DOI:
10.1039/C9RA00041K
(Paper)
RSC Adv., 2019,
9, 6169-6176
A facile synthetic route to tungsten diselenide using a new precursor containing a long alkyl chain cation for multifunctional electronic and optoelectronic applications†
Received
3rd January 2019
, Accepted 10th February 2019
First published on 20th February 2019
Abstract
Single source precursors for coating and subsequent thermal decomposition processes enable a large-scale, low-cost synthesis of two-dimensional transition metal dichalcogenides (TMDs). However, practical applications based on two-dimensional TMDs have been limited by the lack of applicable single source precursors for the synthesis of p-type TMDs including layered tungsten diselenide (WSe2). We firstly demonstrate the simple and facile synthesis of WSe2 layers using a newly developed precursor that allows improved dispersibility and lower decomposition temperature. We study the thermal decomposition mechanism of three types of (Cat+)2[WSe4] precursors to assess the most suitable precursor for the synthesis of WSe2 layers. The resulting chemical and structural exploration of solution-processed WSe2 layers suggests that the (CTA)2[WSe4] may be a promising precursor because it resulted in the formation of high-crystalline WSe2. In addition, this study verifies the capability of WSe2 layers for multifunctional applications in optoelectronic and electronic devices. The photocurrent of WSe2-based photodetectors shows an abrupt switching behavior under periodic illumination of visible or IR light. The extracted photoresponsivity values for WSe2-based photodetectors recorded at 0.5 V correspond to 26.3 mA W−1 for visible light and 5.4 mA W−1 for IR light. The WSe2-based field effect transistors exhibit unipolar p-channel transistor behavior with a carrier mobility of 0.45 cm2 V−1 s−1 and an on-off ratio of ∼10.
Introduction
Layered two-dimensional (2D) transition metal dichalcogenides (TMDs) have opened new prospects for their potential applications owing to their distinctive electronic, optical, and catalytic properties that differ from their bulk counterparts.1–3 Unlike 2D graphene with a zero-overlap semimetal, TMDs possess a layer-dependent bandgap from 1.1 to 1.9 eV, which facilitates the realization of next-generation nanoelectronic devices, such as high performance field-effect transistors (FETs), logic circuits, photodetectors, gas sensors, spintronic devices, and memtransistors.4–9 Despite these extraordinary features, a major challenge for these practical applications based on semiconducting TMDs is the lack of a reliable and facile synthetic route in large-scale TMDs with spatial homogeneity. The large-scale TMDs need to be compatible with top-down lithographic fabrication processes, so they can realize device arrays with the same response. Thus far, early approaches, including mechanical and chemical exfoliation and chemical vapor deposition (CVD), have been implemented to produce atomically thin TMDs.10–13 However, these strategies are technically hindered for industrial applications due to their inability to synthesize large-area TMD films. To resolve this deficiency, an alternative synthetic route was suggested: the synthesis of molybdenum disulfides (MoS2), as the representative member of TMDs, was performed using well-designed two-step thermal decomposition processes of a (NH4)2MoS4 single source precursor, thereby facilitating a simple, large-scale, and low-cost growth of MoS2 layers that could be easily applied to plastic substrates.14,15 Also, previous attempts achieved a facile synthesis of three-dimensional binary and ternary TMD nanostructures by solvothermal routes.16–19 Unfortunately, it is widely recognized that the bottleneck of 2D TMDs in terms of their solution-processed growth is the lack of applicable and commercially-available single source precursors for the synthesis of p-type TMDs including layered tungsten diselenides (WSe2). WSe2 is one of few p-type TMDs and has high carrier mobility, which is a prerequisite for applications in complementary logic circuits, gas sensors, and optoelectronics, because most of the TMDs have n-type semiconducting behavior.20 Although MoTe2 possesses p-type semiconducting conduction, this material is unstable in ambient conditions owing to a relatively small cohesive energy (12.01 eV).21 For the first time, we report the simple and facile synthesis of WSe2 layers using a newly developed precursor that gives improved dispersibility and lower decomposition temperature. In previous studies, (Cat+)2[WSe4] complexes containing (Ph4P)2[WSe4] and (Et4N)2[WSe4] were synthesized.22,23 Notably, there are crucial issues for adopting single source precursors to synthesize WSe2 layers: (i) “dispersibility” in organic solvents and (ii) “low decomposition temperature” are strictly necessary because the solution containing the precursors can be uniformly coated on desired substrates for the formation of atomically thin WSe2 layers and the low synthetic temperature allows the direct application to plastic substrates. We rationally designed an unprecedented precursor, (CTA)2[WSe4] with a long alkyl chain of cetyltrimethylammonium, for the synthesis of WSe2 layers to improve the dispersibility and decrease the decomposition temperature. To assess the most suitable precursor for the synthesis of WSe2 layers, we systematically explored the decomposition mechanism of newly developed (CTA)2[WSe4], (Ph4P)2[WSe4], and (Et4N)2[WSe4] and implemented the pre-synthesis of WSe2 nanoparticles (NPs) using the three types of precursors via thermal decomposition of (Cat+)2[WSe4] with oleylamine as a capping ligand.
Experimental section
Synthesis of Na2Se4
A solid mixture of Na (0.25 g, 10 mmol) and Se (1.58 g, 20 mmol) was placed in a 100 mL side-armed flask equipped with a magnetic spin bar. The flask was also equipped with a bubbler that was charged with 30 mL of liquid ammonia in a dry ice/acetone bath (−78 °C). The reaction mixture was stirred from −72 °C to room temperature to produce dry Na2Se4 under an inert atmosphere.
Synthesis of (Ph4P)2[WSe4] and (Et4N)2[WSe4]
(Ph4P)2[WSe4] and (Et4N)2[WSe4] were prepared by modifying the previously established methods.18,19 (Ph4P)2[WSe4] and (Et4N)2[WSe4] were both synthesized in the same way. A solid mixture of Na2Se4 (1.83 g, 5 mmol) and W(CO)6 (1.76 g, 5 mmol) was placed into a 100 mL flask equipped with a cooling condenser. The reaction mixture was dissolved in 40 mL of N,N-dimethylformamide (DMF) and stirred at 90 °C for 60 min. After cooling to 70 °C, Ph4PCl (3.74 g, 10 mmol) or Et4NCl (1.66 g, 10 mmol) was added to the solution and the reaction was cooled down to room temperature with stirring for 30 min. The mixture was filtered to remove by-products, mainly NaCl. The filtrate was added 40 mL of tetrahydrofuran (THF) and the flask was placed in the freezer (−30 °C) for 24 h to grow a red colored crystalline solid for (Ph4P)2[WSe4] (2.20 g, 37%). Calcd for C48H40P2Se4W (1178.46): C, 48.9; H, 3.42. Found: C, 48.4; H, 3.40. IR (cm−1): 3045, 3002, 754, 723, 689, 526, 300. In addition, red crystalline solid (0.80 g, 21%) for (Et4N)2[WSe4] was collected at the bottom of the flask. Calcd for C16H40N2Se4W (760.18): C, 25.3; H, 5.30; N, 3.69. Found: C, 24.9; H, 5.10; N, 3.8. IR (cm−1): 2974, 2920, 1440.47, 300.
Synthesis of (CTA)2[WSe4]
A solid mixture of Na2Se4 (1.83 g, 5 mmol) and W(CO)6 (1.76 g, 5 mmol) was placed in a 125 mL flask equipped with a cooling condenser. The reaction mixture was dissolved in 40 mL of DMF and allowed to stir at 90 °C for 60 min. After the mixture was cooled to 70 °C, cetyltrimethylammonium chloride (3.20 g, 10 mmol) was added to the solution followed by cooling down to room temperature with stirring for 30 min. The mixture was filtered to remove by-products, mainly NaCl. The filtrate was added to the excess amount of diethyl ether to precipitate. The precipitate was dissolved in THF and filtered, and the solvent was then removed under reduced pressure. A red crystalline solid (2.00 g, 37%) was eventually collected at the bottom of the flask. Calcd for C38H84N2Se4W (1068.77): C, 42.7; H, 7.92; N, 2.62. Found: C, 42.5; H, 7.90; N, 2.80. IR (cm−1): 3000, 2919, 2850, 1468, 301.
Solvothermal decomposition of (Cat+)2[WSe4]
A mixture of oleylamine (50 mL) and (Cat+)2[WSe4] (0.5 g) were placed in a 100 mL flask equipped with a cooling condenser connected with bubbler. The reaction mixture was allowed to stir at 300 °C for 12 h under an inert atmosphere. The resultant was cooled down to room temperature and the excess amount of toluene was added. Then, the mixture was centrifuged to obtain WSe2 nanoparticles and washed three times with toluene to remove free oleylamine.
Results and discussion
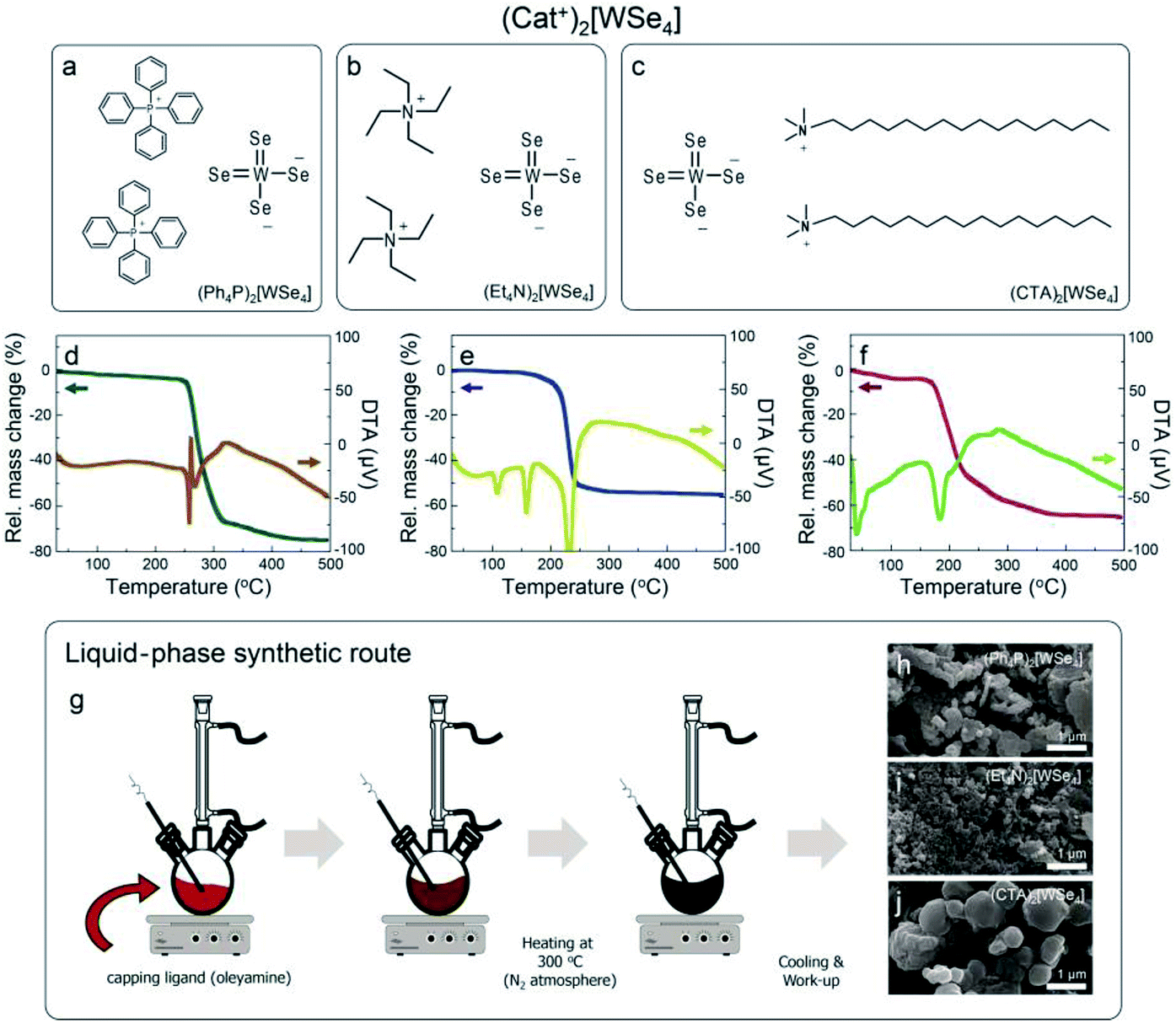
We synthesized the (Ph4P)2[WSe4] and (Et4N)2[WSe4] precursors by modifying previously established methods, as depicted in Fig. 1a and b.22 In the previous methods, K2Se3 was used as a starting material, whereas we adopted Na2Se4 as selenide source for reacting with the W(CO)6.23 In addition, we used (Ph4P)Cl and (Et4N)Cl, which both contain Cl− anions to capture for Na+ cations in as-synthesized Na2WSe4. Based on hard–hard and soft–soft interaction mechanism, it preferably forms NaCl as a by-product when a compound having a Cl− anion is used rather than a compound having a Br− anion, such as (Ph4P)Cl and (Et4N)Cl. Furthermore, when we used K2Se3 as a starting material, a K2Se2 by-product was inevitably formed, and it was difficult to separate it from the resultants for obtaining the pure products because it did not easily dissolve in organic solvents. In order to obtain purer products, we carefully utilized recrystallization using the Na2Se4. The reaction mechanism of the precursors for WSe2 is as follows:| | |
W(CO)6 + Na2Se4 → Na2[WSe4] + 6 CO↑
| (2) |
| | |
Na2[WSe4] + 2 (Cat+)Cl → (Cat+)2[WSe4] + 2 NaCl, (*(Cat+): Ph4P+, Et4N+, and CTA+)
| (3) |
 |
| | Fig. 1 Cationic tungsten selenide complexes, (Cat+)2[WSe4], (a) (Ph4P)2[WSe4], (b) (Et4N)2[WSe4], and (c) (CTA)2[WSe4], (d–f) TG-DTA curves for the all tungsten selenide complexes. (g) Schematic diagram of solvothermal synthesis of WSe2 NPs in a solution phase using the all complexes at 300 °C and (h–j) their morphological features measured by SEM. | |
Based on this mechanism, we synthesized unprecedented (CTA)2[WSe4] using cetyltrimethylammonium salts (CTA+) with long alkyl groups as a precursor for the solution-processable synthesis of WSe2, as seen in Fig. 1c. Unlike the two well-known precursors, it possesses long alkyl groups, which makes it possible to obtain high solubility in various organic solvents. Thus, it seems to be suitable as a precursor for the solution-based synthetic processes. For the synthesis of WSe2 through the basis of thermal properties of the three compounds, thermogravimetric-differential thermal analysis-mass spectroscopic analysis (TG-DTA-Mass) was performed to understand the synthetic mechanism for WSe2 (Fig. 1d–f). We examined thermal behaviors for all the samples from room temperature to 500 °C. First, (Ph4P)2[WSe4] was decomposed in the range of 260.4–270.6 °C, and it was clearly confirmed by the result of DTA that melting and decomposition of the compound occurs in this temperature region, as shown in Fig. 1d. The mass intensity at the region appears at 78 and 234 m/z, which correspond to the molecular weights for selenium and diphenyl selenide, respectively (Fig. S1†). We referred to mass spectrum of National Institute of Standards and Technology (NIST). The mass spectra of diphenyl diselenide showed 78 and 234 m/z exhibiting a similar isotope peaks. Furthermore, the residue was found to be 25% at about 500 °C, which was similar to the ratio of WSe2 (29%) in total molecular weight. A TGA curve of (Et4N)2[WSe4] showed endothermic peaks at 108 and 158 °C, and it was completely melted with decomposition at between 234.8 °C and 239.5 °C, as shown in Fig. 1e. The mass intensity appears at 101 and 218 m/z corresponding to the molecular weight of triethylamine and diethyl diselenide, respectively (Fig. S2†). The mass spectra of trimethylamine showed a same isotope peaks. In addition, the residue at 500 °C in the TGA result was estimated to be 45%, which corresponds to the ratio of WSe2 in total molecular weight. In case of (CTA)2[WSe4], it showed endothermic peak at 40 °C and completely melted and decomposed at the temperature range of 183.8–196.0 °C, which is the lowest temperature comparing with the other two precursors, as displayed in Fig. 1f. The mass intensity appeared at 190 m/z, which coincided with the major peak of mass spectrum of dimethyl diselenide. However, the mass intensity observed at below 110 m/z could not be unambiguously confirmed. Because of the long alkyl groups of (CTA)2[WSe4], a variety of decomposition products could not be completely identified. Thus, it was considered that it was decomposed into several short alkyl chains at below than 110 m/z. The peak observed at 58 m/z in TG-DTA-Mass resulted from the major peak of cetyl dimethylamine. This was monitored mainly by the photoionization (PI) mode of the mass spectrometer with an weak energy intensity (10.2 eV) (Fig. S3†). The residue of (CTA)2[WSe4] at 500 °C was 34%, which is similar to the ratio of WSe2 in the total molecular weight (32%). Based on these results of the TG-DTA-Mass analysis, the mechanism of WSe2 synthesis from (Cat+)2[WSe4] could be summarized as follows:
| | |
(Ph4P)2[WSe4] → 2 Ph3P + Ph–Se–Se–Ph + WSe2
| (4) |
| | |
(Et4N)2[WSe4] → 2 Et3N + Et–Se–Se–Et + WSe2
| (5) |
| | |
(CTA)2[WSe4] → 2 (cetyl)(Me)2N + Me–Se–Se–Me + WSe2
| (6) |
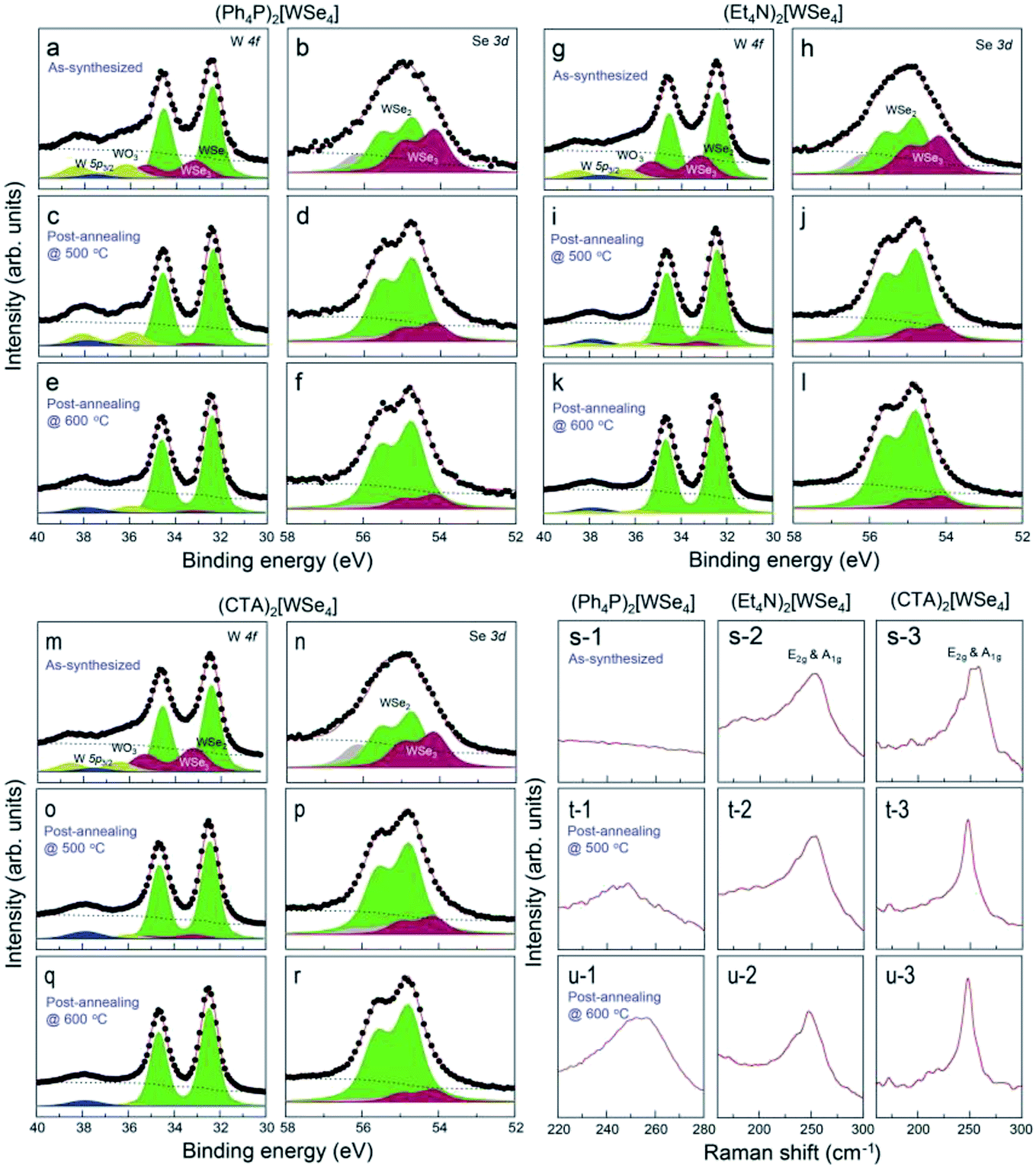
The mechanism of thermal decomposition for all the samples is described in eqn (4) to (6). Based on these results, we preliminarily synthesized WSe2 NPs by direct pyrolysis of (Cat+)2[WSe4] precursors in solution to assess the most suitable precursor for the synthesis of WSe2 layers, as shown in Fig. 1g. Oleylamine is typically employed as a solvent and also acts as a capping ligand to prevent the aggregation of materials.24 In general, the use of specific surface capping ligands is the most important factor in producing thickness-controlled TMDs.25 Furthermore, the redox reaction occurs easily to form WSe2, since the electrons in (Cat+)2[WSe4] easily migrate between W and Se without the addition of an external reducing agent.26 The synthesis of WSe2 with oleylamine was implemented at 300 °C for 12 h. Fig. 1h–j showed representative scanning electron microscopy (SEM) images of WSe2 synthesized using (Ph4P)2WSe4, (Et4N)2[WSe4] and (CTA)2[WSe4] precursors. In the rationally-designed (CTA)2[WSe4] precursor, uniform and larger particles were observed unambiguously. First, we compared the chemical and structural features of WSe2 NPs pre-synthesized using the three types of (Cat+)2[WSe4] precursors ((Ph4P)2[WSe4], (Et4N)2[WSe4], and (CTA)2[WSe4]). The chemical identification of WSe2 NPs synthesized by a solvothermal synthetic route was conducted using X-ray photoelectron spectroscopy (XPS). XPS spectra were acquired with a normal emission geometry using conventional monochromatic Al Kα radiation (hν = 1486.6 eV). Each spectrum was fitted with a Gaussian–Lorentzian function. The XPS W 4f and Se 3d core level spectra extracted from the as-synthesized WSe2 NPs using (Ph4P)2[WSe4] and after post-annealing at 500 and 600 °C, respectively, are displayed in Fig. 2a–f. Fig. 2a, c, and e reveal that the W 4f7/2 and W 4f5/2 doublet peaks corresponding to WSe2 appear at binding energies (EB) of 32.4 and 34.6 eV, respectively. The WSe3-related bonding states (EB = 33.2 and 35.4 eV), WO3-related states (EB = 36.1 and 38.3 eV), and W 5p2/3 (EB = 37.5 eV) are observed simultaneously. With adopting post-annealing and increasing the temperature, the intensity of WSe3 peaks decreased markedly, presumably owing to the heat-driven deselenization process. WO3-related peaks is originated from the fact that the sample was easily oxidized when it was exposed to ambient conditions because of their low cohesive energy (15.45 eV) induced by a small difference in electronegativity between W and Se. However, the oxidation was effectively inhibited due to their high crystallinity. Fig. 2b, d, and f display the Se 3d core level spectra for WSe2 NPs before and after post-annealing, indicating that the intensity of WSe3-related bonding states (EB = 54.1 and 54.9 eV) and Se–C bonding states (EB = 55.4 and 56.2 eV) decreased, whereas the WSe2-related peaks (EB = 54.7 and 55.5 eV) were predominantly observed with increasing post-annealing temperature, which is in agreement with the W 4f core level spectra. The temperature evolution of W 4f and Se 3d core level spectra obtained from the WSe2 NPs using (Et4N)2[WSe4] and (CTA)2WSe4 precursors seemed to follow a similar trend, as displayed in Fig. 2g–r. However, it should be noted that WO3-related and WSe3-related peaks disappeared completely in the W 4f core level spectra for WSe2 NPs synthesized using (CTA)2WSe4, which reflects its effective deselenization and crystallization presumably due to its lower decomposition temperature. To evaluate the crystallinity of WSe2 NPs synthesized by the three types of precursors, Raman spectroscopy analysis was implemented, as shown in Fig. 2s–u. The Raman spectra were recorded at an excitation wavelength of 514 nm. Generally, degenerated E2g (in-plane vibration) and A1g (out-of-plane vibration) phonon modes reflected the existence of high-crystalline WSe2.27 However, the two phonon modes are absent for WSe2 NPs synthesized using the (Ph4P)2[WSe4] precursor before post-annealing, whereas the broad peak at ∼250 cm−1 is observed after the post-annealing at 500 °C and the intensity of the peak becomes higher with increasing post-annealing temperature. Conversely, there are degenerated A1g and E2g phonon modes of the WSe2 NPs synthesized using (Et4N)2[WSe4] and (CTA)2[WSe4], regardless of the adoption of post-annealing. Astonishingly, the full widths at half maximum (FWHM) of the two phonon modes of WSe2 using (CTA)2[WSe4] decreased unambiguously. This finding suggests that (CTA)2[WSe4] is a promising precursor because it yields the synthesis of high-crystalline WSe2. We carefully developed the optimal precursor for synthesis of the WSe2 layer through preliminary studies of chemical and structural features of WSe2 NPs using the three types of (Cat+)2[WSe4] precursors. To synthesize WSe2 layers, a 0.5 wt% (CTA)2[WSe4] single source precursor was stirred in DMF at room temperature for 60 min, as displayed in Fig. 3a. The precursor solution was then drop-casted onto SiO2 (300 nm)/Si(001) substrates with UV-stimulated hydrophilic treatment. After the drop-casting of the precursor solution, the samples were immediately annealed at 100 °C for 1 min to remove the solvent. During this procedure, the few-layered lamellar structure of (CTA)2[WSe4] is formed in the surface of SiO2 substrate. The formation of lamella structure on the surface can be mediated by the electrostatic attraction between (CTA)2[WSe4] and SiO2. In addition, chemical identification of the as-formed lamellar structure on SiO2 was implemented by XPS, reflecting that the as-coated precursor was well-maintained under atmospheric conditions (Fig. S4†). The samples were located into the furnace, after which a two-step thermal decomposition process for the synthesis of WSe2 layers was implemented at 300 °C (1st Step) by introducing Ar (450 sccm) for 30 min and subsequently annealed at 600 °C (2nd Step) under 450 sccm flow of Ar at a pressure of 3.5 torr for 30 min. These synthetic conditions were based on the optimized synthetic parameters for WSe2 NPs. The surface morphology of the WSe2 layers was confirmed by optical microscopy, as seen in Fig. 3b and c. Raman mapping was conducted to evaluate the uniformity of the WSe2 layers, as seen in Fig. 3d, with the result that IE2g+A1g exhibited excellent homogeneity over large areas except for some small structural imperfections. The chemical characterization of the synthesized WSe2 layers was conducted by XPS. The survey spectrum reveals that W, Se, C, O, and Si-related peaks are observed, which implies the successful formation of WSe2 layers on a SiO2/Si substrate, as shown in Fig. 3e. Moreover, the intensity of C 1s peak decreases significantly after the formation of WSe2 layers, which means that the long alkyl chain of (CTA)2[WSe4] is thermally decomposed. Fig. 3f exhibits the W 4f core level spectrum extracted from the WSe2 layers on SiO2/Si, which reveals the successful formation of WSe2 as confirmed by the presence of W 4f7/2 and W 4f5/2 bonding states without non-stoichiometric chemical states or oxidation states, which is in good agreement with the Se 3d core level spectrum, as displayed in Fig. 3g. Raman spectra with an excitation wavelength of 514 nm for the WSe2 layers display degenerated E2g phonon and A1g phonon modes prominently, as seen in Fig. 3h. By comparing the Raman spectrum of WSe2 NPs using (CTA)2[WSe4], a remarkable decrease in the FWHM of the Raman modes can be adequately explained by the improvement in crystallinity induced by the formation of a two-dimensional structure. Additionally, the crystal structure of WSe2 synthesized by (CTA)2WSe4 was explored by X-ray
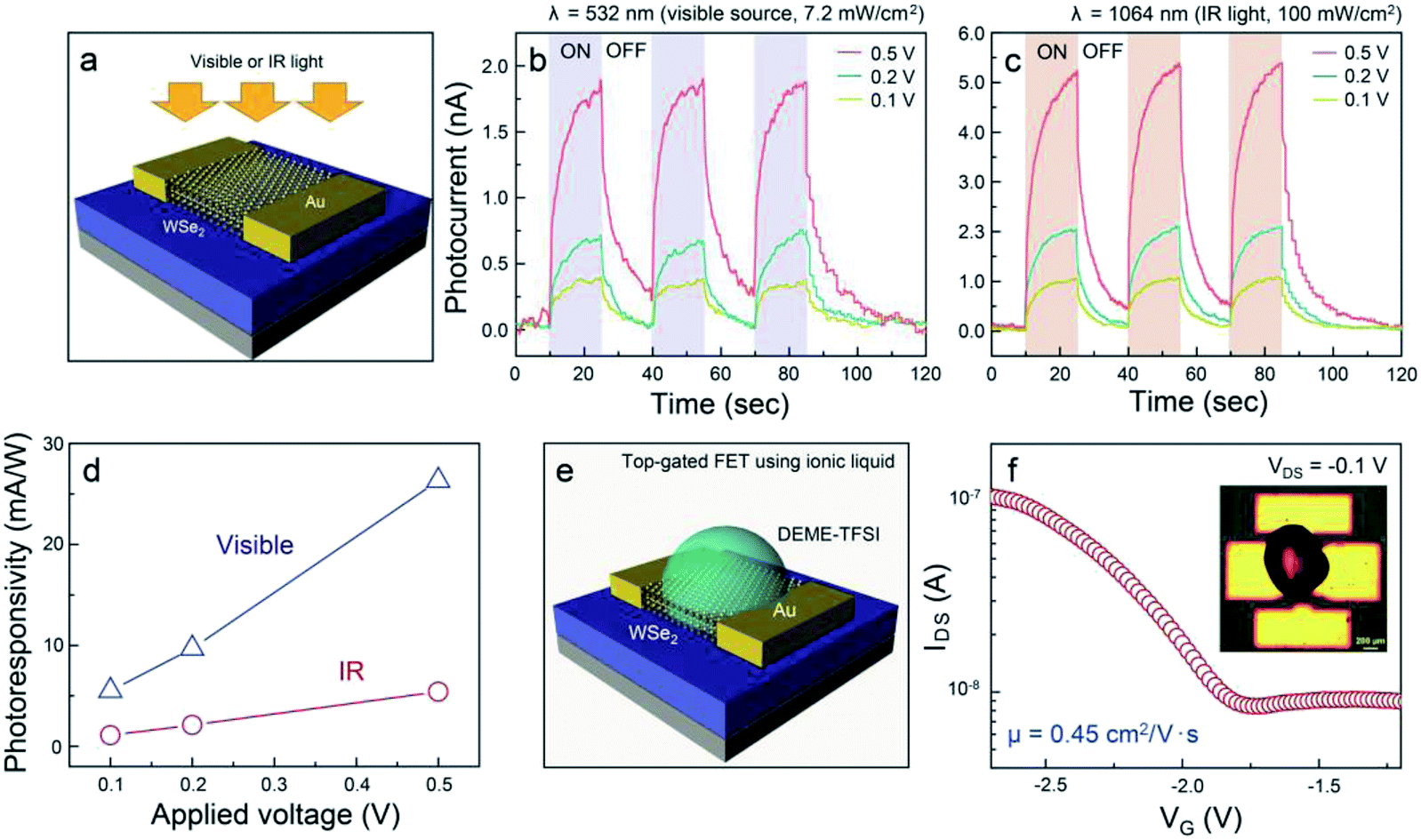
diffraction (XRD). The XRD patterns of WSe2 were displayed (Fig. S5†). The distinct diffraction peaks were located at 13.4, 31.8, 41.1, and 55.7° corresponding to (002), (100), (006), and (008) 2H–WSe2 crystal planes, respectively, in good agreement with a prior report on the crystal structure of 2H–WSe2.28 In general, the synthesis of WSe2 by a conventional chemical vapor deposition technique is implemented using co-evaporation of WO3 and Se powder at higher reaction temperatures of 700–900 °C.29–31 In this process, the volatile WO3−x suboxide reduced by injecting H2 is effectively selenized, resulting in the formation of triangular- or hexagonal-shaped WSe2 flakes, which seems to be technically limited for the realization of the device arrays with the same response. We believe that our approach provides a feasible and reliable synthetic route for obtaining homogeneous WSe2 layers through simultaneous conversion of the single source precursor at a relatively low temperature. To examine the photoelectrical properties of WSe2 layers synthesized by the two-step thermal decomposition process using (CTA)2WSe4, we fabricated WSe2-based photodetectors, as illustrated in Fig. 4a. Metal evaporation with a shadow mask was employed to fabricate 100 nm-thick Au electrodes. The channel length and width corresponded to 10 and 100 μm, respectively. The infrared (IR, 1064 nm wavelength, 100 mW cm−2) and visible (532 nm wavelength, 7.2 mW cm−2) light source were employed for measurement of the photoelectrical properties of the WSe2-based photodetector. Fig. 4b and c display the time-dependent photoconductivity of the WSe2-based photodetector recorded for different bias voltages (0.1, 0.2, and 0.5 V) using visible and IR light sources and indicate the photocurrent with an abrupt switching behavior under periodic light illumination, regardless of the light sources. Based on these results, the response time (90% of the maximum photocurrent) and decay time (10% of the minimum photocurrent) at a 0.5 V bias voltage of the WSe2-based photodetectors using a visible light source were estimated to be 6.8 and 5.6 s, respectively, and the response and decay time of the WSe2-based photodetectors using an IR light source were 5.8 and 7.8 s, which are much greater than those of previously-reported values for solution-processed MoS2-based photodetectors.14 These results suggest that the WSe2 layers synthesized using (CTA)2WSe4 may be an appropriate material to realize flexible broadband photodetectors. Fig. 4d shows a linear dependence of the photoresponsivity on the applied voltage for visible and IR light sources, which is the conventional optoelectronic behavior of photoconductors. In general, the photoresponsivity linearly depends on the illumination power or external applied voltage. When the electron–hole pair is formed under the illumination of the light source, the density of photoexcited electrons is proportional to the illumination power, and the Fermi velocity of the electrons contributing to the photocurrent may be influenced by external applied voltages. The extracted photoresponsivity for WSe2-based photodetectors recorded at 0.5 V corresponds to 26.3 mA W−1 for visible light and 5.4 mA W−1 for IR light. In addition, electrochemically-gated WSe2-based field effect transistors (FETs) were fabricated to evaluate the carrier transport properties, as depicted in Fig. 4e. The devices contain a top-gate configuration, where N,N-diethyl-N-methyl-N-(2-methoxyethyl)ammonium bis(trifluoromethylsulfonyl)imide (DEME-TFSI) and 100 nm-thick Au were used as ionic liquid dielectric and electrodes, respectively. It is widely recognized that DEME-TFSI possesses a large electrochemical stability window (>3 V at room temperature).32 It should be noted that TMDs-based FETs with ionic liquid dielectrics provide a crucial route for achieving flexible, high-performance, and low power consumption devices. The channel length and width were 200 and 500 μm, respectively. Fig. 4f shows that a representative transfer curve at VDS = −0.1 V for WSe2-based FETs reveals unipolar p-type channel behavior with an on-off current ratio and mobility of 10 and 0.45 cm2 V−1 s−1. Since previously-reported WSe2-based FETs have exhibited controversial hole-dominated, ambipolar, and electron-dominated transport behavior, this result paves the way for a universal and effective synthetic methodology for few p-type nanomaterials.33–35
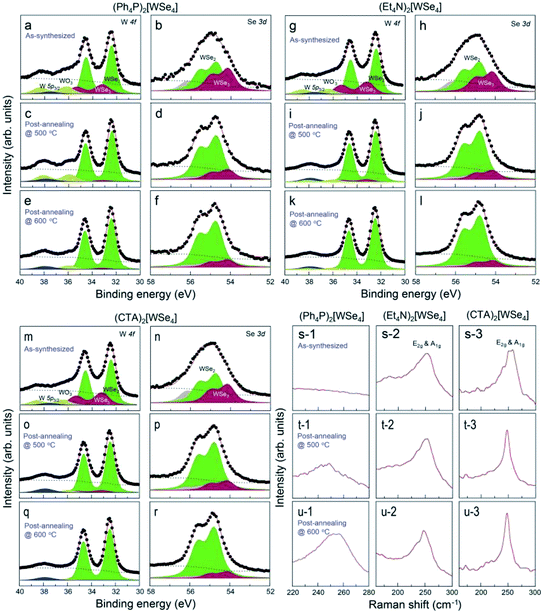 |
| | Fig. 2 XPS W 4f and Se 3d core level spectra for WSe2 NPs grown by a solvothermal synthetic route using (a–f) (Ph4P)2[WSe4], (g–l) (Et4N)2[WSe4], (m–r) (CTA)2[WSe4] before and after post-annealing at 500 °C and 600 °C. Representative Raman spectra with an excitation wavelength of 514 nm of WSe2 NPs using (s-1, t-1, u-1) (Ph4P)2[WSe4], (s-2, t-2, u-2) (Et4N)2[WSe4], and (s-3, t-3, u-3) (CTA)2[WSe4] before and after post-annealing at 500 °C and 600 °C. | |
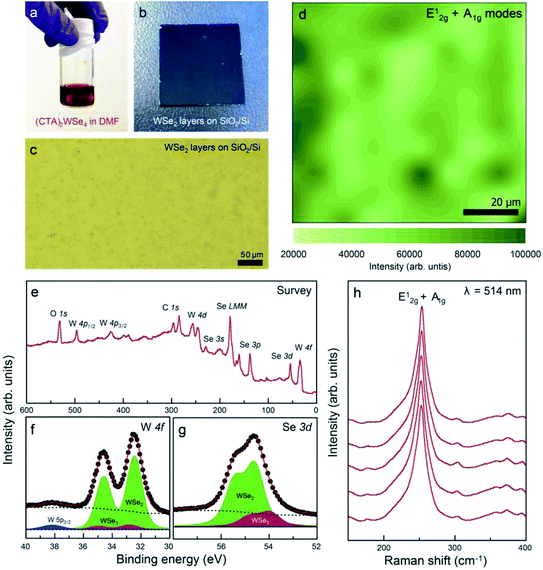 |
| | Fig. 3 Photographic images of (a) 0.5 wt% (CTA)2[WSe4] in DMF and (b) WSe2 layers synthesized on SiO2 (300 nm)/Si(001) using the precursor coating and subsequent thermal decomposition at 600 °C, (c) a optical microscopy image of the WSe2 layers on SiO2/Si, (d) Raman E12g and A1g map recorded with an excitation wavelength of 532 nm for the WSe2 layers on SiO2/Si, (e) XPS survey, (f) W 4f, and (g) Se 3d core level spectra of the WSe2 layers, (h) Raman spectra with an excitation wavelength of 514 nm for the WSe2 layers. Each spectrum was taken from different regions of the sample. | |
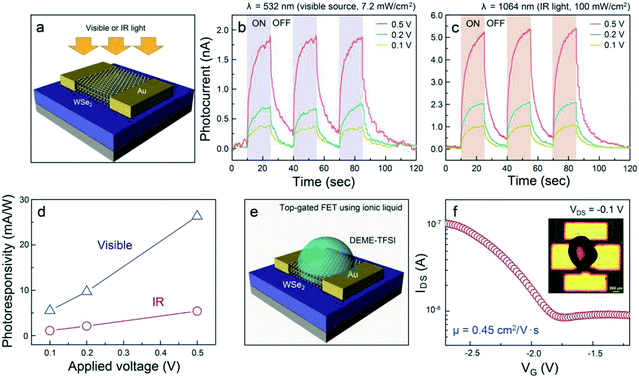 |
| | Fig. 4 (a) Schematic of a WSe2-based photodetector built on 300 nm-thick SiO2/Si with 100 nm-thick Au electrodes, time-dependent photocurrent generated by (b) visible and (c) IR light sources of the WSe2-based photodetector recorded for different bias voltages (0.1, 0.2, and 0.5 V), (d) the photoresponsivity plots for WSe2-based visible and IR photodetectors as a function of the applied voltage. (e) Schematic depiction of an electrochemically gated WSe2-based FET. (f) A transfer curve at VDS = −1 V for the WSe2-based FET. Inset indicates an optical microscopy image of the device structure. | |
Conclusions
In summary, we proposed two crucial solutions for a promising precursor with high dispersibility and low decomposition temperature and a facile synthetic methodology to produce a large-area p-type WSe2 with spatial homogeneity. In contrast to previously reported (Cat+)2[WSe4] complexes, a (CTA)2[WSe4] precursor with long alkyl chain of cetyltrimethylammonium enables the synthesis of high-crystalline WSe2 layers, as confirmed by XPS and Raman spectroscopy. The optoelectrical and electrical properties of the WSe2-based devices consolidate the possibilities of the newly developed precursor and our synthetic route, which can be used in applications for multifunctional devices, such as complementary logic circuits, gas sensors, and optoelectronic devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by a grant (2011-0031636) from the Center for Advanced Soft Electronics under the Global Frontier Research Program of the Ministry of Science, ICT and Future Planning, Korea, and a grant from the development of smart chemical materials for IoT devices project through the Korea Research Institute of Chemical Technology (KRICT) of Republic of Korea (SI1803). We would like to thank the Center for Chemical Analysis at the KRICT for using their facilities for solving the molecular structures.
References
- Q. H. Wang, K. K. Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- P. Miro, M. Audiffred and T. Heine, Chem. Soc. Rev., 2014, 43, 6537–6554 RSC.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- S. Ghatak, A. N. Pal and A. Ghosh, ACS Nano, 2011, 5, 7707–7712 CrossRef CAS PubMed.
- B. Radisavlijevic, M. B. Whitwick and A. Kis, ACS Nano, 2011, 5, 9934–9938 CrossRef PubMed.
- O. L. Sanchez, D. Lembke, M. Kayci, A. Radenovic and A. Kis, Nat. Nanotechnol., 2013, 8, 497–501 CrossRef PubMed.
- D. J. Late, Y. K. Huang, B. Liu, J. Acharya, S. N. Shirodkar, J. Luo, A. Yan, D. Charles, U. V. Waghmare, V. P. Dravid and C. N. R. Rao, ACS Nano, 2013, 7, 4879–4891 CrossRef CAS PubMed.
- T. Cao, G. Wang, W. Han, H. Ye, C. Zhu, J. Shi, Q. Niu, P. Tan, E. Wang, B. Liu and J. Feng, Nat. Commun., 2012, 3, 887 CrossRef PubMed.
- V. K. Sangwan, H. S. Lee, H. Bergeron, I. Balla, M. E. Beck, K. S. Chen and M. C. Hersam, Nature, 2018, 554, 500 CrossRef CAS PubMed.
- H. Li, J. Wu, Z. Yin and H. Zhang, Acc. Chem. Res., 2014, 47, 1067–1075 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- A. M. V. D. Zande, P. Y. Huang, D. A. Chenet, T. C. Berkelbach, Y. You, G. H. Lee, T. F. Heinz, D. R. Reichman, D. A. Muller and J. C. Hone, Nat. Mater., 2013, 12, 554–561 CrossRef PubMed.
- X. Wang, H. Feng, Y. Wu and L. Jiao, J. Am. Chem. Soc., 2013, 135, 5304–5307 CrossRef CAS PubMed.
- Y. R. Lim, W. Song, J. K. Han, Y. B. Lee, S. J. Kim, S. Myung, S. S. Lee, K. S. An, C. J. Choi and J. Lim, Adv. Mater., 2016, 28, 5025–5030 CrossRef CAS PubMed.
- Y. R. Lim, J. K. Han, S. K. Kim, Y. B. Lee, Y. Yoon, S. J. Kim, B. K. Min, Y. Kim, C. Jeon, S. Won, J. H. Kim, W. Song, S. Myung, S. S. Lee, K. S. An and J. Lim, Adv. Mater., 2018, 30, 1705270 CrossRef PubMed.
- K. Guo, S. Cui, H. Hou, W. Chen and L. Mi, Dalton Trans., 2016, 45, 19458–19465 RSC.
- K. Guo, F. Yang, S. Cui, W. Chen and L. Mi, RSC Adv., 2016, 6, 46523–46530 RSC.
- S. Huang, W. Zhang, S. Cui, W. Chen and L. Mi, Inorg. Chem. Front., 2017, 4, 727–735 RSC.
- W. Wei, L. Mi, S. Cui, B. Wang and W. Chen, ACS Sustainable Chem. Eng., 2015, 3, 2777–2785 CrossRef CAS.
- H. Fang, S. Chuang, T. C. Chang, K. Takei, T. Takahashi and A. Javey, Nano Lett., 2012, 12, 3788–3792 CrossRef CAS PubMed.
- J. Kang, S. Tongay, J. Zhou, J. Li and J. Wu, Appl. Phys. Lett., 2013, 102, 012111 CrossRef.
- S. C. O'Neal and J. W. Kolis, J. Am. Chem. Soc., 1988, 110, 1971–1973 CrossRef.
- Q. Zhang, M. Hong, W. Su, R. Cao and H. Liu, Polyhedron, 1997, 16, 1433–1437 CrossRef CAS.
- C. Altavilla, M. Sarno and P. Ciambelli, Chem. Mater., 2011, 23, 3879–3885 CrossRef CAS.
- W. Jung, S. Lee, D. Yoo, S. Jeong, P. Miro, A. Kuc, T. Heine and J. Cheon, J. Am. Chem. Soc., 2015, 137, 7266–7269 CrossRef CAS PubMed.
- Z. Xu, C. Shen, Y. Hou, H. Gao and S. Sun, Chem. Mater., 2009, 21, 1778–1780 CrossRef CAS.
- A. Mohammed, H. Nakamura, P. Wochner, S. Ibrahimkutty, A. Schulz, K. Muller, U. Starke, B. Stuhlhofer, G. Cristiani, G. Logvenov and H. Takagi, Appl. Phys. Lett., 2017, 111, 073101 CrossRef.
- J. Wang, D. Rhodes, S. Feng, M. A. T. Nguyen, K. Watanabe, T. Taniguchi, T. E. Mallouk, M. Terrones and J. Zhu, Appl. Phys. Lett., 2015, 106, 152104 CrossRef.
- J. Chen, B. Liu, Y. Liu, W. Tang, C. T. Nai, L. Li, J. Zheng, L. Gao, Y. Zheng, H. S. Shin, H. Y. Jeong and K. P. Loh, Adv. Mater., 2015, 27, 6722–6727 CrossRef CAS PubMed.
- J. Huang, L. Yang, D. Liu, J. Chen, Q. Fu, Y. Xiong, F. Lin and B. Xiang, Nanoscale, 2015, 7, 4193 RSC.
- B. Liu, M. Fathi, L. Chen, A. Abbas, Y. Ma and C. Zhou, ACS Nano, 2015, 9, 6119–6127 CrossRef CAS PubMed.
- M. M. Perera, M. W. Lin, H. J. Chuang, B. P. Chamlagain, C. Wang, X. Tan, M. M. C. Cheng, D. Tomanek and Z. Zhou, ACS Nano, 2013, 7, 4449–4458 CrossRef CAS PubMed.
- H. Zhou, C. Wang, J. C. Shaw, R. Cheng, Y. Chen, X. Huang, Y. Liu, N. O. Weiss, Z. Lin, Y. Huang and X. Duan, Nano Lett., 2015, 15, 709–713 CrossRef CAS PubMed.
- W. Liu, J. Kang, D. Sarkar, Y. Khatami, D. Jena and K. Banerjee, Nano Lett., 2013, 13, 1983–1990 CrossRef CAS PubMed.
- S. Das and J. Appenzeller, Appl. Phys. Lett., 2013, 103, 103501 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Data of TG-DTA-Mass spectroscopy. See DOI: 10.1039/c9ra00041k |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Bo Keun Parkab,
Jongsun Lim
a,
Bo Keun Parkab,
Jongsun Lim