
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring unsymmetrical diboranes(4) as boryl ligand precursors: platinum(II) bis-boryl complexes†
Wiebke Drescher (née Oschmann),
Corinna Borner,
Daniel J. Tindall and
Christian Kleeberg *
*
Institut für Anorganische und Analytische Chemie, Technische Universität Carolo-Wilhelmina zu Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: ch.kleeberg@tu-bs.de
First published on 30th January 2019
Abstract
A series of five unsymmetrical platinum(II) bis-boryl complexes, bearing two distinct boryl ligands, are obtained by the oxidative addition reaction of unsymmetrical diborane(4) derivatives, bearing either two different dialkoxy or one dialkoxy and one diamino boryl moiety, with [(Ph3P)2Pt(C2H4)]. All five complexes were structurally and spectroscopically characterised. The bis-boryl platinum(II) complexes exhibit slightly distorted square-planar cis-boryl structures with acute B–Pt–B angles, short B⋯B distances of 2.44–2.55 Å and relatively long trans-boryl P–Pt distances around 2.34 Å. The 31P–195Pt NMR coupling constants are indicative for the strongly donating/trans-influencing boryl ligands. Despite the structural and spectroscopic data at hand no finally conclusive order of the donor properties/trans-influence of the boryl ligands can be deduced on the basis of these data. This may be explained by an (residual) interaction of two boryl ligands.
Introduction
Boryl complexes of transition metals have received considerable attention in the last few years, either due to their role as reactive intermediates in various catalytic processes or due to their unique coordination properties and reactivity of their complexes.1,2 In particular a substantial number of boryl platinum(II) complexes have been reported over the last years, amongst these complexes those of the type (R3P)2Pt(boryl)2 are especially well studied.3–6 The latter type of complexes are typically straightforwardly prepared by the oxidative addition reaction of a diborane(4) derivative (1) with a suitable platinum(0) precursor (e.g. [Pt(PPh3)2(C2H4)] (2)) and, most importantly, more than 35 complexes of this type are structurally and spectroscopically characterised.3–6 About half of these complexes are symmetrical complexes of the specific type [(Ph3P)2Pt(boryl)2] (3) with both identical boryl ligands ranging from dialkoxy and diaryloxy boryl ligands but also including ligands such as BCl(NMe2) or BF2 (Scheme 1). Moreover, we have reported the first unsymmetrical congeners with one dialkoxy and one diamino boryl ligand ([(Ph3P)2Pt(Bpin)(B(NR)2C6H4)] with R = Me (3ad) and R = Bn).3 Hence, complexes of the type [(Ph3P)2Pt(boryl)2] (3) suggest themselves as well suited to comparatively study the coordination properties of different boryl ligands due to their abundant data available and their facile synthesis. In particular, complexes of the type cis-[(Ph3P)2PtXY] (X, Y: anionic ligands) have been especially useful to study the cis and the trans influence of different anionic ligands X, Y.7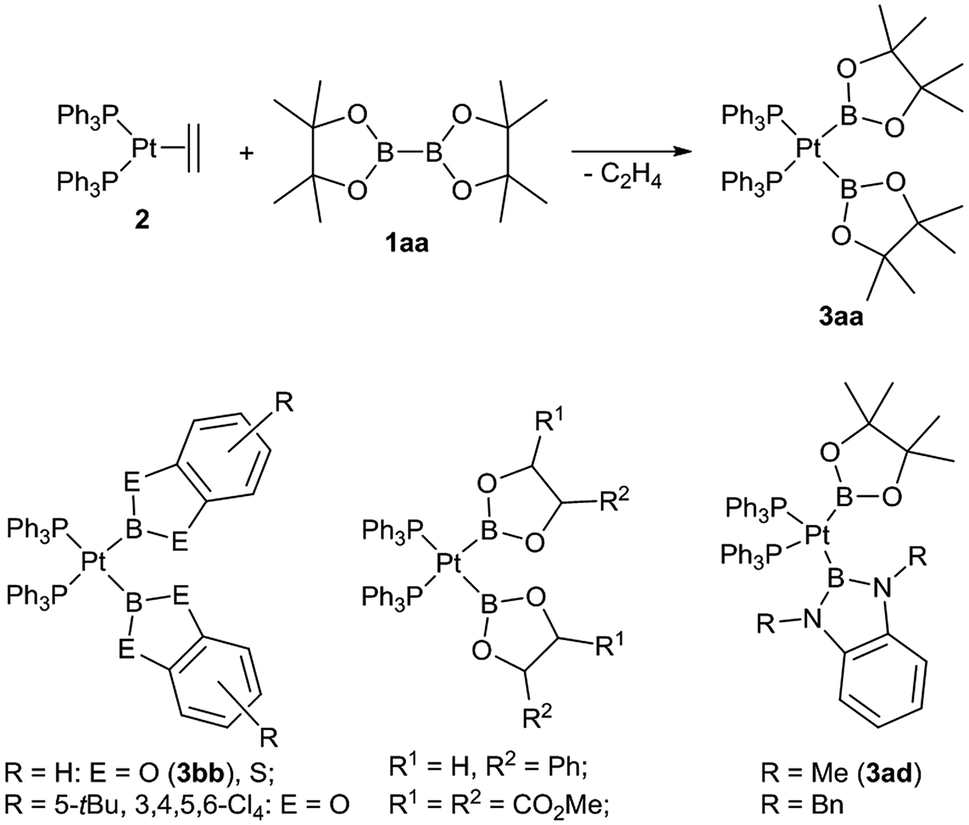 | ||
| Scheme 1 Synthesis of 3aa and examples of complexes [(Ph3P)2Pt(boryl)2].3,4 | ||
We have recently reported the synthesis of a number of unsymmetrical diboranes(4) bearing different dialkoxy as well as one dialkoxy and one diaminoboryl moieties (1) as potential precursors for boryl complexes.8 In this work we endeavour to the study reactivity of these diborane(4) derivatives towards 2 and the coordination properties of the resulting unsymmetrical platinum boryl complexes of the type [(Ph3P)2Pt(boryl)(boryl′)]. The facile availability of this series of unsymmetrical complexes [(Ph3P)2Pt(boryl)(boryl′)] renders a comparative study of sterically and electronically different boryl moieties within one complex fragment for the first time possible.
Experimental
General considerations
The unsymmetrical diboranes(4) 1ab, 1ac, 1ae–ag, 1bd and 1be were prepared according to literature procedures.3,8 The synthesis and analytical data of 1hh are described in the ESI.†9 All other compounds were commercially available and were used as received; their purity and identity was checked by appropriate methods. All solvents were dried using MBraun solvent purification systems, deoxygenated using the freeze–pump–thaw method and stored under purified nitrogen. All manipulations were performed using standard Schlenk techniques under an atmosphere of purified nitrogen or in a nitrogen filled glove box (MBraun). NMR spectra were recorded on Bruker Avance II 300, Avance III HD 300, Avance III 500 or Avance 600 spectrometers. For air sensitive samples NMR tubes equipped with screw caps (WILMAD) were used and the solvents were dried over potassium/benzophenone and degassed. Chemical shifts (δ) are given in ppm, using the (residual) resonance signal of the solvents for calibration (C6D6: 1H NMR: 7.16 ppm, 13C NMR: 128.06 ppm).10,11 B{1H} and 31P{1H} NMR chemical shifts are reported relative to external BF3·Et2O and 85% aqueous H3PO4, respectively. 13C{1H}, 11B{1H} and 31P{1H} NMR spectra were recorded employing composite pulse 1H decoupling. If necessary 2D NMR techniques were employed to assign the individual signals (1H–1H NOESY (1 s mixing time), 1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC and 1H–31P HMBC). Melting points were determined in flame sealed capillaries under nitrogen using a Büchi 535 apparatus and are not corrected. Elemental analyses were performed at the Institut für Anorganische und Analytische Chemie of the Technische Universität Carolo-Wilhelmina zu Braunschweig using an Elementar vario MICRO cube instrument. GC/MS measurements were performed using a Shimadzu GCMS-QP2010SE instrument operating in positive EI mode (70 eV, 60–700 m/z) with the following conditions: injection temperature 250 °C; interface temperature 280 °C; temperature program: start temperature 50 °C for 3 min, heating rate 12 °C min−1, end temperature 300 °C for 8 min or 38 min; column type: ZB-5MS GUARDIAN, 30 m × 0.25 mm, 0.25 μm film thickness; He carrier gas (1.5 mL min−1). High resolution GC/MS measurements were performed by the Analytical Facility at the Technische Universität Carolo-Wilhelmina zu Braunschweig using an Agilent 6890 GC instrument coupled to a AccuTOF GC JMS-T100GC (JOEL) mass spectrometer operating in positive EI mode (70 eV) with the following conditions: injection temperature 250 °C; interface temperature 270 °C; temperature program: start temperature 50 °C for 3 min, heating rate 10 °C min−1, end temperature 310 °C for 3 min; column type: ZB-5MS, 30 m × 0.25 mm, 0.25 μm film thickness; He carrier gas (1.0 mL min−1).| Compound | 3ab(C6H6) | 3ac | 3ae(C4H8O)1.5b | 3bd | 3be(C6H6) | 3cc(C4H8O)c | 3hh(C4H8O) |
|---|---|---|---|---|---|---|---|
| a Obs. criterion: I > 2σ(I).b The crystal is non-merohedrally twinned and contains disordered solvent.9c The crystal is a non-merohedrally three-component twin.9 | |||||||
| Formula | C54H52B2O4P2Pt | C47H52B2O4P2Pt | C52H64B2O3.5P2Pt | C50H44B2N2O2P2Pt | C52H50B2N2O2P2Pt | C50H58B2O5P2Pt | C54H52B2N2O3P2Pt |
| Mr/(g mol−1) | 1043.60 | 959.53 | 1051.70 | 983.52 | 1013.59 | 1017.61 | 1055.62 |
| Crystal shape | Rod | Irregular block | Rhombohedron | Block | Prism | Fragment of rod | Fragment of block |
| Crystal colour | Clear colourless | Clear colourless | Clear colourless | Clear pale yellow | Clear light orange | Clear colourless | Clear light yellow |
| Cryst. dim./mm3 | 0.49 × 0.13 × 0.06 | 0.40 × 0.33 × 0.21 | 0.32 × 0.17 × 0.16 | 0.55 × 0.36 × 0.28 | 0.53 × 0.25 × 0.25 | 0.41 × 0.23 × 0.11 | 0.41 × 0.26 × 0.22 |
| Crystal system | Triclinic | Orthorhombic | Monoclinic | Triclinic | Triclinic | Triclinic | Monoclinic |
| Space group (no.) | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (2) (2) |
Pbca (61) | P21/c (14) | P (2) |
P (2) |
P (2) |
P21/c (14) |
| a/Å | 13.1742(5) | 18.1455(3) | 13.1260(3) | 13.4322(3) | 9.9026(5) | 12.6111(6) | 22.1926(3) |
| b/Å | 15.3010(5) | 14.2463(3) | 12.8989(3) | 13.6477(4) | 11.8550(5) | 14.3974(7) | 10.4677(2) |
| c/Å | 24.3714(13) | 33.2215(7) | 29.1381(6) | 14.5369(4) | 21.5121(9) | 14.9326(7) | 20.6740(3) |
| α | 73.469(4)° | 90° | 90° | 70.017(2)° | 94.470(3)° | 68.803(5)° | 90° |
| β | 89.111(4)° | 90° | 100.612(2)° | 82.459(2)° | 96.674(4)° | 66.072(5)° | 105.849(2)° |
| γ | 87.082(3)° | 90° | 90° | 60.926(3)° | 114.620(4)° | 88.190(4)° | 90° |
| V/Å3 | 4703.6(4) | 8588.0(3) | 4849.0(2) | 2186.9(1) | 2257.8(2) | 2289.4(2) | 4620.1(1) |
| Z, Z′ | 4, 2 | 8, 1 | 4, 2 | 2, 1 | 2, 1 | 2, 1 | 4, 1 |
| Dcalcd./(g cm−3) | 1.474 | 1.484 | 1.441 | 1.494 | 1.491 | 1.476 | 1.518 |
| μ/mm−1 (λ/Å) | 3.097 (0.71073) | 3.384 (0.71073) | 3.005 (0.71073) | 3.319 (0.71073) | 3.221 (0.71073) | 3.180 (0.71073) | 3.153 (0.71073) |
| Absorption corr. | Analytical | Gaussian | Gaussian | Analytical | Analytical | Gaussian | Multi-scan |
| 2θ range (compl.) | 4.7–54.2° (99.4%) | 4.4–56.0° (100%) | 4.5–60.0° (99.9%) | 4.5–68.0°(99.7%) | 4.5–68.0° (99.7%) | 4.4–68.0° (99.4%) | 5.0–70.0° (100%) |
| Refl. Measured | 195![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 947 947 |
144260 |
29889 |
275463 |
65840 |
18989 |
456256 |
| Unique (Rint) | 20759 (0.1095) |
10365 (0.0465) |
29889 |
18533 (0.0426) |
13137 (0.0315) |
18989 |
24974 (0.0315) |
| observeda | 13218 |
9040 | 117184 | 16987 |
12617 |
16575 |
21349 |
| Param./restr. | 1143/0 | 511/0 | 593/70 | 534/0 | 552/0 | 547/0 | 579/0 |
| R1 (obs. rflns.)a | 0.0393 | 0.0.89 | 0.0356 | 0.0195 | 0.0189 | 0.0290 | 0.0262 |
| wR2 (all rflns.) | 0.0838 | 0.0516 | 0.0500 | 0.0444 | 0.0385 | 0.0623 | 0.0562 |
| GooF on F2 | 1.039 | 1.234 | 0.786 | 1.120 | 1.095 | 1.004 | 1.045 |
| max./min. ρ/(e Å−3) | 1.646/−0.947 | 1.009/−0.724 | 2.305/−0.982 | 1.877/−1.095 | 0.518/−1.035 | 1.814/−1.177 | 1.860/−0.953 |
| CCDC no. | 1881540 | 1881536 | 1881534 | 1881538 | 1881541 | 1881537 | 1881542 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C(Ph)(Bpin). 1ad (55 mg, 202 μmol), Ph–C
C(Ph)(Bpin). 1ad (55 mg, 202 μmol), Ph–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ph (47 mg, 264 μmol), 2 (4.3 mg, 5.8 μmol, 3 mol%), toluene (5 mL), reaction time 72 h, off-white solid (44 mg, 98 μmol, 48%). Single crystals suitable for X-ray diffraction were obtained by cooling a hot n-hexane solution to 4 °C. mp 198–200 °C. Found: C, 74.3; H, 7.2; N, 5.95%. Calc. for C28H32B2N2O2: C, 74.7; H, 7.2; N, 6.2%. δ1H (300 MHz, C6D6, rt): 0.76 (12H, s, OC(CH3)2), 3.20 (6H, s, NCH3), 6.84–6.91 (1H, m, CHdmab), 6.93–7.00 (5H, m, CHPh), 7.06–7.13 (2H, m, CHdmab), 7.13–7.21 (4H, m, CHPh), 7.37–7.43 (2H, m, CHPh). δ13C{1H} (75 MHz, C6D6, rt): 24.5 (OC(CH3)2), 29.7 (NCH3), 83.3 (OC(CH3)2), 108.3 (CHdmab), 119.2 (CHdmab), 126.4 (CHPh), 126.5 (CHPh), 128.0 (CHPh), 128.2 (CHPh), 129.7 (CHPh), 130.2 (CHPh), 138.9 (Cdmab), 142.2 (CPh), 143.0 (CPh), the quaternary boron bound carbon atoms were not detected. δ11B{1H} (96 MHz, C6D6, rt): 29.5 (s, Δw1/2 = 484 Hz). m/z (EI+, 70 eV, GC/MS) 450 [M]+, 393, 367, 350, 246, 188, 179, 162, 145, 69.C(Ph)(Bpin) (mixture of isomers). 1ad (100 mg, 368 μmol), Me–CC–Ph (56 mg, 482 μmol), 2 (8.3 mg, 11.0 μmol, 3 mol%), toluene (10 mL), reaction time 122 h, brownish oil (133 mg, 343 μmol, 93%). (dmabB)(Me)C
C–Ph (47 mg, 264 μmol), 2 (4.3 mg, 5.8 μmol, 3 mol%), toluene (5 mL), reaction time 72 h, off-white solid (44 mg, 98 μmol, 48%). Single crystals suitable for X-ray diffraction were obtained by cooling a hot n-hexane solution to 4 °C. mp 198–200 °C. Found: C, 74.3; H, 7.2; N, 5.95%. Calc. for C28H32B2N2O2: C, 74.7; H, 7.2; N, 6.2%. δ1H (300 MHz, C6D6, rt): 0.76 (12H, s, OC(CH3)2), 3.20 (6H, s, NCH3), 6.84–6.91 (1H, m, CHdmab), 6.93–7.00 (5H, m, CHPh), 7.06–7.13 (2H, m, CHdmab), 7.13–7.21 (4H, m, CHPh), 7.37–7.43 (2H, m, CHPh). δ13C{1H} (75 MHz, C6D6, rt): 24.5 (OC(CH3)2), 29.7 (NCH3), 83.3 (OC(CH3)2), 108.3 (CHdmab), 119.2 (CHdmab), 126.4 (CHPh), 126.5 (CHPh), 128.0 (CHPh), 128.2 (CHPh), 129.7 (CHPh), 130.2 (CHPh), 138.9 (Cdmab), 142.2 (CPh), 143.0 (CPh), the quaternary boron bound carbon atoms were not detected. δ11B{1H} (96 MHz, C6D6, rt): 29.5 (s, Δw1/2 = 484 Hz). m/z (EI+, 70 eV, GC/MS) 450 [M]+, 393, 367, 350, 246, 188, 179, 162, 145, 69.C(Ph)(Bpin) (mixture of isomers). 1ad (100 mg, 368 μmol), Me–CC–Ph (56 mg, 482 μmol), 2 (8.3 mg, 11.0 μmol, 3 mol%), toluene (10 mL), reaction time 122 h, brownish oil (133 mg, 343 μmol, 93%). (dmabB)(Me)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Ph)(Bpin) (major component), δ1H (600 MHz, C6D6, rt): 0.74 (12H, s, OC(CH3)2), 1.93 (3H, s, C(Bdmab)CH3), 3.16 (6H, s, NCH3), 7.00–7.02 (2H, m, CHdmab), 7.10–7.15 (1H, m, 4-CHPh), 7.18–7.21 (2H, m, CHdmab), 7.28–7.32 (2H, m, 3-CHPh), 7.41–7.44 (2H, m, 2-CHPh). δ13C{1H} (150 MHz, C6D6, rt): 20.7 (C(Bdmab)CH3), 24.5 (OC(CH3)2), 29.5 (NCH3), 83.0 (OC(CH3)2), 108.1 (CHdmab), 119.0 (CHdmab), 126.2 (4-CHPh), 128.4 (3-CHPh), 129.4 (2-CHPh), 139.0 (Cdmab), 142.3 (CPh), 144 (br., BCCB), 148 (br., BCCB). m/z (EI+, 70 eV, GC/MS) 388 (35) [M]+, 331 (21), 305 (20), 288 (100), 245 (18), 188 (15), 162 (31), 145 (27), 117 (30), 69 (10). m/z (HR-EI+, 70 eV, GC/MS) 388.25114 [M]+, calc. for C23H30B2N2O2: 388.24934 (1.80 mmu). (pinB)(Me)CC(Ph)(Bdmab) (minor component) δH (600 MHz, C6D6, rt): 1.13 (12H, s, OC(CH3)2), 2.21 (3H, s, C(Bdmab)CH3), 3.52 (6H, s, NCH3), 6.94–6.97 (2H, m, CHdmab), 7.14–7.17 (2H, m, CHdmab), 7.44–7.49 (2H, m, 2-CHPh), not all signals could be unambiguously identified. δ13C{1H} (150 MHz, C6D6, rt): 18.5 (C(Bpin)CH3), 25.3 (OC(CH3)2), 30.9 (NCH3), 82.7 (OC(CH3)2), 108.1 (CHdmab), 118.9 (CHdmab), 129.3 (2-CHPh), not all signals could be unambiguously identified. m/z (EI+, 70 eV, GC/MS) 388 (80) [M]+, 331 (74), 305 (40), 288 (100), 245 (30), 188 (90), 162 (43), 145 (42), 117 (66), 69 (33). m/z (HR-EI+, 70 eV, GC/MS) 388.25046 [M]+, calc. for C23H30B2O2: 388.24934 (1.12 mmu). δ11B{1H} (96 MHz, C6D6, rt): 29.8 (s, Δw1/2 = 348 Hz), isomers not resolved.C(Ph)(Bcat) (mixture of isomers). 3ab (100 mg, 407 μmol), Me–CC–Ph (61 mg, 529 μmol), 2 (9.1 mg, 12 μmol, 3 mol%), toluene (10 mL), reaction time 2 h, brownish oil (146 mg, 403 μmol, 99%). (pinB)(Me)CC(Ph)(Bcat) (major component), δ1H (500 MHz, C6D6, rt): 0.95 (12H, s, OC(CH3)2), 2.06 (3H, s, C(Bcat)CH3), 6.81–6.84 (2H, m, CHcat), 6.99–7.04 (1H, m, 4-CHPh), 7.06–7.10 (2H, m, CHcat), 7.10–7.15 (2H, m, 3-CHPh), 7.33–7.38 (2H, m, 2-CHPh). δ13C{1H} (125 MHz, C6D6, rt): 17.7 (C(Bpin)CH3), 24.6 (OC(CH3)2), 84.2 (OC(CH3)2), 112.5 (CHcat), 122.5 (CHcat), 127.0 (4-CHPh), 128.7 (3-CHPh), 128.8 (2-CHPh), 140.6 (CPh), 142 (br., BCCB), 147 (br., BCCB). 149.2 (Ccat). m/z (EI+, 70 eV, GC/MS) 362 (3) [M]+, 305 (28), 262 (7), 220 (5), 158 (7), 143 (17), 128 (11), 115 (12), 84 (100), 69 (28). m/z (HR-EI+, 70 eV, GC/MS) 362.18433 [M]+, calc. for C21H24B2O4: 362.18607 (−1.74 mmu). (catB)(Me)CC(Ph)(Bpin) (minor component) δ1H (500 MHz, C6D6, rt): 0.97 (12H, s, OC(CH3)2), 1.97 (3H, s, C(Bcat)CH3), 6.85–6.89 (2H, m, CHcat), 7.04–7.10 (1H, m, 4-CHPh, overlapping), 7.11–7.15 (2H, m, CHcat, overlapping), 7.19–7.24 (2H, m, 3-CHPh), 7.31–7.36 (2H, m, 2-CHPh, overlapping). δ13C{1H} (125 MHz, C6D6, rt): 18.5 (C(Bcat)CH3), 24.6 (OC(CH3)2, overlapping), 84.1 (OC(CH3)2), 112.6 (CHcat), 122.7 (CHcat), 126.7 (4-CHPh), 128.3 (3-CHPh), 129.0 (2-CHPh), 141.0 (CPh), 142 (br., BCCB), 147 (br., BCCB). 149.1 (Ccat), not all signals could be unambiguously identified. m/z (EI+, 70 eV, GC/MS) 362 (5) [M]+, 305 (100), 291 (9), 262 (7), 220 (10), 158 (25), 143 (70), 128 (37), 115 (73), 105 (33), 84 (25), 69 (32). m/z (HR-EI+, 70 eV, GC/MS) 362.18847 [M]+, calc. for C21H24B2O4: 362.18607 (2.40 mmu). δ11B{1H} (160 MHz, C6D6, rt): 31.7 (s, Δw1/2 = 522 Hz), isomers not resolved.C(Ph)(Bpin) (mixture of isomers). 3ac (100 mg, 417 μmol), Me–CC–Ph (63 mg, 542 μmol), 2 (9.4 mg, 13 μmol, 3 mol%), toluene (10 mL), reaction time 2.5 h, brownish oil (146 mg, 410 μmol, 98%). (neopB)(Me)CC(Ph)(Bpin) (major component), δ1H (500 MHz, C6D6, rt): 0.67 (6H, s, OCH2C(CH3)2), 1.12 (12H, s, OC(CH3)2), 2.05 (3H, s, C(Bneop)CH3), 3.41 (4H, s, OCH2C(CH3)2), 7.03–7.08 (1H, m, 4-CHPh), 7.19–7.25 (2H, m, 3-CHPh), 7.39–7.42 (2H, m, 2-CHPh). δ13C{1H} (125 MHz, C6D6, rt): 18.1 (C(neop)CH3), 21.9 (OCH2C(CH3)2), 25.1 (OC(CH3)2), 31.4 (OCH2C(CH3)2), 72.0 (OCH2C(CH3)2), 83.1 (OC(CH3)2), 126.1 (4-CHPh), 128.3 (3-CHPh), 128.9 (2-CHPh), 143.0 (CPh), 146 (br., BCCB), 151 (br., BCCB). m/z (EI+, 70 eV, GC/MS) 356 (0.3) [M]+, 341 (2) [M–Me]+, 298 (4), 283 (3), 240 (5), 213 (4), 187 (3), 169 (2), 155 (3), 143 (12), 129 (6), 116 (12), 105 (4), 84 (100), 69 (16). m/z (HR-EI+, 70 eV, GC/MS) 341.20789 [M–Me]+, calc. for C19H27B2O4: 341.20954 (−1.66 mmu). (pinB)(Me)CC(Ph)(Bneop) (minor component), δ1H (500 MHz, C6D6, rt): 0.64 (6H, s, OCH2C(CH3)2), 1.16 (12H, s, OC(CH3)2), 2.03 (3H, s, C(Bneop)CH3), 3.43 (4H, s, OCH2C(CH3)2), 7.03–7.08 (1H, m, 4-CHPh, overlapping), 7.19–7.25 (2H, m, 3-CHPh, overlapping), 7.39–7.42 (2H, m, 2-CHPh, overlapping). δ13C{1H} (125 MHz, C6D6, rt): 18.1 (C(Bneop)CH3), 22.0 (OCH2C(CH3)2), 25.1 (OC(CH3)2), 31.4 (OCH2C(CH3)2), 72.2 (OCH2C(CH3)2), 83.3 (OC(CH3)2), 126.1 (4-CHPh), 128.3 (3-CHPh), 128.9 (2-CHPh), 143.0 (CPh). (EI+, 70 eV, GC/MS) 356 (1) [M]+, 341 (5) [M–Me]+, 143 (13), 129 (8), 116 (100), 105 (8), 69 (42). m/z (HR-EI+, 70 eV, GC/MS) 356.23438 [M]+, calc. for C20H30B2O2: 356.23302 (1.37 mmu). δ11B{1H} (96 MHz, C6D6, rt): 27.6 (s, Δw1/2 = 417 Hz), 31.4 (s, Δw1/2 = 424 Hz), isomers not resolved.
C(Ph)(Bpin) (major component), δ1H (600 MHz, C6D6, rt): 0.74 (12H, s, OC(CH3)2), 1.93 (3H, s, C(Bdmab)CH3), 3.16 (6H, s, NCH3), 7.00–7.02 (2H, m, CHdmab), 7.10–7.15 (1H, m, 4-CHPh), 7.18–7.21 (2H, m, CHdmab), 7.28–7.32 (2H, m, 3-CHPh), 7.41–7.44 (2H, m, 2-CHPh). δ13C{1H} (150 MHz, C6D6, rt): 20.7 (C(Bdmab)CH3), 24.5 (OC(CH3)2), 29.5 (NCH3), 83.0 (OC(CH3)2), 108.1 (CHdmab), 119.0 (CHdmab), 126.2 (4-CHPh), 128.4 (3-CHPh), 129.4 (2-CHPh), 139.0 (Cdmab), 142.3 (CPh), 144 (br., BCCB), 148 (br., BCCB). m/z (EI+, 70 eV, GC/MS) 388 (35) [M]+, 331 (21), 305 (20), 288 (100), 245 (18), 188 (15), 162 (31), 145 (27), 117 (30), 69 (10). m/z (HR-EI+, 70 eV, GC/MS) 388.25114 [M]+, calc. for C23H30B2N2O2: 388.24934 (1.80 mmu). (pinB)(Me)CC(Ph)(Bdmab) (minor component) δH (600 MHz, C6D6, rt): 1.13 (12H, s, OC(CH3)2), 2.21 (3H, s, C(Bdmab)CH3), 3.52 (6H, s, NCH3), 6.94–6.97 (2H, m, CHdmab), 7.14–7.17 (2H, m, CHdmab), 7.44–7.49 (2H, m, 2-CHPh), not all signals could be unambiguously identified. δ13C{1H} (150 MHz, C6D6, rt): 18.5 (C(Bpin)CH3), 25.3 (OC(CH3)2), 30.9 (NCH3), 82.7 (OC(CH3)2), 108.1 (CHdmab), 118.9 (CHdmab), 129.3 (2-CHPh), not all signals could be unambiguously identified. m/z (EI+, 70 eV, GC/MS) 388 (80) [M]+, 331 (74), 305 (40), 288 (100), 245 (30), 188 (90), 162 (43), 145 (42), 117 (66), 69 (33). m/z (HR-EI+, 70 eV, GC/MS) 388.25046 [M]+, calc. for C23H30B2O2: 388.24934 (1.12 mmu). δ11B{1H} (96 MHz, C6D6, rt): 29.8 (s, Δw1/2 = 348 Hz), isomers not resolved.C(Ph)(Bcat) (mixture of isomers). 3ab (100 mg, 407 μmol), Me–CC–Ph (61 mg, 529 μmol), 2 (9.1 mg, 12 μmol, 3 mol%), toluene (10 mL), reaction time 2 h, brownish oil (146 mg, 403 μmol, 99%). (pinB)(Me)CC(Ph)(Bcat) (major component), δ1H (500 MHz, C6D6, rt): 0.95 (12H, s, OC(CH3)2), 2.06 (3H, s, C(Bcat)CH3), 6.81–6.84 (2H, m, CHcat), 6.99–7.04 (1H, m, 4-CHPh), 7.06–7.10 (2H, m, CHcat), 7.10–7.15 (2H, m, 3-CHPh), 7.33–7.38 (2H, m, 2-CHPh). δ13C{1H} (125 MHz, C6D6, rt): 17.7 (C(Bpin)CH3), 24.6 (OC(CH3)2), 84.2 (OC(CH3)2), 112.5 (CHcat), 122.5 (CHcat), 127.0 (4-CHPh), 128.7 (3-CHPh), 128.8 (2-CHPh), 140.6 (CPh), 142 (br., BCCB), 147 (br., BCCB). 149.2 (Ccat). m/z (EI+, 70 eV, GC/MS) 362 (3) [M]+, 305 (28), 262 (7), 220 (5), 158 (7), 143 (17), 128 (11), 115 (12), 84 (100), 69 (28). m/z (HR-EI+, 70 eV, GC/MS) 362.18433 [M]+, calc. for C21H24B2O4: 362.18607 (−1.74 mmu). (catB)(Me)CC(Ph)(Bpin) (minor component) δ1H (500 MHz, C6D6, rt): 0.97 (12H, s, OC(CH3)2), 1.97 (3H, s, C(Bcat)CH3), 6.85–6.89 (2H, m, CHcat), 7.04–7.10 (1H, m, 4-CHPh, overlapping), 7.11–7.15 (2H, m, CHcat, overlapping), 7.19–7.24 (2H, m, 3-CHPh), 7.31–7.36 (2H, m, 2-CHPh, overlapping). δ13C{1H} (125 MHz, C6D6, rt): 18.5 (C(Bcat)CH3), 24.6 (OC(CH3)2, overlapping), 84.1 (OC(CH3)2), 112.6 (CHcat), 122.7 (CHcat), 126.7 (4-CHPh), 128.3 (3-CHPh), 129.0 (2-CHPh), 141.0 (CPh), 142 (br., BCCB), 147 (br., BCCB). 149.1 (Ccat), not all signals could be unambiguously identified. m/z (EI+, 70 eV, GC/MS) 362 (5) [M]+, 305 (100), 291 (9), 262 (7), 220 (10), 158 (25), 143 (70), 128 (37), 115 (73), 105 (33), 84 (25), 69 (32). m/z (HR-EI+, 70 eV, GC/MS) 362.18847 [M]+, calc. for C21H24B2O4: 362.18607 (2.40 mmu). δ11B{1H} (160 MHz, C6D6, rt): 31.7 (s, Δw1/2 = 522 Hz), isomers not resolved.C(Ph)(Bpin) (mixture of isomers). 3ac (100 mg, 417 μmol), Me–CC–Ph (63 mg, 542 μmol), 2 (9.4 mg, 13 μmol, 3 mol%), toluene (10 mL), reaction time 2.5 h, brownish oil (146 mg, 410 μmol, 98%). (neopB)(Me)CC(Ph)(Bpin) (major component), δ1H (500 MHz, C6D6, rt): 0.67 (6H, s, OCH2C(CH3)2), 1.12 (12H, s, OC(CH3)2), 2.05 (3H, s, C(Bneop)CH3), 3.41 (4H, s, OCH2C(CH3)2), 7.03–7.08 (1H, m, 4-CHPh), 7.19–7.25 (2H, m, 3-CHPh), 7.39–7.42 (2H, m, 2-CHPh). δ13C{1H} (125 MHz, C6D6, rt): 18.1 (C(neop)CH3), 21.9 (OCH2C(CH3)2), 25.1 (OC(CH3)2), 31.4 (OCH2C(CH3)2), 72.0 (OCH2C(CH3)2), 83.1 (OC(CH3)2), 126.1 (4-CHPh), 128.3 (3-CHPh), 128.9 (2-CHPh), 143.0 (CPh), 146 (br., BCCB), 151 (br., BCCB). m/z (EI+, 70 eV, GC/MS) 356 (0.3) [M]+, 341 (2) [M–Me]+, 298 (4), 283 (3), 240 (5), 213 (4), 187 (3), 169 (2), 155 (3), 143 (12), 129 (6), 116 (12), 105 (4), 84 (100), 69 (16). m/z (HR-EI+, 70 eV, GC/MS) 341.20789 [M–Me]+, calc. for C19H27B2O4: 341.20954 (−1.66 mmu). (pinB)(Me)CC(Ph)(Bneop) (minor component), δ1H (500 MHz, C6D6, rt): 0.64 (6H, s, OCH2C(CH3)2), 1.16 (12H, s, OC(CH3)2), 2.03 (3H, s, C(Bneop)CH3), 3.43 (4H, s, OCH2C(CH3)2), 7.03–7.08 (1H, m, 4-CHPh, overlapping), 7.19–7.25 (2H, m, 3-CHPh, overlapping), 7.39–7.42 (2H, m, 2-CHPh, overlapping). δ13C{1H} (125 MHz, C6D6, rt): 18.1 (C(Bneop)CH3), 22.0 (OCH2C(CH3)2), 25.1 (OC(CH3)2), 31.4 (OCH2C(CH3)2), 72.2 (OCH2C(CH3)2), 83.3 (OC(CH3)2), 126.1 (4-CHPh), 128.3 (3-CHPh), 128.9 (2-CHPh), 143.0 (CPh). (EI+, 70 eV, GC/MS) 356 (1) [M]+, 341 (5) [M–Me]+, 143 (13), 129 (8), 116 (100), 105 (8), 69 (42). m/z (HR-EI+, 70 eV, GC/MS) 356.23438 [M]+, calc. for C20H30B2O2: 356.23302 (1.37 mmu). δ11B{1H} (96 MHz, C6D6, rt): 27.6 (s, Δw1/2 = 417 Hz), 31.4 (s, Δw1/2 = 424 Hz), isomers not resolved.Results and discussion
Synthesis
The unsymmetrical platinum bis-boryl complexes 3ab, 3ac, 3ae, 3bd and 3be were obtained by oxidative addition of the respective unsymmetrical diborane(4) derivatives 1 to the platinum(0) complex 2 (Scheme 2). The procedure involved repeated evacuation of the reaction mixture consisting of equimolar amounts of 1 and 2 in order to remove the formed ethene and shift the equilibrium of the reversible oxidative addition reaction to the product side.3 The unsymmetrical diboranes 1 employed comprise dialkoxy/aryloxy substituents such as Bpin (pin = (OCMe2)2), Bneop (neop = (OCH2)2CMe2), Bcat (cat = 1,2-O2C6H4) as well as diamino substituents such as BMeEn (MeEn = (NMe)2C2H4), Bdmab (dmab = 1,2-(NMe)2C6H4) but also the sterically more demanding congeners BiPrEn (iPrEn = (NiPr)2C2H4) and BtBuEn (tBuEn = (NtBu)2C2H4) (Scheme 2).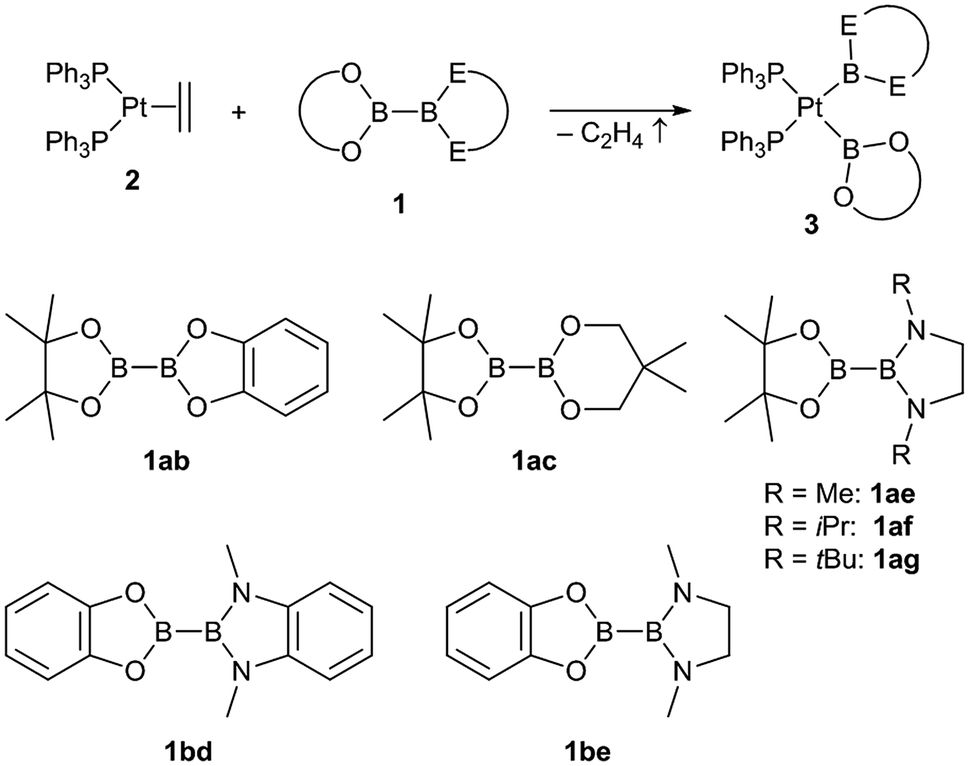 | ||
| Scheme 2 Synthesis of the unsymmetrical complexes (Ph3P)2Pt(boryl)2 3ab, 3ac, 3ae, 3bd and 3be by oxidative addition of the respective unsymmetrical diboranes(4) 1 to [(Ph3P)2Pt(C2H4)] (2). | ||
For the five unsymmetrical diboranes(4) 1ab, 1ac, 1ae, 1bd and 1be the respective unsymmetrical platinum(II) complexes 3ab (58%), 3ac (52%), 3ae (68%), 3bd (58%) and 3be (67%) were obtained as colourless or pale solids in good yields.
In contrast, with the sterically more demanding congeners of 1ae, pinB–BiPrEn (1af) and pinB–BtBuEn (1ag), no platinum(II) complexes could be isolated. However, for the reaction of 1af with 2 in situ NMR spectroscopy indicates that an oxidative addition to the complex [(Ph3P)2Pt(Bpin)(BiPrEn)] (3af) takes place (Fig. 1). According to 1H NMR signals ratios a 1af:3af ratio of 26:100 is obtained after repeated evacuation cycles. The 31P{1H} NMR spectrum indicates also incomplete reaction, as besides the signals assigned to the complex 3af unreacted 2 is observed (Fig. 1).9 In our hands all attempts to isolate 3af resulted in mixtures of 1af, 3af and variable amounts of unidentified decomposition products (Fig. 1).9
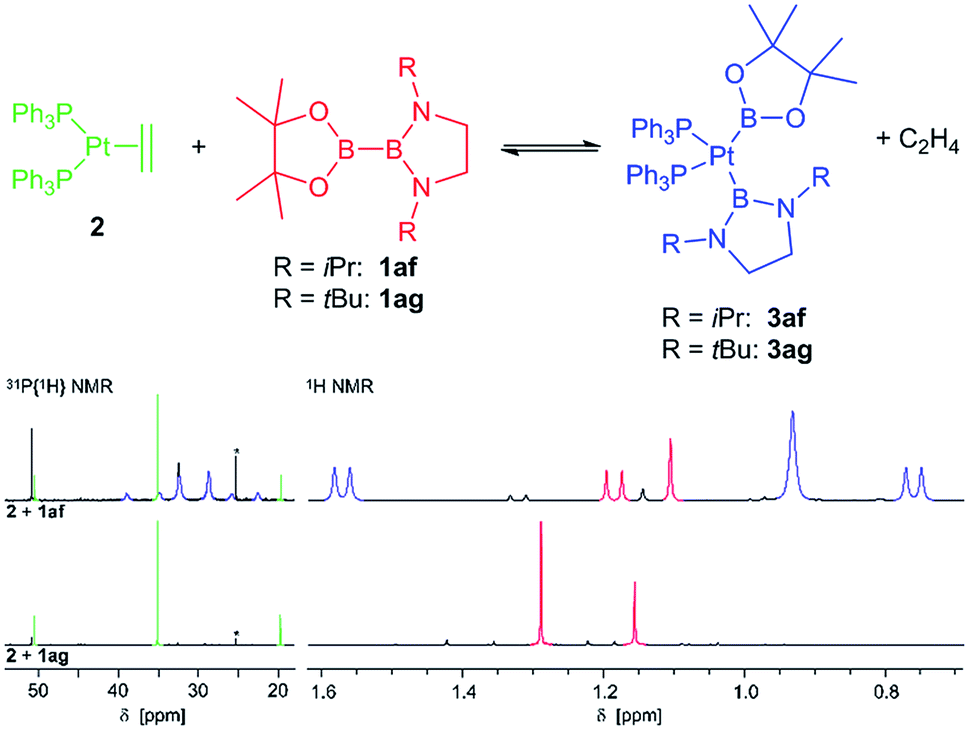 | ||
| Fig. 1 NMR spectra of the reactions of 2 with 1af and 1ag, respectively. Top: 1af + 2, material obtained after eight cycles of evacuation (8 h). Bottom: In situ NMR spectra of 1ag + 2 after three cycles of evacuation (24 h). 300/122 MHz, rt, C6D6, *impurity of OPPh3. | ||
With the more demanding 1ag, however, no reaction is observed by in situ NMR spectroscopy (Fig. 1), as also reported for the related sterically demanding unsymmetrical diborane(4) pinB–Bdtab (tab = 1,2-(N(SiMe3))2C6H4).3,9
In addition to those unsymmetrical complexes the symmetrical complexes [(Ph3P)2Pt(Bneop)2] (3cc) and [(Ph3P)2Pt(Bmap)2] (3hh, map = 1,2-(O)(NMe)C6H4, derived from N-methylaminophenol)9 were analogously obtained from the respective symmetrical diborane(4) derivatives B2neop2 (1cc) and B2map2 (1hh) in yields of 57% and 39%, respectively (Scheme 3).
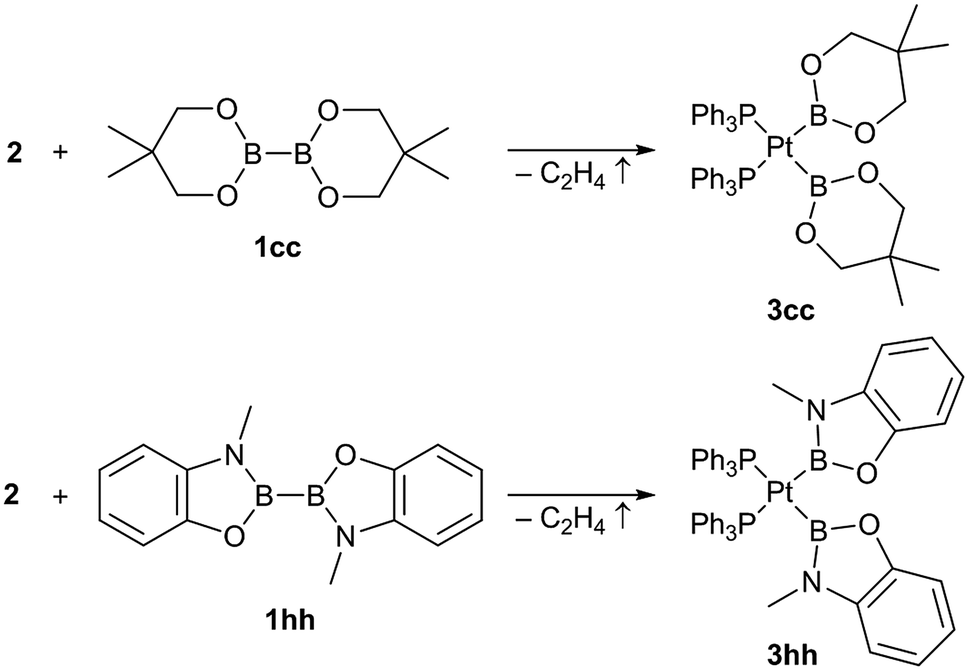 | ||
| Scheme 3 Synthesis of the symmetrical bis-boryl platinum complexes (Ph3P)2Pt(boryl)2 3cc and 3hh. | ||
Crystallography
All five isolated unsymmetrical platinum(II) bis-boryl complexes 3ab, 3ac, 3ae, 3bd and 3be as well as the two symmetrical complexes 3cc and 3hh were structurally characterized by single-crystal X-ray diffraction (Table 1). Well-developed single-crystals suitable for X-ray diffraction studies were typically obtained by vapour diffusion of n-pentane at room temperature into solutions of the complexes in benzene or THF typically within 16–30 h. This technique gave repeatedly well-developed, relatively large crystals, whilst crystallisation from the same or related solvents combinations (toluene instead of benzene) at −40 °C results frequently microcrystalline powders or intergrown crystals. However, as the platinum(II) bis-boryl complexes appear not indefinitely stable in solutions at room temperature (vide infra) comparably fast crystallization at room temperature appears favourable. The complexes 3ab, 3ae, 3be, 3cc and 3hh crystallize as the solvates 3ab(C6H6), 3ae(C4H8O)1.5, 3be(C6H6), 3cc(C4H8O) and 3hh(C4H8O) whereas 3ac and 3bd crystallize without co-crystallization of solvent. 3ab(C6H6), 3bd, 3be(C6H6) and 3cc(C4H8O) crystallize in the triclinic system in the space group type P, whilst 3ae(C4H8O)1.5 and 3hh(C4H8O) were found to crystallise in the monoclinic system (P21/c), whereas 3ac crystallises in the orthorhombic system (Pbca). Common to the solid state structures of all seven complexes is the absence of any molecular symmetry; all complexes are situated on general positions. For the complexes 3ac, 3ae, 3bd, 3be, 3cc and 3hh the asymmetric unit of the respective structures contain one independent complex molecule (Z′ = 1), whereas for 3ab(C6H6) two independent molecules of 3ab as well as C6H6 are found in the asymmetric unit (Z′ = 2). Contrary to the platinum complexes the solvent in 3ae(C4H8O)1.5 and 3be(C6H6) exhibits addition symmetry and resides (partially) on centres of inversion. In 3ae(C4H8O)1.5 two distinct THF moieties are found, one on a centre of inversion and one on a general position, whereas in 3be(C6H6) the two independent molecules of benzene are located on the two distinct types of centres of inversion in P. None of the structures exhibits disorder within the platinum bis-boryl complex, however, positional disorder is observed for one of the co-crystallized solvent molecules in 3ae(C4H8O)1.5.
All described platinum(II) bis-boryl complexes exhibit, as to be expected, a slightly distorted square-planar coordination environments at the platinum(II) (d8) ion with cis-arrangement of the boryl ligands. The sum of angles in the coordination sphere lies in a very narrow range of 360–362°, except for 3bd where a value of 364.5(1)° is found (Fig. 2–5). The distortion from planarity towards a tetrahedron is, as indicated by the angle τ(B,B/P,P) included by the [B,Pt,B] and [P,Pt,P] planes, small but significant (Table 2).
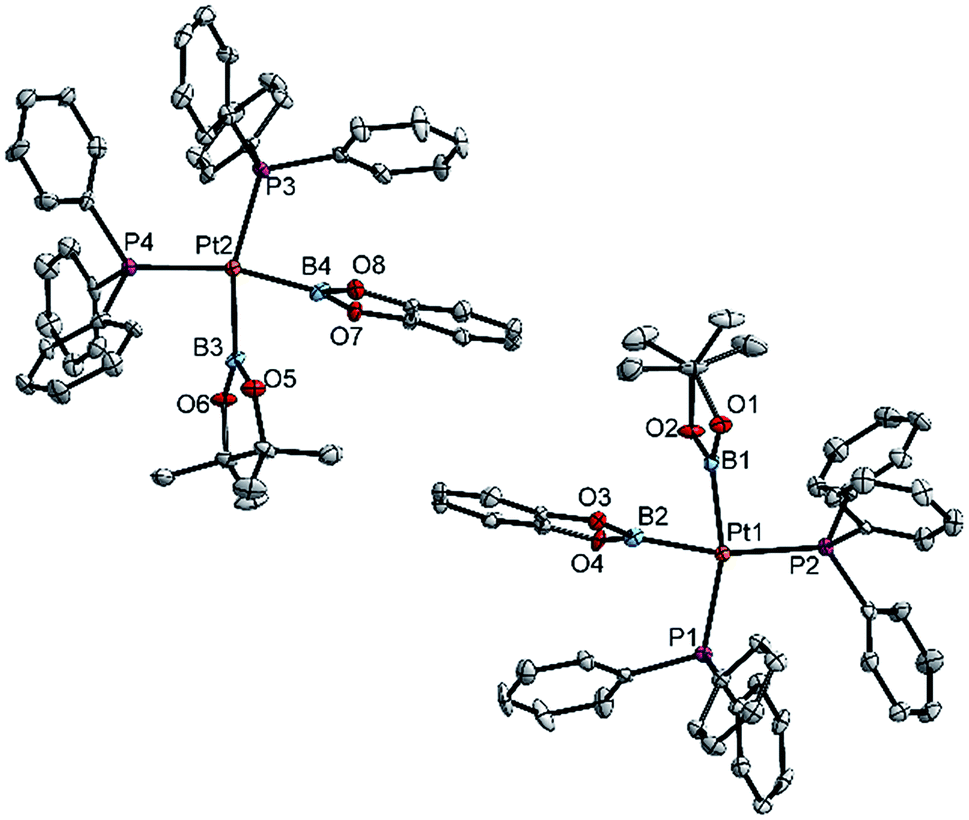 | ||
| Fig. 2 Molecular structures of the two independent molecules of 3ab in the crystal structure of 3ab(C6H6) (Table 3). Selected distances (Å) and angles [°]: Pt1–P1 2.340(1), Pt1–P2 2.344(1), Pt1–B1 2.074(6), Pt1–B2 2.041(6), P1–Pt1–P2 104.97(5), B1–Pt1–B2 72.8(2), B1–Pt1–P2 92.9(2), B2–Pt1–P1 89.6(2), Σ(Pt1) 360.3(2); Pt2–P3 2.340(1), Pt2–P4 2.349(1), Pt2–B3 2.079(6), Pt2–B4 2.042(6), P3–Pt2–P4 105.55(5), B3–Pt2–B4 73.2(2), B3–Pt1–P4 92.1(2), B4–Pt2–P3 89.2(2), Σ(Pt1) 360.1(2). | ||
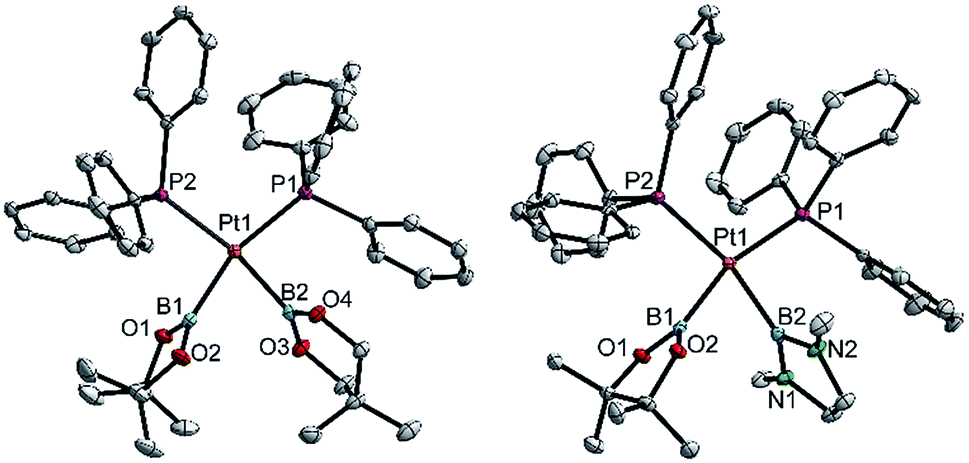 | ||
| Fig. 3 Molecular structures of 3ac (left) and 3ae (right, from the crystal structure of 3ae(C4H8O)1.5, Table 3). Selected distances (Å) and angles [°]: 3ac: Pt1–P1 2.3305(7), Pt1–P2 2.3376(7), Pt1–B1 2.071(3), Pt1–B2 2.084(3), P1–Pt1–P2 103.94(2), B1–Pt1–B2 75.1(1), B1–Pt1–P2 94.48(9), B2–Pt1–P1 88.19(9), Σ(Pt1) 361.7(1). 3ae: Pt1–P1 2.3259(8), Pt1–P2 2.3325(8), Pt1–B1 2.062(4), Pt1–B2 2.088(4), P1–Pt1–P2 103.50(3), B1–Pt1–B2 71.9(1), B1–Pt1–P2 93.8(1), B2–Pt1–P1 90.8(1), Σ(Pt1) 360.0(1). | ||
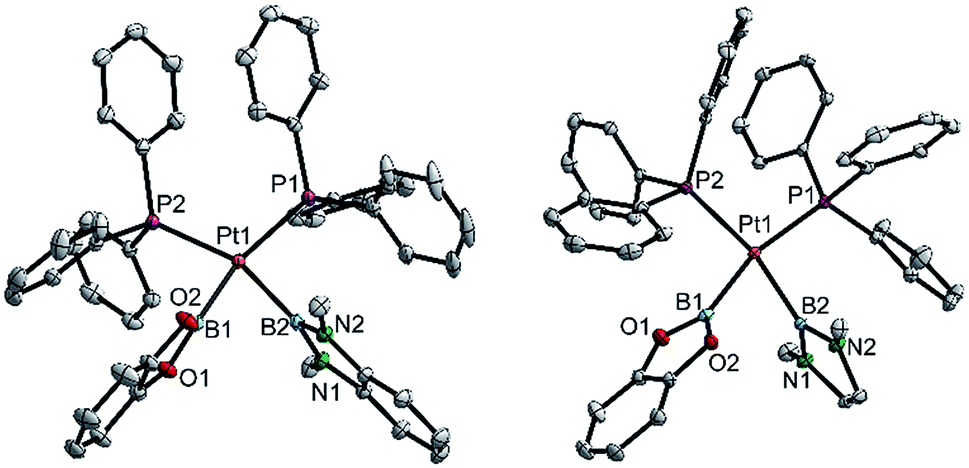 | ||
| Fig. 4 Molecular structures of 3bd (left) and 3be (right, from the crystal structure of 3be(C6H6), Table 3). Selected distances (Å) and angles [°]: 3bd: Pt1–P1 2.3353(3), Pt1–P2 2.3462(3), Pt1–B1 2.041(2), Pt1–B2 2.089(2), P1–Pt1–P2 107.51(1), B1–Pt1–B2 75.55(6), B1–Pt1–P2 89.28(4), B2–Pt1–P1 92.11(4), Σ(Pt1) 364.5(1). 3be: Pt1–P1 2.3328(4), Pt1–P2 2.3547(4), Pt1–B1 2.057(2), Pt1–B2 2.093(2), P1–Pt1–P2 102.98(1), B1–Pt1–B2 73.51(7), B1–Pt1–P2 92.39(5), B2–Pt1–P1 91.15(5), Σ(Pt1) 360.0(1). | ||
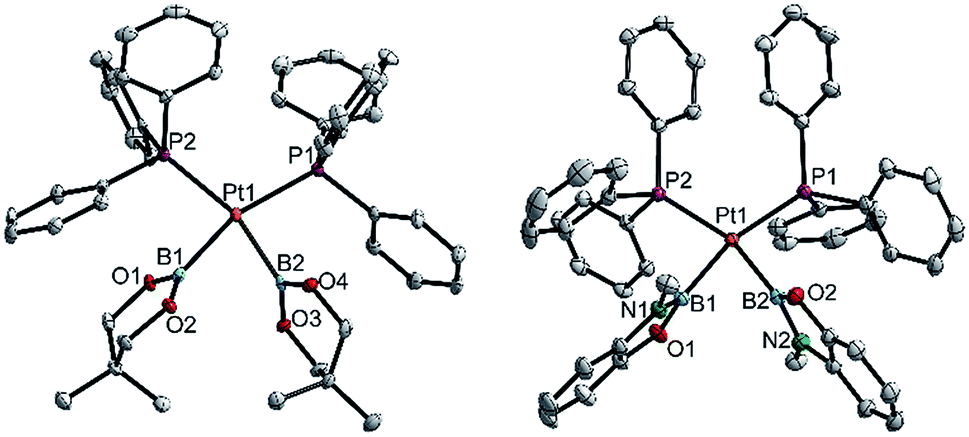 | ||
| Fig. 5 Molecular structures of 3cc (left, from the crystal structure of 3cc(C4H8O)) and 3hh (right, from the crystal structure of 3hh(C4H8O), Table 3). Selected distances (Å) and angles [°]: 3cc: Pt1–P1 2.348(1), Pt1–P2 2.345(1), Pt1–B1 2.085(5), Pt1–B2 2.075(4), P1–Pt1–P2 111.44(3), B1–Pt1–B2 76.18(13), B1–Pt1–P2 86.52(12), B2–Pt1–P1 86.99(12), Σ(Pt1) 361.1(5). 3hh: Pt1–P1 2.3369(3), Pt1–P2 2.3409(3), Pt1–B1 2.073(1), Pt1–B2 2.065(2), P1–Pt1–P2 106.53(1), B1–Pt1–B2 73.80(6), B1–Pt1–P2 87.39(4), B2–Pt1–P1 92.27(4), Σ(Pt1) 360.0(1). | ||
| 3yz17 | B⋯B [Å] | τ(B,B/P,P)a [°] | P(y)–Pt [Å] | P(z)–Pt [Å] | B(y)–Pt [Å] | B(z)–Pt [Å] |
|---|---|---|---|---|---|---|
| a Angle included by the planes [B,Pt,B] and [P,Pt,P].b Σ(Pt1) 359.9(4).c Two independent molecules in the asymmetric unit.d Σ(Pt1) 361.3(2).e Σ(Pt1) 360.2(3). | ||||||
| 3aab | 2.54(1) | 3.6(2) | 2.351(2) | 2.353(1) | 2.076(8) | 2.077(6) |
| 3abc | 2.443(8) | 6.3(2) | 2.340(1) | 2.344(1) | 2.074(6) | 2.041(6) |
| 2.457(8) | 2.3(2) | 2.340(1) | 2.349(1) | 2.079(6) | 2.042(6) | |
| 3ac | 2.532(5) | 14.60(8) | 2.3305(7) | 2.3376(7) | 2.071(3) | 2.084(3) |
| 3add | 2.448(5) | 13.09(9) | 2.3398(7) | 2.3322(7) | 2.078(3) | 2.067(3) |
| 3ae | 2.430(6) | 6.5(1) | 2.3259(8) | 2.3325(8) | 2.062(4) | 2.088(4) |
| 3bbe | 2.554(9) | 9.3(2) | 2.354(1) | 2.347(1) | 2.040(5) | 2.058(6) |
| 3bd | 2.443(2) | 13.21(6) | 2.3353(3) | 2.3462(3) | 2.041(2) | 2.089(2) |
| 3be | 2.484(2) | 2.09(5) | 2.3328(4) | 2.3547(4) | 2.057(2) | 2.093(2) |
| 3cc | 2.566(8) | 11.4(1) | 2.348(1) | 2.345(1) | 2.085(5) | 2.075(4) |
| 3hh | 2.485(2) | 6.25(4) | 2.3369(3) | 2.3409(3) | 2.073(1) | 2.065(2) |
All P–Pt distances are in a quite narrow range of 2.326–2.355 Å. These comparably long P–Pt distances are evidence for the generally strong trans-influence of boryl ligands, e.g. [(Ph3P)2PtCl2] 2.26 Å, [(Ph3P)2Pt(nBu)2] 2.30 Å, [(Ph3P)2Pt(SiR2H)2] (R = Me, Ph) 2.34–2.37 Å.5
It has been proposed that the P–Pt distances are a suitable measure for the donor properties of a ligand in a complex [(Ph3P)2PtXY] (X, Y: anionic ligand), following the reasoning that a shortening of a trans-boryl P–Pt bond in dependence of a particular ligand indicates a relatively lower trans-influence of this ligand compared to a reference ligand, and analogously for the cis-influence of this ligand.7 However, the differences in P–Pt distances between the individual boryl ligands are quite small and appear not especially indicative for a comparison of the trans- and cis-influences of the individual boryl ligands given in Table 2. If at all, it may be stated that, taking 3aa as a reference compound, all other boryl ligands have a relatively lower trans- and cis-influence.7 However, this contradicts experimental and computational studies suggesting a significantly stronger trans-influence of diamino boryl than dialkoxy boryl ligands.12
The Pt–B distances vary significantly and apparently systematically with the type of the individual boryl ligand (Table 2). Within the set of structures discussed here (Table 2) three types of boryl ligands may defined: (i) (alkylO)2B, such as Bpin and Bneop, (ii) aryloxyboryl ligands, here Bcat, (iii) and diaminoboryl ligands, such as Bdmab and BMeEn. The first class of ligands exhibits comparably long Pt–B distances in a range of 2.062–2.085 Å (0.003–0.008 Å esd) with an arithmetic mean of 2.076 Å (Table 2). Although slightly shorter values were found for individual B–Pt distances in the (alkylO)2B complexes [(Ph3P)2Pt(B(OCH(COOMe))2)2] and [(Ph3P)2Pt(B(OCH2C(H)PhO)2)2] (<2.06 Å) this range appears typical for this class of ligands.4d In contrast the aryloxy substituted Bcat ligand leads to significantly shorter B–Pt distances in a range of 2.040–2.058 Å (0.002–0.006 Å esd) (arithmetic mean 2.047 Å). This agrees also with the Pt–B distances found for the catechol derived boryl ligands in [(Ph3P)2Pt(B(O2C6Cl4))2] (2.03(2), 2.04(2) Å) and [(Ph3P)2Pt(B(O2C6H3-4-tBu)2] (2.046(13), 2.047(10) Å).4c,g Finally, diaminoboryl ligands result in B–Pt distances comparable to those in dialkoxy boryl complexes but on the long side of those in aryloxy boryl complexes, as evidenced by the complexes 3ad,3 3ae, 3bd and 3be exhibiting B–Pt distances in a range of 2.067–2.093 Å (0.002–0.003 Å esd) with an arithmetic mean of 2.084 Å. The latter agrees also with the distances reported for [(Ph3P)2Pt((NBn)2C6H4))2] (2.092(2), 2.080(2) Å).3
The B⋯B distances are for all bis-boryl platinum(II) complexes of the type (Ph3P)2Pt(boryl)2, reported in this work (Table 2) or in the literature, much shorter, <2.62 Å, than the doubled van-der-Waals radius of boron (3.84 Å).3,4,14a However, those B⋯B distances are still much longer than those in the parent diborane(4) derivatives (averaged 1.7 Å, double covalent radii 1.68 Å).3,8,14b,15 However, those B⋯B distances are also longer than those found in bis-boryl cobalt complexes (2.19–2.27 Å) and a bis-boryl iridium complex (2.22 Å) where the presence of a three-centre metal–boron–boron interaction is supported by DFT computations.6d,16 Nonetheless, the short B⋯B distances still suggest an residual B⋯B interaction and, hence, the non-independence of the two boryl ligands within this series of complexes (vide infra).
NMR spectroscopy
The 11B{1H} spectra of 3ac, 3ae, 3af (in situ NMR data, vide supra), 3be, 3cc and 3hh exhibit very broad signals (1200–2500 Hz FWHM) around 47 ppm in agreement with the chemical shifts reported for related complexes.3,4 However, for none of the unsymmetrical complexes 3ac, 3ae, 3af and 3be (and also 3ad and [(Ph3P)2Pt((NBn)2C6H4)2] (45 ppm, 3200 Hz)) two discernible signals for the two individual boryl ligands were observed.3 For the complexes 3ab and 3bd, however, no 11B{1H} NMR signal could be detected, presumably due to the very broad line shape of the quadrupolar 11B nucleus in a low-symmetry environment and overlapping of the signals of the two three coordinate boron nucleus in the unsymmetrical boryl complexes. The narrow range and the averaged nature of these signals render an assignment of individual chemical shifts to specific boryl ligands impossible.All new complexes 3ab, 3ac, 3ae, 3af, 3bd, 3be, 3cc and 3hh were also characterised in solution by 1H, 13C{1H}, and 31P{1H} NMR spectroscopy. Beyond this, the complexes 3ab, 3ac and 3ae allowed a more detailed characterisation by 1H–1H NOESY and 1H–31P HMBC NMR spectroscopy allowing the assignment of the 31P{1H} NMR signals trans to a specific boryl ligand (Table 3).3 For the other new unsymmetrical complexes 3bd and 3be this was not unambiguously possible for a lack of indicative 1H–1H NOESY contacts,9 whilst 3af was not isolated in pure form and was characterised only in situ in the reaction mixture by NMR spectroscopy (vide supra).
| 3yz17 | δBa [ppm] | δP(y) [ppm] | JP–Pt(y) [Hz] | δP(z) [ppm] | JP–Pt(z) [Hz] |
|---|---|---|---|---|---|
| a Δw1/2 (FWHM) [Hz] in parentheses.b Measured at 96 MHz (11B{1H}) and 162 MHz (31P{1H}).c Not isolated, in situ NMR data. Measured at 96 MHz (11B{1H}) and 122 MHz (31P{1H}).d No assignment to individual ligands conducted. | |||||
| 3aa | 46.0 | 27.8 | 1515 | ||
| 3ab | 35.5 | 1565 | 28.7 | 1690 | |
| 3ac | 51 (2500) | 33.7 | 1585 | 27.5 | 1455 |
| 3ad | 47.5 (1680)b | 34.5b | 1590b | 31.8b | 1715b |
| 3ae | 47 (1600) | 36.1 | 1615 | 31.1 | 1590 |
| 3af | 45 (1200)c | 28.7/32.4c | 1495/1600c | ||
| 3bb | 47.0 (br)4c | 30.2 (ref. 4b)/28.7 (ref. 4c) | 1610 (ref. 4b)/1640 (ref. 4c) | ||
| 3bd | 31.0/33.7d | 1650/1715d | |||
| 3be | 46 (2135) | 30.9/35.6d | 1520/1745d | ||
| 3cc | 45 (1500) | 31.1 | 1440 | ||
| 3hh | 47 (1740) | 32.8 | 1610 | ||
The 31P{1H} NMR chemical shifts of the phosphorous atom trans to a particular boryl ligand shows no systematic trend and varies e.g. for Bpin in a range of 4 ppm around 33.5 ppm (Table 3). However, for the 1JPt–P coupling constants a certain systematic is discernible. The 1JPt–P coupling trans to a Bpin ligand varies in a range of 100 Hz between 1515 Hz and 1615 Hz (this includes also [(Ph3P)2Pt((NBn)2C6H4)(Bpin)]: JP–Pt(trans-Bpin) 1530 Hz, JP–Pt(trans-B((NBn)2C6H4)) 1590 Hz), whereas for the closely related dialkoxy boryl ligand Bneop lower values of 1440 Hz (3cc) and 1455 Hz (3ac) are observed.3,12 For the diaryloxy ligand Bcat larger values above 1600 Hz are found, however, the restricted data set and the not unambiguously assigned data for 3bd and 3be renders this assignment tentative. Moreover, it should be noted that for the symmetrical 3bb two distinct JPt–P coupling constants are reported.4b,c From the data available it is clear that the 1JPt–P coupling constant is not a viable measure of the trans-influence of the trans-boryl ligand.7,18 It is clear, as suggested earlier, that the ligand in cis position has an appreciable influence on the trans 1JPt–P coupling constants (cis influence).3
Stability and decomposition
It has been earlier reported for 3ad and [(PCy3)2Pt(B(OMe)2)2] that bis-boryl platinum(II) complexes exist in solution in equilibrium with their reductive elimination products, the parent diborane(4) and a platinum(0) complex [(R3P)2Pt].3,6a This appears to hold also for the complexes 3ac, 3ae and 3bd, as in the 1H NMR spectra of freshly prepared samples of these complexes signals indicative for the respective parent diborane(4) derivatives (Fig. S2a, S3b and S4a†) were observed in minute amounts (3ac:1ac 25:1, 3ae:1ae 30:1, 3bd:1bd 100:1).9 However, only for 3bd unambiguous exchange signals were detected by 1H–1H NOESY spectroscopy (Fig. S4a†).9 Nonetheless, it must be emphasised that these complexes are not indefinitely stable in solution at room temperature and one of the decomposition products is the parent diborane(4) derivative itself.
Freshly prepared C6D6 solutions of the bis-boryl platinum(II) complexes are virtually colourless but over time an intense reddish or orange colouration is observed.19 Exemplarily the decomposition of 3cc was monitored by NMR spectroscopy over 38 h at ambient temperature in C6D6 and indicates the reductive elimination of the diborane(4), B2neop2 (1cc), along with the formation of so far unidentified platinum phosphine complexes (Fig. S7†).9
From solutions of the bis-boryl platinum(II) complexes 3 repeatedly intense orange crystals of [((Ph3P)Pt-μ-PPh2)2] and [(Ph3P)2Pt3(μ-PPh2)3Ph] (as their THF solvates) were obtained upon crystallisation at room temperatures after several days (vide supra) and were characterised crystallographically (Fig. S8 and S9†).9 These two complexes have been described previously as thermal decomposition products of 2.20
It should be noted that we could not observe any evidence for scrambling of the boryl ligands, hence the formation of symmetrical bis-boryl platinum complexes or symmetrical diborane(4) derivatives. Considering that this could proceed e.g. via a bimolecular ligand exchange between two unsymmetrical bis-boryl platinum(II) complexes, a reaction of a bis-boryl platinum(II) complexes and a free boryl ligand (e.g. from dissociation from a bis-boryl platinum(II) complexes) or via an oxidative addition/reductive elimination pathway of a bis-boryl platinum(II) complex and a diborane(4) derivative. All these reactions are apparently not favourable for the square-planar bis-boryl platinum(II) complexes considered here.
Catalysis
Since its introduction by Suzuki and co-worker in 1993 the platinum catalysed bis-borylation of alkynes with diborane(4) derivatives has been established as a facile and widely used access to 1,2-bis-borylated alkenes.2 For these borylation reactions bis-boryl platinum complexes are established reactive intermediates.2,4a More recently Suginome and co-worker have shown that the unsymmetrical diborane(4) pinB–Bdan (dan = naphthalene-1,8-diaminato) may be used as reagent in the regioselective bis-borylation of unsymmetrical alkynes.21The unsymmetrical diborane(4) derivatives 1ab, 1ac and 1ad were exemplarily used to borylate the unsymmetrical alkyne Ph–CC–Me (Scheme 4) in the presence of 2 as pre-catalyst. The bis-borylation proceeds smoothly with all three diboranes, yielding the respective unsymmetrically borylated alkenes 5 in excellent yields (Scheme 4). However, the dialkoxy diamino diboron reagent 1ad reacts significantly slower than the unsymmetrical tetra alkoxy diboranes(4) 1ac and 1ad (Scheme 4). The sluggish reaction of 1ad is also apparent in the bis-borylation of symmetrical diphenyl acetylene to give 4ad (Scheme 4).
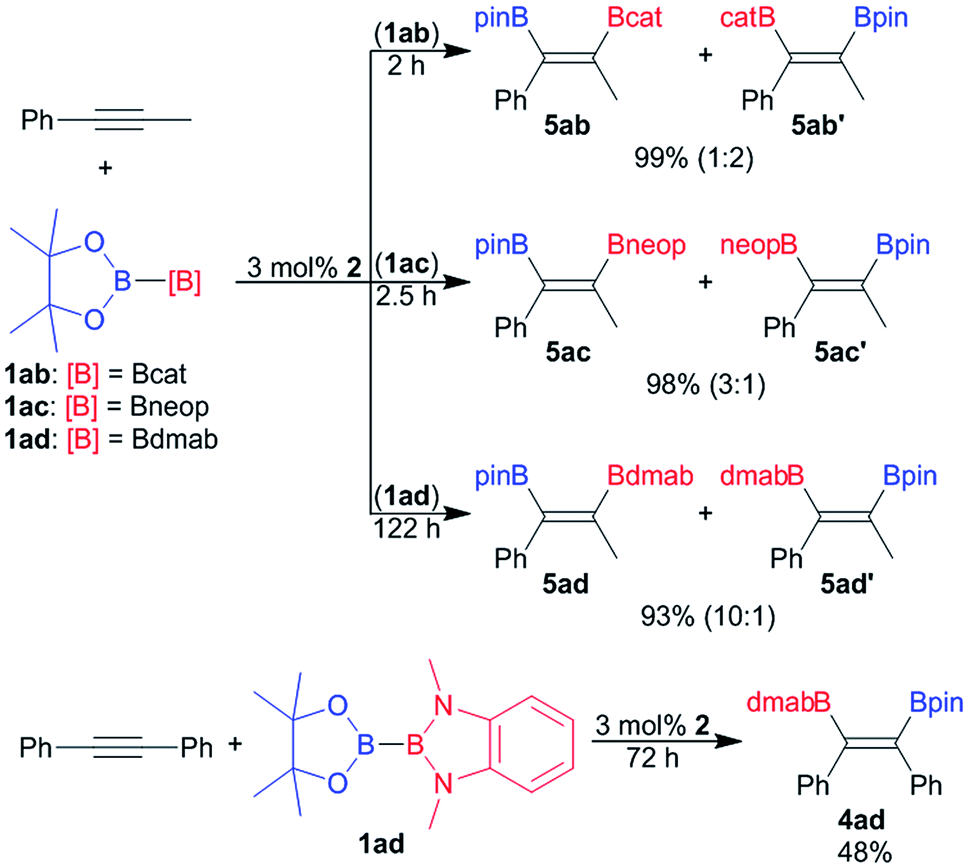 | ||
| Scheme 4 Exemplary platinum catalysed bis-borylation of alkynes with unsymmetrical diboranes(4) 1ab, 1ac and 1ad. | ||
In the case of unsymmetrically substituted acetylene as substrates the bis-borylation gives mixtures of the regioisomers of the unsymmetrically bis-borylated alkenes (Scheme 4). The different regioisomeric bis-borylation products were assigned by 1H–1H NOESY NMR spectroscopy and the isomeric ratio were determined on the basis of the ratio of the characteristic methyl group signals. The alkene 4ad was also characterised by single crystal X-ray diffraction (Fig. S11†).9
The bis-borylation of Ph–CC–Me proceeds for all diboranes 1ab, 1ac and 1ad with some degree of selectivity (Scheme 4). For the dialkoxy diamino borane(4) 1ad a selectivity of 1:10 in favour of the isomer 5ad was observed, notably, the same preference, the Bpin moiety adjacent to the phenyl ring, was also reported for pinB–Bdan as borylating reagent.21 In contrast to that the selectivity with 1ab and 1ac is significantly lower, and for 1ab even reversed. Hence, the isomer 5ab′ with the Bpin moiety located at the carbon atom remote from the phenyl ring becomes predominant. It may be speculated that the selectivity origins for steric reasons – the sterically less demanding boryl moiety favours the more encumbered position adjacent to the phenyl ring. However, more data are certainly needed to derive final conclusions.
Conclusions
The facile access to unsymmetrical diborane(4) derivatives enables for the first time the synthesis of a series of unsymmetrical platinum(II) bis-boryl complexes (Ph3P)2Pt(boryl)(boryl′). The diborane(4) compounds used comprises seven combinations of dialkoxy boryl (Bpin, Bcat, Bneop) and diamino boryl (Bdmab, BMeEn, BiPrEn, BtBuEn) moieties. From these diborane(4) precursors five bis-boryl complexes (Ph3P)2Pt(boryl)(boryl′) were obtained; the sterically demanding ligands BiPrEn and BtBuEn did not allow the isolation of the corresponding boryl complexes.This series of complexes obtained was characterised spectroscopically as well as structurally. The most prominent feature of the distorted square-planar cis-bis-boryl platinum complexes is the short B⋯B distance of 2.44–2.55 Å, >1.2 Å shorter than the doubled van-der-Waals radius of boron. The trans-boryl P–Pt distances are comparably long but in a narrow range around 2.34 Å, as expected for the strongly trans-influencing boryl ligands. However, no distinct variations in the P–Pt distances within this series of boryl ligands is observed that allow to draw conclusions on the relative donor strength of the different boryl ligands. Similar is true for the 31P–195Pt NMR coupling constants, that are indicative for the strongly donating/trans-influencing boryl ligands, but exhibit no systematic variations to allow conclusions on the coordination properties. These findings may be rationalised by the interaction of the two boryl ligands: whilst the complexes may well be described as platinum(II) bis-boryl complexes there is (residual) interaction between the two boryl ligands. In other words, the boryl ligands exhibit not only a significant trans-influence, but also a significant cis-influence. Hence, a specific pair of boryl ligands may be best considered as entity and not as two individual boryl ligands.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. K., W. D. and C. B. gratefully acknowledges support by a Research Grant (KL 2243/5-1) of the Deutsche Forschungsgemeinschaft (DFG) and by the Fonds der Chemischen Industrie. We acknowledge support by the German Research Foundation and the Open Access Publication Funds of the Technische Universität Braunschweig. The authors thank AllyChem Co. Ltd. for a generous gift of diborane(4) reagents.Notes and references
- (a) For an overview on boryl complexes see: L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987–3995 RSC; (b) G. J. Irvine, M. J. G. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, E. G. Robins, W. R. Roper, G. R. Whittell and L. J. Wright, Chem. Rev., 1998, 98, 2685–2722 CrossRef CAS PubMed; (c) H. Braunschweig and M. Colling, Coord. Chem. Rev., 2001, 223, 1–51 CrossRef CAS; (d) S. Aldridge and D. L. Coombs, Coord. Chem. Rev., 2004, 248, 535–559 CrossRef CAS; (e) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed; (f) H. Braunschweig, Angew. Chem., Int. Ed., 1998, 37, 1786–1801 CrossRef CAS.
- (a) For an overview on borylation catalysis see: R. Barbeyron, E. Benedetti, J. Cossy, J.-J. Vasseur, S. Arseniyadis and M. Smietana, Tetrahedron, 2014, 70, 8431–8452 CrossRef CAS; (b) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271–280 CrossRef CAS PubMed; (c) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (d) T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63–73 CrossRef CAS.
- C. Borner and C. Kleeberg, Eur. J. Inorg. Chem., 2014, 2486–2489 CrossRef CAS.
- (a) T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki and N. Miyaura, Organometallics, 1996, 15, 713–720 CrossRef CAS; (b) C. N. Iverson and M. R. Smith III, J. Am. Chem. Soc., 1995, 117, 4403–4404 CrossRef CAS; (c) G. Lesley, P. Nguyen, N. J. Taylor, T. B. Marder, A. J. Scott, W. Clegg and N. C. Norman, Organometallics, 1996, 15, 5137–5154 CrossRef CAS; (d) W. Clegg, T. R. F. Johann, T. B. Marder, N. C. Norman, A. G. Orpen, T. M. Peakman, M. J. Quayle, C. R. Rice and A. J. Scott, J. Chem. Soc., Dalton Trans., 1998, 1431–1438 RSC; (e) A. Kerr, T. B. Marder, N. C. Norman, A. G. Orpen, M. J. Quayle, C. R. Rice, P. L. Timms and G. R. Whittell, Chem. Commun., 1998, 319–320 RSC; (f) D. Curtis, M. J. G. Lesley, N. C. Norman, A. G. Orpen and J. Starbuck, J. Chem. Soc., Dalton Trans., 1999, 1687–1694 RSC; (g) W. Clegg, F. J. Lawlor, G. Lesley, T. B. Marder, N. C. Norman, A. G. Orpen, M. J. Quayle, C. R. Rice, A. J. Scott and F. E. S. Souza, J. Organomet. Chem., 1998, 550, 183–192 CrossRef CAS.
- (a) H.-K. Fun, S. Chantrapromma, Y.-C. Liu, Z.-F. Chen and H. Liang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, E62, m1252–m1254 CrossRef; (b) H. Arii, M. Takahashi, A. Noda, M. Nanjo and K. Mochida, Organometallics, 2008, 27, 1929–1935 CrossRef CAS; (c) M. S. Hannu-Kuure, A. Wagner, T. Bajorek, R. Oilunkaniemi, R. S. Laitinen and M. Ahlgren, Main Group Chem., 2005, 4, 49–68 CrossRef CAS.
- (a) H. Braunschweig and A. Damme, Chem. Commun., 2013, 49, 5216–5218 RSC; (b) H. Braunschweig, P. Brenner, R. D. Dewhurst, F. Guethlein, J. O. C. Jimenez-Halla, K. Radacki, J. Wolf and L. Zöllner, Chem.–Eur. J., 2012, 18, 8605–8609 CrossRef CAS PubMed; (c) H. Braunschweig, A. Damme and T. Kupfer, Chem.–Eur. J., 2013, 19, 14682–14686 CrossRef CAS PubMed; (d) H. Schubert, W. Leis, H. A. Mayer and L. Wesemann, Chem. Commun., 2014, 50, 2738–2740 RSC; (e) H. Braunschweig, M. Lutz, K. Radacki, A. Schaumlöffel, F. Seeler and C. Unkelbach, Organometallics, 2006, 25, 4433–4435 CrossRef CAS; (f) H. Braunschweig, M. Kaupp, C. J. Adams, T. Kupfer, K. Radacki and S. Schinzel, J. Am. Chem. Soc., 2008, 130, 11376–11393 CrossRef CAS PubMed; (g) S. Pospiech, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 6967–6974 CrossRef CAS; (h) H. Braunschweig, M. Lutz and K. Radacki, Angew. Chem., Int. Ed., 2005, 44, 5647–5651 CrossRef CAS PubMed; (i) J. P. H. Charmant, C. Fan, N. C. Norman and P. G. Pringle, Dalton Trans., 2007, 114–123 RSC; (j) H. Braunschweig, R. Bertermann, P. Brenner, M. Burzler, R. D. Dewhurst, K. Radacki and F. Seeler, Chem.–Eur. J., 2011, 17, 11828–11837 CrossRef CAS PubMed; (k) H. Braunschweig, M. Fuss, S. K. Mohapatra, K. Kraft, T. Kupfer, M. Lang, K. Radacki, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Eur. J., 2010, 16, 11732–11743 CrossRef CAS PubMed; (l) H. Braunschweig, T. Kupfer, M. Lutz, K. Radacki, F. Seeler and R. Sigritz, Angew. Chem., Int. Ed., 2006, 45, 8048–8051 CrossRef CAS PubMed; (m) N. Lu, N. C. Norman, A. G. Orpen, M. J. Quayle, P. L. Timms and G. R. Whittell, J. Chem. Soc., Dalton Trans., 2000, 4032–4037 RSC; (n) J. H. Muessig, D. Prieschl, A. Deißenberger, R. D. Dewhurst, M. Dietz, J. O. C. Jiménez-Halla, A. Trumpp, S. R. Wang, C. Brunecker, A. Haefner, A. Gärtner, T. Thiess, J. Böhnke, K. Radacki, R. Bertermann, T. B. Marder and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 13056–13063 CrossRef CAS PubMed.
- L. Rigamonti, A. Forni, M. Manassero, C. Manassero and A. Pasini, Inorg. Chem., 2010, 49, 123–135 CrossRef CAS PubMed.
- W. Oschmann, C. Borner and C. Kleeberg, Dalton Trans., 2018, 47, 5318–5327 RSC.
- See ESI for details.†.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- (a) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC; (b) CrysAlisPro, Version 1.171.36.28 – 1.171.40.21, Agilent Technologies, 2010–2012 Search PubMed; (c) G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 Search PubMed; (d) G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 Search PubMed; (e) G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed; (f) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS; (g) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS; (h) D. Kratzert, J. J. Holstein and I. Krossing, J. Appl. Crystallogr., 2015, 48, 933–938 CrossRef CAS PubMed; (i) C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS; (j) Diamond 4.2.2. – Crystal and Molecular Structure Visualization, Crystal Impact, H. Putz and K. Brandenburg GbR, Bonn, Germany, 2016 Search PubMed.
- (a) J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed; (b) H. Braunschweig, P. Brenner, A. Müller, K. Radacki, D. Rais and K. Uttinger, Chem.–Eur. J., 2007, 13, 7171–7176 CrossRef CAS PubMed.
- Data of (3bb(C6D6)) from ref. 4c; in ref. 4b another solvate (3bb(C7H8)) of 3bb is reported. However, the latter data agree well with the one of 3bb(C6D6), albeit with significant larger errors.
- (a) M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed; (b) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- M. Eck, S. Würtemberger-Pietsch, A. Eichhorn, J. H. J. Berthel, R. Bertermann, U. Paul, H. Schneider, A. Friedrich, C. Kleeberg, U. Radius and T. B. Marder, Dalton Trans., 2017, 46, 3661–3680 RSC.
- (a) C. J. Adams, R. A. Baber, A. S. Batsanov, G. Bramham, J. P. H. Charmant, M. F. Haddow, J. A. K. Howard, W. H. Lam, Z. Lin, T. B. Marder, N. C. Norman and A. G. Orpen, Dalton Trans., 2006, 1370–1373 RSC; (b) C. Dai, G. Stringer, J. F. Corrigan, N. J. Taylor, T. B. Marder and N. C. Norman, J. Organomet. Chem., 1996, 513, 273–275 CrossRef CAS.
- B(y) refers to properties of the boron atom in the boryl group y as given in the compound number 3yz, referring to the naming scheme given in Schemes 2 and 3. Accordingly, P(y) refers to properties of the phosphorous atom trans to the boryl group y, this applies also to the NMR parameters δP(y) and JP–Pt(y). An analogous naming scheme applies with respect to the boryl group z.
- An attempt to differentiate between trans and cis influence implying an additive relation as suggested by Pasini and co-workers was not fruitful.7.
- It should be mentioned that even analytically pure samples of complexes 3 are often slightly coloured, presumably due to decomposition during preparation. However, a deepening of the reddish/orange colour of their solutions is clearly observed nonetheless.
- (a) N. J. Taylor, P. C. Chieh and A. J. Carty, J. Chem. Soc., Chem. Commun., 1975, 448–449 RSC; (b) W. Petz, C. Kutschera and B. Neumüller, Organometallics, 2005, 24, 5038–5043 CrossRef CAS; (c) R. Bender, P. Braunstein, A. Dedieu, P. D. Ellis, B. Huggins, P. D. Harvey, E. Sappa and A. Tiripicchio, Inorg. Chem., 1996, 35, 1223–1234 CrossRef CAS PubMed; (d) R. Bender, P. Braunstein, A. Tiripicchio and M. Tiripicchio Camellini, Angew. Chem., Int. Ed., 1985, 24, 861–862 CrossRef.
- N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548–2549 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental, NMR spectroscopic and crystallographic data. CCDC 1881532–1881542. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra00170k |
| This journal is © The Royal Society of Chemistry 2019 |