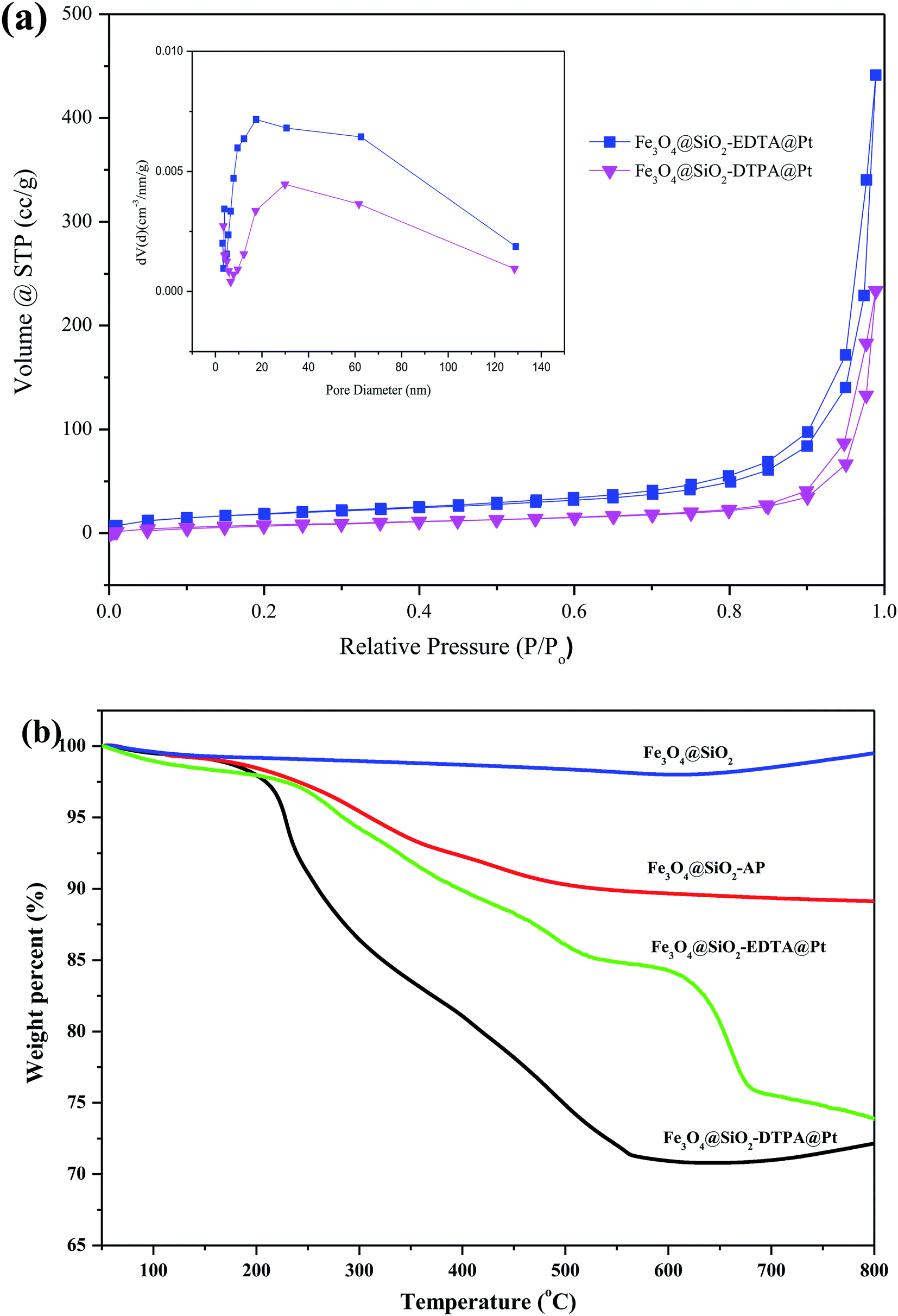 | ||
| Fig. 6 (a) Nitrogen adsorption–desorption isotherms and (inset) the corresponding pore-size distribution curves of Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt catalysts, (b) TGA curves of Fe3O4@SiO2, Fe3O4@SiO2-AP, SiO2@Fe3O4-EDTA@Pt and SiO2@Fe3O4-DTPA@Pt. | ||
To test the thermal stability and the grafting amount of carboxyl groups on the magnetic silica nanoparticles, TGA analysis was performed for Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt nanoparticles (Fig. 6b.). As shown in the TGA curve of the prepared nanoparticles, the weight loss below 200 °C was attributed to the removal the water, and the organic parts were decomposed completely at about 700 °C. The TGA curve of bare Fe3O4@SiO2 nanoparticles showed that the weight loss over a temperature range from 50 to 800 °C was about 4%, which was attributed to the escape of physically adsorbed water or/and structural water on the surface.64 Moreover, the weight increases slightly in the temperature range of 700–800 °C, which might be caused by the slight oxidation of Fe3O4.65 For Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt nanoparticles, the weight loss at temperatures below 200 °C was about 3%, which was related to the release of moisture and structural water on the surface of the silica layer. However, apart from the water loss, organic components inside the particles began to degrade rapidly at a temperature higher than 200 °C, and almost completely decomposed at 550 °C for Fe3O4@SiO2-EDTA@Pt and 700 °C for Fe3O4@SiO2-DTPA@Pt, respectively. Therefore, the weight loss from 200 to 800 °C in the TGA curves of Fe3O4@SiO2-EDTA@Pt (about 28 wt%) and Fe3O4@SiO2-DTPA@Pt (about 30 wt%) could be used to estimate the weight proportions of EDTA or DTPA-modified on Fe3O4@SiO2. This phenomenon suggested that the weight proportion of multi-carboxyl groups on Fe3O4@SiO2-DTPA@Pt (30 wt%) was higher than that on Fe3O4@SiO2-EDTA@Pt (28 wt%). It was consistent with one more carboxyl group in DTPA than EDTA.
The key for magnetic materials is the sufficient magnetic property. Therefore the magnetic properties of these nanoparticles were analyzed by VSM (Fig. 7.). The saturation magnetization was found to be 74.7, 48.4, 44.4, 25.4 and 23.0 emu g−1 for Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt, respectively. All of them showed superparamagnetic properties due to the presence of nanometer-sized magnetic Fe3O4 particles in the core.66 Although the saturation magnetization values of Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt were lower than that of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2-AP, they could be readily separated from their dispersion in 1 min with an external magnetic field owing to their high magnetite content and thus good magnetic response (Fig. 7, inset). These results indicated that the magnetization of Fe3O4 decreases considerably after coated with SiO2 due to the weight contribution from the nonmagnetic SiO2. Nevertheless, both of the catalysts exhibited enough magnetic response to meet the need of magnetic separation.
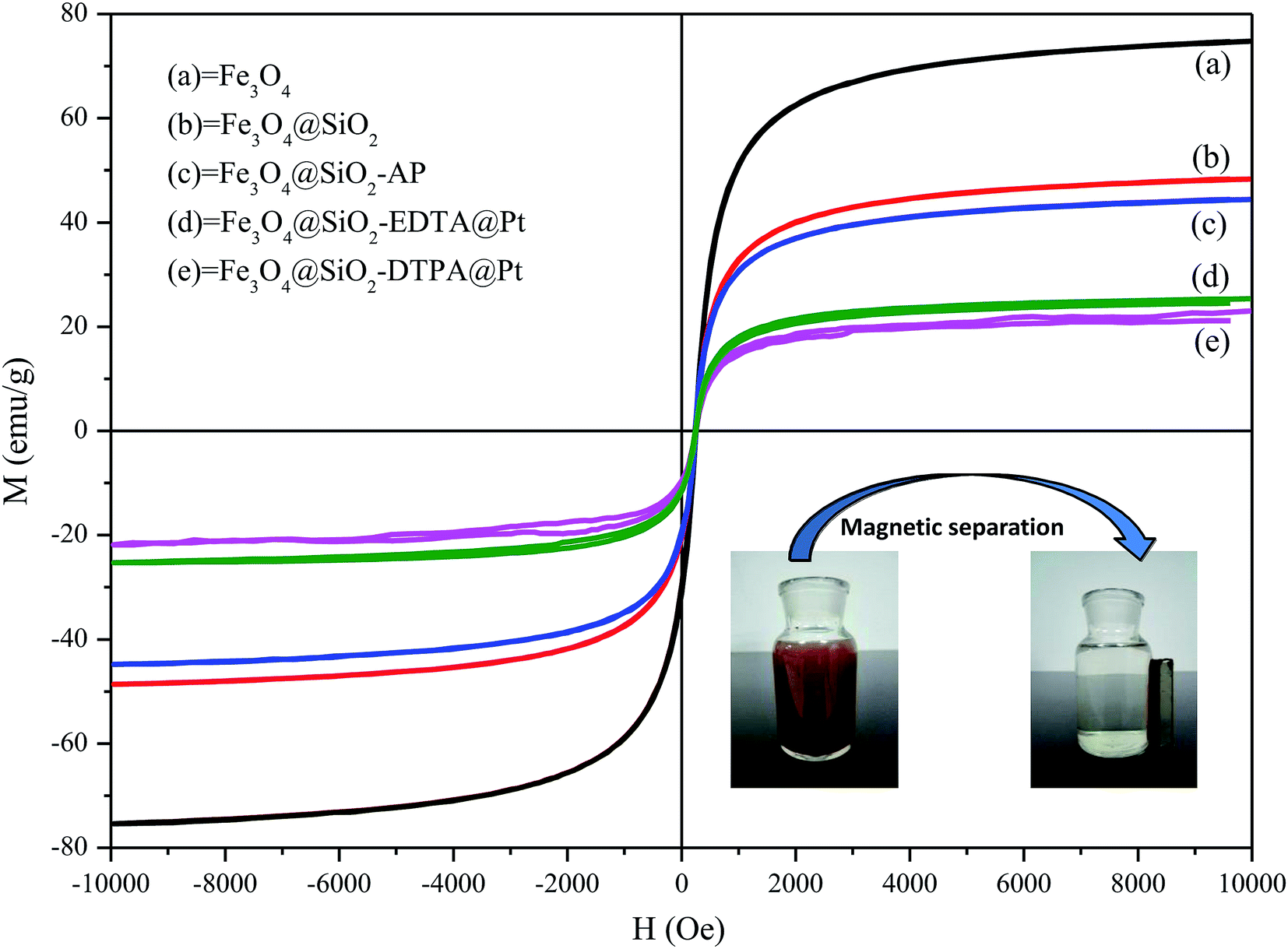 | ||
| Fig. 7 VSM magnetization curves of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-AP, Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt. Inset: magnetic separation of Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt from the aqueous suspensions. | ||
3.3. Catalyst performances for hydrosilylation
Hydrosilylation conditions are important for yield and regioselectivity. To exhibit well the activities of two catalysts, we optimized hydrosilylation conditions of 1-hexene and methyldichlorosilane, including reaction temperatures (30, 40, 50, 60, 70, and 80 °C), times (0.5, 1, 2, 3, 4, and 5 h), the molar ratio of methyldichlorosilane and 1-hexene (0.5:1.0, 1.0:1.0, 1.2:1.0, 1.5:1.0, 1.8:1.0, and 2.0:1.0) and the additive sequence of 1-hexene and methyldichlorosilane (1-hexene added to methyldichlorosilane after methyldichlorosilane mixing with the catalyst for 30 min, simultaneous addition of 1-hexene and methyldichlorosilane, methyldichlorosilane added to 1-hexene after mixing with the catalyst for 30 min). The detail results were summarized in support information (Fig. 1S†). Results indicated both Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt (containing 2.01 × 10−3 mmol Pt in each catalyst) could efficiently catalyze the hydrosilylation. The yields were up to 99.4% and 98.77% when 19.2 mmol methyldichlorosilane was added to 11.8 mmol 1-hexene after mixing with the catalyst for 30 min and maintained the reaction 3 h at 50–60 °C. As a comparison, the yield only was 35.46% when Fe3O4@SiO2@Pt was used as catalyst.
In research of platinum catalyst, the turnover number (TON) of Pt often is used as criteria. Thus, we calculated the TON approached 3628 and 3550 when Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt with 2.60 μmol Pt catalyzed 11.2 mmol of 1-hexene in 4 h. To further increase the TON for hydrosilylation, amount of Pt catalyst was gradually decreased to 2.10, 1.82, 1.47, 0.87, even 0.34 μmol. Results indicated the hydrosilylation between 1-hexene and methyldichlorosilane with low amount of Pt could be catalyzed and the TON quickly increased to 4721, 5435, 6744, 10905 and 16006 for Fe3O4@SiO2-EDTA@Pt, 4750, 5464, 6794 and 10789 for Fe3O4@SiO2-DTPA@Pt. Furthermore, we catalyzed the hydrosilylation of methyldichlorosilane (180 mmol) and 1-hexene (110 mmol) using only 0.3 mg Fe3O4@SiO2-EDTA@Pt (0.0052 μmol Pt) or 0.3 mg Fe3O4@SiO2-DTPA@Pt (0.057 μmol Pt) in 12 h. Remarkably, the region-selectivity was still around 99% and TON increased to 662733 for Fe3O4@SiO2-EDTA@Pt, 579947 for Fe3O4@SiO2-DTPA@Pt, respectively.
Usually, the stability of a catalyst is an important characteristic before it can be used in an industrial application. Therefore, the reusability of Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt catalysts were evaluated through repetitively catalyzing the hydrosilylation of 1-hexene with methyldichlorosilane. A series of 12 consecutive hydrosilylation reactions were carried out under optimal reaction conditions. After each catalytic reaction, the two platinum catalysts were held in the original system using a magnet and catalyzed next hydrosilylation without any other process. The results were showed in Fig. 8. Data showed a satisfactory yield of 1-heptylmethyldichlorosilane (about 90% after 12 catalytic runs for both of Pt catalysts) which was apparent better than the Pt catalysts immobilized on non-magnetic SiO2 supports (only 80% after 12 or 13 runs).36,37 After 12 runs, the magnetism both of Pt catalysts were similar and the platinum amount was 155.78 mmol g−1 for Fe3O4@SiO2-EDTA@Pt and 177.62 mmol g−1 for Fe3O4@SiO2-DTPA@Pt. There were only 10.4% Pt loss in Fe3O4@SiO2-EDTA@Pt and 7.9% Pt loss in Fe3O4@SiO2-DTPA@Pt. The average loss of Pt was 0.87% for Fe3O4@SiO2-EDTA@Pt and 0.66% for Fe3O4@SiO2-DTPA@Pt in each reaction which was apparent better than homogeneous Pt catalyst (about 10 ppm) and Pt catalysts immobilized on non-magnetic support. The data were listed in Table S1.† In order to identify the active species of Pt catalysts, the state of Pt catalysts in high-resolution XPS after recycle experiments was tested. The data were listed in Fig. S3 and Table S2.† The total amount of Pt2+ and Pt0 was 71.1% (before recycle) and 76.1% (after recycle) for Fe3O4@SiO2-EDTA@Pt. The total amount of Pt2+ and Pt0 was 73.6% (before recycle) and 79.6% (after recycle) for Fe3O4@SiO2-DTPA@Pt. According to the data, there wasn't the apparent state change of the recycled Pt catalyst in both Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt. This also indicated both of catalysts, especially Fe3O4@SiO2-DTPA@Pt, were very stable, which maybe result from strong coordination interaction between carboxyl group and the metallic Pt. The residual four carboxyl groups of DTPA in Fe3O4@SiO2-DTPA maybe produce a stronger interaction with Pt than the residual three carboxyl groups of EDTA in Fe3O4@SiO2-EDTA (Table 2).
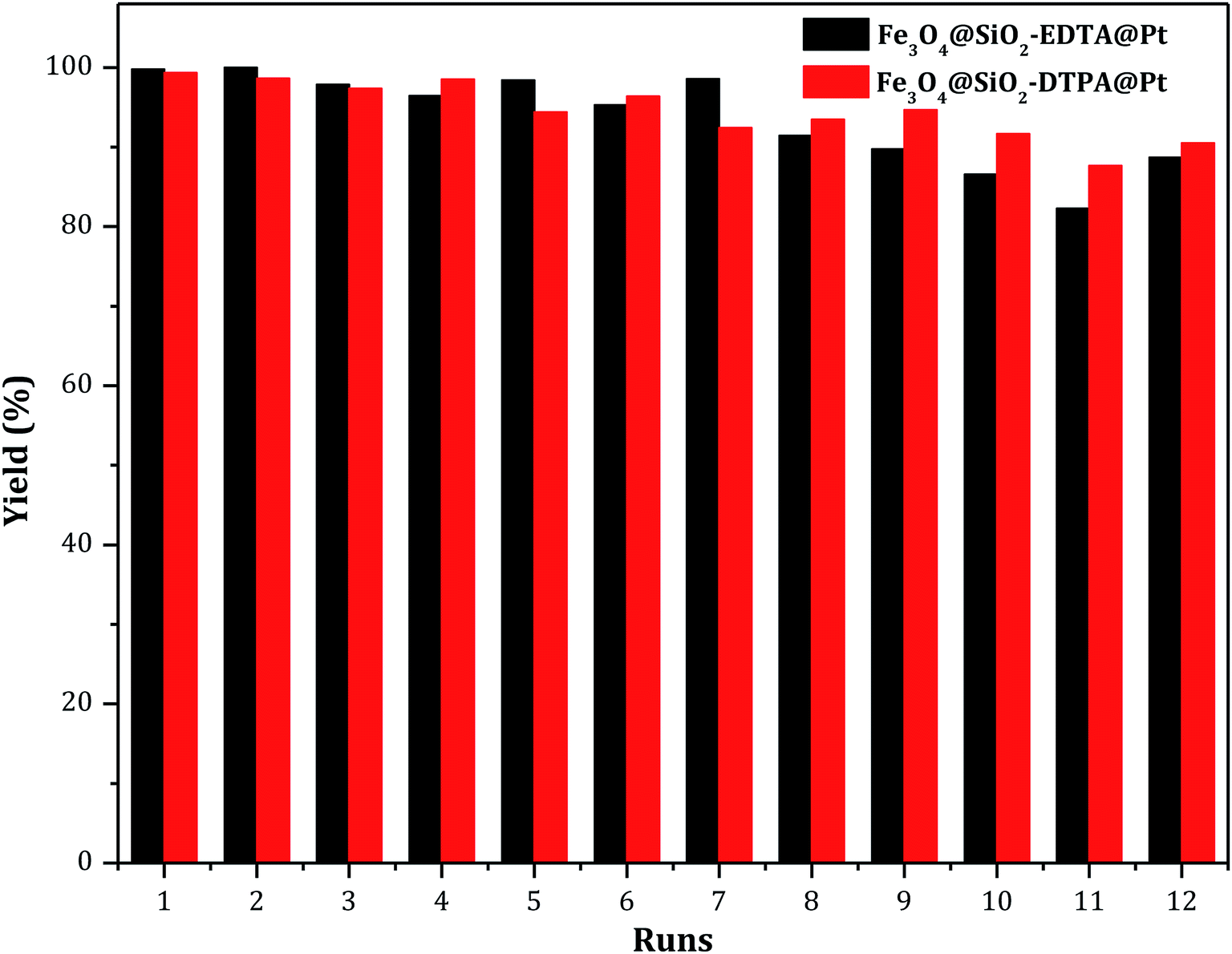 | ||
| Fig. 8 Catalyst recycle experiments for the hydrosilylation of 1-hexene with methyldichlorosilane. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Alkene | Yieldb (%) | Product | β/α | ||
| a | b | a | b | |||
| a Conditions: alkenes, 10 mmol; methyldichlorosilane, 15 mmol; 8 h; a, Fe3O4@SiO2-EDTA@Pt as catalyst; b Fe3O4@SiO2-DTPA@Pt as catalyst.b yield were determined by GC using n-decane as an internal standard. | ||||||
| 1 | 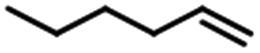 |
97 | 99 | 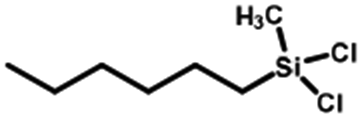 |
>20:1 |
>20:1 |
| 2 |  |
87 | 88 | >20:1 |
>20:1 |
|
| 3 | 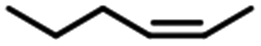 |
95 | 98 | >20:1 |
>20:1 |
|
| 4 |  |
78 | 84 | >20:1 |
>20:1 |
|
| 5 | 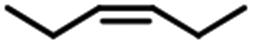 |
83 | 87 | >20:1 |
>20:1 |
|

|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Alkene | Structure | Time/h | Conv.b (%) | TOFc (103 h−1) | ||
| a | b | a | b | ||||
| a Conditions: 15 mmol methyldichlorosilane, 10 mmol alkene; temperature: 60 °C; a, Fe3O4@SiO2-EDTA@Pt as catalyst (1.74 × 10−3 mmol Pt); b, Fe3O4@SiO2-DTPA@Pt as catalyst (1.93 × 10−3 mmol Pt).b The conversion was based on the amount of alkene consumed.c Initial TOFs were calculated by GC after 30 min of reaction time. | |||||||
| 1 | Cyclohexene | 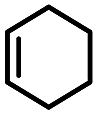 |
4 | 2 | 13 | <0.1 | <0.1 |
| 2 | N,N-Dimethylacrylamide | 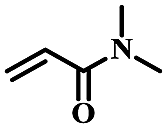 |
4 | 96 | 98 | 8.9 | 8.2 |
| 3 | 1,2-Epoxy-4-vinylcyclohexane | 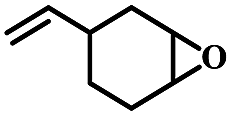 |
4 | 99 | 98 | 9.5 | 9.3 |
| 4 | Norbornylene | 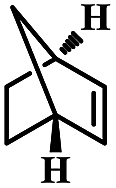 |
4 | 97 | 99 | 7.1 | 6.8 |
| 5 | Methyl methacrylate | 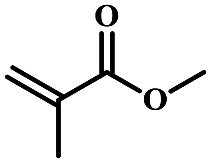 |
5 | 58 | 67 | 4.9 | 5.1 |
| 6 | Butyl acrylate | 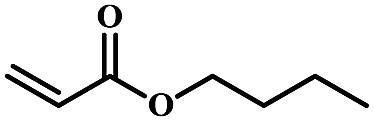 |
5 | 57 | 87 | 4.1 | 3.8 |
| 7 | Ethane-1,2-diyl bis(2-methylacrylate) | 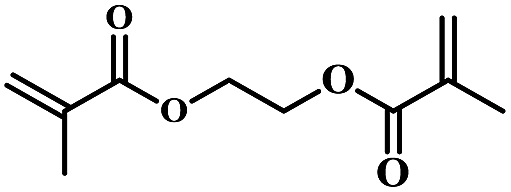 |
5 | 72 | 81 | 5.1 | 5.8 |
| 8 | Crotonic acid ethyl ester | 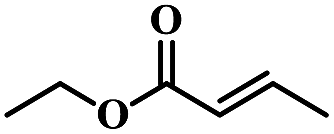 |
5 | 69 | 87 | 6.6 | 6.1 |
| 9 | trans-2-Hexenyl acetate | 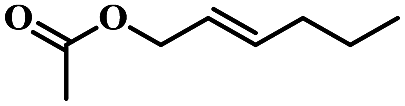 |
5 | 68 | 97 | 5.6 | 4.9 |
| 10 | trans-2-Hexen-1-yl phenylacetate | 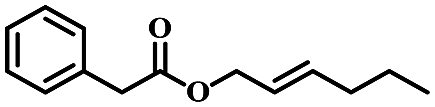 |
6 | 61 | 68 | 7.0 | 8.2 |
| 11 | 1,2-Epoxy-7-octene | 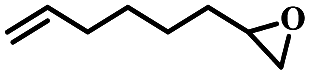 |
6 | 98 | 99 | 8.7 | 9.1 |
| 12 | Allyl glycidyl ether | 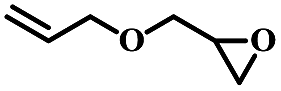 |
6 | 84 | 98 | 5.1 | 4.4 |
| 13 | 5-Hexen-2-one |  |
6 | 86 | 97 | 8.2 | 7.6 |
| 14 | Allylchloride |  |
4 | 58 | 64 | 6.2 | 6.1 |
The yield of 1-hexylmethyldichlorosilane changed in hydrosilylation of different hexane and methyldichlorosilane. When Fe3O4@SiO2-EDTA@Pt was used as catalyst, the highest yield (97%) was from 1-hexene. The yield of 1-hexylmethyldichlorosilane decreased to 95% for cis-2-hexene and 83% for cis-3-hexene. When cis-2-hexene and cis-3-hexene were changed to trans-2-hexene and trans-3-hexene, the yield further decreased corresponding to 87% and 78%. The similar phenomenon was observed when Fe3O4@SiO2-DTPA@Pt was used as catalyst. It indicated the hydrosilylation was harder for internal hexane than 1-hexene and trans-hexene than cis-hexane. The results also verified both of Pt catalysts could be used for the remote functionalization of alkenes, which has been shown to be a remarkable step in organic synthesis.71,72
4. Conclusions
Two novel magnetic Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt catalysts were successfully prepared. The saturation magnetization was 25.4 and 23.0 emu g−1 for Fe3O4@SiO2-EDTA@Pt and Fe3O4@SiO2-DTPA@Pt, respectively. The Pt amount of the 30–50 nm nanoparticles was corresponding to 173.80 and 192.77 μmol g−1, which the predominant valence was Pt2+ and Pt0. In catalyzing hydrosilylation of 1-hexene and methyldichlorosilane, the yield of 1-heptylmethyldichlorosilane was about 99% for both of catalysts and the TON were up to 662733 for Fe3O4@SiO2-EDTA@Pt and 579947 for Fe3O4@SiO2-DTPA@Pt, respectively. The two catalysts, especially Fe3O4@SiO2-DTPA@Pt, showed good activity in hydrosilylation of 5 isomerous hexanes and 14 other alkene with different substituent. Fe3O4@SiO2-DTPA@Pt even could catalyze the hydrosilylation of cyclohexene. After 12 catalytic runs for both of Pt catalysts, yield of 1-heptylmethyldichlorosilane was still up to 90% and the magnetism was similar. The average loss of Pt in each reaction was 0.87% for Fe3O4@SiO2-EDTA@Pt and 0.66% for Fe3O4@SiO2-DTPA@Pt. The unprecedented activity and selectivity of the two Pt nanoparticle catalysts are prospective and potential in catalyzing more reactions and replacing the current homogeneous Pt catalyst industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to appreciate the financial supports from the National Natural Science foundation of China (No. 21605112).References
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrence and Uses, VCH Publications, Weinheim, New York, 1996 Search PubMed.
- S. Xuan, Y. X. Wang, J. C. Yu and K. C. Leung, Langmuir, 2009, 25, 11835–11843 CrossRef CAS PubMed.
- B. Gaihre, M. S. Khil, D. R. Lee and H. Y. Kim, Int. J. Pharm., 2009, 365, 180–189 CrossRef CAS PubMed.
- N. Kohler, C. Sun, A. Fichtenholtz, J. Gunn, C. Fang and M. Zhang, Small, 2006, 2, 785–792 CrossRef CAS PubMed.
- H. Lee, M. K. Yu, S. Park, S. Moon, J. J. Min, Y. Y. Jeong, H. W. Kang and S. Jon, J. Am. Chem. Soc., 2007, 129, 12739–12745 CrossRef CAS PubMed.
- H. Liu, Z. Jia, S. Ji, Y. Zheng, M. Li and H. Yang, Catal. Today, 2011, 175, 293–298 CrossRef CAS.
- A. M. Donia, A. A. Atia and K. Z. Elwakeel, J. Hazard. Mater., 2008, 151, 372–379 CrossRef CAS PubMed.
- N. Nuryono, N. M. Rosiati, B. Rusdiarso, S. C. W. Sakti and S. Tanaka, SpringerPlus, 2014, 3, 515–526 CrossRef PubMed.
- S. Hokkanen, E. Repo, S. Lou and M. Sillanpaa, Chem. Eng. J., 2015, 260, 886–894 CrossRef CAS.
- X. Sun, Q. Li, L. Yang and H. Liu, Particuology, 2016, 26, 79–86 CrossRef CAS.
- A. Khazaei, M. Khazaei and M. Nasrollahzadeh, Tetrahedron, 2017, 73, 5624–5633 CrossRef CAS.
- Y. Fang, Y. Chen, X. Li, X. Zhou, J. Li, W. Tang, J. Huang, J. Jin and J. Ma, J. Mol. Catal. A: Chem., 2014, 392, 16–21 CrossRef CAS.
- B. Panella, A. Vargas and A. Baiker, J. Catal., 2009, 261, 88–93 CrossRef CAS.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2010, 12, 144–149 RSC.
- M. Amir, U. Kurtan and A. Baykal, Chin. J. Catal., 2015, 36, 1280–1286 CrossRef CAS.
- J. Sun, Z. Dong, X. Sun, P. Li, F. Zhang, W. Hu, H. Yang, H. Wang and R. Li, J. Mol. Catal. A: Chem., 2013, 367, 46–51 CrossRef CAS.
- J. J. Martíneza, E. X. Aguileraa, J. Cubillosa, H. Rojasa, A. Gómez-Cortésb and G. Díaz, Mol. Catal., 2019, 465, 54–60 CrossRef.
- S. A. Theofanidis, V. V. Galvita, H. Poelman, R. Batchu, L. C. Buelens, C. Detavernier and G. B. Marin, J. Mol. Catal. B: Enzym., 2018, 239, 502–512 CAS.
- Z. Mohamed, V. D. Dasireddy, S. Singh and H. B. Friedrich, Int. J. Hydrogen Energy, 2018, 43, 22291–22302 CrossRef CAS.
- B. Cheng, W. Liu and Z. Lu, J. Am. Chem. Soc., 2018, 140, 5014–5017 CrossRef CAS PubMed.
- B. Panella, A. Vargas and A. Baiker, J. Catal., 2009, 261, 88–93 CrossRef CAS.
- M. Walczak, K. Stefanowska, A. Franczyk, J. Walkowiak, A. Wawrzynczak and B. Marciniec, J. Catal., 2018, 367, 1–6 CrossRef CAS.
- T. Yang, Y. Huo, Y. Liu, Z. Rui and H. Ji, Appl. Catal., B, 2017, 200, 543–551 CrossRef CAS.
- H. A. Khan, P. Natarajan and K. Jung, Appl. Catal., B, 2018, 231, 151–160 CrossRef CAS.
- H. Zai, Y. Zhao, S. Chen, L. Ge and C. Chen, Nano Res., 2018, 61, 2544–2552 CrossRef.
- Q. Li, S. Ji, M. Li and X. Duan, Sci. China Mater., 2018, 11, 1339–1344 CrossRef.
- M. Mahmoudi, S. Sant, B. Wang, S. Laurent and T. Sen, Adv. Drug Delivery Rev., 2011, 63, 24–46 CrossRef CAS PubMed.
- R. T. Yang, Adsorbents: Fundamentals and Applications, John Wiley & Sons, USA, 2003 Search PubMed.
- A. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- G. Zeng, Y. Pang, Z. Zeng, L. Tang, Y. Zhang, Y. Liu, J. Zhang, X. Lei, Z. Li, Y. Xiong and G. Xie, Langmuir, 2012, 28, 468–473 CrossRef CAS PubMed.
- J. Ramo, M. Sillanpaa, V. Vickackaite, M. Orama and L. Niinisto, J. Pulp Pap. Sci., 2000, 26, 125–131 CAS.
- J. Ramo and M. Sillanpaa, J. Cleaner Prod., 2001, 9, 191–195 CrossRef.
- M. E. T. Sillanpaa and J. H. P. Ramo, Environ. Sci. Technol., 2001, 35, 1379–1384 CrossRef CAS PubMed.
- M. Sillanpaa, M. Orama, J. Ramo and A. Oikari, Sci. Total Environ., 2001, 267, 23–31 CrossRef CAS PubMed.
- K. Pirkanniemi, S. Metsarinne and M. Sillanpaa, J. Hazard. Mater., 2007, 147, 556–561 CrossRef CAS PubMed.
- F. Li and Y. Li, J. Mol. Catal. A: Chem., 2016, 420, 254–263 CrossRef CAS.
- D. Shao and Y. Li, RSC Adv., 2018, 8, 20379–20393 RSC.
- Z. Lu, G. Wang, J. Zhuang and W. Yang, Colloids Surf., A, 2006, 278, 140–143 CrossRef CAS.
- Y. S. Kang, S. Risbud, J. F. Rabolt and P. Stroeve, Chem. Mater., 1996, 8, 2209–2211 CrossRef CAS.
- S. Zhang, H. Niu, Y. Zhang, J. Liu, Y. Shi, X. Zhang and Y. Cai, J. Chromatogr. A, 2012, 1238, 38–45 CrossRef CAS PubMed.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- M. J. Jacinti, O. H. Santos, R. Landers, P. K. Kiyohara and L. M. Rossi, Appl. Catal., B, 2009, 90, 688–692 CrossRef.
- Z. Ma, Y. Guan and H. Liu, J. Magn. Magn. Mater., 2006, 301, 469–477 CrossRef CAS.
- Q. Yuan, N. Li, Y. Chi, W. Geng, W. Yan, Y. Zhao, X. Li and B. Dong, J. Hazard. Mater., 2013, 254–255, 157–165 CrossRef CAS PubMed.
- X. Sun, Q. Li, L. Yang and H. Liu, Particuology, 2016, 26, 79–86 CrossRef CAS.
- M. J. Jacinto, P. K. Kiyohara, S. H. Masunaga, R. F. Jardim and L. M. Rossi, Appl. Catal., A, 2008, 338, 52–57 CrossRef CAS.
- M. Tülü and K. E. Geckeler, Polym. Int., 1999, 48, 909–914 CrossRef.
- Y. Liu, M. Chen and Y. Hao, Chem. Eng. J., 2013, 218, 46–54 CrossRef CAS.
- Y. Shiraishi, G. Nishimura, A. T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 5065–5070 CrossRef CAS.
- E. Repoa, J. K. Warcholc, T. A. Kurniawana and M. E. T. Sillanpää, Chem. Eng. J., 2010, 161, 73–82 CrossRef.
- M. A. Hughes and E. Rosenberg, Sep. Sci. Technol., 2007, 42, 261–283 CrossRef CAS.
- G. Y. Li, K. L. Huang, Y. R. Jiang, D. L. Yang and P. Ding, Int. J. Biol. Macromol., 2008, 42, 405–412 CrossRef CAS PubMed.
- F. J. Arriagada and K. J. Osseo-Asare, J. Colloid Interface Sci., 1999, 211, 210–220 CrossRef CAS PubMed.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875–5879 CrossRef CAS PubMed.
- H. Pan, X. Li, D. Zhang, Y. Guan and P. Wu, J. Mol. Catal. A: Chem., 2013, 277, 108–114 CrossRef.
- S. Sayin and M. Yilmaz, J. Chem. Eng. Data, 2011, 56, 2020–2029 CrossRef CAS.
- Z. C. Li, H. T. Fan, Y. Zhang, M. X. Chen, Z. Y. Yu, X. Q. Cao and T. Sun, Chem. Eng. J., 2011, 171, 703–710 CrossRef CAS.
- H. Chen, C. Deng and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 607–611 CrossRef CAS PubMed.
- R. Qin, F. Li, M. Chen and W. Jiang, Appl. Surf. Sci., 2009, 256, 27–32 CrossRef CAS.
- A. I. Frenkel, C. W. Hills and R. G. Nuzzo, J. Phys. Chem. B, 2001, 105, 12689–12703 CrossRef CAS.
- G. Neri, C. Milone, S. Galvagno, A. P. J. Pijpers and J. Schwank, Appl. Catal., A, 2002, 227, 105–115 CrossRef CAS.
- R. Nie, D. Liang, L. Shen, J. Gao, P. Chen and Z. Hou, Appl. Catal., B, 2012, 127, 212–220 CrossRef CAS.
- Z. Yan, Z. Xu, Z. Yang, L. Yue and L. Huang, Appl. Surf. Sci., 2019, 467–468, 277–285 CrossRef CAS.
- J. Wang, S. Zheng, Y. Shao, J. Liu, Z. Xu and D. Zhu, J. Colloid Interface Sci., 2010, 349, 293–299 CrossRef CAS PubMed.
- F. Ge, M. M. Li, H. Ye and B. X. Zhao, J. Hazard. Mater., 2012, 211–212, 366–372 CrossRef CAS PubMed.
- C. J. Tan and Y. W. Tong, Anal. Chem., 2007, 79, 299–306 CrossRef CAS PubMed.
- D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154–19166 CrossRef CAS PubMed.
- W. Hu, H. Xie, H. Yue, P. Prinsen and R. Luquee, Catal. Commun., 2017, 97, 51–55 CrossRef CAS.
- C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 12108–12118 CrossRef CAS PubMed.
- I. Buslov, F. Song and X. Hu, Angew. Chem., Int. Ed., 2016, 55, 12295–12299 CrossRef CAS PubMed.
- A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209–219 CrossRef CAS PubMed.
- R. Ciriminna, V. Pandarus, G. Gingras, F. Béland and M. Pagliaro, ACS Sustainable Chem. Eng., 2013, 1, 249–253 CrossRef CAS.
- E. A. Chernyshev, Z. V. Belyakova, L. K. Knyazeva and N. N. Khromykh, Russ. J. Gen. Chem., 2007, 77, 55–61 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00375d |
| This journal is © The Royal Society of Chemistry 2019 |