
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic synthesis of chloroacetates with thermoregulated ionic liquids based on vanadium-substituted polyoxometalate
Jingsen Yana,
Zeqing Wanga,
Yongsheng Ea,
Fengwei Hea,
Danfeng Zhanga and
Qingyin Wu *ab
*ab
aSchool of Biomedical & Chemical Engineering, Liaoning Institute of Science and Technology, Benxi 117004, Liaoning, P. R. China. E-mail: qywu@zju.edu.cn
bDepartment of Chemistry, Zhejiang University, Hangzhou, 310027, P. R. China
First published on 14th March 2019
Abstract
A series of polyoxometalate-based ionic liquid (POM-IL) catalysts with functional sulfonic acid groups, [TEAPS]3+nPW12−nVnO40 (n = 1, 2, 3) were synthesized and characterized by nuclear magnetic resonance spectroscopy (NMR), Fourier transform infrared spectrophotometry (FT-IR), UV-Vis spectrophotometry (UV), potentiometric titration and thermogravimetry-differential scanning calorimetry (TG-DSC). The catalytic ability and reusability of the POM-IL catalysts were evaluated on esterification of chloroacetic acid and n-amyl alcohol. The optimum reaction conditions, 0.2 g of the catalyst amount, 10 mL of water carrier, 140 °C of reaction temperature, and 1.2/1 of the molar ratio of alcohol/acid, were obtained by an orthogonal test. [TEAPS]5PW10V2O40 was found to be the best active catalyst with an esterification rate of 98.75% and could be reused five times without significant decrease in activity. The ionic liquid acted as a temperature-responsive catalyst, forming a homogeneous mixture with the reactants at reaction temperature, and could be precipitated and separated from products when the reaction ends at ambient temperature. Therefore, an environmentally friendly and highly efficient approach for the synthesis of chloroacetates is provided.
1. Introduction
Chloroacetates are important organic synthesis intermediates in medicine, pesticides, plasticizers, and preparation of the esters is mostly achieved through acid-catalyzed reaction processes with mineral acids, such as H2SO4, HCl, H3PO4.1–3 However, these acids as homogenous catalysts are hardly separated and reused. Furthermore, these acids are highly corrosive and need to be neutralized at the end of the esterification reaction.4–6 Thus, various heterogeneous catalysts, such as polyoxometalates (POM), acidic resins, solid superacids have been explored as alternatives.7–9 However, these catalysts also have some disadvantages such as easy deactivation and high mass transfer resistance, which limit their application.In response to both the advantages and disadvantages of homogeneous and heterogeneous catalysts, phase-transfer catalysts and temperature regulated catalysts have attracted much attention.10,11 Among them ionic liquids (ILs) have been regarded as green reaction media or promise catalyst owing to their excellent thermal stability, outstanding solubility, negligible volatility, diversified structure and physiochemical properties.12–14 Cations and anions of ILs can be fashioned to bind specific functional group for specific chemical applications.15,16 Cole et al. firstly synthesized Brønsted acidic ILs that bear an alkyl sulfonic acid group in imidazolium cations or triphenylphosphine cations.17 These Brønsted acidic ionic liquids exhibited temperature-controlled liquid–solid separation as solvent/catalysts for Fischer esterification and the pinacol rearrangement. Then, there were many articles on the functionalized ILs used in different esterification, the esterification system is homogeneous at reaction temperature and heterogeneous at the end of reaction.18–20 However the ionic liquid catalysts is also not an excellent candidate owing to high content of ionic liquid needed in the reaction and relatively long reaction time.
In order to resolve this problem, the design and synthesis of polyoxometalate-based ionic liquid catalysts (POM-ILs) have been applied in esterification reaction.21–24 However, the regular anion in POM-ILs is binary polyoxometalate rather than ternary, only a few reports focus on ternary polyoxometalate. In fact, ternary polyoxometalates usually show more perfect properties than binary ones such as reversible thermal response and electrochemical performance.25–28 Herein, a series of novel polyoxometalate ionic liquid catalysts were prepared by using an organic ammonium, 1-(3-sulfonic group) grafted triethylamine (TEAPS) and vanadium-substituted ternary heteropolyacids. The catalysts have good activity and reusability in esterification system owing to their acidity and thermoregulated property. Their structures, activities in esterification, reversible thermal responsive properties and reaction mechanism are also investigated.
2. Experimental section
2.1 Instrumentation and reagents
1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE 500 MHz spectrometer in D2O. Solid-state 31P MAS NMR spectra was carried out on a Varian 400 InfinityPlus spectrometer at relaxation delay of 4 s. Infrared spectra was measured by Beijing Beifen-Ruili WQF-510A FT/IR spectrometer during the wavenumber range 400–4000 cm−1 using KBr pellet. UV absorption spectra was monitored by a Beijing Persee Specord TU-1901 UV-Vis spectrophotometer during scanning range 190–400 nm. X-ray powder diffraction analysis was conducted on a BRUKER D8 ADVANCE X-ray diffractometer using a Cu tube operated at 40 kV and 40 mA in the range of 2θ = 4–40° at a rate of 0.02° s−1. The thermal stability of samples was investigated on a SHIMADZU thermal analyser in the range of 25–550 °C at a rate of 10 °C min−1. The reaction products were measured by Agilent 1200 high performance liquid chromatography (HPLC). Melting point of samples was measured using WRS-1B digital microscopic melting point apparatus. The acidity of samples was determined using potentiometric titration. For the potentiometric titration, 0.1 g of POM-ILs solid was dissolved in menthol (25 mL). The solution was titrated with 0.01 mol L−1 solution of n-butylamine in acetonitrile. The potential variation was measured by Shanghai Leici pHS-2F digital acidity meter using pH composite electrode.All the chemicals were of analytical grade and used without further purification.
2.2 Synthesis of the POM-ILs catalysts
Synthesis procedure of the POM-ILs was expressed in the following schematic Fig. 1. TEAPS was synthesized according to the literature,21 and the detailed procedure was as follows: 1,3-propane sultone (0.066 mol) and triethylamine (0.066 mol) were dissolved in acetone(13 mL). Then they were mixed and stirred at 50 °C for 12 h. A white precipitate (TEAPS) was filtered, washed with acetone three times, and dried at 60 °C in a vacuum. A series of polyoxometalate (POM), H4[PW11VO40], H5[PW10V2O40], H6[PW9V3O40] and the POM-ILs were synthesized according to the literatures.29–32 The synthesis method of POM-ILs catalysts are as follows: the pre-synthesized TEAPS and POM, H4[PW11VO40], H5[PW10V2O40], H6[PW9V3O40] were taken in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 5:1 and 6:1 molar ratio to give one mole of [TEAPS]4PW11VO40, [TEAPS]5PW10V2O40, and [TEAPS]6PW9V3O40. TEAPS was added into an aqueous solution of various POM, respectively. The mixture was stirred for 12 h at room temperature. Water was firstly evaporated in a water bath at 50 °C and then removed under vacuum at 60 °C to give the final products. The obtained [TEAPS]4PW11VO40, [TEAPS]5PW10V2O40 and [TEAPS]6PW9V3O40 were pale yellow powder, yellow quasi solid and brown gel-type solid, and their melting points were determined to be 145.0 °C, 105.4 °C and 88.1 °C, respectively.
:1, 5:1 and 6:1 molar ratio to give one mole of [TEAPS]4PW11VO40, [TEAPS]5PW10V2O40, and [TEAPS]6PW9V3O40. TEAPS was added into an aqueous solution of various POM, respectively. The mixture was stirred for 12 h at room temperature. Water was firstly evaporated in a water bath at 50 °C and then removed under vacuum at 60 °C to give the final products. The obtained [TEAPS]4PW11VO40, [TEAPS]5PW10V2O40 and [TEAPS]6PW9V3O40 were pale yellow powder, yellow quasi solid and brown gel-type solid, and their melting points were determined to be 145.0 °C, 105.4 °C and 88.1 °C, respectively.
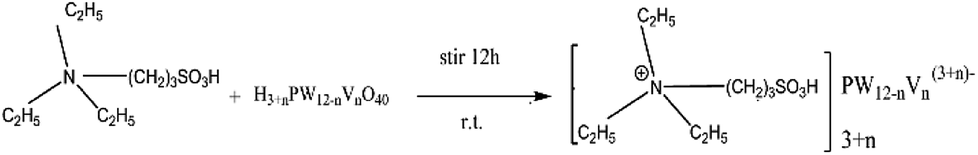 | ||
| Fig. 1 Scheme of POM-ILs synthesis procedure. | ||
2.3 Procedure for esterification reactions
The typical esterification reaction process was as follows: chloroacetic acid (0.16 mmol), 1-pentanol (0.192 mmol), POM-ILs catalysts (0.5 g) and benzene (10 mL) were added into a three-necked flask with a water segregator. The reaction was refluxed at 140 °C with stirring until the water separated in the water segregator no longer increases. The products were analysed by HPLC with external standard method. The determination conditions were as follows: Agilent ZORBAX XDB-C18 column (4.6 × 150 mm), UV absorbance detector at 213 nm, and the mobile phase was ethanol and water (65:35, v/v), flow rate at 1.0 mL min−1 and column temperature at 20 °C. Esterification rate was calculated by conversion of chloroacetic as follows:wherein C0 was initial content of chloroacetic acid and CR was chloroacetic acid content at the end of reaction.
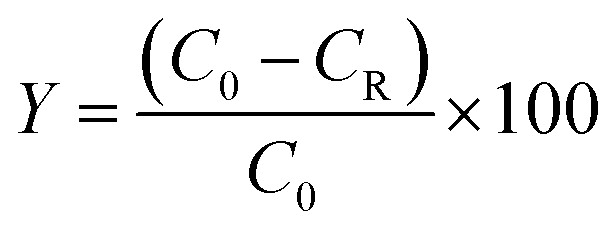
3. Results and discussion
3.1 NMR analysis
Fig. 2 shows the 1H NMR spectrum (Fig. 2a), 13C NMR (Fig. 2b) and 31P MAS NMR of [TEAPS]5PW10V2O40. The 1H NMR chemical shift values (ppm) are: δ 1.33 (t, 9H), 2.15 (m, 2H), 2.98 (t, 2H), 3.17 (t, 2H), and 3.36 (m, 6H), corresponding to the structural formula ([(C2H5)3N(CH2)3SO3H]PW10V2O40). Peaks at 1.33 ppm and 3.36 ppm correspond to methyl and methylene in ethyl group, respectively. Peaks at 2.15 ppm, 2.98 ppm and 3.17 ppm correspond to three methylene linking to N and S atoms.21 The 13C NMR chemical shift values (ppm) for [TEAPS]5PW10V2O40 are: δ 9.61, δ 20.16, δ 50.08, δ 55.77, and δ 57.75, representing carbon atoms in five different chemical environments, respectively. For the solid 31P MAS NMR spectra (Fig. 2c), the sharp resonance at −15.4 ppm was assigned to the chemical shift of phosphorus, and the peak value is a little more negative compared to the corresponding signal from HPW(δp = −15.0 ppm).33 It may be a result of the interaction between the Keggin tungstophosphate anion and the V5+ ion after the synthesis of [TEAPS]5PW10V2O40. The results indicated that the heteropoly acid and TEAPS were successfully assembled. | ||
| Fig. 2 1H NMR (a), 13C NMR (b) and 31P MAS NMR spectra of [TEAPS]5PW10V2O40. | ||
3.2 FTIR and UV analysis
Fig. 3 shows FT-IR spectra of H5[PW10V2O40], fresh [TEAPS]5PW10V2O40 and recycled [TEAPS]5PW10V2O40. The wavenumber of major bands of the compounds are given in the Table 1. As seen from Fig. 3 and Table 1, there were four characteristic vibrational bands which were assigned to νas(P–Oa), νas(M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od), νas(M–Ob–M) and νas(M–Oc–M), (MW, V) in the region between 700 and 1100 cm−1. Those four well-known characteristic bands proved the presence of Keggin-type heteropoly anion.34,35
Od), νas(M–Ob–M) and νas(M–Oc–M), (MW, V) in the region between 700 and 1100 cm−1. Those four well-known characteristic bands proved the presence of Keggin-type heteropoly anion.34,35
 | ||
| Fig. 3 FT-IR spectra of H5[PW10V2O40] (a), fresh [TEAPS]5PW10V2O40 (b) and recycled [TEAPS]5PW10V2O40 (c). | ||
| Vibrations/cm−1 | PW10V2 | Fresh [TEAPS]5PW10V2O40 | Recycled [TEAPS]5PW10V2O40 |
|---|---|---|---|
| P–Oa stretching | 1074 | 1066 | 1064 |
| MOd stretching |
975 | 968 | 964 |
| M–Ob–M stretching | 883 | 888 | 890 |
| M–Oc–M stretching | 794 | 804 | 800 |
| SO bending |
— | 1160 | 1158 |
| –CH2 stretching | — | 2977 | 2981 |
| –CH2 scissoring | — | 1486 | 1489 |
Compared with pure heteropolyacids, the M–Od and P–Oa vibration frequencies of POM-ILs were decreased when sulfo-group grafted ammonium ions were added to POM. This phenomenon could be explained that the electrostatic between the organic cation and heteropolyacid anions were weakened as the anion–anion distance increases.34 Furthermore, some other characteristic peaks at 1160 cm−1, 2977 cm−1 were assigned to νSO and νC–H, respectively.34 Since the characteristic peaks still existed, we can infer that these compounds still retained Keggin-type structure of heteropolyanion without decomposition. At the same time they maintained structure of sulfonic-functionalized triethylamine cation when TEAPS was added. This further proved the heteropoly acid and TEAPS were successfully assembled. Compared with the fresh catalyst, the infrared spectrum of the recycled catalyst was almost unchanged, indicating that Keggin-type structure of the catalyst remained stable after catalytic reaction.
The UV absorption spectrum of these compounds are shown in Fig. 4. As shown Fig. 4, we can find that the absorption bands of [TEAPS]5PW10V2O40, fresh [TEAPS]5PW10V2O40 and recycled [TEAPS]5PW10V2O40 appeared at about 191 nm and 252 nm. The absorption bands were attributed to the charge transfer between oxygen and coordinate metal.30,34 This can further provide an evidence that Keggin-type structure was still present in fresh [TEAPS]5PW10V2O40 and recycled [TEAPS]5PW10V2O40.
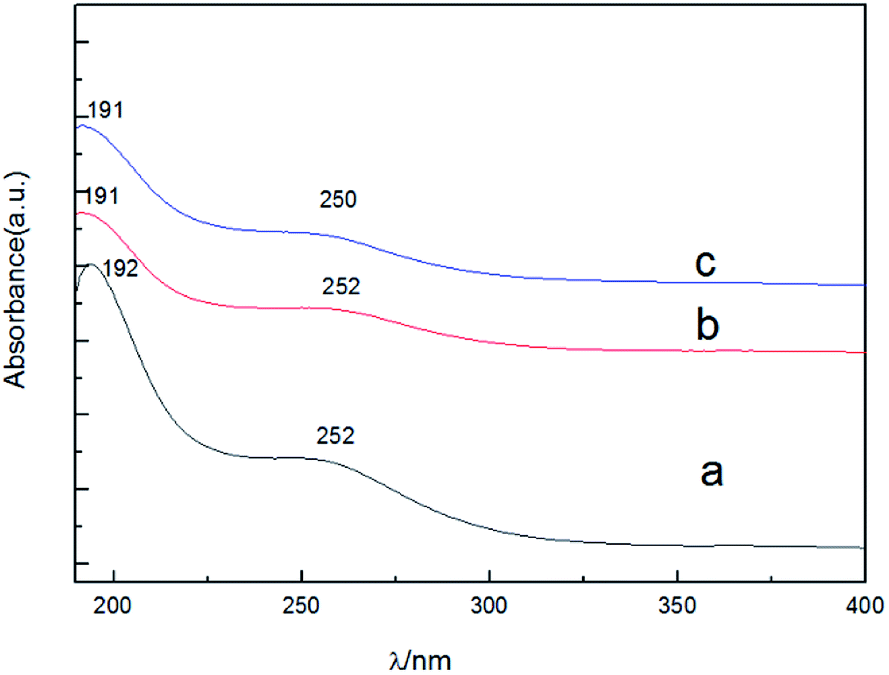 | ||
| Fig. 4 UV spectra of H5[PW10V2O40] (a), fresh [TEAPS]5PW10V2O40 (b) and recycled [TEAPS]5PW10V2O40 (c). | ||
3.3 XRD analysis
The phase and structure of the heteropoly compounds were further identified, as shown in Fig. 5. It is clear that the XRD pattern of [TEAPS]5PW10V2O40 was distinctly different from that of the pure heteropoly acid. For H5[PW10V2O40], there were typical and strong diffraction peaks in the range of 2θ = 7–12°, 18–24°, 24–36°, which revealed its crystalline state. The strong diffraction peaks at 7–12° can be considered as the POM anion structure, they were also observed in the XRD pattern of [TEAPS]5PW10V2O40 in the small angle area, suggesting that the POM anion structure existed in the hybrid material.30,34 However, [TEAPS]5PW10V2O40 exhibited a broad diffraction peak in the range of 2θ = 12–40°, which revealed its amorphous state, which was consistent with the gel-type phase at room temperature. It is reported that an organized layer-type structure existed in this series of compounds, and the weak connections of the layers leaded to the appearance of smectic gel-type phase.30,34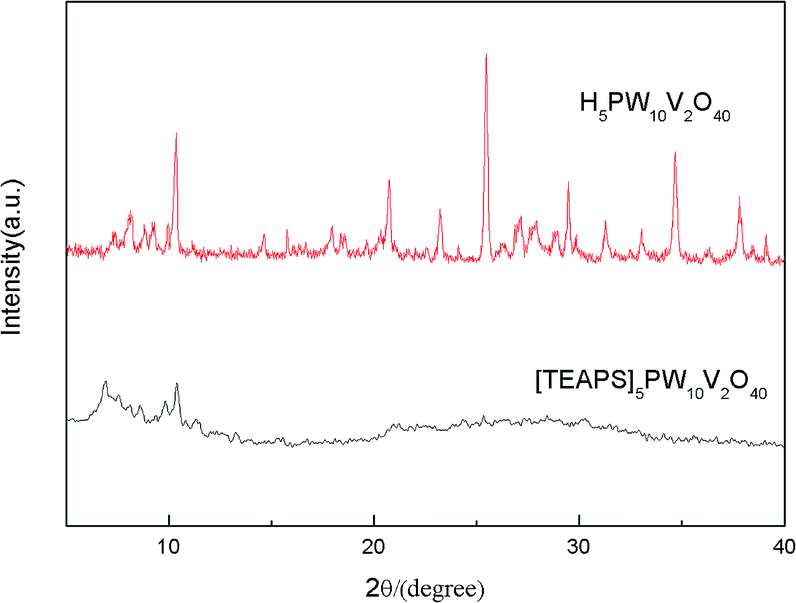 | ||
| Fig. 5 XRD patterns of H5PW10V2O40 and [TEAPS]5PW10V2O40. | ||
3.4 TG-DSC analysis
TG-DSC curves of typical samples, H5PW10V2O40 and [TEAPS]5PW10V2O40, were shown in Fig. 6, which displayed three steps weight loss for the H5PW10V2O40 at 144.7 °C, 220–400 °C and 400–550 °C. The weight loss below 400 °C corresponded to loss of surface adsorption water and protonated water. The primary weight loss between 400–550 °C corresponded to the loss of structure water and decomposition of Keggin-type structure. For [TEAPS]5PW10V2O40, there was an obvious endothermic peak at 105.4 °C, which corresponded to its melting point. The large weight loss beginning at 350 °C was due to the decomposition of organic cation, suggesting that [TEAPS]5PW10V2O40 was stable below 350 °C.30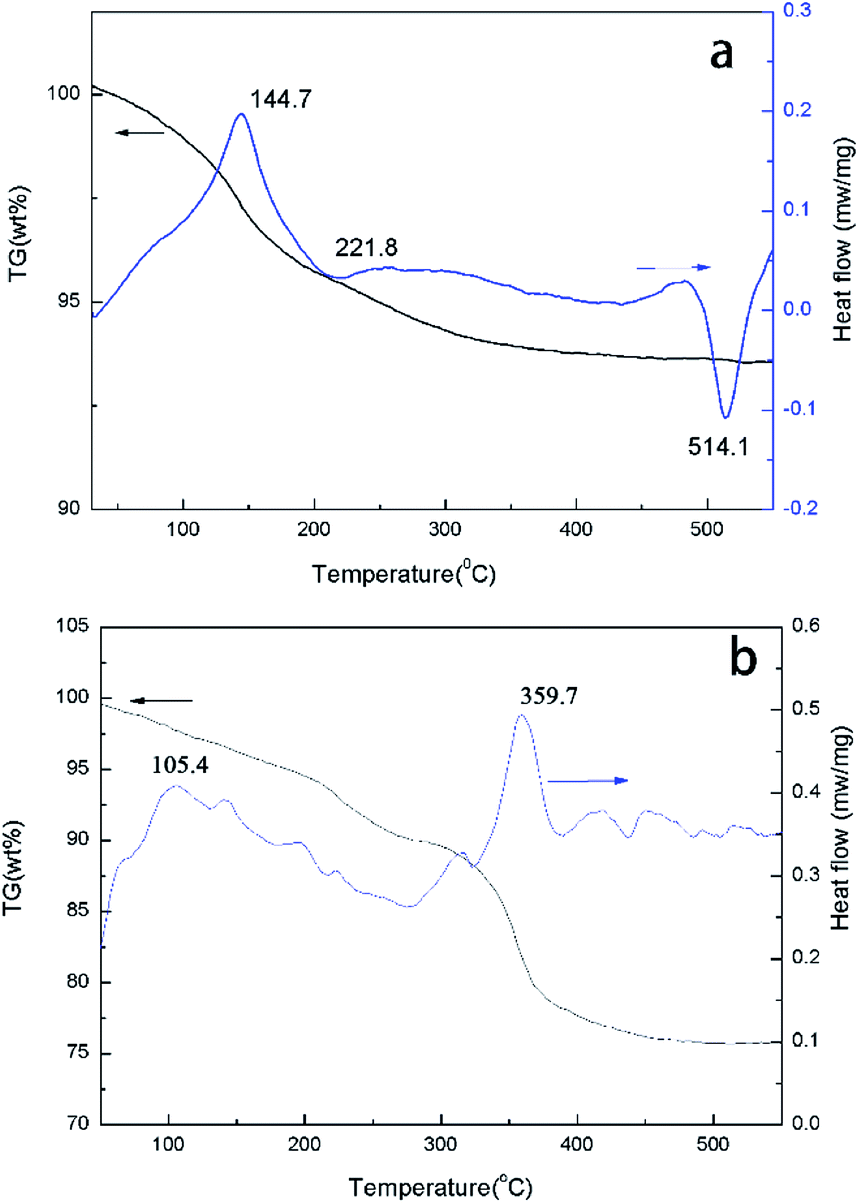 | ||
| Fig. 6 TG-DSC plots of H5PW10V2O40 (a) and [TEAPS]5PW10V2O40 (b). | ||
3.5 Determination of acidity of POM-ILs catalysts
Esterification is a typical acid-catalyzed reaction and so it is necessary to measure the acidity of catalysts. The acidity of POM-ILs were determined by potentiometric titration with n-butylamine. Potentiometric titration curves of different POM-ILs were shown in Fig. 7. The initial electrode potential (Ei) or pH before the titration demonstrates the maximum strength of the acid sites. At the end of titration, the value from which the plateau is reached (mmol amine per g catalyst) indicates the total number of acid sites.36,37 The acid strength of heteropoly acid can be judged by initial titration potential (Ei). It is defined as very strong acid (Ei > 100 mV), strong acid (0 mV < Ei < 100 mV), and weak acid (−100 mV < Ei < 0 mV).37 As shown in Fig. 7, the initial Ei value of all POM-ILs catalysts, [TEAPS]4PW11VO40 = 341 mV (pH = 1.25), [TEAPS]5PW10V2O40 = 263 mV (pH = 2.56), [TEAPS]6PW9V3O40 = 229 mV (pH = 3.13), which meant that acid strength of POM-ILs catalysts are very strong acid. The order of acid strength was as follows: [TEAPS]4PW11VO40 > [TEAPS]5PW10V2O40 > [TEAPS]6PW9V3O40, whose order is in accordance with their initial pH values. The number of acid sites was in the following order: [TEAPS]5PW10V2O40 > [TEAPS]4PW11VO40 > [TEAPS]6PW9V3O40. | ||
| Fig. 7 Potentiometric titration curves of different POM-ILs. | ||
3.6 Analysis of catalytic esterification reaction process
Conventional homogenous catalyst for esterification is difficult to be separated and reused from product. However, [TEAPS]5PW10V2O40 catalyst can easily overcome the above-mentioned problems due to a characteristic of temperature-controlled liquid–solid separation. Its reaction process with temperature change was shown in Fig. 8. At the beginning of the reaction, the catalyst was undissolved in the reactant mixture and formed a heterogeneous system (Fig. 8a). At reaction temperature, the catalyst was dissolved in the mixture, the system transferred from heterogeneous to homogeneous and became yellow owing to the colour of catalyst itself (Fig. 8b). At the end of the reaction, with temperature decreased, the produced colourless ester was in the upper level of the resulting mixture, and the catalyst gradually precipitated at bottom (Fig. 8c). Therefore the catalyst could be easily recycled from the products by simple filtration. | ||
| Fig. 8 Photographs of the esterification of chloroacetic acid and n-pentyl alcohol over [TEAPS]5PW10V2O40 catalyst. (a) [TEAPS]5PW10V2O40 (light yellow solid at bottom), chloroacetic acid (white solid in the middle), and n-pentyl alcohol (liquid in the upper level) before reaction; (b) homogeneous yellow mixture during the reaction; (c) the catalyst has precipitated at bottom, the colourless product is in the upper level at the end of the reaction. | ||
Table 2 listed the results of esterification reaction over various catalysts. The esterification of chloroacetic acid with n-pentyl alcohol system can be self-catalytic and reach only 75% of esterification rate because of its strong acidity (entry 1). The catalytic activity of conventional catalyst, such as sulfuric acid, presented unsatisfactory esterification rate of 87.47% (entry 2). Although serial heteropolyacid catalysts exhibited high esterification rate (nearly 95%, entry 3–5), it is hardly separated from the product.36 However, it is noted that POM-ILs catalysts not only could be separated from the product conveniently but also exhibited notable catalytic activity (95.3–98.75%, entry 6–8).
| Entry | Catalyst | Phenomenon | Esterification rate (%) |
|---|---|---|---|
| a 1, 2 represent the catalyst state during reaction and after reaction, respectively. | |||
| 1 | Without catalyst | Homogenous1,2 | 75.00 |
| 2 | H2SO4 | Homogenous1,2 | 87.47 |
| 3 | H4PW11VO40 | Homogenous1,2 | 94.52 |
| 4 | H5PW10V2O40 | Homogenous1,2 | 94.63 |
| 5 | H6PW9V3O40 | Homogenous1,2 | 95.00 |
| 6 | [TEAPS]4PW11VO40 | Partly dissolved1, phase seperation2 | 95.30 |
| 7 | [TEAPS]5PW10V2O40 | Homogenous1, phase separation2 | 98.75 |
| 8 | [TEAPS]6PW9V3O40 | Homogenous1, phase separation2 | 97.54 |
Esterification reaction mechanism of chloroacetic acid using [TEAPS]5PW10V2O40 catalyst was proposed in Fig. 9. Carbonyl of chloroacetic acid was protonated by hydrogen ions which were provided by functional group TEAPS in the ionic liquid catalysts and n-pentyl alcohol attack carbocation of carbonyl in chloroacetic acid. The ester was generated by dehydrate process.
 | ||
| Fig. 9 Esterification reaction mechanism of chloroacetic acid using [TEAPS]5PW10V2O40 catalyst. | ||
We measured the acidity of POM-ILs by potentiometric titration with n-butylamine to analyze any relationship between acidic properties and catalytic activity. As previous mentioned, the order of acid strength was as follows: [TEAPS]4PW11VO40 > [TEAPS]5PW10V2O40 > [TEAPS]6PW9V3O40. The number of acid sites was in the following order: [TEAPS]5PW10V2O40 > [TEAPS]4PW11VO40 > [TEAPS]6PW9V3O40.
As esterification reaction is a typical acid-catalyzed mechanism, generally, the higher acid strength of catalysts, the higher catalytic activities. However, the esterification rate of [TEAPS]4PW11VO40 was less than [TEAPS]5PW10V2O40 or [TEAPS]6PW9V3O40. This result could be attributed to their different melting point and solubility in reaction system. [TEAPS]4PW11VO40 catalyst has higher melting point (145.0 °C) and partly dissolved in the reactants at reaction temperature, and that it was more likely to result in lower activity. Another reason may be resulted from its less number of acid sites. From the result of esterification reaction over various catalysts and their phenomenon, it is assumed that the sulfonic-functionalized triethylamine cation provides the acid site responsible for the high activity in esterification, and the static interaction between organic cation and polyoxometalate anion is crucial to temperature responsive characteristics, which is responsible for the solid–liquid–solid phase transformation and catalyst separation.
The effect of various reaction parameters, including the amount of catalyst, water carrier volume, reaction temperature, the ratio of alcohol to acid, were investigated by an orthogonal experiment method using (TEAPS)5PW10V2O40 as catalyst. The above-mentioned four reaction parameters and three orthogonal levels were reasonably designed using L9(34) orthogonal table. The effect of every experiment factor and level on esterification rate was shown in Table 3. Range analysis showed that the reaction temperature was the dominant factor, next were water carrier volume, the amount of catalyst and the ratio of alcohol to acid in descending order. The optimized solution was that the amount of catalyst was 0.2 g, water carrier was 10 mL, reaction temperature was 140 °C, and the ratio of alcohol to acid was 1.2:1.
| No. | A (g) | B (mL) | C (°C) | D | Esterification rate (%) |
|---|---|---|---|---|---|
| a A, B, C, D represent the amount of catalyst, water carrier volume, reaction temperature, the ratio of alcohol to acid, respectively. The numbers in brackets represent orthogonal level order, K1, K2, K3 represent the sum of the experimental results at each level, respectively. R represents range. | |||||
| 1 | 1(0.1) | 1(5) | 1(100) | 1(1.1) | 80.21 |
| 2 | 1(0.1) | 2(10) | 2(120) | 2(1.2) | 91.82 |
| 3 | 1(0.1) | 3(15) | 3(140) | 3(1.3) | 94.26 |
| 4 | 2(0.2) | 1(5) | 2(120) | 3(1.3) | 89.09 |
| 5 | 2(0.2) | 2(10) | 3(140) | 1(1.1) | 97.78 |
| 6 | 2(0.2) | 3(15) | 1(100) | 2(1.2) | 85.56 |
| 7 | 3(0.3) | 1(5) | 3(140) | 2(1.2) | 93.14 |
| 8 | 3(0.3) | 2(10) | 1(100) | 3(1.3) | 84.26 |
| 9 | 3(0.3) | 3(15) | 2(120) | 1(1.1) | 87.03 |
| K1 | 266.29 | 262.44 | 250.03 | 265.02 | |
| K2 | 272.43 | 273.86 | 267.94 | 270.52 | |
| K3 | 264.43 | 266.85 | 285.18 | 267.61 | |
| R | 8.00 | 11.42 | 35.15 | 5.5 | |
| Factor order: C > B > A > D | |||||
| Optimal solution: A2B2C3D2 | |||||
3.7 Reusability of [TEAPS]5PW10V2O40 catalyst
As the catalyst could be easily recovered by simple filtration at the end of the reaction, the catalyst without any regeneration was reused for the next run. The reusability of [TEAPS]5PW10V2O40 in the esterification reaction system was shown in the Fig. 10. The esterification rate decreased from 98.75% for the first run to 90.97% for the fifth run. The IR and UV spectra for the recycled catalyst was well consistent with the fresh catalyst (see Fig. 3 and 4), which meant that structure of the catalyst was stable. Thus, the slight decrease in catalytic activity result from the slight loss of catalyst during dissolution process.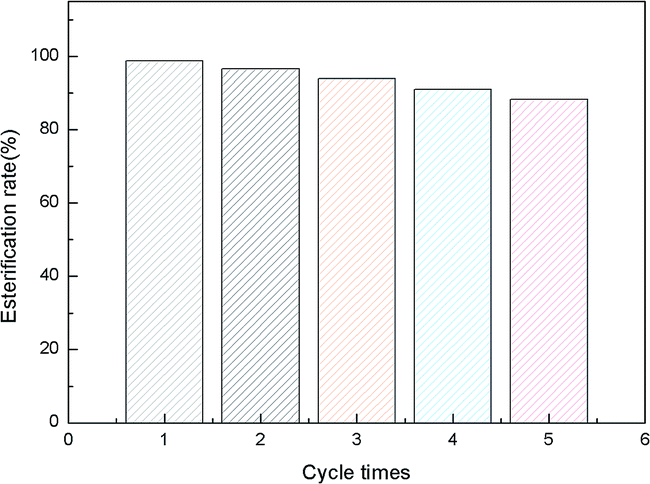 | ||
| Fig. 10 Reusability of [TEAPS]5PW10V2O40 catalyst on the esterification reaction. | ||
4. Conclusions
A series of thermo-regulated ionic liquids catalysts were synthesized by ionic self-assembly method with sulfonic-functionalized triethylamine cation and vanadium-substituted heteropolyacids. The ionic liquid, [TEAPS]5PW10V2O40 exhibited the highest catalytic activity with esterification rate of 98.75% under the optimized conditions. The sulfonic-functionalized triethylamine cation provided the acid site responsible for the high activity in esterification, and the static interaction between organic cation and polyoxometalate anion led to temperature responsive characteristics, which was responsible for the solid–liquid–solid phase transformation and separation. The catalyst could be easily recycled by simple filtration and reused without significant decrease.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was financially supported by the key projects of Liaoning Provincial Natural Science Foundation of China (No. 20170540475, 201602404 & 20180550114), the Zhejiang Provincial Natural Science Foundation of China (No. LY18B010001) and PhD Research Startup Foundation of Liaoning Institute of Science and Technology (No. 1810B08 & 1810B07).Notes and references
- G. D. Yadav and M. B. Thathagar, React. Funct. Polym., 2002, 52, 99–110 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS PubMed.
- A. Alsalme, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2010, 390, 219–224 CrossRef CAS.
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustainable Chem. Eng., 2014, 2, 1338–1352 CrossRef.
- P. Mäki-Arvela, T. Salmi, M. Sundell, K. Ekman, R. Peltonen and J. Lehtonen, Appl. Catal., A, 1999, 184, 25–32 CrossRef.
- A. Morone, M. Apte and R. A. Pandey, Renewable Sustainable Energy Rev., 2015, 51, 548–565 CrossRef CAS.
- Z. R. Xie, H. Wu, Q. Y. Wu and L. M. Ai, RSC Adv., 2018, 8, 13984–13988 RSC.
- M. Li, D. Y. Chen and X. F. Zhu, Chin. J. Catal., 2013, 34, 1674–1682 CrossRef CAS.
- X. T. Hu, Z. Zhou, D. F. Sun, Y. T. Wang and Z. B. Zhang, Catal. Lett., 2009, 133, 90–96 CrossRef CAS.
- E. Rafiee and S. Eavani, J. Mol. Liq., 2014, 199, 96–101 CrossRef CAS.
- T. Ooi and K. Maruoka, Angew. Chem., 2007, 119, 4300–4345 CrossRef.
- G. A. N. Anwar, F. Laffir, C. Dickinson, M. Vagin and T. Mccormac, Electrochim. Acta, 2018, 265, 254–258 CrossRef.
- Y. Y. Li, X. F. Wu, Q. Y. Wu, H. Ding and W. F. Yan, Ind. Eng. Chem. Res., 2014, 53, 12920–12926 CrossRef CAS.
- A. B. Bourlinos, K. Raman, R. Herrera, Q. Zhang, L. A. Archer and E. P. Giannelis, J. Am. Chem. Soc., 2004, 126, 15358–15359 CrossRef CAS PubMed.
- C. N. Dai, J. Zhang, C. P. Huang and Z. G. Lei, Chem. Rev., 2017, 117, 6929–6983 CrossRef CAS PubMed.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44–60 CrossRef CAS.
- A. C. Cole, J. L. Jensen, I. Ntai, K. L. T. Tran, K. J. Weaver, D. C. Forbes and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 5962–5963 CrossRef CAS PubMed.
- C. X. Xie, H. L. Li, L. Li, S. T. Yu and F. S. Liu, J. Hazard. Mater., 2008, 151, 847–850 CrossRef CAS PubMed.
- M. Bonamico, A. Medici, C. Chiesa and F. Carpino, Green Chem., 2012, 14, 1736–1742 RSC.
- A. L. Zhu, M. Y. Wang, L. J. Li and J. J. Wang, RSC Adv., 2015, 5, 73974–73979 RSC.
- Y. Leng, J. Wang, D. R. Zhu, X. R. Ren, H. Q. Ge and L. Shen, Angew. Chem., Int. Ed., 2009, 48, 168–171 CrossRef CAS PubMed.
- J. J. Chen, J. C. Ye, X. G. Zhang, M. D. Symes, S. C. Fan, D. L. Long, M. S. Zheng, D. Y. Wu, L. Cronin and Q. F. Dong, Adv. Energy Mater., 2018, 8, 1701021 CrossRef.
- J. Azizullah, M. Al-Rashida, A. Haider, U. Kortz, S. A. Joshi and J. Iqbal, ChemistrySelect, 2018, 3, 1472–1479 CrossRef.
- Z. Chen, W. B. Bu, D. L. Ni, C. J. Zuo, C. Chao, Q. Li, L. L. Zhang, Z. Wang and J. L. Shi, J. Am. Chem. Soc., 2016, 138, 8156–8164 CrossRef PubMed.
- T. P. Huang, Z. R. Xie, Q. Y. Wu and W. F. Yan, Dalton Trans., 2016, 3958–3963 RSC.
- Z. R. Xie, Q. Y. Wu, W. S. Dai and F. W. He, Int. J. Electrochem. Sci., 2018, 13, 11684–11690 CrossRef CAS.
- Z. R. Xie, Q. Y. Wu and L. M. Ai, Funct. Mater. Lett., 2018, 11, 1850059 CrossRef CAS.
- L. M. Ai, Z. Q. Wang, F. W. He and Q. Y. Wu, RSC Adv., 2018, 8, 34116–34120 RSC.
- X. Tong, N. Q. Tian, W. M. Zhu, Q. Y. Wu, F. H. Cao and W. F. Yan, J. Alloys Compd., 2012, 544, 37–41 CrossRef CAS.
- X. F. Wu, W. Wu, Q. Y. Wu and W. F. Yan, Langmuir, 2017, 33, 4242–4249 CrossRef CAS PubMed.
- (a) X. Tong, N. Q. Tian, W. Wu, W. M. Zhu, Q. Y. Wu, F. H. Cao, W. F. Yan and A. B. Yaroslavtsev, J. Phys. Chem. C, 2013, 117, 3258–3263 CrossRef CAS; (b) Q. Y. Wu, X. Tong and X. F. Wu, J. Xuzhou Inst. Technol., Nat. Sci. Ed., 2011, 26, 1–8 Search PubMed.
- (a) X. F. Wu, Ph. D. thesis, Zhejiang University, China, 2017; (b) X. F. Wu, Y. Y. Li, Q. Y. Wu, H. Ding and W. F. Yan, Phys. Chem. Chem. Phys., 2014, 16, 24598–24603 RSC.
- I. V. Kozhevnikov, K. R. Kloetstra, A. Sinnema, H. W. Zandbergen and H. V. Bekkum, J. Mol. Catal. A: Chem., 1996, 114, 287–298 CrossRef CAS.
- X. F. Wu, T. P. Huang, X. Tong, Z. R. Xie, W. X. Chen, Q. Y. Wu and W. F. Yan, RSC Adv., 2015, 5, 21973–21977 RSC.
- X. F. Wu, X. Tong, Q. Y. Wu, H. Ding and W. F. Yan, J. Mater. Chem. A, 2014, 2, 5780–5784 RSC.
- E. Rafiee and F. Mirnezami, J. Mol. Struct., 2017, 1130, 296–322 CrossRef CAS.
- E. Rafiee, M. Joshaghani, S. Eavani and S. Rashidzadeh, Green Chem., 2008, 10, 982–989 RSC.
| This journal is © The Royal Society of Chemistry 2019 |