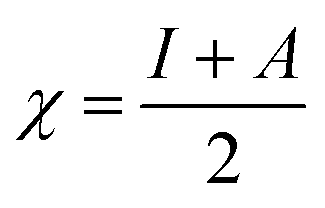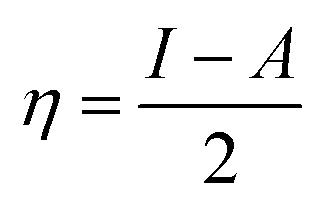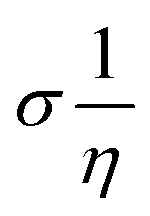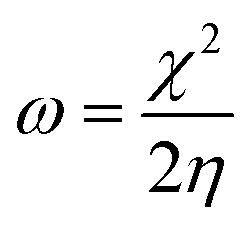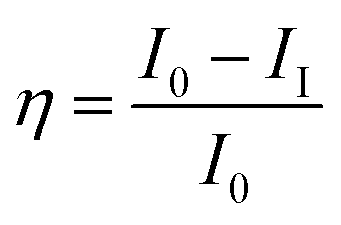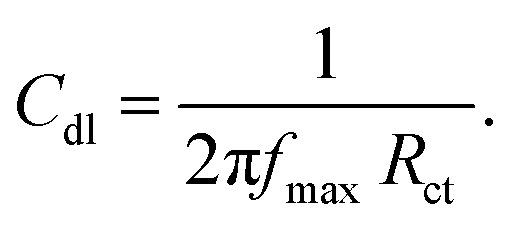DOI:
10.1039/C9RA00106A
(Paper)
RSC Adv., 2019,
9, 8537-8545
Neodymium-decorated graphene oxide as a corrosion barrier layer on Ti6Al4V alloy in acidic medium†
Received
5th January 2019
, Accepted 20th February 2019
First published on 14th March 2019
Abstract
Ti6Al4V alloy is light weight and is used in construction, oil industries and airbus, automobile, and bio implant materials. The native oxide layers of the alloy are not stable at high temperatures and strong mineral acid environments. The conventional epoxy-based layers are porous and the alloy finally fails in the harsh environment in the long term. Therefore, the carbon-based functional materials are being proposed as coating materials to overcome the alloy degradation. In the present contribution, we have used the neodymium-decorated graphene oxide as the corrosion inhibiting barrier for the Ti6Al4V alloy. As a novelty, we found that the few-layer graphene decorated with neodymium acts as a self-cleaning coating. The Nd-decorated graphene oxide were studied by XRD, TEM, FESEM, FTIR, UV, and Raman spectroscopy. The corrosion inhibition efficiency was studied by electrochemical methods.
1. Introduction
Recently titanium-based alloys, specifically Ti6Al4V, have been used to prepare body parts in airbus, automobile industry, nuclear fuel plants and bio-implants due to their strong corrosion resistance in the environment. In the past decades, epoxy coating has been used for corrosion inhibition in the acidic and salt environments. The problem of epoxy coating is the large pore size that allows the ions to penetrate, letting the alloy surface be damaged in the long term. Early reports have shown that Pd-, Pt-, and Ru-based titanium alloys have a strong corrosion resistance in the acidic environment,1 but noble metals are economically unfavourable for making the industrial components. Moreover, several authors reported that the hydroxyl derivatives and heterocyclic compounds can act as corrosion inhibitors in the acidic and salt media.2,3 However, the organic inhibitors do not achieve long-term corrosion inhibition efficiency in the acidic media. Hence, the carbon-based functional materials have been proposed to overcome the long-term corrosion inhibition failure in the acidic medium.4 The carbon-based materials can achieve excellent adsorption on the metal surface, which allows this family of materials to act as corrosion inhibition barrier in several environments. Recently, graphene oxide-functional materials are becoming significant for surface protection because the electrolytes are not able to penetrate through the layered structure of graphene oxide.5 Singhbabu et al. reported a green, oil-based graphene oxide corrosion inhibitor for mild steel, whose performance increased as it improved the hydrophobicity of the coated surface.6 Dutta et al. reported a graphene oxide-polymer composite material that reinforced the corrosion inhibition in the mild steel, which was attributed to the improved hydrophobicity due to the polymer matrix.7 Furthermore, Huh et al. reported that the acetone-derived graphene oxide acts as a corrosion inhibitor on copper in a 3.5% NaCl medium.8 Maia et al. reported that silica nano particles have been used as a corrosion barrier for an aluminium alloy in salt environments.9 Liu et al. reported the investigation of an imidazole-based ionic liquid functionalized graphene oxide as a corrosion inhibitor in NaCl solution on the mild steel;10 the authors attributed the enhanced corrosion inhibition efficiency to the improved electron transfer between the coating surface and the aggressive ions, favoured by the anionic and cationic parts of the ionic liquids. In addition, several authors reported the corrosion inhibition barrier layers in the 3.5% NaCl environment, prepared from graphene oxides functionalized with pyridine as well as composites of graphene oxide with polystyrene, metronidazole, polyvinylene and lignin.11–18 Moreover, the rare earth elements have been reported as green corrosion inhibitors because of their biocompatibility and the ability to transfer electrons to the vacant metal d orbitals. It is also known that the lanthanide series elements can improve the long-term corrosion inhibition efficiency because they have multiple oxidation states. Several authors have reported lanthanide series composite materials and additives for various alloys to improve their corrosion resistance in salt environments. The lanthanides such as Nd, Gd and Ce have been recently found to improve the corrosion resistance in acidic environments.19–23 The addition of La, Sm, Tb or Dy as a microstructure filler to different alloys for the enhanced corrosion resistance has been also investigated. In other reports, La3+ was incorporated into the polymer matrix and Ce-modified coating, Nd and Ce additives, imidazole derivatives and N-functionalized graphene oxide have been demonstrated to improve the corrosion resistance in a 3.5% NaCl solution.24–31 Recently, we investigated the praseodymium-decorated graphene oxide as an inhibition barrier layer on magnesium AZ13 alloy in strong acidic environments, and showed that GO-Pr improved the inhibition efficiency because the corrosive species could not interact with the alloy surface.32 Even though the corrosion inhibition activity of the neodymium-decorated graphene oxide has not been studied to the best of our knowledge, their slightly minor ionic ratio (Nd3+ 112 pm, Pr3+ 112 pm) and the presence of the 2+ and 3+ states in comparison with Pr that preferentially displays the 3+ state could be beneficial for the redox-aided inhibition as mentioned above. Thus, in the present work, we investigated the effect of a neodymium-decorated graphene oxide layer to assess its corrosion inhibition activity in acidic environments in the Ti6Al4V alloy.
2. Experimental
Natural graphite powder, sodium nitrate, H2O2, KMnO4, and neodymium(III) nitrate hexahydrate (Nd(NO3)3·6H2O) were purchased from Alfa Aesar and used without further purification. The GO synthesis was carried out by the modified Hummers method. According to our previous work,32 1 g of natural graphite powder, 50 mL of H2SO4, and 40 mL of H3PO4 were added into a 250 mL round bottom flask and the flask was kept in an ice container, followed by the slow addition of 3 g of the oxidation agent KMnO4 to the mixture. The mixture temperature was increased and kept at 60 °C during 12 h of reflux. Afterwards, the mixture was allowed to cool to room temperature and then poured into 1000 mL of ice water for fine dispersion. Then, 10 mL of H2O2 was added to the mixture to terminate the reaction. The product was washed three times with 1 M HCl for the removal of reaction by-products. The GO was washed several times with distilled water till the solution became neutral. The final product was dried at 80 °C in a vacuum oven.
2.1. GO–neodymium decoration
For decoration, 100 mg of neodymium(III) nitrate hexahydrate and 200 mg of graphene oxide were mixed in a round bottom flask containing 100 mL of distilled water. The mixture was sonicated for 1 h for fine dispersion in a SB-300DTYMULI Ultrasonic cleaner. Then, 10 mL of 1 M NaOH was added to the mixture as an activator. The mixture was stirred for 24 h at room temperature. The product was washed with distilled water until a neutral solution was obtained. The GO + Nd final product was dried at 80 °C in a vacuum oven.
2.2. Material characterization
The UV-visible absorption measurements were performed on a Shimadzu UV-VIS 3600 spectrometer. The molecular structure of the prepared materials was studied by the KBr pellet method on a Spectrum 65 Fourier Transform Infrared Spectrometer (FTIR) from Perkin Elmer. The Raman spectra were acquired from a WITec confocal Raman spectrometer, using a 532 nm laser as the excitation source. The crystal structure was studied by X-ray diffraction analysis performed on a D8 Advance diffractometer from Bruker, using Cu Kα radiation (λ = 1.5406 Å) in the Bragg–Brentano geometry. The morphology was analysed by a Carl Zeiss FESEM Marline microscope. The microstructure of graphene oxide and neodymium-decorated graphene oxide were confirmed by the Transmission Electron Microscopy (TEM) on a FEI Techani G2 Twin 500. The coated films on the Ti6Al4V alloy surface were studied by Attenuated Total Reflectance (ATR) on a Thermo Fisher ATR-FTIR iD7 Nicolet S5 Model spectrometer. The coating thickness was analysed by ellipsometry, with the incident light at 70° and in a range from 0.6500 to 6.500 eV with 0.05 eV increment on a HORIBA UVI Cell2 Ellipsometer.
The theoretical study to model the interactions between the GO + Nd coating and the Ti6Al4V surface to assess the corrosion inhibition activity was carried out using a 03 Gaussian programme package B3LYP 312*G basis set solvent methods. The electronegativity (χ), chemical hardness (η), softness (σ), electrophilicity (ω), and quantum chemical parameters were calculated by following equations:
| |
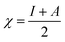 | (1) |
| |
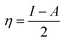 | (2) |
| |
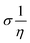 | (3) |
| |
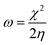 | (4) |
2.3. Corrosion study
The titanium Ti6Al4V alloy was cut into 1 cm × 1 cm pieces. The alloy was connected with a copper wire for electrical conduction and was mounted with cold cure material on one side to block the alloy surface. The exposed area of the alloy was polished with emery paper with 600 to 1200 grits, rinsed with soap solution and distilled water, and finally washed with acetone. The material was kept in a desiccator before use. The coating material was prepared with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 wt% ratio of hardener (polyamine) epoxy and GO + Nd, dispersed in ethanol and sonicated for 1 h in a SB-300DTYMULI ultrasonic cleaner. A colloid solution was formed and used as the coating material. This was carefully applied onto the polished TI6Al4V surface using a 0.2 mm paint brush. The Ti6Al4V-coated coupons were dried at 80 °C for 24 h in a vacuum oven before the corrosion inhibition study. The corrosion inhibition study was done using a CHI 920D electrochemical station and the coated coupons of the TI6Al4V alloy as the working electrode. A platinum foil was used as the counter electrode and an Ag/AgCl electrode was used as the reference electrode. The corrosion inhibition efficiency was calculated by the following equation:
:2 wt% ratio of hardener (polyamine) epoxy and GO + Nd, dispersed in ethanol and sonicated for 1 h in a SB-300DTYMULI ultrasonic cleaner. A colloid solution was formed and used as the coating material. This was carefully applied onto the polished TI6Al4V surface using a 0.2 mm paint brush. The Ti6Al4V-coated coupons were dried at 80 °C for 24 h in a vacuum oven before the corrosion inhibition study. The corrosion inhibition study was done using a CHI 920D electrochemical station and the coated coupons of the TI6Al4V alloy as the working electrode. A platinum foil was used as the counter electrode and an Ag/AgCl electrode was used as the reference electrode. The corrosion inhibition efficiency was calculated by the following equation:| |
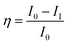 | (5) |
| |
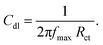 | (6) |
3. Results and discussion
3.1. UV-vis and infrared characterization
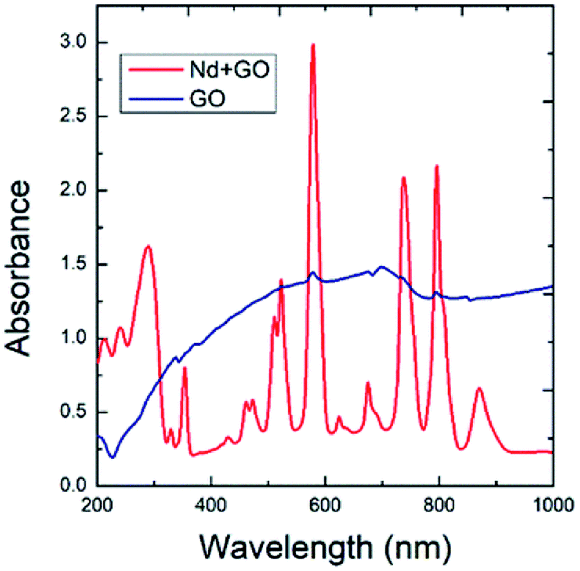
The optical absorption spectra of GO and GO + Nd are shown in Fig. 1. The optical absorption has been used to investigate the electronic transitions of GO and GO + Nd composite materials. The GO absorbs in the range from 200 nm to 250 nm, which is attributable to the electronic transition between π and π*.33,34 However, GO + Nd spectrum presents sharp peaks between 300 and 350 nm due to the n–π electron transition in neodymium. The further absorption peaks observed in GO + Nd spectrum at 500 nm, 650 nm, 750 nm, 800 nm, and 900 nm are due to the valence electron transitions in neodymium.35
 |
| | Fig. 1 Optical absorption spectra of GO and GO + Nd materials. | |
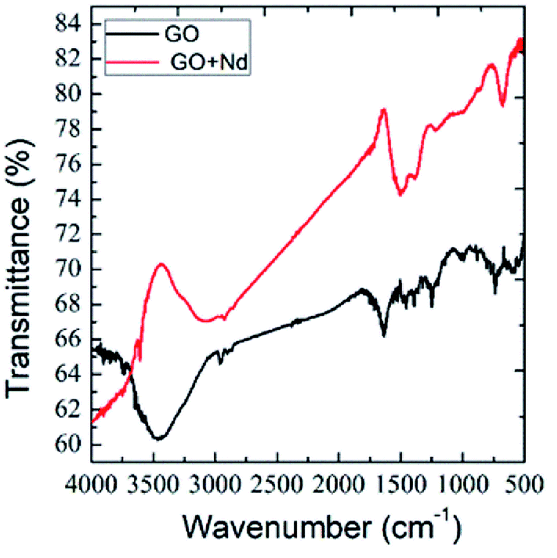
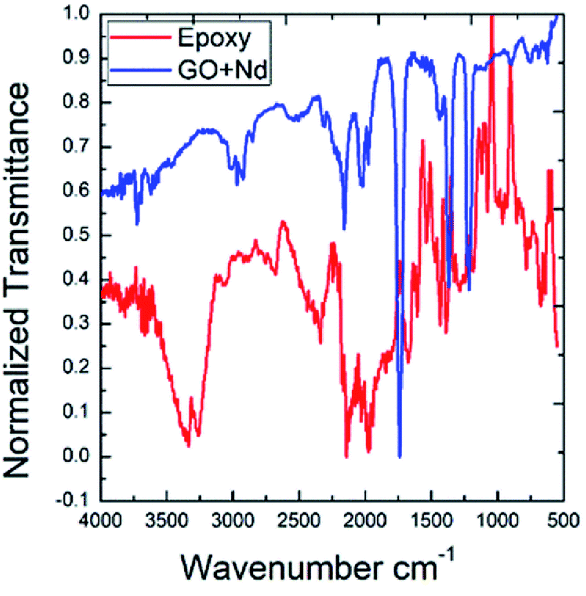
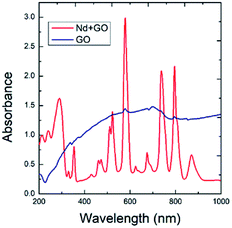
Fig. 2 presents the FTIR spectra of GO and GO + Nd obtained by the KBr pellet method. The GO spectrum displays features related with the carboxyl group's stretching frequency that appears around 1740 cm−1.36 The GO carbonyl groups stretching frequency is around 1060 cm−1, while the edge hydroxyl groups stretching frequency is around 1265 cm−1. Correspondingly, the FTIR spectrum of the neodymium-decorated graphene oxide presents the bond stretching frequency at 1600 cm−1 related with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond,37 suggesting a strong reduction of GO upon decoration, as reported before for Pr-doped GO.32
C bond,37 suggesting a strong reduction of GO upon decoration, as reported before for Pr-doped GO.32
 |
| | Fig. 2 FTIR spectra of GO and GO + Nd. | |
3.2. Structure
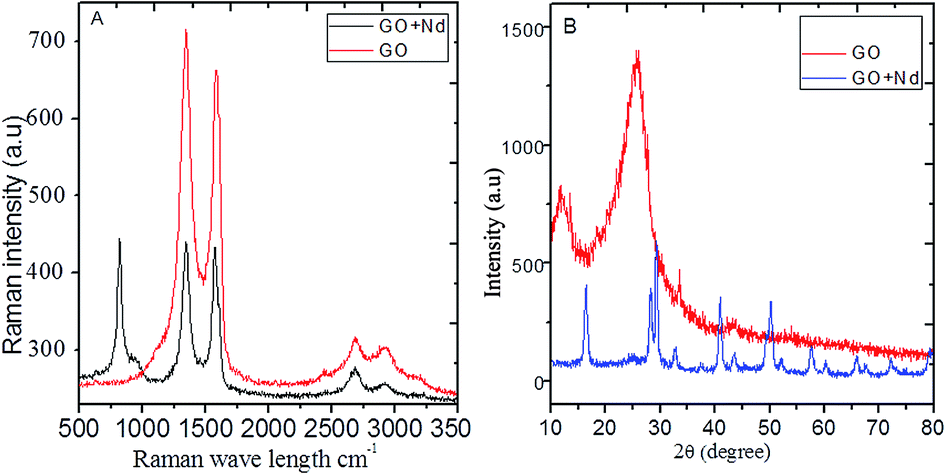
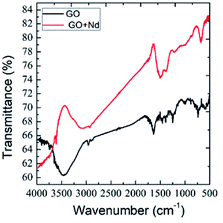
The Raman spectra of the prepared materials are shown in Fig. 3A. The GO spectrum features the characteristic D, G and 2G bands attributed to the disorder originating from the sp3 peaks, carbon symmetric vibrations and high order vibrations of the carbon skeleton43 at around 1360 cm−1, 1580 cm−1, and 2600 cm−1, respectively. GO + Nd spectrum presents the same bands with a noticeable higher intensity G band as well as a peak at around 780 cm−1 related to Nd(OH)3 nanorods, corroborating the strong GO reduction suggested by the results from FTIR spectroscopy.44,45
 |
| | Fig. 3 (A) Raman spectra of GO and GO + Nd; (B) X-ray diffractograms of GO and GO + Nd materials. | |
The X-ray diffractograms of the neodymium-decorated graphene oxide and GO are shown in Fig. 3B. The GO diffractogram shows a peak at 2θ = 15° associated with the GO (002) plane and a peak at 2θ = 25° attributed to the graphitic carbon. In the diffractogram of GO + Nd, several peaks were indexed to the (104), (004), (105), (200), (211) and (213) crystal planes of the hexagonal phase of Nd(OH)3 nanorods.38–42
3.3. Morphology and composition
Fig. 4 displays the morphology study of GO and GO + Nd. Fig. 4A presents a GO micrograph, where the stacked GO sheets can be clearly observed. Fig. 4C presents the corresponding micrograph of the GO + Nd sample, where it can be observed that the sheets are covered with a powdery-like deposit. Fig. 4B presents the morphology of a selected region and the EDS spectrum, where Nd can be observed, indicating the Nd interaction with the graphene oxide sheet.46–50 The TEM image presented in Fig. 4D shows the defined rods with a hexagonal structure as inferred from the electron diffraction pattern shown in Fig. 4D inset. The morphology and structure agrees with the XRD and Raman data. As shown in Fig. 5, the EDS elemental mapping of GO + Nd indicates that Nd3+ cation is uniformly distributed on the graphene oxide sheet.
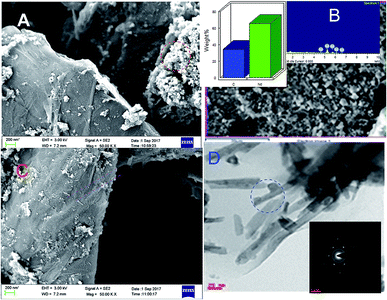 |
| | Fig. 4 FESEM micrographs of (A) GO and (C) GO + Nd. (B) FESEM micrograph and EDS spectrum of the GO + Nd sample and (D) TEM micrograph with electron diffraction pattern in the inset. | |
 |
| | Fig. 5 Elemental mapping of GO + Nd. | |
3.4. Proposed mechanism for neodymium decoration
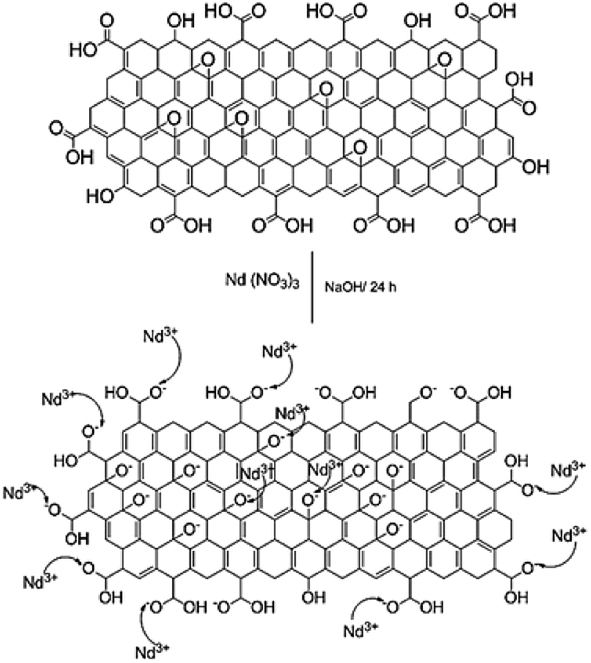
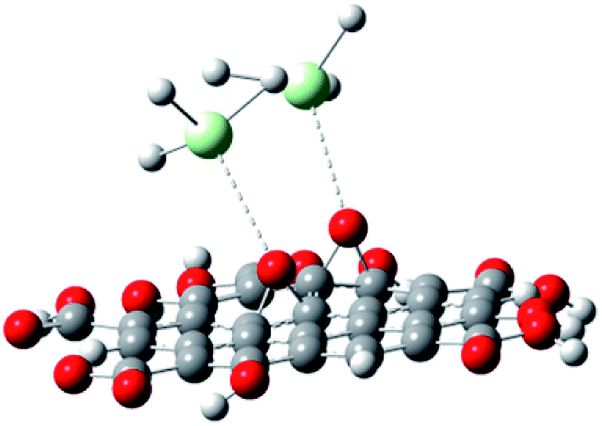
With the experimental evidence of FTIR spectroscopy, Raman spectroscopy, XRD and TEM/FESEM, a mechanism to explain the Nd decoration in GO sheet is proposed. As shown in Fig. 6, Nd3+ in basic media interacts with the negatively charged graphene oxide sheet with the epoxy moieties.51 Fig. 7 presents a model of interaction between the neodymium ions with carboxyl oxygens. The out-of-the-plane epoxy groups in the graphene oxide sheet could be the most efficient to be co-ordinated with neodymium Nd3+. Therefore, in the alkaline conditions prevalent during the reaction, the growth of out-of-the-plane Nd(OH)3 rods is favoured.
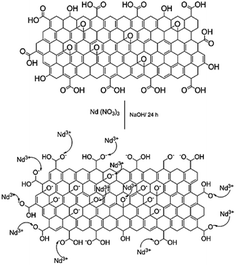 |
| | Fig. 6 Proposed reaction mechanism of the reaction between Nd3+ and the graphene oxide epoxy groups. | |
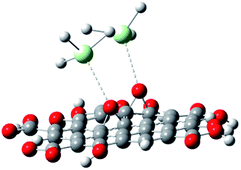 |
| | Fig. 7 Neodymium cation coordination with GO epoxy moieties. | |
3.5. Redox activity
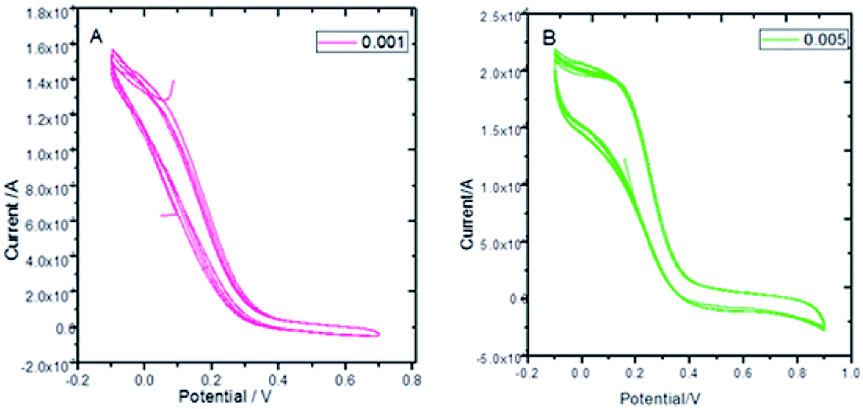
Fig. 8 presents the cyclic voltammograms of the GO + Nd coating on the Ti6Al4V coupons to study the redox behaviour of the coating using 50 μM K4Fe(CN)6 solution as a probe and 1 M H2SO4 as an electrolyte. Upon potential cycling, the electrode featured almost no electrochemical response at potentials above 0.4 V, while a large anodic response was evident when the potential was swept between 0.4 and −0.2 V, suggesting some penetration of the coating. However, the current remained stable during the 5 cycles, demonstrating the coating stability. This indicated that the GO + Nd coating is being restored during the cycling, protecting the Ti6Al4V surface.52–55 Fig. S1† presents the ellipsometry spectra of the GO + Nd coating and the reference epoxy coating. The calculated coating thicknesses from spectra-fitting are shown in Table 1. The epoxy and GO + Nd coating thicknesses are 10.952 μm and 14.872 μm, respectively.56–59
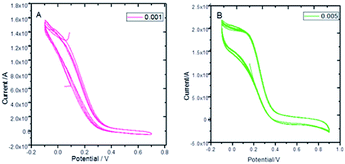 |
| | Fig. 8 Cyclic voltammetry of GO + Nd at different scan rates: (A) 0.001 mV s−1, (B) 0.005 mV s−1 using a K4Fe(CN)6 redox probe in 1 M H2SO4 electrolyte. | |
Table 1 Corrosion inhibition efficiency values of epoxy and GO + Nd coated Ti6Al4V alloys and film thickness values
| S. no. |
−Ecorr (mV) |
−Icorr (mA) |
(%) |
Rct (Ω cm2) |
Cdl μF |
χ2 |
(Å) |
| Epoxy |
122 |
6.179 |
|
480 |
3.7046 |
1.7149 |
1.0952 |
| GO + Nd |
850 |
2.201 |
64.5 |
970 |
2.7333 |
0.7670 |
1.4872 |
3.6. Electrochemical study of corrosion inhibition
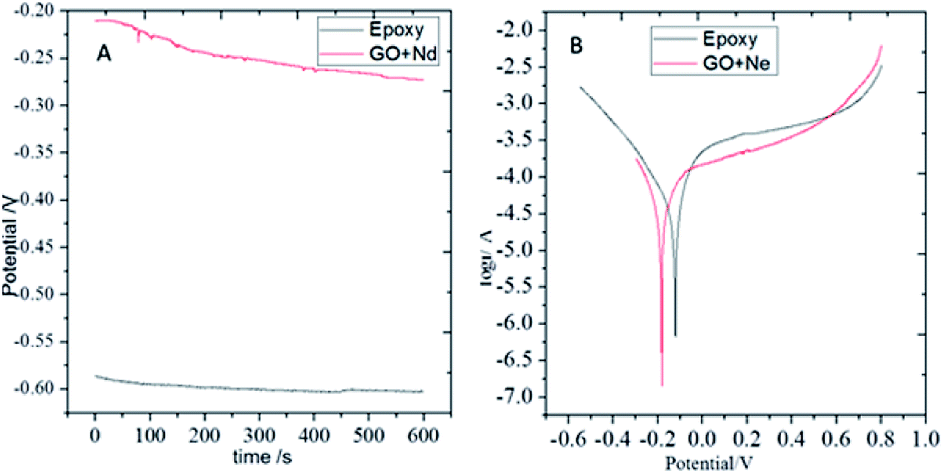
Open circuit potential (OCP) measurements are shown in Fig. 9. The epoxy coating potential has an OCP value of around −0.6 V, indicating the easiness of corrosion of the alloy. The epoxy coating is porous and the aggressive ions can penetrate to the alloy surface. Moreover, the GO + Nd coating OCP values largely shifted to the less negative potentials with values ranging from −0.20 to −0.25 V, indicating that the coating does not allow the corrosive ions to penetrate to the alloy surface.60–65 In Fig. 9, the potentiodynamic polarization behaviour of the epoxy and GO + Nd coatings are shown. The epoxy coating corrosion potential decreased and the corrosion current increased because of the porosity and poor adhesion of the epoxy coating on the metal alloy surface.66–75 Correspondingly, for the GO + Nd coated alloy, the cathodic hydrogen evolution was in control and the anodic metal dissolution decreased because of the strong impermeable barrier layer on the alloy, leading to an increase in the corrosion potential and the decrease, by one order of magnitude, in the corrosion current.
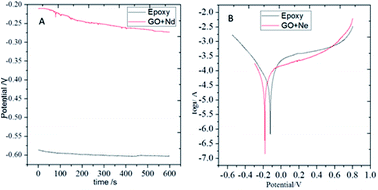 |
| | Fig. 9 (A) OCP of epoxy and GO + Nd coated Ti6Al4V alloys and (B) potentiodynamic plots of epoxy and GO + Nd coated Ti6Al4V alloy. | |
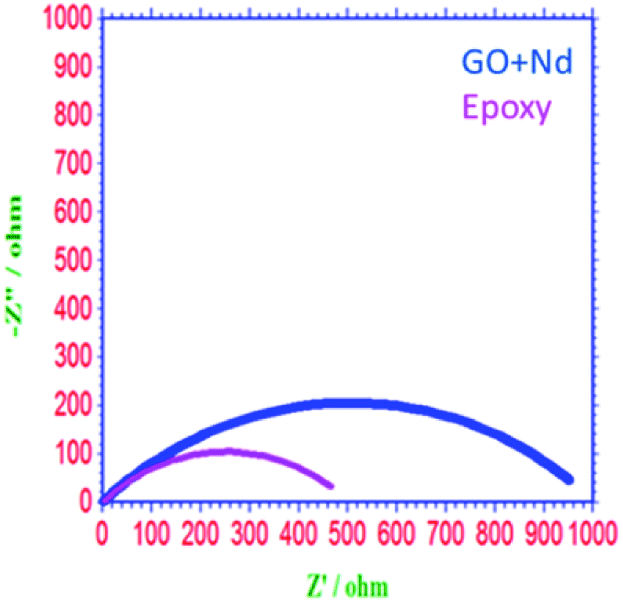
As shown in Fig. 10, the Nyquist spectrum of the epoxy coating presents a suppressed semicircle due to pit formation on the alloy surface and coating instability in the acidic environment.69–75 In the case of the GO + Nd coating, the Nyquist plot shows an enlarged semicircle because of the barrier layer adsorbed on the alloy surface. According to the charge transfer values of GO + Nd coating, the value increased to 980 Ω compared to the 470 Ω of the reference epoxy coating, which is result of the alloy porosity. The double layer capacity of the epoxy coating is higher than that of the GO + Nd because the epoxy coating could not control the ion diffusion.30–33 The corrosion inhibition efficiency is 64.5% higher for the GO + Nd coating, demonstrating that it is an excellent corrosion resistance coating material in the acidic environment for the Ti6Al4V alloy.
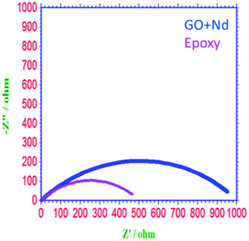 |
| | Fig. 10 Nyquist plot of epoxy and GO + Nd-coated Ti6Al4V alloys. | |
3.7. Surface studies of the coated Ti6Al4V alloys
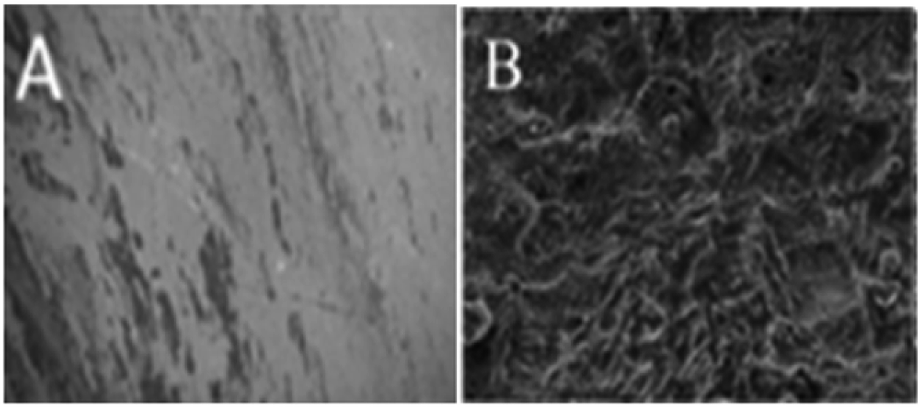
Fig. 11 presents the morphology of the (A) GO + Nd coated and (B) epoxy coated Ti6Al4V alloys. As shown in Fig. 11, the GO + Nd coating has a homogeneous surface because the barrier layer does not allow corrosive ions into the coating surface,76,77 indicating the strong physisorption on the alloy surface of the GO + Nd-based coating. The alloy dissolution has been controlled by the impermeable GO + Nd sheet on the alloy surface as shown in the electrochemical characterization. As shown in Fig. 11, the epoxy coated titanium alloy surface exhibits pitting, and localized corrosion initiated due to the porosity of the barrier layer and aggressive ion attack.78,79 The atomic force microscopy images and roughness analysis (ESI, Fig. S2–S4†) show the effects of the penetration of aggressive ions into the epoxy layer, while confirming the corrosion inhibition of the GO + Nd coating.80,81
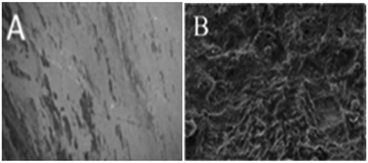 |
| | Fig. 11 After corrosion treatment surface morphology of the GO + Nd-coated Ti6Al4V alloy (A) and epoxy-coated Ti6Al4V titanium alloy (B). | |
As shown in Fig. 12, the ATR-FTIR spectra after the exposure to the acidic environment of the epoxy and GO + Nd coatings onto the Ti6Al4V alloy was studied.
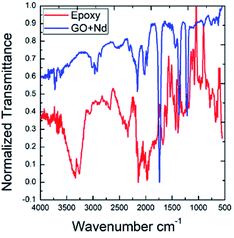 |
| | Fig. 12 ATR-FTIR normalized spectra of the epoxy and GO + Nd coated Ti6Al4V alloy surfaces after exposure to the corrosion conditions. | |
The epoxy coated alloy surface stretching frequency at 3500 cm−1 is associated with the hydroxyl groups adsorbed.82,83 The peak that appears at 2000 cm−1 is related to the alkyl chain vibration. The peaks at 1600 cm−1 and 1700 cm−1 could be attributed to the epoxy groups adsorbed on the alloy surface. In the spectrum of GO + Nd coating, peaks appear at 2900 cm−1 and 2800 cm−1 attributed to the C–C, CC graphite carbon chain, while the peak at 2300 cm−1 is associated with the COOH adsorbed on the alloy surface.84,85 The peaks at 1388 cm−1 and 1720 cm−1 are attributed to C–O and CO, respectively. The peak at 920 cm−1 is associated with the Ti–O–Ti bond vibration, indicating the possible formation of a Ti oxide surface layer.
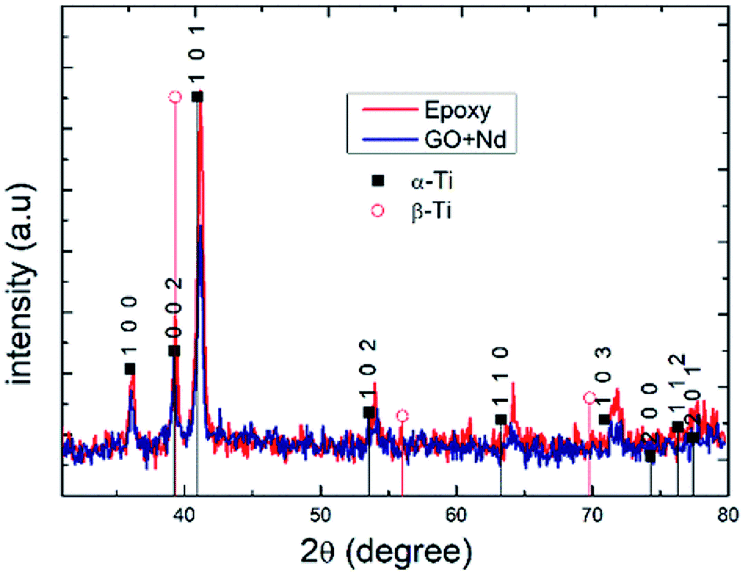
Fig. 13 presents the X-ray diffractograms of the Ti6Al4V-coated coupons after being exposed to the acidic environment to assess the possible presence of the corrosion products or structure modification due to aggressive ions diffusion.86–90 The peaks in both diffractograms were indexed to the alfa Ti phase, and no clear presence of the beta-Ti phase, and none of Ti oxides or other corrosion products was found, although ATR-FTIR results suggest the formation of Ti–O–Ti bond that could arise from surface TiO2.91–95 The relatively higher peak intensities in the spectrum of epoxy reference coating arises from its lower thickness (ca. 10 micrometers) compared to that of the GO + Nd (ca. 14 micrometers).
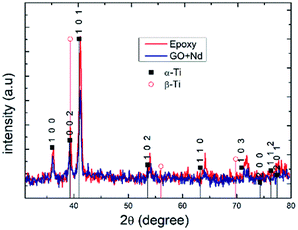 |
| | Fig. 13 X-ray diffractograms of epoxy and GO + Nd-coated Ti6Al4V alloy surfaces after exposure to corrosive media. | |
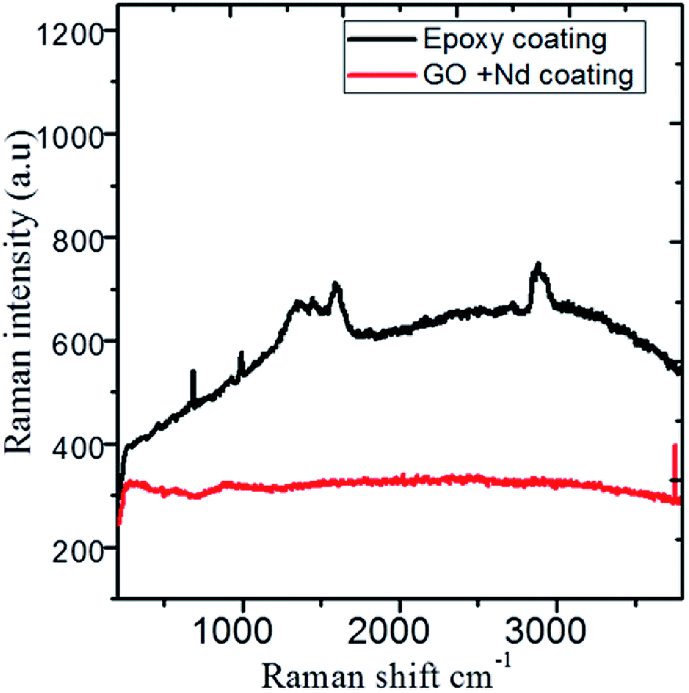
The Raman spectra shown in Fig. 14 correspond to the epoxy and GO + Nd coated alloy surfaces after corrosion treatment. The alloy microstructure was investigated after 5 day immersion in the 1 M H2SO4 medium. The spectra of the epoxy coating on alloy surface has peaks at 410 cm−1, 500 cm−1, 640 cm−1 and 970 cm−1 related with the anatase phase, although due to the thickness, is not detected by XRD and is formed because the epoxy layer could not withstand the corrosive medium for a long time. The coating layer was ruptured due to the diffusion of the corrosion species in the alloy surface.96–100 The Raman spectrum of the GO + Nd-based coating onto the Ti6Al4V alloy shows the characteristic D and G band of graphene oxide, and has a very small evidence of the oxide layer with weak bands at 410 cm−1 and 970 cm−1, indicating the strong interaction of the GO + Nd based coating with the alloy surface that prevents extensive corrosion.
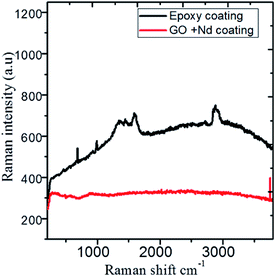 |
| | Fig. 14 Raman spectra of the epoxy and GO + Nd coated Ti6Al4V alloy surfaces after corrosion treatment. | |
3.8. Quantum chemical studies
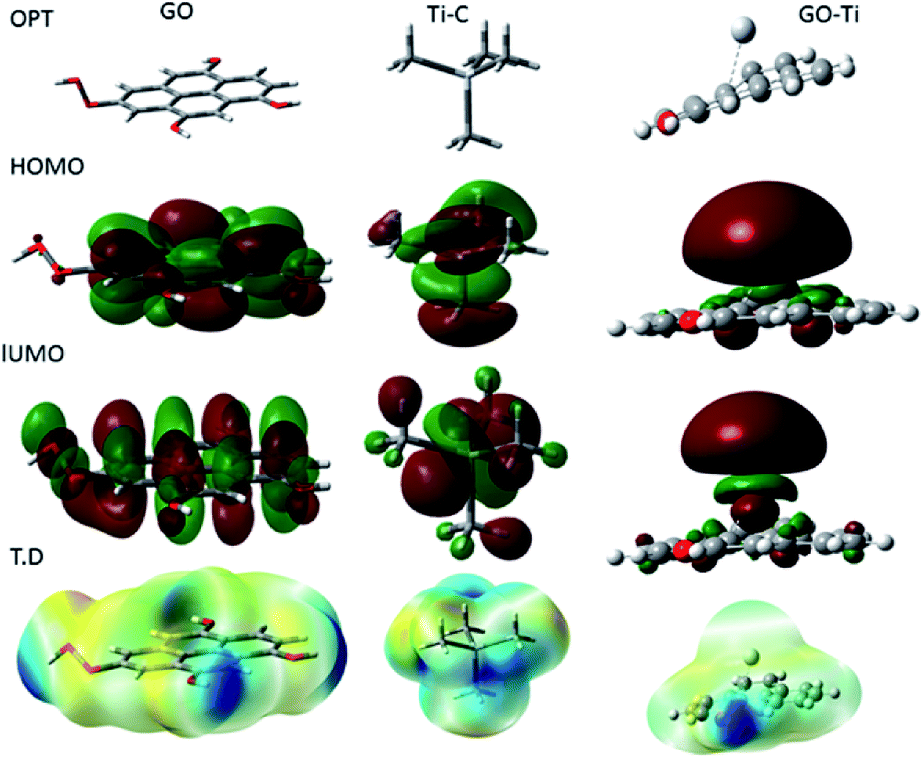
Fig. 15 shows the DFT modelling of the GO onto a Ti surface. The graphene oxide model comprised 30 atoms, including one carboxyl group and three hydroxyl groups, and graphene oxide adsorption on titanium surface has been investigated for the possible electron transfer between the alloy surfaces and the inhibition barrier layer.101,102 The negatively charged GO surface favours the chemisorbed guest molecules on the plane surface. However, from the DFT studies, we suggest that neodymium-modified graphene oxide sheet is adsorbed on the alloy surface. Hence, the GO functional groups such as hydroxyl, carbonyl, and carboxyl could improve the chemisorption of the graphene oxide layer onto the Ti alloy. The electronic values of HOMO (highest unoccupied molecular orbital) is ca. 5 eV and that of LUMO (lowest unoccupied molecular orbital) is ca. 1.3 eV for GO. The GO electronegativity, electrophilicity, and the softness and hardness values indicate a strong physisorption of the coating on the alloy surface, possibly because of the electron transfer between the coating surface and the polarized state of the metal surface. In the Ti–C model, the HOMO value increases to 7.32 eV, while the LUMO value increases to 1.84 eV, as well as the values for rest of the parameters except σ. With respect to the electron transfer between the graphene oxide sheet and the Ti oxide layer, the energy level of HOMO value decreases to 3.37 eV while the LUMO energy level increases to 1.95 eV compared with those of GO, suggesting that the titanium alloy could accept and donate electrons from the functionalized graphene oxide.103,104 The electronic parameters such as chemical hardness, softness, and electronegativity values contributed to the corrosion barrier activity on the alloys surface. The quantum chemical parameter values are presented in Table 2.
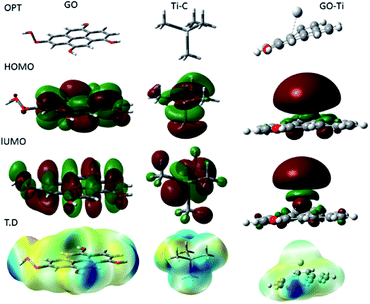 |
| | Fig. 15 Quantum chemical frontier molecular orbital structure of GO, C–Ti and GO–Ti, and HOMO and LUMO electronic structures. | |
Table 2 Quantum chemical values of GO and GO–Ti, model, rutile TiO2
| S. no. |
HOMO (eV) |
LUMO (eV) |
ΔE (eV) |
χ (eV) |
η (eV) |
σ (eV) |
ω |
| GO |
5.0327 |
1.3436 |
3.6894 |
3.1881 |
1.8445 |
0.5421 |
2.7551 |
| Ti–C |
7.3236 |
1.8405 |
5.4831 |
4.5820 |
2.7415 |
0.3641 |
3.8291 |
| GO–Ti |
3.3774 |
1.9518 |
1.4256 |
2.4015 |
0.7128 |
1.4256 |
4.0450 |
4. Conclusions
From the obtained results, the following conclusions can be stated: the GO + Nd consists of Nd oxide nanorods in the hexagonal phase, attached to the GO sheet by the epoxy groups. The GO + Nd + epoxy coating onto the Ti6Al4V alloy showed a 65% increment in the corrosion inhibition activity with respect to the reference epoxy coating, as studied by the Tafel plots and the electrochemical impedance spectroscopy. The voltammetric analysis indicates that the GO + Nd coating is stable and it can be regenerated even after voltage cycling in the acidic media. The Raman and ATR-FTIR spectra of the coated Ti6Al4V coupons after exposure to the acid medium suggested the formation of a TiOx layer, which was not visible in XRD. The corresponding morphology analysis showed pit formation on the alloy with the epoxy coating, while GO + Nd coated surface was not affected by the aggressive ions, presenting a uniform surface. Finally, the modelling of the GO–Ti interactions showed the GO chemisorption on the alloy surface, confirming that GO + Nd is an excellent barrier layer on titanium alloy in the acidic medium.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
NP would like to thank Central University of Gujarat Central Instrument Facility and Non-Net fellowship. NP would like to thank Professor M. V. Rajasekahran for the support provided for this work completion under the UGC Network Resource Centre Hyderabad, University of Hyderabad. FCB acknowledges financial support from SIP-IPN through 20194931 project. Professor K. R. Justin Thomas Department of Chemistry, Indian Institute of Technology Roorkee for supporting electron microscopy studies.
References
- E. B. Tamarit, A. Igual-Munoz, J. G. Anton and D. M. Garcia-Garcia, Corros. Sci., 2009, 5, 11095–11102 Search PubMed.
- C. S. Brossia and G. A. Cragnolino, Corros. Sci., 2004, 46, 1693–1711 CrossRef CAS.
- U. Eduok and J. Szpunar, Ultrason. Sonochem., 2018, 44, 288–298 CrossRef CAS PubMed.
- N. T. Kirkland, T. Schiller, N. Medhekar and N. Birbilis, Corros. Sci., 2012, 56, 1–4 CrossRef CAS.
- M. Breedon, M. C. Per, I. S. Cole and A. S. Barnard, J. Mater. Chem. A, 2014, 2, 16660–16668 RSC.
- Y. N. Singhbabu, B. Sivakumar, J. K. Singh, H. Bapari, A. K. Pramanick and R. K. Sahu, Nanoscale, 2015, 7, 8035–8047 RSC.
- D. Dutta, A. N. F. Ganda, J. K. Chih, C. C. Huang, C. J. Tsenga and C. Y. Su, Nanoscale, 2018, 10, 12612–12624 RSC.
- J. H. Huh, S. H. Kim, J. H. Chu, S. Y. Kim, J. H. Kim and S. Y. Kwon, Nanoscale, 2014, 6, 4379–4386 RSC.
- F. Maia, J. Tedim, A. D. Lisenkov, A. N. Salak, M. L. Zheludkevich and M. G. S. Ferreira, Nanoscale, 2012, 4, 1287–1298 RSC.
- C. Liu, S. Qiu, P. Du, H. Zhao and L. Wang, Nanoscale, 2018, 10, 8115–8124 RSC.
- R. K. Gupta, M. Malviya, C. Verma, N. K. Gupta and M. A. Quraishi, RSC Adv., 2017, 7, 39063–39074 RSC.
- Z. Qiu, R. Wang, J. Wu, Y. Zhang, Y. Qu and X. Wu, RSC Adv., 2015, 5, 44149–44159 RSC.
- Y. Liu, J. Zhang, S. Li, Y. Wang, Z. Hana and L. Ren, RSC Adv., 2014, 4, 45389–45396 RSC.
- Z. Yu, L. Lv, Y. Ma, H. Dia and Y. He, RSC Adv., 2016, 6, 18217–18226 RSC.
- Y. K. Xiao, W. F. Ji, K. S. Chang, K. T. Hsu, J. M. Yeh and W. R. Liu, RSC Adv., 2017, 7, 33829–33836 RSC.
- M. J. Nine, M. A. Cole, D. N. H. Tran and D. Losic, J. Mater. Chem. A, 2015, 3, 12580–12602 RSC.
- Y. Deng, W. Bai, J. Chen, X. Zhang, S. Wang, J. Lin and Y. Xu, RSC Adv., 2017, 7, 45034–45044 RSC.
- J. Di, S. Guo, L. Chen, P. Yi, G. Ding, K. Chen, M. Li, D. Lee and A. Yan, J. Rare Earths, 2018, 36, 826–831 CrossRef CAS.
- Z. Xichun, S. Xiaoyan and L. Zhongwu, J. Rare Earths, 2016, 34, 889 CrossRef.
- Z. Qiaoying, L. Gang, L. Zhuang, G. Shuai, Y. Aru, L. Don and L. Jianzhong, J. Rare Earths, 2016, 34, 152 CrossRef.
- Z. Dan, S. Jie, Z. Lili, T. Yong and L. Ji, J. Rare Earths, 2010, 28, 371–374 CrossRef.
- Z. Enyao, C. Yungui and T. Yongbai, J. Rare Earths, 2012, 30, 269 CrossRef.
- H. Jie, F. Song, H. Yan, L. Yi and X. Jianing, J. Rare Earths, 2016, 34, 283 CrossRef.
- W. Daogao, Y. Shihong, W. Zhiqiang, Z. Zhiqi, M. Ruiying, Z. Xiaowei and C. Dehong, J. Rare Earths, 2014, 32, 663 CrossRef.
- Y. Hongchuan, Z. Shirong, Y. Dunbo, L. Kuoshe, H. Quanxia, Y. Yuanfei, Z. Kun and L. Hongwei, J. Rare Earths, 2015, 33, 629 CrossRef.
- B. Guangli, J. Jing, Z. Fan, F. Daqing, L. Yuandong, M. Ying and H. Yuan, J. Rare Earths, 2016, 34, 931 CrossRef.
- Q. Yingjie, L. Wenpeng, W. Guixiang and Z. Xiaohong, J. Rare Earths, 2015, 33, 647 CrossRef.
- Z. Mahidashti and B. Ramezanzadeh, J. Rare Earths, 2018, 36, 1112–1120 CrossRef CAS.
- H. Baojun, G. Dongdong, H. Qiong, Z. Xiaolian, P. Guanghuai and Y. Chubin, J. Rare Earths, 2016, 34, 731 CrossRef.
- S. Ren, M. Cui, W. Li, J. Pu, Q. Xuea and L. Wang, J. Mater. Chem. A, 2018, 6, 24136–24148 RSC.
- U. Eduok, O. Fayea and J. Szpunara, RSC Adv., 2016, 6, 108777–108790 RSC.
- N. Palaniappan, I. S. Cole, F. C. Briones, K. Balasubaramanian and C. Lal, RSC Adv., 2018, 8, 34275–34286 RSC.
- S. M. Derazkola, S. Z. Ajabshira and M. S. Niasar, RSC Adv., 2015, 5, 56666–56676 RSC.
- M. Guzik, E. Tomaszewicz, Y. Guyot, J. Legendziewicza and G. Boulonc, J. Mater. Chem., 2012, 22, 14896–14906 RSC.
- P. A. Bozkurt, Ultrason. Sonochem., 2017, 397–404 CrossRef PubMed.
- B. Shahmoradi, K. Soga, S. Ananda, R. Somashekar and K. Byrappa, Nanoscale, 2010, 2, 1160–1164 RSC.
- S. Anandana, A. M. Asiric and M. Ashokkumar, Ultrason. Sonochem., 2014, 21, 920–923 CrossRef PubMed.
- J. Ma, Y. Gu, L. Shi, L. Chen and Y. Qian, Solid State Commun., 2004, 132, 743–745 CrossRef CAS.
- A. Hadia, J. Zahirifara, J. K. Sabet and A. Dastbaz, Ultrason. Sonochem., 2018, 44, 204–214 CrossRef PubMed.
- B. Shahmoradi, K. Soga, S. Ananda, R. Somashekar and K. Byrappa, Nanoscale, 2010, 2, 1160–1164 RSC.
- T. Saito, H. Sato and T. Motegi, J. Alloys Compd., 2006, 425, 145–147 CrossRef CAS.
- S. K. Ponnaiah, P. Prakash and B. Vellaichamy, Ultrason. Sonochem., 2018, 44, 196–203 CrossRef CAS PubMed.
- V. M. R. Betancourtt and D. Nattland, Phys. Chem. Chem. Phys., 2005, 77, 173–179 RSC.
- V. Vinoth, J. J. Wu, A. M. Asiric and S. Anandan, Ultrason. Sonochem., 2017, 39, 363–373 CrossRef CAS PubMed.
- R. Chen, Y. Wang, Y. Liu and J. Li, RSC Adv., 2015, 5, 85065–85072 RSC.
- S. Sasikaladevi, J. Aravind, V. Eswaraiaha and S. Ramaprabhu, J. Mater. Chem., 2011, 21, 17094–17097 RSC.
- D. M. Tobaldi, R. A. S. Ferreira, R. C. Pullar, M. P. Seabra, L. D. Carlos and J. A. Labrincha, J. Mater. Chem. C, 2015, 3, 4970–4986 RSC.
- W. Yue, Z. Lin, S. Jiang and X. Yang, J. Mater. Chem., 2012, 22, 16318–16323 RSC.
- A. Arunchander, M. Vivekanantha, S. G. Peera and A. K. Sahu, RSC Adv., 2016, 6, 95590–95600 RSC.
- A. Dorris, C. Sicard, M. C. Chen, A. B. McDonald and C. J. Barrett, ACS Appl. Mater. Interfaces, 2011, 3, 3357–3365 CrossRef CAS PubMed.
- M. Kaplan, Z. Yuan, J. D. Benck, A. G. Rajan, X. S. Chu, Q. H. Wang and M. S. Strano, Chem. Soc. Rev., 2017, 46, 4530–4571 RSC.
- C. Xu, B. Xu, Y. GU, Z. Xiong, J. Sun and X. S. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 RSC.
- S. Szunerits and R. Boukherrou, Electrochemistry, 2014, 12, 211–242 CAS.
- D. W. Chang, H. J. Choi, A. Filer and J. B. Baek, J. Mater. Chem. A, 2014, 2, 12136–12149 RSC.
- M. T. Lee, C. Y. Fan, Y. C. Wang, H. Y. Li, J. K. Chang and C. M. Tseng, J. Mater. Chem. A, 2013, 1, 3395–3405 RSC.
- N. Palaniappan, L. R. Chowhan, J. Sathiskumar, I. G. Bosco and I. S. Cole, Surf. Interfaces, 2017, 6, 237–246 CrossRef CAS.
- Y. Zhou, S. Xu, L. Guo, S. Zhang, H. Lu, Y. Gong and F. Gao, RSC Adv., 2015, 5, 14804–14813 RSC.
- I. Tissot, O. C. Monteiro, M. A. Barreiros, V. Corregidor, J. Correia and M. F. Guerra, RSC Adv., 2016, 6, 51856–51863 RSC.
- K. Fushimi, K. Kurauchi, Y. Yamamoto, T. Nakanishi, Y. Hasegawa and T. Ohtsuka, Electrochim. Acta, 2014, 144, 56–63 CrossRef CAS.
- D. Dutta, A. N. F. Ganda, J. K. Chih, C. C. Huang, C. J. Tseng and C. Y. Su, Nanoscale, 2018, 10, 12612–12624 RSC.
- J. R. Chen and W. T. Tsai, Electrochim. Acta, 2011, 56, 1746–1751 CrossRef CAS.
- F. Yu, L. Camilli, T. Wang, D. M. A. Mackenzie, M. Curioni, R. Akid and P. Boggild, Carbon, 2018, 132, 78–84 CrossRef CAS.
- Y. N. Singhbabu, B. Sivakumar, J. K. Singh, H. Bapari, A. K. Pramanick and R. K. Sahu, Nanoscale, 2015, 7, 8035–8047 RSC.
- J. Hoon Huh, S. H. Kim, J. H. Chu, S. Y. Kim, J. H. Kim and S. Y. Kwon, Nanoscale, 2014, 6, 4379–4386 RSC.
- S. K. Aneja, S. Bohm, A. S. Khanna and H. L. M. Bohm, Nanoscale, 2015, 7(7), 17879–17888 RSC.
- J. H. Huh, S. H. Kim, J. H. Chu, S. Y. Kim, J. H. Kim and S. Y. Kwon, Nanoscale, 2014, 6, 4379–4386 RSC.
- C. Liu, S. Qiu, P. Du, H. Zhao and L. Wang, Nanoscale, 2018, 10, 8115–8124 RSC.
- E. Godlewska, M. Mitoraj and K. Leszczynska, Corros. Sci., 2014, 78, 63–70 CrossRef CAS.
- Z. B. Wang, H. X. Hu, Y. G. Zheng, W. Ke and Y. X. Qiao, Corros. Sci., 2016, 103, 50–65 CrossRef CAS.
- Y. Sia and Z. Guo, Nanoscale, 2015, 7, 5922–5946 RSC.
- H. Lu, S. Zhang, W. Li, Y. Cui and T. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 4034–4043 CrossRef CAS PubMed.
- W. Sun, L. Wang, T. Wu, M. Wang, Z. Yang, Y. Pan and G. Liu, Chem. Mater., 2015, 27, 2367–2373 CrossRef CAS.
- Y. P. Hsieh, M. Hofmann, K. W. Chang, J. G. Jhu, Y. Y. Li, K. Y. Chen, C. C. Yang, W. S. Chang and L. C. Chen, ACS Nano, 2014, 8, 443–448 CrossRef CAS PubMed.
- D. Prasai, J. C. Tuberquia, R. R. Harl, G. K. Jennings and K. I. Bolotin, ACS Nano, 2012, 6, 1102–1108 CrossRef CAS PubMed.
- A. M. Fekry, RSC Adv., 2016, 6, 20276 RSC.
- N. Lin, Q. Liu, J. Zou, D. Li, S. Yuan, Z. Wang and B. Tang, RSC Adv., 2017, 7, 13517–13535 RSC.
- M. A. Surmeneva, A. Vladescu, R. A. Surmenev, C. M. Pantilimon, M. Brai and C. M. Cotrut, RSC Adv., 2016, 6, 87665–87674 RSC.
- L. Jinlong, Z. Huasheng and W. Yongxin, RSC Adv., 2016, 6, 105854–105861 RSC.
- M. S. Hussein and A. M. Fekry, ACS Omega, 2019, 4, 73–78 CrossRef CAS.
- J. Rong Chen and W. Ta Tsai, Electrochim. Acta, 2011, 56, 1746–1751 CrossRef.
- C. C. Hoa and S. J. Ding, J. Mater. Chem. B, 2015, 3, 2698 RSC.
- V. Raj and M. S. Mumjitha, Electrochim. Acta, 2015, 153, 1–11 CrossRef CAS.
- B. Rikhari, S. Pugal Mani and N. Rajendran, RSC Adv., 2016, 6, 80275–80285 RSC.
- V. Jokanovic, M. VilotijevI, B. Jokanovic, M. Jenko, I. Anzel, D. Stamenkovic, V. Lazic and R. Rudolf, Corros. Sci., 2014, 82, 180–190 CrossRef CAS.
- A. C. Alves, F. Wenger, P. Ponthiaux, J. P. Celis, A. M. Pinto, L. A. Rocha and J. C. S. Fernandes, Electrochim. Acta, 2017, 234, 16–27 CrossRef CAS.
- A. U. Chaudhry, B. Mansoor, T. Mungole, G. Ayoub and D. P. Field, Electrochim. Acta, 2018, 264, 69–82 CrossRef CAS.
- T. M. Chiu, M. Mahmoudi, W. Dai, A. Elwany, H. Liang and H. Castaneda, Electrochim. Acta, 2018, 279, 143–151 CrossRef CAS.
- M. F. Lopez, J. A. Jimene and A. Gutierrez, Electrochim. Acta, 2003, 48, 1395–1401 CrossRef CAS.
- W. Simkaa, A. Sadkowski, M. Warczak, A. Iwaniak, G. Dercz, J. Michalska and A. Maciej, Electrochim. Acta, 2011, 56, 8962–8968 CrossRef.
- J. Lin, S. Ozan, K. Munir, K. Wang, X. Tong, Y. Li, G. Li and C. Wen, RSC Adv., 2017, 7, 12309 RSC.
- N. D. Alqarni, J. Wysocka, N. El-Bagoury, J. Ryl, M. A. Amin and R. Boukherroub, RSC Adv., 2018, 8, 19289–19300 RSC.
- N. Lin, Q. Liu, J. Zou, D. Li, S. Yuan, Z. Wang and B. Tanga, RSC Adv., 2017, 7, 13517–13535 RSC.
- B. Wua, Z. Pana, S. Lib, D. Cuiuria, D. Ding and H. Lia, Corros. Sci., 2018, 137, 176–183 CrossRef.
- C. C. I. Popa, P. M. Brossard, D. Monceau and S. Chevalier, Corros. Sci., 2016, 110, 91–104 CrossRef.
- V. Prabu, P. Karthick, A. Rajendran, D. Natarajan, M. S. Kiran and D. K. Pattanayak, RSC Adv., 2015, 5, 50767–50777 RSC.
- P. Jianga, L. Lin, F. Zhanga, X. Dong, L. Rena and C. Lin, Electrochim. Acta, 2013, 107, 16–25 CrossRef.
- L. Mohan, C. Anandan and N. Rajendran, Electrochim. Acta, 2015, 155, 411–420 CrossRef CAS.
- Z. J. Liua, X. Z. H. Liu, I.-L. Tsai, U. Donatus and G. E. Thompson, Electrochim. Acta, 2015, 182, 482–492 CrossRef.
- E. Matykina, J. M. H. Lopez, A. Conde, C. Domingo, J. J. de Damborenea and M. A. Arenas, Electrochim. Acta, 2011, 56, 2221–2229 CrossRef CAS.
- D. W. Boukhvalov, RSC Adv., 2013, 3, 7150 RSC.
- S. T. Skowron, I. V. Lebedeva, M. Popovic and E. Bichoutskaia, Chem. Soc. Rev., 2015, 44, 3143–3176 RSC.
- S. Nigar, Z. Zhou, H. Wang and M. Imtiaze, RSC Adv., 2017, 7, 51546–51580 RSC.
- Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541–4583 RSC.
- M. Pykal, P. Jurecka, F. Karlickya and M. Otyepka, Phys. Chem. Chem. Phys., 2016, 18, 6351–6372 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00106a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
I. S. Cole
*a,
I. S. Cole