
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-confined polyoxometalate-ionic liquid in silicic framework for environmentally friendly asymmetric di-hydroxylation of olefins†
Dong Liang‡
 a,
Yan Wang‡b,
Sifan Wanga,
Chengkun Songc,
Yonghe Shia,
Qinghao Liua,
Hailin Zhua,
Xia Lia,
Laishuan Liu*a and
Na Zhu*d
a,
Yan Wang‡b,
Sifan Wanga,
Chengkun Songc,
Yonghe Shia,
Qinghao Liua,
Hailin Zhua,
Xia Lia,
Laishuan Liu*a and
Na Zhu*d
aSchool of Chemical Engineering and Technology, North University of China, Taiyuan, People's Republic of China 030051. E-mail: Liulaishuan@nuc.edu.cn
bCollege of Chemistry and Chemical Engineering, Taiyuan University of Technology, Taiyuan, People's Republic of China 030024
cSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing, People's Republic of China 211189
dCollege of Environment and Resource, Shanxi University, Taiyuan, People's Republic of China 030006. E-mail: zhuna@sxu.edu.cn
First published on 19th February 2019
Abstract
Chiral 1,2-diols with a high yield could be directly prepared from asymmetric di-hydroxylation of olefins via an eco-friendly and enduring catalyst, in which abundant “chiral pools” of polyoxometalate-ionic liquid were target-designed into the silicic framework (POM-ILS) and well stabilized in aqueous media.
1 Introduction
Optically active 1,2-diols can provide substrates and intermediates for chiral drugs, pesticides and cosmetics. Previously, they were often synthesized via asymmetric hydrolysis of epoxides and di-hydroxylation of olefins.1 In particular, for the latter transformation, progressive data have been attained for osmium catalysts; however, they are still not close to the industrial applications owing to the high toxicity, volatility and secondary contamination of osmium species.2 A few researchers have proposed a greener alternative route, in which modified polyoxometalates (POMs) with chiral ligands or ionic liquids can present desirable enantioselectivity for di-hydroxylation.3 The complex stereo-catalytic systems can also be smoothly applied in the asymmetric cross-aldol reaction, Mannich reaction, Michael addition, Diels–Alder cycloaddition, etc.4Nevertheless, it must be pointed out that the high solubilities of organic ligands, ionic liquids and polyoxometalates in water and solvents make them unstable for use. To address this issue, considerable efforts have been devoted to stabilizing these complex “chiral pools”. The first and most popular attempt is screening different types of polymeric and inorganic carriers, pre-anchoring the organic groups on surfaces or encapsulating them into frameworks, and then combining them with polyoxometalates.5 As proved by some concrete examples, particularly oxidation and esterification, the stability of complex catalysts remains at a satisfactory level even after several recycles.6
Another route involves the current in situ technologies such as metal–organic frameworks (MOFs), periodic mesoporous organosilica (PMO) and polymerizable ionic liquids (PILs),7 which indicate that functional catalytic precursors can develop with the growth of the skeleton; thus, their catalytic capacity can be deeply promoted by the surrounding environment. Also, with more content of POMs and paired organics, their catalytic capacity and efficiency will be much better. Inspired by the above-mentioned works, we developed a simple in situ process to obtain toughness of hybrids in aqueous media.
2 Experimental
2.1 Preparation of catalysts
Inspired by the above works, we developed a simple in situ process to obtain toughness of hybrids in aqueous media. In brief, dark-yellow chiral ionic-liquid silicone (ILS) was first pre-synthesized with a substitution reaction between 3-chloropropyltriethoxysilane and transparent S-nicotine at equimolar amount under reflux in methanol for 24 h. This natural alkaloid is one of important by-products of the tobacco industry and it may be evolved into a chiral solvent or catalyst.8 Then, certain amounts of 12-tungstophosphoric acid (TPA) and 75% H3PO4 were dissolved in methanol, incorporated into the above IL solution and cultured for another 24 h till the formation of a tough framework. The molar ratios of 12-TPA, ILS, H3PO4 and methanol were separately controlled as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:100, 1:2:1:100, and 1:3:1:100. The final pink precipitates were centrifuged, washed, dried and denoted as POM-ILS (1/1), POM-ILS (1/2), and POM-ILS (1/3).
:1:1:100, 1:2:1:100, and 1:3:1:100. The final pink precipitates were centrifuged, washed, dried and denoted as POM-ILS (1/1), POM-ILS (1/2), and POM-ILS (1/3).
Non-silica POM-IL and non-IL POM-S catalysts were required as contrasts to prove the reaction mechanism. For the former, transparent S-nicotine was directly precipitated with TPA anions. The yellow solid was produced and named as a POM-IL catalyst. For the latter, 3-aminopropyl-triethoxysilane was used in place of the above CIL solution for precipitation with TPA. The white solid was produced and denoted as the POM-S catalyst.
2.2 Characterization of catalysts
FTIR spectra were recorded on a Fourier transform infrared spectrometer (Nicolet Nexus 470) using the conventional KBr pellet method. Thermogravimetric (TG) analysis was conducted on a Mettler Toledo TGA/SDTA851 analyzer (Switzerland) from 80 °C to 800 °C (10 °C min−1) under nitrogen (20 mL min−1). The BET surface area of the catalyst was measured using nitrogen adsorption at −196 °C with a Quantachrome Autosorb analyzer. SEM was performed using a Philips FEI Quanta 200 instrument equipped with EDX capability.2.3 Di-hydroxylation of olefins
The above five catalysts were all applied for di-hydroxylation of styrene. In every attempt, 50 mg catalyst, 100 μL styrene and 150 μL 30% H2O2 were added into 10 mL H2O at 55–60 °C for 2.5 h. After completion of the reaction, the catalyst was recycled by centrifugation, washed with EtOH (3 × 5 mL) and dried in vacuum. Excess H2O2 was eliminated using MnO2. The final liquid was extracted with CH2Cl2 (3 × 5 mL) and the extract was concentrated to 1 mL. Quantitative analysis of the products was performed with Agilent-6890N GC using a capillary column (HP-5, 30 m × 0.25 mm × 0.53 μm) with an FID detector. Optical products were identified by Agilent-1200 HPLC using a chiral column (Chiralcel OB-H) and eluted with hexane/isopropanol = 8:2 (v/v) at a flow rate of 0.5 mL min−1, with retention times of 13.5 min for S-1-phenyl-2-ethanediol and 16.7 min for R-1-phenyl-2-ethanediol.
Di-hydroxylation of cyclohexene is another prototypical reaction. POM-ILS (1/3) was preferred for examining the endurance and applicability of catalysts. In every attempt, 50 mg of catalyst, 100 μL of cyclohexene and 150 μL of 30% H2O2 were added into 10 mL of H2O at 55–60 °C for 2.5 h. After the reaction, the catalyst was centrifuged, washed and dried in vacuum for subsequent use. The final products were extracted and analyzed by GC. The optical products were identified by HPLC and were eluted with hexane/isopropanol = 8:2 (v/v) at a flow rate of 0.5 mL min−1, with retention times of 7.4 min for cis-1,2-cyclohexanediol and 8.1 min for trans-1,2-cyclohexanediol.
3 Results and discussion
The characteristic composition and properties of the studied materials are presented in Fig. 1. The FT-IR spectrum shows the parent peaks of polyoxymetalates and organic ionic liquids. IR (KBr disc, cm−1): 3540, 3064, 2970, 2891, 2700, ν(N–H, C–H); 1638, 1566, 1508, 1460, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN); 1078, 980, 893, 802, 520, ν(P–O, W–O, W–O–W, W–O–O).9 The BET surface area was 68 cm3 g−1 and the average pore diameter was 1.85 nm. The TG analysis revealed the composition of volatile components. A significant weight loss of 11.7% was observed in the range of 400–600 °C due to the organic content and after that, 4.4% weight loss was observed, which could be attributed to POx in POM. As shown in SEM images, the POM-ILS (1/3) nanoparticles displayed relatively narrow size distribution with diameter of 180–220 nm. The amount of tungsten loaded was 2.54 mmol g−1, which was calculated from the EDX spectrum.
C, CN); 1078, 980, 893, 802, 520, ν(P–O, W–O, W–O–W, W–O–O).9 The BET surface area was 68 cm3 g−1 and the average pore diameter was 1.85 nm. The TG analysis revealed the composition of volatile components. A significant weight loss of 11.7% was observed in the range of 400–600 °C due to the organic content and after that, 4.4% weight loss was observed, which could be attributed to POx in POM. As shown in SEM images, the POM-ILS (1/3) nanoparticles displayed relatively narrow size distribution with diameter of 180–220 nm. The amount of tungsten loaded was 2.54 mmol g−1, which was calculated from the EDX spectrum.
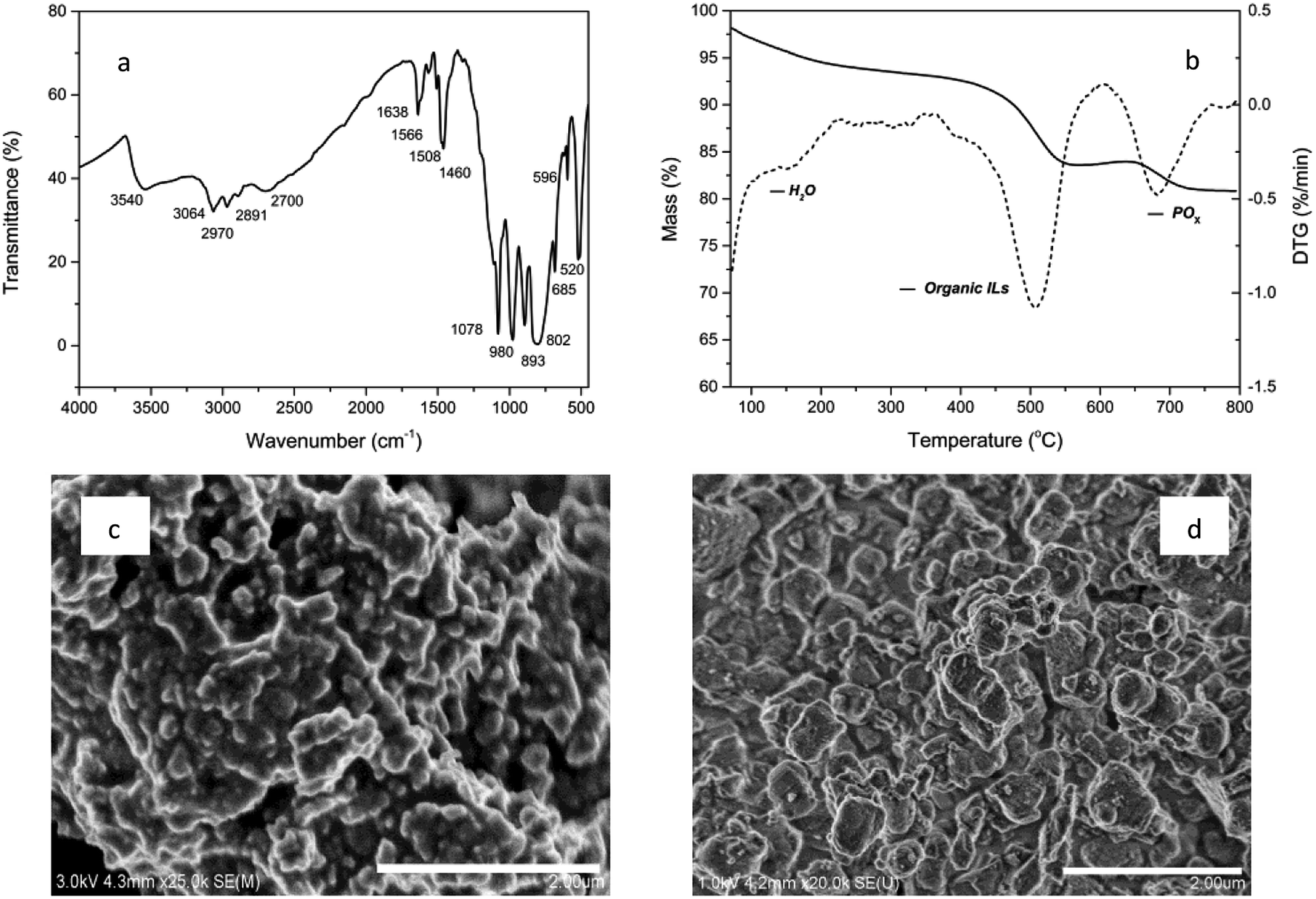 | ||
| Fig. 1 FTIR, TG, SEM of POM-ILS(3/1) (a, b and c) and SEM of POM (d). | ||
The collaborative confinement of POM anions by ionic liquids and silicic frameworks is considered to be mainly responsible for the stability of complex catalysts. To prove this, the ratios of POM anions to ILS in structure were controlled to 1/2 and 1/1, corresponding to POM-ILS (1/2) and POM-ILS (1/1). Then, the dissolution of TPA into water was examined using a UV spectrophotometer at a wavelength of 253 nm. The data in Fig. 2 show that the stability of the catalyst gradually declined with the reduction in ionic liquid content. We found 5.4%, 19.6% and 33.8% TPA in three catalysts, which separately leached into water after being treated for 3 h at 55–60 °C. Moreover, nearly 40% of TPA in POM-IL dissolved in water after 3 h without protection of the silicic framework. After replacing 3-aminopropyltriethoxysilane with the above-mentioned ionic-liquid silicone, the catalyst was prepared and named as POM-S, in which the majority of TPA could not be stably reserved in the final structure.
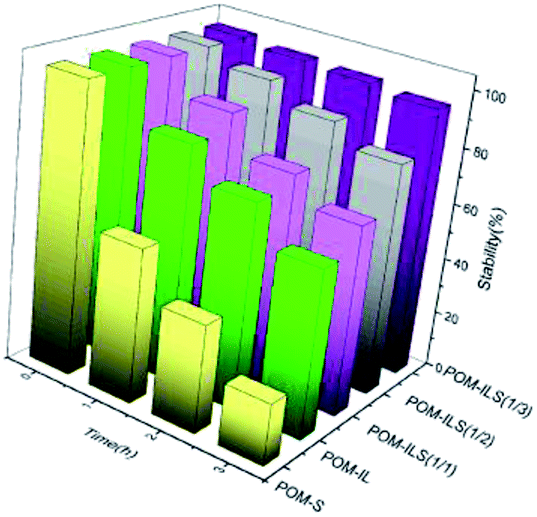 | ||
| Fig. 2 TPA stability of catalysts in hot water. | ||
Initially, di-hydroxylation of styrene was selected as a prototypical reaction to examine the catalytic activity in neat water. The results are summarized in Table 1. It was found that the complex catalyst was efficient for the asymmetric transformation of styrene with H2O2. In contrast, the control POM-S without ionic liquids yielded a small quantity of racemic diols (Table 1, entry 1). The conversion of styrene increased with the employment of ionic liquids and optically active products were achieved on POM-IL (Table 1, entry 2). POM-IL without silicic components may be considered as a “pseudo” homogeneous catalyst because it can be dissolved into a hot H2O2 solution and precipitated until the medium is cooled and the oxidants run off.10 Also, the catalyst could realize higher efficiency than heterogeneous catalysts, but the 1,2-diols were not the dominant products. When the POM anions were surrounded with abundant chiral ionic liquids, the selectivity of vicinal diols remarkably increased from 57% to 90% and the enantiomeric excess of R/S isomers remained at a satisfactory level of 82–95% (Table 1, entries 3–5). After seven recycles, POM-ILS (1/3) still exhibited better activity than other catalysts and slight decrease could be observed (Table 1, entry 6).
| Catalysts | Conv.% | Sel.%BA | Sel.%diols | e.e.%(R/S) |
|---|---|---|---|---|
| a Benzaldehyde (BA) is the main by-product. | ||||
| POM-S | 58 | 41 | 59 | 0 |
| POM-IL | 99 | 55 | 45 | 78 |
| POM-ILS(1/1) | 77 | 43 | 57 | 82 |
| POM-ILS(1/2) | 83 | 18 | 82 | 87 |
| POM-ILS(1/3) | 96 | 10 | 90 | 95 |
| POM-ILS(1/3)7 | 86 | 12 | 88 | 94 |
The difference in their catalytic properties might be further understood via characterization of 29Si MAS NMR and 31P MAS NMR. Fig. 3a shows the observed resonances in 29Si NMR spectra. T2 (−57 ppm) and T3 (−64 ppm) represented the Si atom environments of –CH2–Si(OR)(OSi)2 and –CH2–Si(OSi)3, respectively.11 In addition to the increase in ionic liquid-silicone, silicate thoroughly condensed and the percentage content of T3 peak in total peaks was found to be slightly higher than that of T2 peak. Fig. 3b presents the chemical resonances in 31P NMR spectra. The peaks (from −14 ppm to −16 ppm) could be attributed to 12-phosphotungstic acid.12 There was also a negligible shift of 1–2 ppm probably because of the electrostatic impacts of acidic–basic principles. These results suggested that the ionic liquid-based silicic framework was realized as expected, and the catalytic capacity of POMs was improved by the neighbouring chiral ligands.
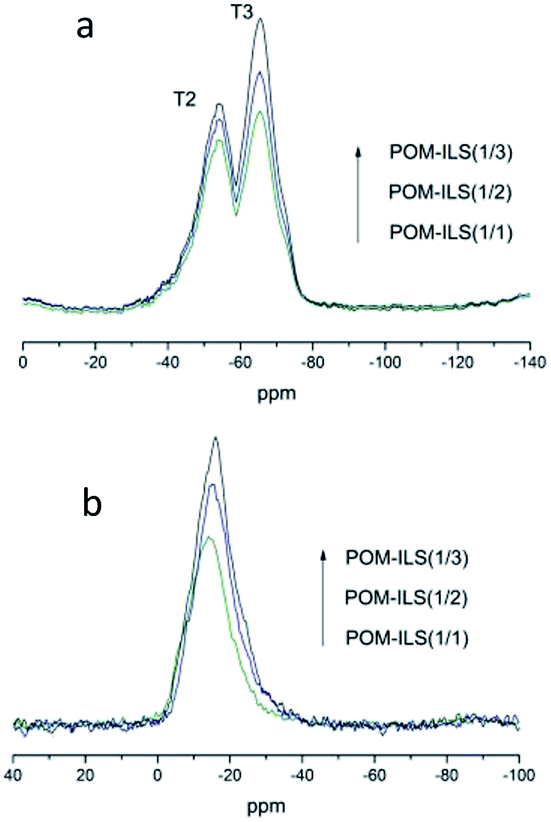 | ||
| Fig. 3 29Si-NMR (a) and 31P-NMR (b) of POM-ILS catalysts. | ||
Furthermore, another example was carried out to explore the scope of our protocol for asymmetric di-hydroxylation. Cyclohexene was employed under similar conditions using the POM-ILS (1/3) and H2O2 system. The reusability of catalyst was then established with the preferred catalysts after each run (Table 2). After the first use, the catalyst was separated, washed, dried and subjected to another fresh cycle. Seven consecutive preparation processes of trans/cis-1,2-cyclohexanediol showed no significant loss in yield, indicating that the activity of recovered catalysts did not change significantly compared with that of the fresh catalyst. Also, no significant loss in stereo-selectivity was observed and the trans-products were always the dominant products.
| a Only a small amount of cyclohexanone was formed. | |||||||
|---|---|---|---|---|---|---|---|
| Cycles | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Conv.% | 96 | 95 | 96 | 94 | 95 | 93 | 93 |
| Sel.%diols | 92 | 92 | 90 | 91 | 90 | 89 | 89 |
| e.e.%(trans/cis) | 93 | 95 | 92 | 94 | 91 | 92 | 90 |
| W(mmol g−1) | 2.53 | 2.53 | 2.51 | 2.48 | 2.45 | 2.44 | 2.42 |
Undoubtedly, enantiomerically pure alkaloids such as nicotine, cinchonidine, quinidine and ephedrine as well as various amino acids can be utilized as origins of chiral ionic liquids and can provide a favorable and soft pool for asymmetric transformations.13 Now, the dark phosphotungstic anions can be engaged to immobilize blue chiral ILs (Fig. 4); this is similar to how they rapidly freeze vulnerable cells and micelles in experiments related to biochemistry and colloid polymer science. Meanwhile, the yellow organic silicates grafted on ionic liquids undergo polycondensation under acidic conditions and afford toughness of the hybrid structure.
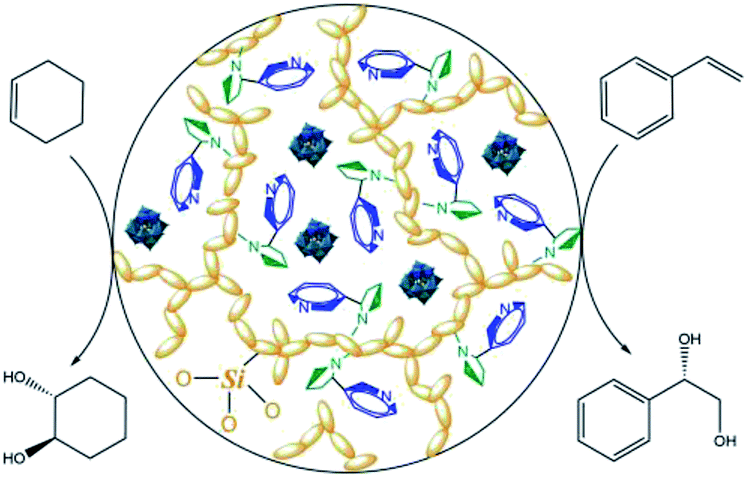 | ||
| Fig. 4 Schematic of double-locked POM catalysts by silicic frameworks and chiral ionic liquids. | ||
4 Conclusions
In summary, polyoxometalate anions might be double-locked with the silicic framework and ionic liquid, which profoundly confirmed that the heterogenous catalysts could survive in hot H2O2 solution. Apart from this, catalytic POM-IL hybrids were introduced from the starting step and presented a richer content than any other previous material (Table S1†).14 The in situ-planted abundant “chiral pools” might be responsible for the exceptional activity and enantioselectivity of the catalyst in this study.Conflicts of interest
The authors declare that there is no conflict of interests regarding the publication of this full article.Acknowledgements
Thanks for the financial supports by the National Natural Science Foundation of China (No. 21406211, 21407101, 21605133, 51701188) and the Natural Science Foundation of Shanxi Province (No. 201701D221040, 201701D221196).Notes and references
- A. Duarah, A. Goswami and T. C. Bora, et al., Appl. Biochem. Biotechnol., 2013, 170, 1965 CrossRef CAS PubMed; Y. C. He, C. L. Ma and X. Zhang, et al., Appl. Biochem. Biotechnol., 2013, 97, 7185 Search PubMed; S. Kobayashi and M. Sugiura, Adv. Synth. Catal., 2006, 348, 1496 CrossRef.
- L. C. Branco and C. A. M. Afonso, J. Org. Chem., 2004, 69, 4381 CrossRef CAS PubMed; S. M. Reddy, M. Srinivasulu and Y. V. Reddy, et al., Tetrahedron Lett., 2006, 47, 5285 CrossRef; D. Choi, S. Han and E. Kwueon, et al., Adv. Synth. Catal., 2006, 348, 2560 CrossRef; B. Balagam, R. Mitra and D. E. Richardson, Tetrahedron Lett., 2008, 49, 1071 CrossRef; R. Akiyama, N. Matsuki and H. Nomura, et al., RSC Adv., 2012, 2, 7456 RSC.
- D. Y. Du, L. K. Yan and Z. M. Su, et al., Coord. Chem. Rev., 2013, 257, 702 CrossRef CAS; D. Liang, N. Zhu and H. L. Zhu, et al., Catal. Commun., 2016, 79, 49 CrossRef.
- S. Z. Luo, J. Y. Li and H. Xu, et al., Org. Lett., 2007, 9, 3675 CrossRef CAS PubMed; Q. Gao, S. M. Lu and Y. Liu, et al., Tetrahedron Lett., 2011, 52, 3779 CrossRef; Q. Gao, Y. Liu and S. M. Lu, et al., Green Chem., 2011, 13, 1983 RSC; Q. Chen, C. Xin and L. L. Lou, et al., J. Inorg. Organomet. Polym., 2013, 23, 467 CrossRef.
- L. T. A. Sofia, A. Krishnan and M. Sankar, et al., J. Phys. Chem. C, 2009, 113, 21114 CrossRef CAS; R. Hajian, S. Tangestaninejad and M. Moghadam, et al., J. Coord. Chem., 2011, 64, 4134 CrossRef; J. Zhu, P. Wang and M. Lu, RSC Adv., 2012, 2, 8265 RSC; M. Li, M. Zhang and A. Wei, et al., J. Mol. Catal. A: Chem., 2015, 406, 23 CrossRef; M. Zhang, M. Li and Q. Chen, et al., RSC Adv., 2015, 5, 76048 RSC; Y. Mei and B. Yan, Colloid Polym. Sci., 2015, 293, 817 CrossRef; S. Herrmann, L. De Matteis and J. M. De La Fuente, et al., Angew. Chem., Int. Ed., 2017, 56, 1667 CrossRef.
- K. Yamaguchi, C. Yoshida and S. Uchida, et al., J. Am. Chem. Soc., 2005, 127, 530 CrossRef CAS PubMed; J. Kasai, Y. Nakagawa and S. Uchida, et al., Chem.–Eur. J., 2006, 12, 4176 CrossRef PubMed; A. Bordoloi, S. Suman and F. Lefebvre, et al., J. Catal., 2008, 259, 232 CrossRef; S. Doherty, J. G. Knight and J. R. Ellison, et al., Green Chem., 2012, 14, 925 RSC; R. Hajian, S. Tangestaninejad and M. Moghadam, et al., C. R. Chim., 2012, 15, 975 CrossRef; R. Tan, C. Liu and N. Feng, et al., Microporous Mesoporous Mater., 2012, 158, 77 CrossRef; H. Zhao, L. Zeng and Y. Li, et al., Microporous Mesoporous Mater., 2013, 172, 67 CrossRef; L. Hua, J. Chen and C. Chen, et al., New J. Chem., 2014, 38, 3953 RSC; Y. Leng, J. Wu and P. Jiang, et al., Catal. Sci. Technol., 2014, 4, 1293 RSC; S. Xun, D. Zheng and S. Yin, et al., RSC Adv., 2016, 6, 42402 RSC; W. Jiang, D. Zheng and S. Xun, et al., Fuel, 2017, 190, 1 CrossRef.
- M. A. Neouze, J. L. Bideau and P. Gaveau, et al., Chem. Mater., 2006, 18, 3931 CrossRef CAS; R. N. Biboum, F. Doungmene and B. Keita, et al., J. Mater. Chem., 2011, 22, 319 RSC; Y. Leng, J. Liu and P. Jiang, et al., RSC Adv., 2012, 2, 11653 RSC; J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009 CrossRef; M. H. Yang, B. G. Choi and S. C. Jung, et al., Adv. Funct. Mater., 2015, 24, 7301 CrossRef.
- T. Kitazume, US Pat., US 20010031875A1, 2001 CrossRef CAS; T. Heckel, A. Winkel and R. Wilhelm, Tetrahedron: Asymmetry, 2013, 24, 1127 CrossRef CAS; A. R. Hajipour, Y. Heidari and G. Kozehgary, RSC Adv., 2015, 5, 61179 RSC.
- S. Bareyt, S. Piligkos and B. Hasenknopf, et al., J. Am. Chem. Soc., 2005, 127, 6788 CrossRef CAS PubMed; P. M. Rao, A. Wolfson and S. Kababya, et al., J. Catal., 2005, 232, 210 CrossRef; L. N. Yang, Y. T. Qi and X. D. Yuan, et al., J. Mol. Catal. A: Chem., 2005, 229, 199 CrossRef.
- N. Mizuno and K. Yamaguchi, Chem. Rec., 2010, 6, 12 CrossRef PubMed; S. Ivanova, ChemInform, 2016, 47, 240 Search PubMed.
- V. Lahootun, C. Besson and R. Villanneau, et al., J. Am. Chem. Soc., 2007, 129, 7127 CrossRef CAS PubMed; H. Chen, W. L. Dai and X. L. Yang, et al., Appl. Catal., A, 2006, 309, 62 CrossRef; S. Zhang, et al., J. Mol. Catal. A: Chem., 2008, 289, 22 CrossRef.
- E. Lindner, A. Baumann and P. Wegner, et al., J. Mater. Chem., 2000, 10, 1655 RSC; X. Zhang, D. Wang and N. Zhao, et al., Catal. Commun., 2009, 11, 43 CrossRef CAS.
- C. Baudequin, J. Baudoux and J. Levillain, et al., Tetrahedron: Asymmetry, 2003, 14, 3081 CrossRef CAS; H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40, 1122 CrossRef PubMed; J. Ding and D. W. Armstrong, Chirality, 2010, 17, 281 CrossRef PubMed; M. Vasiloiu and K. Bica. Chiral Ionic Liquids in Separation Sciences. 2016 Search PubMed.
- A. Mandoli, D. Pini and M. Fiori, et al., Eur. J. Org. Chem., 2005, 7, 1271 CrossRef CAS; D. S. Choi, S. S. Han and E. K. Kwueon, et al., Adv. Synth. Catal., 2010, 348, 2560 CrossRef; L. C. Branco, A. Serbanovic and N. D. P. Manuel, et al., ACS Catal., 2011, 1, 1408 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10544h |
| ‡ The first two authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2019 |