
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of ultrathin MnO2 decorated graphene/carbon nanotube nanocomposites as efficient sulfur hosts for high-performance lithium–sulfur batteries†
Nan Wang ,
Sikan Peng,
Xiang Chen,
Jixian Wang,
Chen Wang,
Xin Qi,
Shenglong Dai and
Shaojiu Yan*
,
Sikan Peng,
Xiang Chen,
Jixian Wang,
Chen Wang,
Xin Qi,
Shenglong Dai and
Shaojiu Yan*
Beijing Institute of Aeronautical Materials (BIAM), Beijing, 100095, P. R. China. E-mail: shaojiuyan@126.com
First published on 21st February 2019
Abstract
Lithium–sulfur batteries are attracting significant attention due to their high theoretical specific capacity and low cost. However, their applications are hindered by the poor conductivity of sulfur and capacity fading caused by the shuttle effect. Here, ultrathin manganese dioxide decorated graphene/carbon nanotube nanocomposites are designed as sulfur hosts to suppress the shuttle effect and improve the adsorption efficiency of polysulfides. The graphene/carbon nanotube hybrids, with extraordinary conductivity and large surface area, function as excellent channels for electron transfer and lithium ion diffusion. The ultrathin manganese dioxide nanosheets enable efficient chemical interaction with polysulfides and promote the redox kinetics of polysulfides. As a result, an ultrathin manganese dioxide decorated graphene/carbon nanotube sulfur composite with high sulfur content (81.8 wt%) delivers a high initial specific capacity of 1015.1 mA h g−1 at a current density of 0.1C, high coulombic efficiency approaching 100% and high capacity retention of 84.1% after 100 cycles. The nanocomposites developed in this work have promising applications in high-performance lithium–sulfur batteries.
1. Introduction
The lithium–sulfur (Li–S) battery is a promising candidate for next-generation commercial rechargeable batteries because of its high theoretical specific capacity (1675 mA h g−1) and superior specific energy (2600 W h kg−1).1–3 It has great potential applications in electric vehicles and renewable energy storage. Moreover, sulfur, one of the most abundant elements on Earth, is environmentally friendly and inexpensive. However, the commercialization of the Li–S battery is hindered by two major challenges: (1) S and its discharge products (Li2S2/Li2S) are intrinsically non-conducting, which lowers the S utilization and limits the battery rate performance; (2) the dissolution of lithium polysulfide species (LiPSs) in the electrolyte results in the shuttle effect and uncontrolled deposition of Li2S2 and Li2S, which is responsible for low coulombic efficiency and severe capacity fading of the battery.4,5 In an attempt to tackle these issues, various carbon materials such as activated carbon,6 mesoporous carbon,7 hollow carbon sphere,8 carbon nanotubes (CNTs)9,10 and graphene11 have been employed as sulfur host and conductive framework to suppress shuttle effect and enhance redox kinetics of LiPSs. The carbon materials can accelerate the electron transfer, however, the intrinsically nonpolar carbon hosts exhibit rather weak physisorption toward LiPSs, which cannot efficiently suppress the polysulfides shuttle effect and accomplish the controlled deposition of Li2S2 and Li2S.12,13 Although LiNO3 was added in the electrolyte to protect anode by forming solid electrolyte interphase (SEI) layer, its result is still limited in long-term cycle. Therefore, materials with strong chemical adsorption toward LiPSs are expected to achieve stable cycle performance for the Li–S batteries.Recently, metal oxides such as TiO2,1 MnO2,14 Mn3O4,15 Co3O4,16 VO2,17 and V2O5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 have been found to effectively mitigate LiPSs shuttling via strong chemical interaction with the LiPSs, because of their inherently hydrophilic surfaces.19 The cycling performance of Li–S batteries has been significantly improved by chemical adsorption of metal oxides in immobilizing LiPSs.20 Nevertheless, the poor conductivity of metal oxides inevitably lowers the LiPSs redox reactions and compromises the rate capability of Li–S batteries.21,22 Recently, the performance of Li–S batteries has been improved by coupling polar metal oxides with conductive carbon materials, which enhance the conductivity of carbon–metal oxide composite as sulfur host. As an effective trapper for LiPSs, MnO2 has been widely applied in Li–S batteries because of its earth abundance, low-cost and environmental friendliness. Nazar and co-workers recently reported that MnO2 exhibited strong chemical trapping of LiPSs, with thiosulfate (S2O32−) serving as a surface-bound intermediate arising from the LiPSs reacting on the MnO2 surface and promoted the controlled deposition of Li2S2/Li2S.14 Subsequently, they proved that MnO2 with a redox potential E of 3.05 V lies in the target window (2.4 V < E ≤ 3.05 V) directly correlated to the chemical interaction of polysulfides.17 Liu and co-workers proved the enhanced electrochemical kinetics by using carbon nanofibers supported MnO2 composite.23 Ultrathin MnO2 was employed in Li–S batteries and achieved a high capacity retention of 76.1% at 0.2C after 100 cycles.24 Furthermore, large surface area, and uniform distribution of MnO2 were also confirmed to enhance the adsorption ability towards LiPSs and facilitate the lithium ion diffusion.21,24,25
18 have been found to effectively mitigate LiPSs shuttling via strong chemical interaction with the LiPSs, because of their inherently hydrophilic surfaces.19 The cycling performance of Li–S batteries has been significantly improved by chemical adsorption of metal oxides in immobilizing LiPSs.20 Nevertheless, the poor conductivity of metal oxides inevitably lowers the LiPSs redox reactions and compromises the rate capability of Li–S batteries.21,22 Recently, the performance of Li–S batteries has been improved by coupling polar metal oxides with conductive carbon materials, which enhance the conductivity of carbon–metal oxide composite as sulfur host. As an effective trapper for LiPSs, MnO2 has been widely applied in Li–S batteries because of its earth abundance, low-cost and environmental friendliness. Nazar and co-workers recently reported that MnO2 exhibited strong chemical trapping of LiPSs, with thiosulfate (S2O32−) serving as a surface-bound intermediate arising from the LiPSs reacting on the MnO2 surface and promoted the controlled deposition of Li2S2/Li2S.14 Subsequently, they proved that MnO2 with a redox potential E of 3.05 V lies in the target window (2.4 V < E ≤ 3.05 V) directly correlated to the chemical interaction of polysulfides.17 Liu and co-workers proved the enhanced electrochemical kinetics by using carbon nanofibers supported MnO2 composite.23 Ultrathin MnO2 was employed in Li–S batteries and achieved a high capacity retention of 76.1% at 0.2C after 100 cycles.24 Furthermore, large surface area, and uniform distribution of MnO2 were also confirmed to enhance the adsorption ability towards LiPSs and facilitate the lithium ion diffusion.21,24,25
These results inspired us to design and construct a hybrid structure with ultrathin MnO2 nanosheets uniformly distributed on the highly conductive carbon materials. Graphene/carbon nanotube (G/CNT) hybrids with CNTs covalently anchored on the graphene sheets, which have been proven to possess large surface areas, high porosity and outstanding conductivity,26,27 are selected as conductive framework. In particular, G/CNT hybrids have been applied for Li–S batteries, which exhibit a high rate performance.28 Until now, significant improvements on long-term cycling performance have been obtained by introducing MnO2 as the trapper of LiPSs, however, a rapid capacity fading in the initial several cycles caused by insufficient adsorption are still worth of further investigation. In addition, high addition of MnO2 will lower the energy density of the whole battery which is unsuitable for application of Li–S batteries. In this respect, more attentions should be paid to improving the adsorption efficiency which are identified by the strong interaction with sufficient adsorption sites as well as the fast conversion of LiPSs.
In this study, we report ultrathin MnO2 nanosheets decorated G/CNT nanocomposites with efficient immobilization and improved redox kinetics of LiPSs as sulfur host for Li–S batteries. Several advantages can be expected for Li–S batteries. Firstly, G/CNT hybrids are selected as the framework because of their large specific surface area and porous structures, which should be beneficial for uniform distribution of MnO2 nanosheets and high sulfur loading. Secondly, the composites demonstrate extraordinary electrical conductivity due to the unique three-dimensional (3D) conductive network, which should significantly enhance the charge transfer and facilitate the conversion of LiPSs. Finally, ultrathin MnO2 nanosheets distributed uniformly on the surface of the carbon framework serve as anchoring points for LiPSs. That is, the ultrathin MnO2 nanosheets with large surface areas maximize the chemical interaction between MnO2 and LiPSs, which is expected to remarkably suppress shuttling effect and improve the adsorption efficiency. The G/CNT@MnO2 composites as sulfur host prepared by this facile method is expected to demonstrate a promising performance of Li–S batteries.
1.1. Synthesis of G/CNT@MnO2 and pure MnO2
G/CNT obtained from Beijing Beifang Guoneng New Material Co., Ltd., were used as received, and the typical preparing procedures can be referenced to the method as reported previously.28 First, 100 mg G/CNT was dispersed in 500 ml of distilled water. This was followed by a 10 min ultrasonic treatment to form a stable dispersion solution, which was then transferred for magnetic stirring. Excessive ethanol was added dropwise under vigorous stirring. Finally, 27 mg of KMnO4 was added to the above dispersion solution. The reaction was performed at ambient temperature for 12 h. The obtained precipitate was filtered with vacuum and washed, first with distilled water and then ethanol. Finally, the purified precipitation was desiccated at 80 °C for 12 h in a thermoelectric thermostat drying oven. Hence, a dark powder was obtained. Pure MnO2 was synthesized through the same method without adding G/CNT hybrids.1.2. Synthesis of G/CNT@S, G/CNT@MnO2@S composites
The carbon–S composites were prepared via a typical melt diffusion strategy. The as-prepared samples (G/CNT, G/CNT@MnO2) were first grinded with pure sulfur powder with a C/S mass ratio of 1:5. Subsequently, the mixture was transferred into a stainless-steel autoclave and sealed in a glovebox. Then, the autoclave was maintained at 155 °C for 24 h.
1.3. Preparation of Li2S6 additive electrolyte
The experiment was conducted in a glove box filled with argon (Ar). First, a 0.04 mol L−1 Li2S6 solution was prepared by adding 92 mg (2 mol) of Li2S and 320 mg (10 mol) sublimed sulfur to 50 ml dimethoxyethane (DME) solution with vigorous stirring. Then, 1 ml of 0.04 mol L−1 Li2S6 solution was diluted into 0.004 mol L−1 using 9 ml of DME. Next, 40 mg G/CNT and G/CNT@MnO2 composites were added to different samples of diluted Li2S6 solution, followed by stirring for 2 h. The supernatant was used for test in each case.1.4. Characterization
The morphologies of the as-made samples were characterized using field emission scanning electron microscopy (FESEM; FEI NOVA NanoSEM 450). Further, transmission electron microscopy (TEM) and selected area electron diffraction (SAED) observations were executed utilizing a transmission electron microscope (FEI Tecnai G2 F30) at an accelerating voltage of 100 kV, which was equipped with a high-resolution accessory. Powder X-ray diffraction (XRD) patterns were obtained using a diffractometer (Bruker D8 advance) with copper K radiation (30 kV, 30 mA, λ = 1.5406 Å). In addition, Raman scattering spectra were taken at room temperature using a LabRAM HR800 Raman spectrometer (HORIBA, Japan). The thermal decomposition was investigated with a differential scanning calorimeter (DSC, NETZSCH) using an aluminum (Al) crucible. The specific surface area and pore size distribution were calculated based on the nitrogen (N) physical adsorption with a Kubo-X1000. XPS measurement was performed on a Thermo ESCALAB 250Xi with a monochromatic Al Kα source. Ultraviolet/visible (UV/vis) spectra were obtained using a Perkin-Elmer Lambda 950 spectrometer.1.5. Preparation of electrodes and coin cells
The working electrode films were prepared by casting a uniform slurry of the active material (G/CNT@S, G/CNT@MnO2@S), polyvinylidene difluoride, and HS-100 acetylene black (Denka, Japan) at a weight ratio of 80:10:10 in an N-methyl-2-pyrrolidinone solvent onto carbon-coated Al foil via the doctor blade casting method. The electrode film was dried thoroughly at 60 °C for 24 h in a vacuum oven and cut into round disks with 14 mm diameter. Then, 2016-type button cells were assembled, with Li foil as the anode, Celgard® 2325 membrane as the separator, and 1.0 mol L−1 Li bis(trifluoromethanesulfonyl)imide (LiTFSI) in DME/1,3-dioxolane (DOL) (1:1 v/v, with 1.0 wt% LiNO3) as the electrolyte with electrolyte/sulfur ratio of 12 μl mg−1. The final sulfur loading in the cathode is calculated to be about 65.4%. The areal sulfur loading was kept between 1.5 and 2.0 mg cm−2.
1.6. Electrochemistry
The cycling performance and rate tests were performed using a LAND battery program-control test system (CT2001 A, Wuhan LAND Electronic Co., Ltd., China) within the voltage range of 1.7–2.8 V. The specific capacity values were calculated according to the mass of sulfur in the cathode. Cyclic voltammetry (CV) measurements were conducted using a CHI1000C instrument (Chenhua, China) between 1.7 and 2.8 V vs. Li+/Li at a scanning rate of 0.1, 0.2, 0.3, 0.4, 0.6, 0.8 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were performed using an electrochemical workstation (Zahner-Zennium, Germany) under the open circuit voltage over a frequency range of 100 kHz to 100 mHz. The amplitude of the alternating-current (AC) voltage was 5 mV.2. Results and discussion
The synthesis process of the G/CNT@MnO2@S composites is illustrated in Fig. 1. For comparison, G/CNT@S composites without MnO2 decorating were also synthesized, as mentioned above. Firstly, the G/CNT hybrids fabricated via CVD method were selected as hosts for MnO2 and sulfur.28 Subsequently, ultrathin MnO2 nanosheets were anchored in situ on the surface of G/CNT hybrids through redox deposition to ensure their uniform distribution. The proposed redox reactions are as follows:29–31| 4MnO4− + 2OH− + 3C → 3CO32− + H2O + 4MnO2↓ | (1) |
| 4MnO4− + H+ + 3CH3CH2OH → 3CH3COO− + 5H2O + 4MnO2↓ | (2) |
 | ||
| Fig. 1 Schematic illustration of G/CNT@MnO2@S nanocomposite synthetic process. | ||
Note that carbon acts as the reducing agent and the substrate for the heterogeneous nucleation of MnO2. Further, the excessive ethanol protects the carbon material from over-oxidation and reduces potassium permanganate to form MnO2. Finally, sulfur nanoparticles were coated uniformly on the surface of the G/CNT@MnO2 composites using the melt diffusion method.28
Fig. 2 shows SEM and TEM images of typical G/CNT hybrids and G/CNT@MnO2 composites. The G/CNT hybrids, which have crumpled sheet-like porous structures, are composed of few-layer graphene and multiwalled CNTs (Fig. 2a–c). The CNTs covalently anchored on both sides of graphene flakes constructed a 3D architecture, which could inhibit the aggregation of both CNTs and graphene flakes and possess high surface area, porous structure and high conductivity (Fig. 2 and S1†). The G/CNT hybrids exhibit specific surface area of 1519.9 m2 g−1, with a large total pore volume of 2.56 cm3 g−1. The large specific surface area and sufficient porosity of the G/CNT hybrids indicate their feasibility for efficient sequestration and adsorption of highly loaded sulfur as well as uniform deposition of MnO2 nanosheets.32 Moreover, the crumpled sheet-like graphene and CNT construct a 3D conductive network which could provide large amount of charge transfer channels and Li+ diffusion channels, thus suggesting high rate performance and rapid conversion of LiPSs.
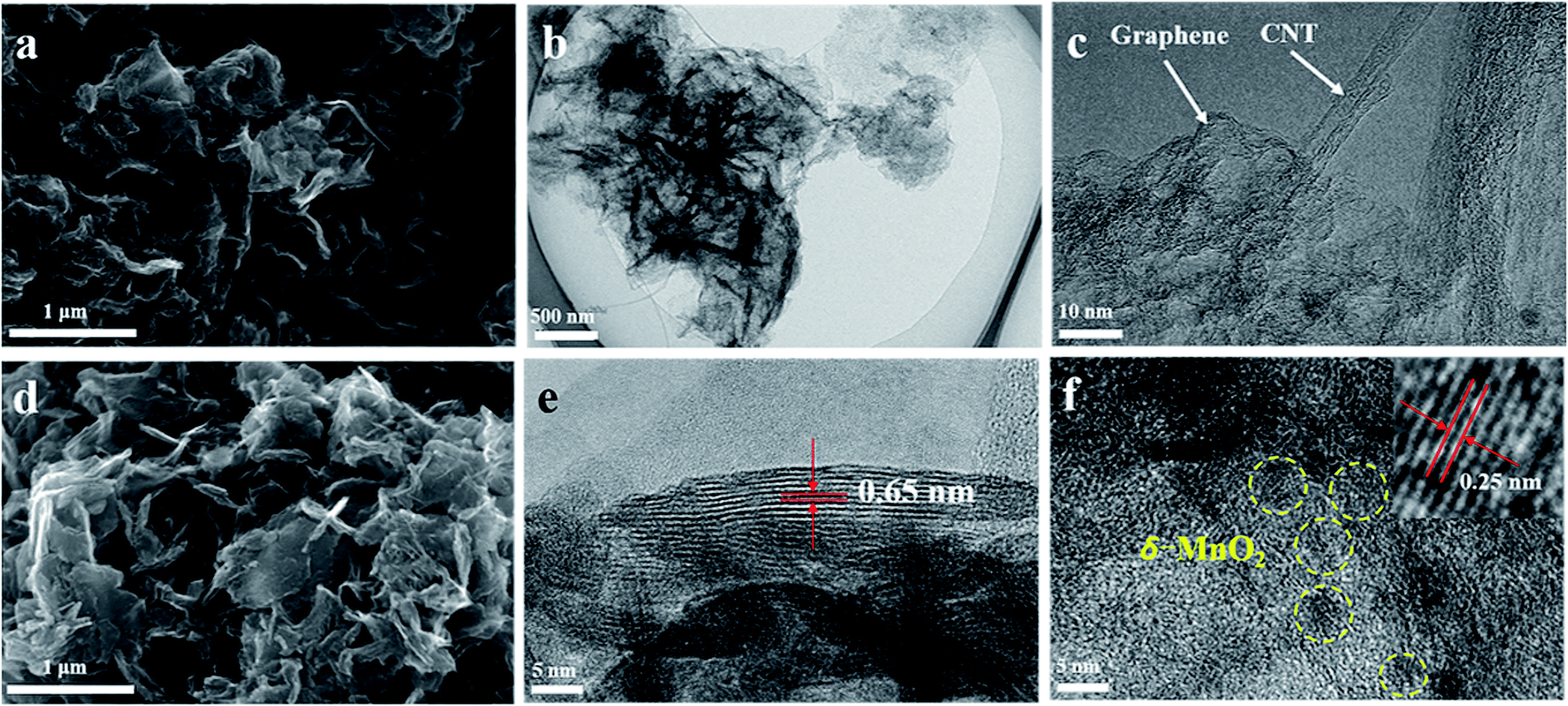 | ||
| Fig. 2 (a) FESEM image of G/CNT. (b and c) TEM images of G/CNT. (d) FESEM image of G/CNT@MnO2. (e and f) TEM images of G/CNT@MnO2. | ||
In situ redox deposition was adopted to obtain ultrathin MnO2 nanosheets and their uniform distribution. No obvious bulk MnO2 were observed in the SEM image Fig. 2d indicates that MnO2 nanosheets were uniformly anchored on the surface of G/CNT hybrids. Further, TEM was employed to explore the structure details of MnO2. The ultrathin MnO2 nanosheets with a lamellar structure and flexuous shape are observed in Fig. 2e, which are typical characteristics viewed along a direction parallel to the (001) plane.33–35 The thickness of MnO2 nanosheets is measured to be 4 nm on average, for which the stripe spacing of (001) plane is further measured to be 0.65 nm. In Fig. 2f, the lattice fringes having an interplanar spacing of 0.25 nm correspond to a δ-MnO2 crystal which shows uniform distribution on the surface of G/CNT. The corresponding element mapping further confirms the uniform distribution of MnO2 in the composites of G/CNT@MnO2, which consistent with the results of SEM and TEM (Fig. S2†). The formation of ultrathin MnO2 nanoflakes is likely due to the self-assembly of δ-MnO2 nano-grains, which is controlled by a Coulomb interaction and driven by the demand for surface energy reduction.33 It is reported that the Li2Sx capture is dominated by monolayer chemisorption.21 Therefore, ultrathin MnO2 nanosheets with large surface area provide sufficient interfaces for chemical adsorption and reduce the addition of MnO2, suggesting an enhancement of adsorption efficiency and improvement of specific energy of the batteries.
XRD measurements were conducted to determine the phase structure of the MnO2 anchoring on the surface of G/CNT hybrids. Peaks centered at 12.5°, 25.2°, 37.3° and 65.6° were found, which match well with the (001), (002), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11) and (020) reflections of monoclinic birnessite, δ-MnO2 (JCPDS no. 80-1098). This result reveals that the as-prepared δ-MnO2 anchoring on the G/CNT is almost X-ray amorphous, indicating that δ-MnO2 is composed of crystalline and amorphous parts, as reported previously (Fig. 3a).29 It has also been reported that poorly crystalline form of manganese oxide will occur under low-temperature synthesis.36
11) and (020) reflections of monoclinic birnessite, δ-MnO2 (JCPDS no. 80-1098). This result reveals that the as-prepared δ-MnO2 anchoring on the G/CNT is almost X-ray amorphous, indicating that δ-MnO2 is composed of crystalline and amorphous parts, as reported previously (Fig. 3a).29 It has also been reported that poorly crystalline form of manganese oxide will occur under low-temperature synthesis.36
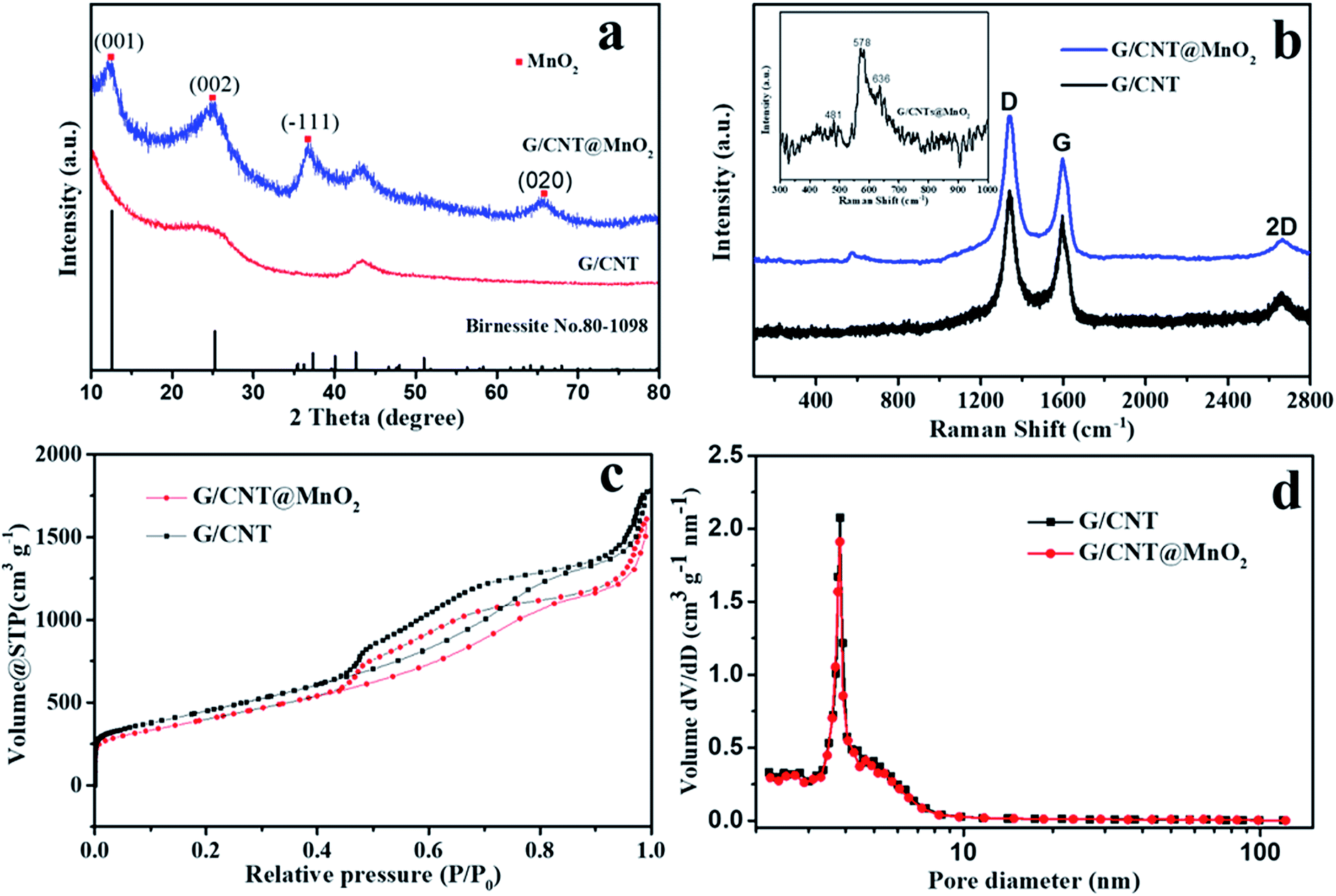 | ||
| Fig. 3 (a) XRD patterns of G/CNT and G/CNT@MnO2. (b) Raman spectra of G/CNT and G/CNT@MnO2, inset image is magnification Raman shift of G/CNT@MnO2. (c) N2 adsorption/desorption isotherms for G/CNT and G/CNT@MnO2. (d) BJH pore size distribution for G/CNT, G/CNT@MnO2. | ||
Raman technique is useful for analyzing the local structures of manganese dioxides, especially for samples with poor crystallinity.37 Therefore, Raman technique was applied to analyze the as-prepared G/CNT@MnO2 composites (Fig. 3b). Raman bands at 1346, 1597 and 2666 cm−1 are related to the D-band, G-band and 2D band of carbon materials in G/CNT hybrids. Furthermore, the G/CNT@MnO2 composites demonstrate higher intensity ratio of D-band and G-band ID/IG (1.40) compared with G/CNT hybrids (1.19). This result indicates that the anchoring of MnO2 on the surface of G/CNT caused more disordered carbon generated from the reaction of carbon and MnO4−.38,39 As shown in the inset of Fig. 3b, three Raman bands located at 481, 578, and 636 cm−1 are in agreement with the three major vibration features of δ-MnO2, which proves the presence of δ-MnO2 and is in good accord with the result of XRD.40 Further, the results of thermogravimetric analysis (TGA) demonstrate that the MnO2 content in the G/CNT@MnO2 composites is approximately 21 wt%, including 7 wt% water contained within the δ-MnO2 (Fig. S3†). As reported in the literature, water molecules and cationic species are located between the sheets of MnO6 octahedra to balance the charge.41 The optimized MnO2 content of 21 wt% in G/CNT@MnO2 composites is in aim to balance the adsorption ability and the conductivity.
An N2 adsorption analysis was performed to study the porosity of the G/CNT hybrids and G/CNT@MnO2 composites (Fig. 3). A typical type-IV isotherm with a type-H3 hysteresis loop was obtained from the results of G/CNT and G/CNT@MnO2 composites, indicating that the two composites are mainly composed of a mesoporous structure (Fig. 3c). The specific surface area of the G/CNT@MnO2 was calculated to be 1431.8 m2 g−1 via the Brunauer–Emmett–Teller (BET) method, being slightly lower than that of the G/CNT hybrids (1519.3 m2 g−1). To evaluate the specific surface area of MnO2 nanosheets on the surface of G/CNT, pure MnO2 nanosheets were also revealed by BET method for reference which was calculated to be 114.3 m2 g−1 (Fig. S4†). In addition, a typical type-III isotherm was obtained from the pure MnO2 nanosheets indicating a nonporous structure which consists well with the results observed from the TEM images. Concluded from the BET results of G/CNT@MnO2 composites and pure MnO2, the MnO2 nanosheets on the surface of G/CNT possess an ultrathin structure and large surface area which will provide sufficient adsorption sites, thus enhance the adsorption efficiency of LiPSs on the surface of MnO2 nanosheets. The total pore volume of G/CNT@MnO2 was calculated to be 2.49 cm3 g−1 nm−1 and its Barrett–Joyner–Halenda (BJH) pore size distribution is shown in Fig. 3d. The result shows slightly pore volume decreasing after MnO2 coating compared with that of G/CNT (2.56 cm3 g−1 nm−1), but identical pore size distribution. The obtained results indicate that the G/CNT@MnO2 composites still maintain a large surface area, pore volume and pore size distribution for high-content sulfur loading.
Next, sulfur was introduced into the G/CNT@MnO2 composites by melt-diffusion method. The SEM and TEM images (Fig. 4a and b) show that sulfur particles were uniformly distributed, and no agglomerated sulfur particles were observed on the surfaces of the G/CNT@MnO2@S composites which was further confirmed by the element mapping shown in Fig. S2.† Also, nanocrystal sulfur particles with an average diameter of 10 nm were observed in Fig. 4b. In addition, high-resolution TEM (HRTEM) imagery revealed that lattice fringes corresponding to interplanar spacings of 0.477 and 0.1789 nm is consistent with the (200) and (![[7 with combining macron]](https://www.rsc.org/images/entities/char_0037_0304.gif) 1) planes of orthorhombic sulfur, respectively (Fig. 4c). The presence of nano-size sulfur particles and their uniform distribution is favorable for improving the utilization of sulfur and reinforcing the cycling stability and rate performance of Li–S batteries.42
1) planes of orthorhombic sulfur, respectively (Fig. 4c). The presence of nano-size sulfur particles and their uniform distribution is favorable for improving the utilization of sulfur and reinforcing the cycling stability and rate performance of Li–S batteries.42
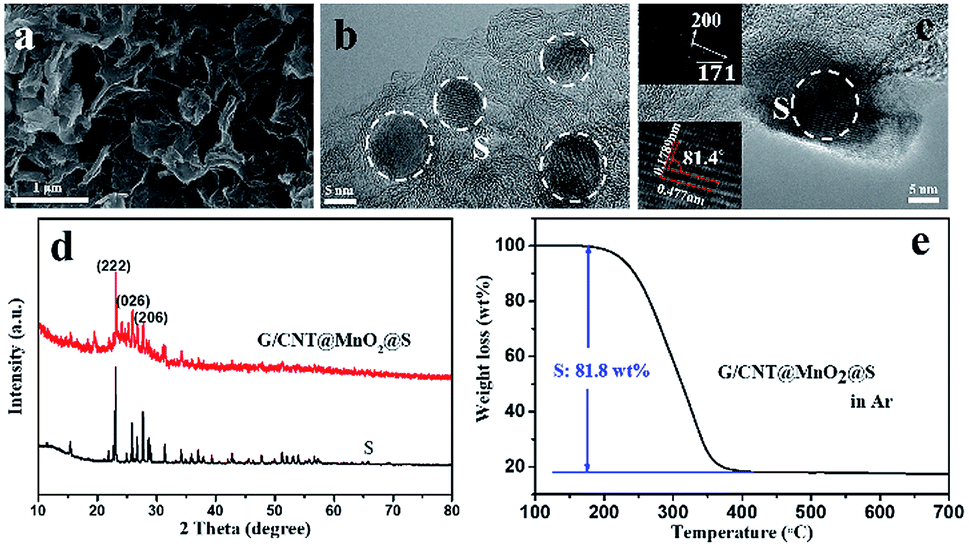 | ||
| Fig. 4 (a) FESEM image. (b and c) TEM images. (d) XRD patterns and (e) TGA of G/CNT@MnO2@S. | ||
The XRD pattern of the G/CNT@MnO2@S composites shows that the sulfur in the composites has the same orthorhombic structure as sulfur powder (Fig. 4d). Further, the Raman bands of the G/CNT@MnO2@S composites are in good accordance with the three major bands of pure sulfur, also confirming the presence of sulfur in the composites (Fig. S5†). The surface chemical state of the G/CNT@MnO2@S composites were further revealed by X-ray photoelectron spectroscopy (XPS). The survey spectrum confirms that the main elements of Mn, O, C, S coexist in the G/CNT@MnO2@S composites (Fig. S6a†). In the C 1s spectrum (Fig. S6b†), the main peak at 284.8 eV is corresponding to sp2 carbon (C–C or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in graphene and carbon nanotubes. The peaks at 285.9 and 289.2 eV can be ascribed to carbon in C–O/C–S bounds and the carboxyl carbon (O–CO) group, respectively.43 The small amount of carbon species may be generated during carbon oxidation. As shown in Fig. S6c,† the peaks centered at 642.6 and 654.1 eV can be assigned to Mn 2p2/3 and Mn 2p1/2, which are both contributed by Mn4+ and reconfirm the existence of MnO2.43 In the S 2p spectrum (Fig. S6d†), the peaks located at 164.1 and 165.3 are corresponding to S–S group, whereas the peak at 169.1 eV can be attributed to the bond of sulfate species. The sulfate species may be generated from the redox interaction between S and MnO2, which will facilitate the immobilization of LiPSs during cycling of Li–S batteries.25 The sulfur loading of the G/CNT@MnO2@S composites was determined to be approximately 81.8 wt% via TGA (Fig. 4e). And the sulfur content in the composite was further confirmed using elemental analysis (Table S1†). The high sulfur loading indicates a promising performance for practical application of Li–S batteries. After sulfur infiltration, the isotherm changed from type-IV to type-III, and the specific surface area and pore volume of the G/CNT@MnO2@S composites sharply decreased to 18.4 m2 g−1 and 0.21 cm3 g−1 nm−1 respectively, suggesting that S8 molecules infiltrated the mesoporous parts of the G/CNT@MnO2 composites (Fig. S7a and b†).
C) in graphene and carbon nanotubes. The peaks at 285.9 and 289.2 eV can be ascribed to carbon in C–O/C–S bounds and the carboxyl carbon (O–CO) group, respectively.43 The small amount of carbon species may be generated during carbon oxidation. As shown in Fig. S6c,† the peaks centered at 642.6 and 654.1 eV can be assigned to Mn 2p2/3 and Mn 2p1/2, which are both contributed by Mn4+ and reconfirm the existence of MnO2.43 In the S 2p spectrum (Fig. S6d†), the peaks located at 164.1 and 165.3 are corresponding to S–S group, whereas the peak at 169.1 eV can be attributed to the bond of sulfate species. The sulfate species may be generated from the redox interaction between S and MnO2, which will facilitate the immobilization of LiPSs during cycling of Li–S batteries.25 The sulfur loading of the G/CNT@MnO2@S composites was determined to be approximately 81.8 wt% via TGA (Fig. 4e). And the sulfur content in the composite was further confirmed using elemental analysis (Table S1†). The high sulfur loading indicates a promising performance for practical application of Li–S batteries. After sulfur infiltration, the isotherm changed from type-IV to type-III, and the specific surface area and pore volume of the G/CNT@MnO2@S composites sharply decreased to 18.4 m2 g−1 and 0.21 cm3 g−1 nm−1 respectively, suggesting that S8 molecules infiltrated the mesoporous parts of the G/CNT@MnO2 composites (Fig. S7a and b†).
Next, the electrochemical performance of the G/CNT@MnO2@S nanocomposites as cathode material for Li–S batteries was evaluated. To demonstrate the advantages of the MnO2 anchoring effect, a similar G/CNT@S nanocomposites with sulfur loading of approximately 81.4 wt% was also synthesized and evaluated as a comparison. The CV profiles of the G/CNT@MnO2@S cathode at 0.1 mV S−1 scan rate in the potential range of 1.7–2.8 V vs. Li/Li+ for the first four cycles are shown in Fig. 5a. Two typical reduction peaks at 2.31 and 2.04 V were observed, because of the multiple reduction of sulfur. The peak at 2.31 V is related to the reduction of elemental S (S8) to high-order polysulfides (Li2Sx, x ≥ 4), while the peak at 2.04 V is attributed to the further reduction of high-order polysulfides to low-order polysulfides (Li2Sx, 1 < x < 4) and Li2S.44 The oxidation peaks at 2.38 V correspond to the reverse oxidation of polysulfide (Li2S/Li2S2) to the neutral S8. In the four cycles, both the CV peak positions and area remained almost identical, suggesting good capacity stability and high reversibility of the G/CNT@MnO2@S cathode.
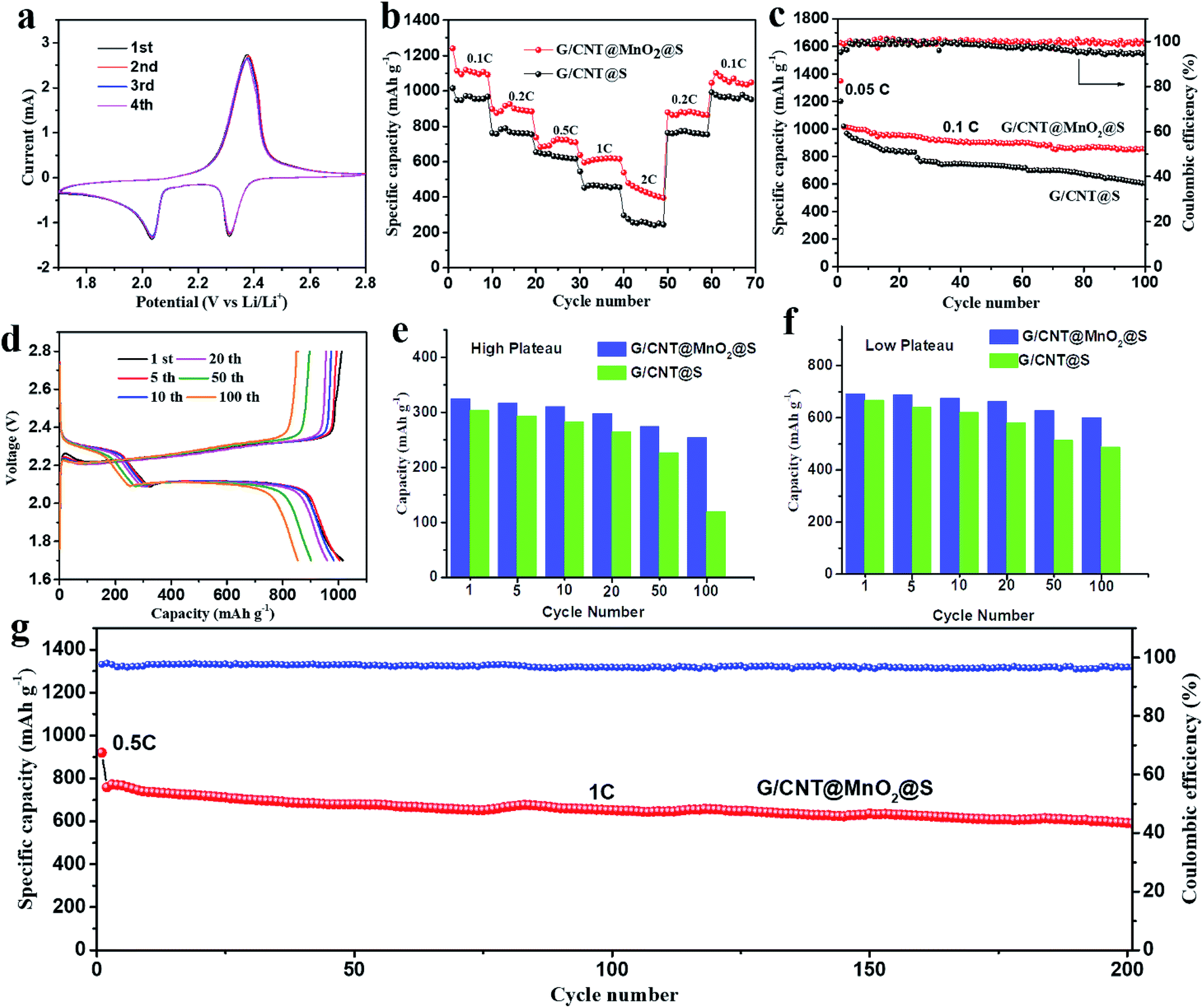 | ||
| Fig. 5 (a) Cyclic voltammetry curves of G/CNT@MnO2@S cathode swept in voltage range of 1.7–2.8 V vs. Li/Li+ at a 0.1 mV s−1 sweep rate. (b) Rate performance of G/CNT@MnO2@S cathode at C rates of 0.1–2C. (c) Cycle capacity and coulombic efficiency of G/CNT@MnO2@S in comparison with G/CNT@S at 0.1C current density. (d) Voltage profiles of G/CNT@MnO2@S at 0.1C. (e and f) High and low plateau discharge capacity for G/CNT@MnO2@S and G/CNT@S. (g) Cycle capacity and coulombic efficiency of G/CNT@MnO2@S cathode at 1C current density. | ||
The rate capability of the G/CNT@MnO2@S cathode was evaluated by cycling at various current rates from 0.1 to 2C every 10 cycles, followed by recovery to 0.2C and 0.1C (Fig. 5b). This electrode delivered capacities of 1240.6, 897.5, 737.3, 638.1, and 538.3 mA h g−1 at 0.1C, 0.2C, 0.5C, 1C, 2C rates, respectively. Notably, after the C-rate is lowered back to 0.2C and 0.1C, the cathode can recover to 879.0 and 1045.7 mA h g−1 with stabilized capacity, which indicates the excellent electronic/ionic transport properties and reaction kinetics. In contrast, the discharge capacity of G/CNT@S electrode decreases rapidly with the increase of discharging rates. Furthermore, the galvanostatic charge–discharge voltage profiles of G/CNT@MnO2@S electrode evaluated at various current rates (from 0.1C to 2C) are shown in Fig. S8a.† Two typical discharge plateaus at ≈2.33 and 2.08 V were observed in the discharge profile, corresponding to the production of high-order polysulfides and the subsequent deposition of low-order Li–S compounds, which agree well with the CV results described above. The galvanostatic charge–discharge voltage profiles of the G/CNT@S electrode are shown in Fig. S8b† (from 0.1C to 2C). Notably, the G/CNT@S cathode shows short discharge plateaus under a high rate of 2C compared to G/CNT@MnO2@S cathode, indicating a higher polarization occurs.
Fig. 5c shows the cycling performance of the G/CNT@S and G/CNT@MnO2@S cathodes at a current density of 0.1C (1C = 1675 mA h g−1) in the voltage range of 1.7–2.8 V. The first cycle was charged and discharged at a current density of 0.05C and delivered a high capacity of 1349 mA h g−1, which is 80.5% of theory capacity, indicating a high sulfur utilization. The G/CNT@MnO2@S cathode delivered a high initial discharge capacity of 1015.1 mA h g−1 at a current density of 0.1C and remained at a capacity of 853.6 mA h g−1 after 100 cycles, corresponding to a high capacity retention of 84.1%. The coulombic efficiency was consistently near 100%, indicating that the shuttle effect was effectively restrained through the anchoring effect of the MnO2 on the LiPSs. In contrast, the G/CNT@S electrode obtained a discharge capacity of 1021.4 mA h g−1 in the first cycle and suffered from a rapid decay after 100 cycles, with a remaining capacity of 605.1 mA h g−1 and a capacity retention of only 59.2%. The coulombic efficiency decreases rapidly after 50 cycles. Furthermore, the capacity retention of G/CNT@MnO2@S cathode remains as high as 94% after 20 cycles and shows a smooth change compare with that of G/CNT@S cathode, which confirm a high adsorption efficiency of ultrathin MnO2 nanosheets to LiPSs (Fig. S9†). The high capacity retention of G/CNT@MnO2@S cathode could be ascribed to ultrathin MnO2 nanosheets with large surface area which tender sufficient adsorption sites for LiPSs. Although LiNO3 was added in the electrolyte to protect lithium anode from being consumed by forming SEI film, its role of suppressing shuttle effect is limited on long-term and high rate cycling due to the additive consumption.45,46 However, the positive effect of LiNO3 on improving safety of the batteries for application is worth considering.47 The MnO2 contribution to the capacity could be ignored, because of this material's low specific capacity and low ratio in the composites.45,48
The galvanostatic charge–discharge voltage profiles of G/CNT@MnO2@S during cycles at 0.1C are shown in Fig. 5d. The discharging/charging plateaus are clearly discerned in the voltage-capacity results. The first three discharge curves (1st, 5th and 10th cycle) nearly overlapped with each other and the capacity only slightly faded in the first 20 cycles, which all suggest an enhanced reversibility of G/CNT@MnO2@S cathode compared with G/CNT@S cathode (Fig. S10a†). This result is in good agreement with the observation in the CV profile. In addition, the discharging/charging plateaus of G/CNT@MnO2@S cathode at the 20th cycle shows smaller polarization (0.17 V), longer voltage plateau and smaller overpotentials for nucleation of Li2S2 when compared with G/CNT@S cathode (Fig. S10b†), which exhibits accelerated electrodeposition kinetics from Li2S4 to Li2S2/Li2S.22,49 To evaluate superiority of the G/CNT@MnO2 composite in the battery, discharge capacity of G/CNT@MnO2@S and G/CNT@S cathodes were divided into high plateau and low plateau capacity separately as plotted in Fig. 5e and f. The high plateau capacity for G/CNT@MnO2@S shows higher cycling stability compared with that of G/CNT@S, which confirms the effective suppression of LiPSs (Li2Sx, x ≥ 4) dissolution due to the high adsorption efficiency of ultrathin MnO2 nanosheets. In the process of conversion from Li2S4 to Li2S2/Li2S, the low plateau capacity for G/CNT@MnO2@S is more stable than that of G/CNT@S, indicating the enhanced kinetics during the conversion of LiPSs to lithium sulfides. Therefore, the G/CNT@MnO2 could not only immobilize the LiPSs through strong chemical interaction but also facilitate fast conversion of LiPSs and lithium sulfides.
The high rate cycling stability of the G/CNT@MnO2@S cathode was further evaluated at a current density of 1C, as shown in Fig. 5g. The discharging and charging current density of the first cycle was 0.5C. Further, the initial discharging capacity of the G/CNT@MnO2@S at a current density of 1C was 758.1 mA h g−1, yielding a high capacity retention of 77.9% after 200 cycles with a high coulombic efficiency above 97%. The cycling performance of G/CNT@MnO2@S cathode was compared with other related state-of-the-art cathodes based on carbon/MnO2 host materials (Table S2†).19,23,25,41,43,46,50–52 Apparently, the G/CNT@MnO2@S cathode exhibits excellent cycling stability. Especially, the cathode exhibits a high capacity retention of 84.1% and 77.9% at a rate of 0.1C and 1C after cycling, respectively, which is comparable to or even better than almost all of previously reported results listed in Table S2.† Furthermore, some results shows good long-term cycling performance, however, the rapid capacity fading in the first several cycles has not yet been solved, which may be due to the low adsorption efficiency of MnO2.
To evaluate the adsorption ability of the G/CNT@MnO2 composites with regard to the LiPSs, the same amount of G/CNT and G/CNT@MnO2 were added to the LiPSs solution. The LiPSs solution (mainly Li2S6) was prepared in accordance with the process described in previous reports and stirred for 2 h.53,54 As shown in Fig. 6, the Li2S6 solution changed color from dark brown initially to yellow or colorless with the addition of the G/CNTs or G/CNT@MnO2, respectively, suggesting the G/CNT@MnO2 composites exhibit stronger adsorption ability for Li2S6 polysulfides compared with G/CNT. The adsorption ability of G/CNT@MnO2 for LiPSs was further characterized through UV-vis spectroscopy. As shown in Fig. 6, the typical absorption peak at 278 nm of Li2S6 in the DME solution confirms that the solution was mainly composed of Li2S6.55 Further, the adsorption peak intensity of the Li2S6 decreases with the addition of G/CNT and G/CNT@MnO2, corresponding well to the color change of the Li2S6 solution. The observations are in good agreement with the results of cycling performance discussed above and thereby re-confirming the significant role of MnO2 in trapping the LiPSs and their great contribution to the cycling stability.
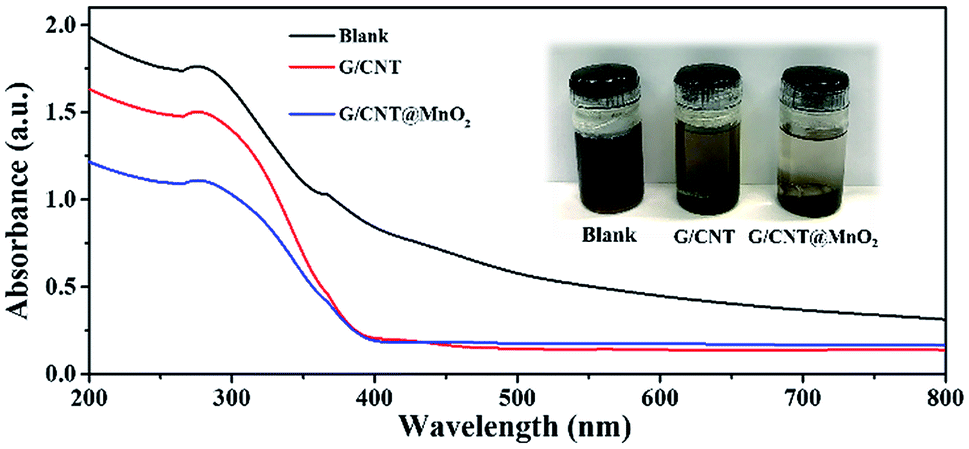 | ||
| Fig. 6 UV-vis spectra of bare Li2S6 solution, G/CNT–Li2S6 solution, and G/CNT@MnO2– Li2S6 solution, inset image is photograph of color changes after addition of G/CNTs and G/CNT@MnO2 to Li2S6 solution with 2 h stirring. | ||
To further evaluated the charge transfer and cycling stability of the cathodes, interfacial resistance at different cycles of G/CNT@MnO2@S and G/CNT@S cathodes were investigated by electrochemical impedance spectroscopy (EIS). As shown in Fig. 7, all the results show a single semicircle in high frequency, and an oblique line in the low frequency, respectively, which are attributed to the charge transfer process (Rct) and lithium ion diffusion in the cathode.43 The two cathodes both exhibit a low resistance before cycling, of which G/CNT@MnO2@S cathode yields a little higher resistance (Fig. 7). This is due to the poor conductivity of MnO2, which slightly reduce the conductivity of G/CNT@MnO2@S cathode. As shown in Fig. 7a, a stable and low resistance after 10, 50, and even the 100 cycles of G/CNT@MnO2@S cathode indicates the strong anchoring effect and enhanced redox conversion of LiPSs. On the contrary, a remarkable resistance increasing after 10, 50, and 100 cycles occurred in G/CNT@S cathode, which could be due to the formation of uncontrolled precipitation of nonconductive Li2S2/Li2S films on surface of G/CNT@S cathode (Fig. 7b).
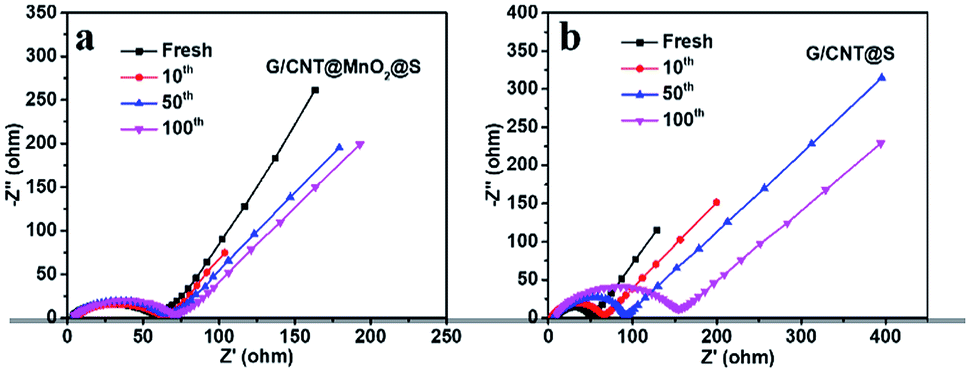 | ||
| Fig. 7 Nyquist plots of (a) G/CNT@MnO2@S electrode and (b) G/CNT@S electrode after different stages of discharge/charge cycling at the rate of 1C. | ||
CV measurements at various scanning rates were carried out to determine the lithium ion diffusion coefficient in the sulfur redox process. As shown in Fig. 8, all cathodic and anodic peak currents are linear with the square root of scan rates, indicating that the reactions are diffusion controlled.23 The lithium ion diffusion coefficient can be calculated from classical Randles–Sevcik equation:
| Ip = 2.69 × 105n1.5AD0.5Cν0.5 |
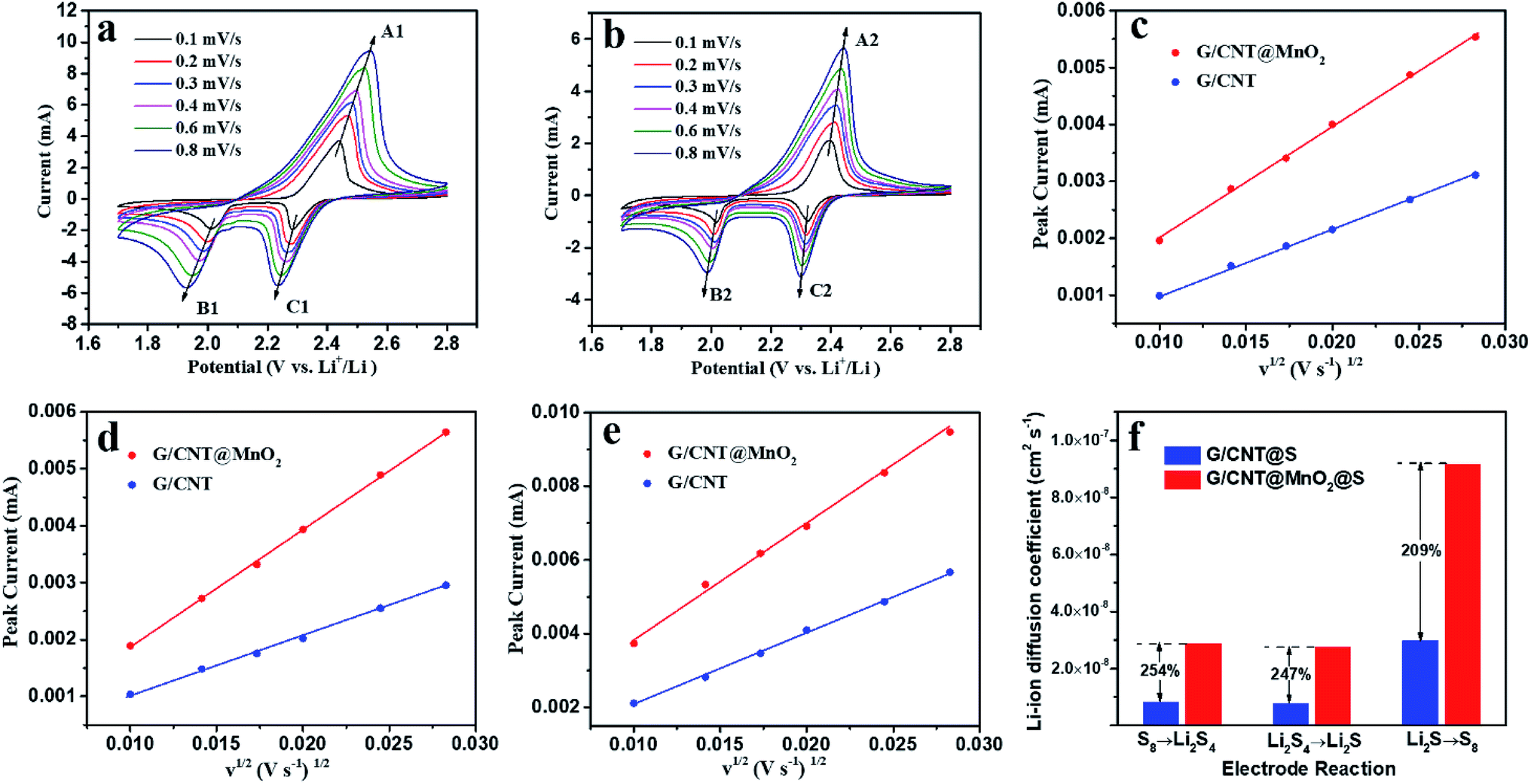 | ||
| Fig. 8 (a and b) CV curves measured at various scan rates for G/CNT@MnO2@S electrode and G/CNT@S electrode. (c–e) Relationship between peak currents and sweep rates of the cathodic reaction of S8 → Li2S4, the cathodic reaction of Li2S4 → Li2S and the anodic reaction of Li2S → S8. (f) Calculated diffusion coefficient of lithium ion. | ||
| Diffusion coefficient (cm2 s−1) | Cathodic reaction, S8 → Li2S4 | Cathodic reaction, Li2S4 → Li2S | Anodic reaction, Li2S → S8 |
|---|---|---|---|
| G/CNT@MnO2@S | 2.87 × 10−8 | 2.74 × 10−8 | 9.17 × 10−8 |
| G/CNT@S | 8.28 × 10−9 | 7.74 × 10−9 | 2.965 × 10−8 |
With the above discussions, the G/CNT@MnO2 composite as sulfur host is therefore proven to present effective polysulfide retention and redox conversion enhancement. On the one hand, ultrathin MnO2 nanosheets can strongly interact with polysulfide species owing to the intrinsically polar property. As previously reported, S2O32− generated from the reaction between long chain LiPSs and MnO2 serves as an internal mediator to anchor the LiPSs, by forming insoluble polythionate and lower LiPSs.14,17 On the other hand, the G/CNT@MnO2 composites provide sufficient surface for charge transfer and lithium ion diffusion to implement the rapid electrochemical reaction. The excellent cycling stability can be attributed to the unique features of G/CNT@MnO2@S nanocomposites. (i) The large surface area of G/CNT hybrids provide adequate space for MnO2 nanosheets and sulfur particles which is beneficial to the uniform distribution of MnO2 and high sulfur loading. (ii) The 3D architecture of G/CNT@MnO2 composites provide large amount of charge transfer channels and Li+ diffusion channels yielding a high rate performance and enhanced redox kinetics of LiPSs. (iii) Ultrathin MnO2 nanosheets uniformly distributed on the surface of G/CNT hybrids enhance the chemical adsorption efficiency between MnO2 and LiPSs, resulting in excellent cycling stability. (iv) The presence of nano-sized sulfur particles and their uniform distribution make best of sulfur and reinforce the cycling stability and rate performance of the fabricated Li–S batteries.
3. Conclusions
In summary, ultrathin MnO2 decorated G/CNT nanocomposites used as S hosts were designed and synthesized through a facile redox deposition method. With the integrated structure design, G/CNT hybrids with superior conductivity and ultrathin MnO2 nanosheets with uniform distribution enhanced the adsorption ability and redox reaction of LiPSs efficiently, thus exhibited stable cycling and high rate performance. As a result, a G/CNT@MnO2@S nanocomposite with high sulfur loading of 81.8 wt% delivers a high initial specific capacity of 1015.1 mA h g−1 at a current density of 0.1C, with superior coulombic efficiency consistently near 100%. In particular, the G/CNT@MnO2@S cathode exhibits high capacity retention of 84.1% and 77.9% at a rate of 0.1C and 1C, respectively. This facile method of fabricating G/CNT@MnO2 nanocomposites constitutes a useful strategy for the design and synthesis of scalable, high-performance Li–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Program of Graphene Special Innovative Fund of AECC Beijing Institute of Aeronautical Materials.Notes and references
- M. P. Yu, J. S. Ma, H. Q. Song, A. J. Wang, F. Y. Tian, Y. S. Wang, H. Qiu and R. M. Wang, Energy Environ. Sci., 2016, 9, 1495–1503 RSC.
- X. Fang and H. S. Peng, Small, 2015, 11, 1488–1511 CrossRef CAS PubMed.
- M. A. Pope and I. A. Aksay, Adv. Energy Mater., 2015, 5, 1500124 CrossRef.
- J. Yan, X. Liu and B. Li, Adv. Sci., 2016, 3, 1600101 CrossRef PubMed.
- S. Y. Lang, Y. Shi, Y. G. Guo, D. Wang, R. Wen and L. J. Wan, Angew. Chem., 2016, 55, 15835–15839 CrossRef CAS PubMed.
- S. T. Zhang, M. B. Zheng, Z. X. Lin, N. W. Li, Y. J. Liu, B. Zhao, H. Pang, J. M. Cao, P. He and Y. Shi, J. Mater. Chem. A, 2014, 2, 15889–15896 RSC.
- W. Z. Bao, Z. Zhang, W. Chen, C. K. Zhou, Y. Q. Lai and J. Li, Electrochim. Acta, 2014, 127, 342–348 CrossRef CAS.
- C. F. Zhang, B. W. Hao, C. Z. Yuan, Z. P. Guo and X. W. Lou, Angew. Chem., 2012, 124, 9730–9733 CrossRef.
- J. C. Guo, Y. H. Xu and C. S. Wang, Nano Lett., 2011, 11, 4288–4294 CrossRef CAS PubMed.
- K. Mi, Y. Jiang, J. K. Feng, Y. T. Qian and S. L. Xiong, Adv. Funct. Mater., 2016, 26, 1571–1579 CrossRef CAS.
- J. Zhang, Z. M. Dong, X. L. Wang, X. Y. Zhao, J. P. Tu, Q. M. Su and G. H. Du, J. Power Sources, 2014, 270, 1–8 CrossRef CAS.
- C. J. Hart, M. Cuisinier, X. Liang, D. Kundu, A. Garsuch and L. F. Nazar, Chem. Commun., 2015, 51, 2308–2311 RSC.
- C. Li, Z. C. Xi, D. X. Guo, X. J. Chen and L. W. Yin, Small, 2018, 14, 1701986 CrossRef PubMed.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- J. Guo, X. Zhang, X. Du and F. Zhang, J. Mater. Chem. A, 2017, 5, 6447–6454 RSC.
- Z. Chang, H. Dou, B. Ding, J. Wang, Y. Wang, X. Hao and D. R. MacFarlane, J. Mater. Chem. A, 2017, 5, 250–257 RSC.
- X. Liang, C. Y. Kwok, F. Lodi-Marzano, Q. Pang, M. Cuisinier, H. Huang, C. J. Hart, D. Houtarde, K. Kaup, H. Sommer, T. Brezesinski, J. Janek and L. F. Nazar, Adv. Energy Mater., 2016, 6, 1501636 CrossRef.
- W. Li, J. Hicks-Garner, J. Wang, J. Liu, A. F. Gross, E. Sherman, J. Graetz, J. J. Vajo and P. Liu, Chem. Mater., 2014, 26, 3403–3410 CrossRef CAS.
- S. Rehman, T. Tang, Z. Ali, X. Huang and Y. Hou, Small, 2017, 13, 1700087 CrossRef PubMed.
- X. Liu, J. Q. Huang, Q. Zhang and L. Q. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed.
- X. Tao, J. Wang, C. Liu, H. Wang, H. Yao, G. Zheng, Z. W. Seh, Q. Cai, W. Li, G. Zhou, C. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed.
- Z. Liu, B. Liu, P. Guo, X. Shang, M. Lv, D. Liu and D. He, Electrochim. Acta, 2018, 269, 180–187 CrossRef CAS.
- Y. Li, D. Ye, W. Liu, B. Shi, R. Guo, H. Zhao, H. Pei, J. Xu and J. Xie, ACS Appl. Mater. Interfaces, 2016, 8, 28566–28573 CrossRef CAS PubMed.
- X. Zhao, H. Wang, G. Zhai and G. Wang, Chemistry, 2017, 23, 7037–7045 CrossRef CAS PubMed.
- X. C. Dong, B. Li, A. Wei, X. H. Cao, M. B. Chan-Park, H. Zhang, L.-J. Li, W. Huang and P. Chen, Carbon, 2011, 49, 2944–2949 CrossRef CAS.
- J. Chen, J. H. Walther and P. Koumoutsakos, Adv. Funct. Mater., 2015, 25, 7539–7545 CrossRef.
- M. Q. Zhao, X. F. Liu, Q. Zhang, G. L. Tian, J. Q. Huang, W. C. Zhu and F. Wei, ACS Nano, 2012, 6, 10759–10769 CrossRef CAS PubMed.
- X. K. Huang, H. J. Yue, A. Attia and Y. Yang, J. Electrochem. Soc., 2007, 154, 26–33 CrossRef.
- S.-B. Ma, K.-Y. Ahn, E.-S. Lee, K.-H. Oh and K.-B. Kim, Carbon, 2007, 45, 375–382 CrossRef CAS.
- X. B. Jin, W. Z. Zhou, S. W. Zhang and G. Z. Chen, Small, 2007, 3, 1513–1517 CrossRef CAS PubMed.
- Y. F. Dong, S. H. Liu, Z. Y. Wang, Y. Liu, Z. B. Zhao and J. S. Qiu, Nanoscale, 2015, 7, 7569–7573 RSC.
- X. Chen, S. J. Yan, N. Wang, S. K. Peng, C. Wang, Q. H. Hong, X. Y. Zhang and S. L. Dai, RSC Adv., 2017, 7, 55734–55740 RSC.
- S. Liu, Y. Zhu, J. Xie, Y. Huo, H. Y. Yang, T. Zhu, G. Cao, X. Zhao and S. Zhang, Adv. Energy Mater., 2014, 4, 1301960 CrossRef.
- N. Yu, H. Yin, W. Zhang, Y. Liu, Z. Tang and M.-Q. Zhu, Adv. Energy Mater., 2016, 6, 1501458 CrossRef.
- Y. Munaiah, B. G. Sundara Raj, T. Prem Kumar and P. Ragupathy, J. Mater. Chem. A, 2013, 1, 4300–4306 RSC.
- C. Julien, M. Massot, R. Baddour-Hadjeanc, S. Frangerd, S. Bachd and J. P. Pereira-Ramosd, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- S. Yang, X. Song, P. Zhang and L. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 3317–3322 CrossRef CAS PubMed.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- R. Xu, J. Lu and K. Amine, Adv. Energy Mater., 2015, 5, 1500408 CrossRef.
- Z. Li, J. Zhang and X. W. Lou, Angew. Chem., 2015, 54, 12886–12890 CrossRef CAS PubMed.
- H. W. Chen, C. H. Wang, W. L. Dong, W. Lu, Z. L. Du and L. W. Chen, Nano Lett., 2015, 15, 798–802 CrossRef CAS PubMed.
- G. H. Yuan, H. F. Jin, Y. Z. Jin and L. Z. Wu, J. Solid State Electrochem., 2017, 22, 693–703 CrossRef.
- L. B. Ni, Z. Wu, G. J. Zhao, C. Y. Sun, C. Q. Zhou, X. X. Gong and G. W. Diao, Small, 2017, 13, 1603466 CrossRef PubMed.
- J. Zhang, Y. Shi, Y. Ding, W. K. Zhang and G. H. Yu, Nano Lett., 2016, 16, 7276–7281 CrossRef CAS PubMed.
- K. Cao, H. Liu, Y. Li, Y. Wang and L. Jiao, Energy Storage Materials, 2017, 9, 78–84 CrossRef.
- M. Sun, X. Wang, J. Wang, H. Yang, L. Wang and T. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 35175–35183 CrossRef CAS PubMed.
- X. Liang and L. F. Nazar, ACS Nano, 2016, 10, 4192–4198 CrossRef CAS PubMed.
- F. Y. Fan and Y.-M. Chiang, J. Electrochem. Soc., 2017, 164, A917–A922 CrossRef CAS.
- X. K. Huang, K. Y. Shi, J. Yang, G. Mao and J. H. Chen, J. Power Sources, 2017, 356, 72–79 CrossRef CAS.
- M. Yan, Y. Zhang, Y. Li, Y. Q. Huo, Y. Yu, C. Wang, J. Jin, L. H. Chen, T. Hasan, B. J. Wang and B. L. Su, J. Mater. Chem. A, 2016, 4, 9403–9412 RSC.
- J. Shen, J. Liu, Z. Liu, R. Hu, J. Liu and M. Zhu, Chemistry, 2018, 24, 4573–4582 CrossRef CAS PubMed.
- L. B. Ni, G. J. Zhao, G. Yang, G. S. Niu, M. Chen and G. W. Diao, ACS Appl. Mater. Interfaces, 2017, 9, 34793–34803 CrossRef CAS PubMed.
- L. B. Ni, G. J. Zhao, Y. T. Wu, Z. Wu, W. Wang, Y. Y. Liao, G. Yang and G. W. Diao, Chem. - Asian J., 2017, 12, 3128–3134 CrossRef CAS PubMed.
- C. Barchasz, F. Molton, C. Duboc, J. C. Leprêtre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84, 3983–3980 CrossRef PubMed.
- M. Chen, Q. Lu, S. Jiang, C. Huang, X. Wang, B. Wu, K. Xiang and Y. Wu, Chem. Eng. J., 2018, 335, 831–842 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00292h |
| This journal is © The Royal Society of Chemistry 2019 |