
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-decorated, porous carbons derived from waste cow manure as efficient catalysts for the selective capture and conversion of CO2†
Weiping Kong * and
Jing Liu
* and
Jing Liu
School of Teacher Education, Shaoxing University, Shaoxing, Zhejiang Province 312000, China. E-mail: kongweiping0111@163.com
First published on 8th February 2019
Abstract
The catalytic conversion of CO2 is a promising solution to the greenhouse effect and simultaneously recycles the carbon sources to produce high value-added chemicals. Herein, we demonstrated a class of nanoporous carbons, which were synthesized by the direct carbonization of bio-waste cow manure, followed by activation with KOH and NaNH2. Various characterizations indicate that the resultant nanoporous carbons have abundant nanopores and nitrogen sites. As a result, their performances for the capture and catalytic conversion of CO2 were investigated. The synthesized nanoporous carbons exhibited superior properties for the selective capture and catalytic cycloaddition of CO2 to propylene oxide as compared to various solid materials.
1. Introduction
Energy harvesting processes, especially the combustion of fossil fuels and industrial hydrogen production, produce large amounts of CO2.1,2 CO2 emitted into the atmosphere severely disrupts the natural carbon cycle; this has resulted in a significant increase in the CO2 concentration in the atmosphere over the past century.3 The excess CO2 accumulated in the atmosphere would prevent the radiation of heat from the surface of earth to the space. This may be a disaster for the environment and eco-system on the earth because it would cause an increase in temperature on the earth and accelerate the rising of the sea level and desertification of land.3 Controlling the emission of CO2 from industrial activities has therefore become an urgent mission for scientists across the world.4 On the other hand, CO2 is an important source of carbon for the construction of a variety of fine chemicals such as cyclic carbonates, urea derivatives, carboxylic acids, and formamides.5–7 Among the available routes for CO2 conversion, the production of cyclic carbonates is particularly attractive owing to the 100% atom efficiency of the cycloaddition of CO2 to epoxides.8 The obtained cyclic carbonates can be used as low-volatile aprotic polar solvents, fine chemical intermediates, precursors of polymeric materials, and even electrolytes for lithium-ion batteries.5 Therefore, the simultaneous capture and conversion of CO2 has been proposed as an effective solution to the CO2 emission-related issues currently confronting human beings.7Due to the thermodynamically stable nature of CO2, advanced catalysts are required to promote the conversion of CO2 under mild conditions. To date, many kinds of catalysts, including homogeneous and heterogeneous catalysts, have been developed for the catalytic conversion of CO2. Typical homogeneous catalysts designated for CO2 conversion are metal complexes,9 ionic liquids (ILs),10 N-heterocyclic carbenes (NHCs),11 superbases,12 and frustrated Lewis pairs (FLPs).13 Typical heterogeneous catalysts designated for CO2 conversion are porous carbons,14–16 porous silicas,17 zeolites,18 metal–organic frameworks (MOFs),19 and porous organic polymers (POPs).20–22 Compared to these homogeneous catalysts, the heterogeneous catalysts are more preferred due to their facile recycling. Particularly, porous carbon materials are believed to be one of the most promising platforms for the fabrication of heterogeneous catalysts for CO2 conversion because they have many unique features such as low cost, large surface area, high thermal stability and strong hydrophobicity.23,24 In this respect, some progresses have been made. For example, Cao et al. synthesized a N-doped porous carbon monolith as a heterogeneous catalyst from alginic acid in the presence of pyrrole, ethylenediamine and glutaraldehyde for the production of chloropropene carbonate from CO2 and epichlorohydrin.14 Hu et al. synthesized N-doped porous carbon nanofiber webs from pre-synthesized polypyrrole nanofiber webs as a heterogeneous catalyst for the production of cyclic carbonates from CO2 and epoxides.15 Overall, many kinds of N-doped porous materials have been fabricated, which have been successfully employed as heterogeneous catalysts or supports for the production of cyclic carbonates from CO2 and epoxides.16
Recently, a lot of interest has been paid to the transformation of bio-waste into carbon materials with the aim to reduce the production cost of carbon materials and improve the utilization of bio-waste. For example, studies have been reported on the synthesis of carbon materials from microalgae residue,25 corn straw,26 banana peel,27 leaves,28 etc. The development of modern green and sustainable chemistry has also emphasized the advantages of using bio-waste for the production of value-added materials. In this study, we report the synthesis of porous carbon materials from cow manure, which is quite abundant in local farms and used for the preparation of carbon quantum dots.29 Fresh cow manure contains a variety of organic and inorganic matter such as protein, fat, fiber and salts. Although cow manure can be used for the production of organic fertilizers, its utilization is still insufficient. The environmental pollution caused by the disposal of cow manure is a significant issue in rural areas. To date, there are very few studies on the use of cow manure for the synthesis of advanced materials.30 Herein, porous carbon materials were synthesized by the direct carbonization of cow manure followed by chemical activation. The synthesized cow manure-derived carbons (CMCs) have a BET surface area as high as 1106 m2 g−1, hierarchical nanoporosity, and nitrogen sites with abundant pyridinic characteristics successfully doped into their networks. As a result, the CMCs display competitive performance in the selective capture of CO2. More interestingly, the synthesized CMCs also display excellent performance for the catalytic cycloaddition of CO2 to epoxides after metallization. This study describes a facile transformation of bio-waste into N-doped porous carbons, which have been successfully used in the selective capture and conversion of CO2; this promotes the widespread application of N-doped porous carbons in various research fields.
2. Experimental
2.1 Chemicals
Fresh cow manure was obtained from local farms. It was dried at 120 °C for 12 h before use. CO2 (99.99 v/v%) and N2 (99.99 v/v%) were supplied by APK Gas Co. Ltd., China. KOH, NaNH2, CoCl2·6H2O, Zn(OAc)2·2H2O, propylene oxide (PO) and tetrabutylammonium bromide (TBAB) were supplied by Sinopharm Chemical Reagent Co. Ltd., China. All chemicals were of analytical grade and used as received without further purification.2.2 Synthesis
Typically, 10.0 g of dried cow manure was loaded into a tube furnace, heated to 800 °C at the rate of 10 °C min−1 under the protection of N2, and maintained at the target temperature for 4 h. After being naturally cooled down to room temperature, the obtained head-product was obtained with the yield of 42.1% and denoted as CMC.The CMC was then subjected to chemical activation to increase its porosity. Herein, two different chemical activation agents (KOH and NaNH2) were used. In a typical run, 1 g of CMC and 3 g of chemical activation agent were mixed thoroughly, loaded into a tube furnace, heated to 600 °C at the rate of 10 °C min−1 under the protection of N2, and maintained at the target temperature for 1 h (as shown in Scheme 1). After being naturally cooled down to room temperature, the activated CMC was obtained with a yield of 37.8%. The final products were denoted as CMC-A-1 (when the chemical activation agent was KOH) and CMC-A-2 (when the chemical activation agent was NaNH2).
 | ||
| Scheme 1 Synthetic processes of CMC-A. | ||
The CMC-based catalysts were prepared by supporting Co2+ or Zn2+ on the surface of the activated CMC. In a typical run, 0.80 g of CMC-A-1 and 0.81 g of CoCl2·6H2O were added to 150 mL of degassed DMF, and the mixture was vigorously stirred at 120 °C under the protection of N2 for 12 h. The resultant solid was separated by filtration and washed thoroughly with hot DMF. After drying at 60 °C and 0.1 kPa for 48 h, Co@CMC-A-1 was obtained with a yield of 82.6%. The final contents of the metal ions in the CMC-based catalysts were determined by ICP.
2.3 Characterization
Before characterization, the samples were pretreated at 200 °C with a N2 flow for 12 h. The N2 adsorption isotherms were obtained at −196 °C using the Micromeritics Tristar II 3020 system to determine the textural properties. The specific surface areas were calculated using the BET equation in the relative pressure range of 0.05–0.2. The micropore volumes were estimated by the t-plot method in the thickness range of 0.45–0.6 nm. The total pore volumes were estimated from the amount of N2 adsorbed at the relative pressure of 0.97. The pore size distributions were calculated using the BJH model. Elemental analysis was performed using the Elementar Vario EL II system. The SEM images were obtained using the Zeiss Auriga Crossbeam electron microscope at an acceleration voltage of 5 kV. The TEM images were obtained using the JEOL JEM-2100F field-induced electron microscope at an acceleration voltage of 200 kV. The XRD patterns were obtained via the PANalytical powder diffractometer using Cu Kα radiation at a voltage of 40 kV and current of 40 mA. Raman spectra were obtained using the Princeton Acton TriVista 555 spectrometer with λ = 532 nm laser excitation. The XPS spectra were obtained using the Thermo Scientific EscaLab 250Xi spectrometer with Al-Kα radiation, and the binding energies were calibrated using the C1s peak at 284.9 eV. The ICP analysis was performed using the Jarrel-AshJ-A1100 spectrometer.2.4 Gas adsorption
Before measurements, the samples were pretreated at 200 °C with a N2 flow for 12 h. The CO2 and N2 adsorption isotherms at 0 and 23 °C were obtained using the same Micromeritics Tristar II 3020 system.2.5 CO2 conversion
The reaction was performed in a stainless steel autoclave with a Teflon tube (inner volume: 25 mL). The reactor was purged with CO2 to evacuate the air and maintained at 0 °C for 10 min. In a typical run, 20 mmol of PO, 1.44 mmol of TBAB and 30 mg of catalyst were quickly loaded into the reactor. As CO2 was charged to 1.5 MPa, the autoclave was sealed, and the reactants were stirred at 100 °C for 1.5 h. After the completion of the reaction, the excess CO2 was vented at 0 °C. The yields of the products were determined by the Agilent 7890A chromatograph equipped with a flame ionization detector (FID). For catalyst recycling, the catalyst was separated by filtration and washed thoroughly with methanol several times. After drying at 60 °C and 0.1 kPa for 48 h, the recycled catalyst was directly used for a consecutive run.3. Results and discussion
3.1 Structural results
The textural properties of the synthesized CMCs were determined using N2 as the probing gas. Fig. 1 shows the N2 adsorption isotherms at −196 °C and pore size distributions of the CMCs. The N2 adsorption isotherms display type II profiles, with a significant increase in N2 uptake when the relative pressure approaches 1.0.31 This result indicates the presence of combined micro–mesopores in the samples.32 The pore size distributions suggest disordered characteristics, with the dV/d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(W) pore volume covering a wide range of 0–15 nm. The calculated textural properties are presented in Table S1.† It can be seen that the pristine CMC has poor porosity, with a specific surface area of 60 m2 g−1 and total pore volume of 0.05 cm3 g−1. However, the activated CMCs (CMC-A-1 and CMC-A-2) have significantly improved porosity, with the specific surface areas of 713 and 1106 m2 g−1 and total pore volumes of 0.56 and 0.93 cm3 g−1, respectively. Obviously, the ability of NaNH2 to activate pristine CMC is stronger than that of KOH. The elemental analysis reveals that the pristine CMC contains ∼0.743 wt% of N. However, the activated CMCs (CMC-A-1 and CMC-A-2) contain less N (0.369 and 0.391 wt%, respectively). The change in N content suggests that some N species in pristine CMC are lost during the chemical activation process.
log(W) pore volume covering a wide range of 0–15 nm. The calculated textural properties are presented in Table S1.† It can be seen that the pristine CMC has poor porosity, with a specific surface area of 60 m2 g−1 and total pore volume of 0.05 cm3 g−1. However, the activated CMCs (CMC-A-1 and CMC-A-2) have significantly improved porosity, with the specific surface areas of 713 and 1106 m2 g−1 and total pore volumes of 0.56 and 0.93 cm3 g−1, respectively. Obviously, the ability of NaNH2 to activate pristine CMC is stronger than that of KOH. The elemental analysis reveals that the pristine CMC contains ∼0.743 wt% of N. However, the activated CMCs (CMC-A-1 and CMC-A-2) contain less N (0.369 and 0.391 wt%, respectively). The change in N content suggests that some N species in pristine CMC are lost during the chemical activation process.
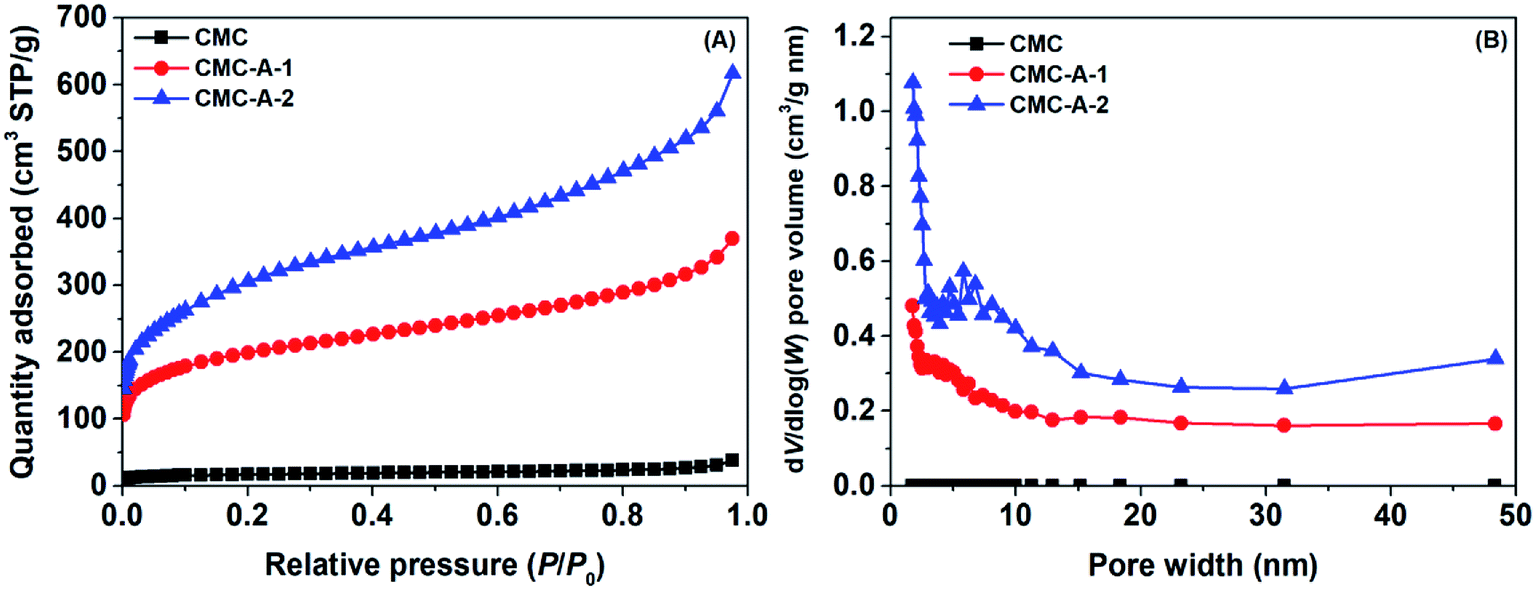 | ||
| Fig. 1 N2 adsorption isotherms at −196 °C (A) and pore width distributions (B) of CMCs. | ||
The morphology of the synthesized CMCs was examined by an electron microscope. Fig. 2 shows the SEM images of the CMCs and TEM images of the activated CMCs. The SEM images show the disordered shape and rough surface of CMCs. In the TEM images of the activated CMCs, a highly micro–mesoporous structure is observed, which is consistent with the textural properties calculated from the N2 adsorption isotherms.
 | ||
| Fig. 2 SEM images of CMC (A), CMC-A-1 (B) and CMC-A-2 (C); TEM images of CMC-A-1 (D–F) and CMC-A-2 (G–I). | ||
The chemical structure of the synthesized CMCs was further characterized by X-ray powder diffraction and various kinds of spectroscopic methods. Fig. 3 shows the XRD patterns, Raman spectra and XPS spectra of the CMCs. In the XRD patterns, the pristine CMC displays the characteristic peaks of CaCO3, which is a component of cow manure; however, these peaks disappear after activation; this suggests that CaCO3 in the pristine CMC is converted to other non-crystalline Ca compounds during the chemical activation process.33 The XRD patterns of the activated CMCs display a weak and broad peak corresponding to the graphitic (002) plane at around 25.6°, suggesting the partially graphitized structure of the activated CMCs.34 In the Raman spectra, two peaks corresponding to the D and G bands are observed at around 1354 and 1587 cm−1. The D band associated with the sp3 carbon is weaker in intensity than the G band associated with sp2 carbon.35 Furthermore, the intensity ratio of the D/G band does not show any obvious change after activation; this suggests that the defective structure of the pristine CMC does not change during the chemical activation process.36 In the full-range XPS survey, a series of peaks associated with C1s (284.16 eV, 73.29 at%), O1s (531.34 eV, 16.4 at%), N1s (399.70 eV, 2.50 at%), Si2p (102.58 eV, 1.91 at%), Ca2p (347.27 eV, 1.68 at%), Al2p (74.06 eV, 1.27 at%), P2p (132.91 eV, 1.18 at%) and Mg1s (1303.52 eV, 1.14 at%) can be observed. The N1s spectra can be deconvoluted into two parts centered at 399.8 and 397.5 eV, corresponding to N in imine/amine/amide and pyridine, respectively.37 However, the percentage of pyridine is much higher in pristine CMC than that in the activated CMCs; this suggests that pyridine in the pristine CMC is lost during the chemical activation process. This is in agreement with the change in N content according to the elemental analysis.
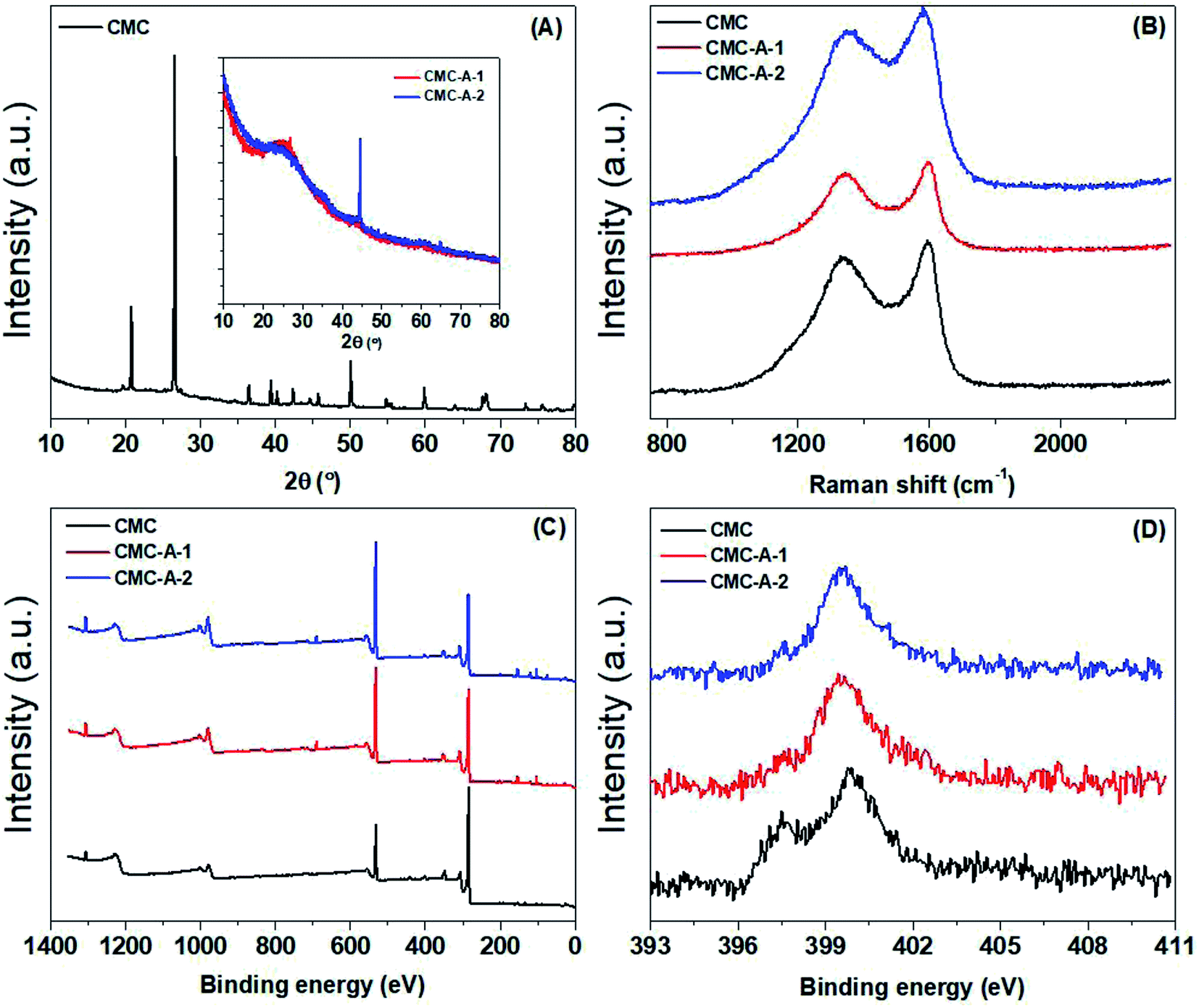 | ||
| Fig. 3 XRD patterns (A), Raman spectra (B), full-range XPS survey (C) and N1s spectra (D) of CMCs. | ||
3.2 CO2 adsorption and conversion
The CO2 capture performance of the synthesized CMCs was then investigated. Fig. 4 shows the CO2 and N2 adsorption isotherms of CMCs at 0 and 23 °C. The CO2 capacities at 0.15 bar and IAST selectivities38 for a mixture of CO2 and N2 (0.15:0.85 v/v) at 1 bar are summarized in Table S2,† which are relevant to the selective capture of CO2 from dilute sources (e.g., flue gas). It is found that the pristine CMC can adsorb only limited amount of CO2 (0.51 mmol g−1 at 0 °C and 0.41 mmol g−1 at 23 °C) because of its poor porosity; however, the activated CMCs can adsorb much more CO2 (0.81 mmol g−1 at 0 °C and 0.63 mmol g−1 at 23 °C for CMC-A-1, 1.44 mmol g−1 at 0 °C and 0.73 mmol g−1 at 23 °C for CMC-A-2) owing to their significantly improved porosity.39 The sequence of CO2 capacities is consistent with the sequence of the specific surface areas (CMC-A-2 > CMC-A-1 > CMC). The CO2 capacities of CMC-A-2 are comparable to or higher than those of many other porous carbon materials reported in the literature.40 The N2 capacities are a magnitude lower than the CO2 capacities. The CO2/N2 selectivities of the CMCs are in the range of 14.9–51.0 at 0 °C and 21.1–41.4 at 23 °C. The sequence of CO2/N2 selectivities is also well consistent with the sequence of the specific surface areas (CMC-A-2 > CMC-A-1 > CMC). This can be explained by the fact that the chemical activation process introduces significant micropores, which enable the size sieving of CO2 against N2.41 As a result, the activated CMCs display highly efficient and selective capture of CO2.
 | ||
| Fig. 4 CO2 (A and B) and N2 (C and D) adsorption isotherms of CMCs at 0 °C (A and C) and 23 °C (B and D). | ||
The cycling property of CMCs in the CO2 capture was also an important factor for their practical applications. Fig. S1† depicts the cycling property of CMC-A-2 in the capture of CO2 at 23 °C. Notably, the absorbed CO2 could be completely released after treating CMC-A-2 with N2 at 75 °C, and the regenerated CMC-A-2 was directly used for the next run. The CO2 capacities of CMC-A-2 did not decrease after 8 cycles, and the capacities of the fresh and regenerated CMC-A-2 for CO2 were around 1.5 mmol g−1. This result is well consistent with the results obtained from the CO2 isotherms. The abovementioned results confirm very good stability of CMCs for the selective capture of CO2.
The activated CMCs were finally supported with metal ions and used as heterogeneous catalysts for CO2 conversion. The cycloaddition of CO2 to PO was used as the model reaction. Table 1 lists the yields, selectivities and TOFs of the activated CMC-based catalysts for the cycloaddition of CO2 to PO under different reaction conditions. As can be seen, Co/CMC-A-1 and Co/CMC-A-2 display comparable performances in the catalytic conversion of CO2, with yields of >95%, selectivities of >99% and TOFs of ∼700 h−1 at 100 °C and 0.5 MPa for 1.5 h (entries 1 and 2). If the loaded Co2+ is changed to Zn2+, the resultant Zn/CMC-A-1 displays comparable performance to Co/CMC-A-1 for the catalytic conversion of CO2 (entry 3). However, the yields decreased to <50% if no TBAB, catalyst or metal ions were used (entries 4–6); this suggested the essential roles of the three components in catalyzing the conversion of CO2. Even after being recycled for 5 times, Co/CMC-A-1 still displayed the yield of 95%, selectivity of 99% and TOF of 719 h−1 (entry 7), comparable to those of the fresh catalyst. This result confirms the high stability of the activated CMC-based catalysts during recycling. The performance of Co/CMC-A-1 for catalyzing the conversion of CO2 at an elevated pressure of 1.0 MPa for a shortened time of 0.5 h (entry 8) or at a room temperature of 25 °C for a long time of 24 h (entry 9) was also investigated. It is found that the temperature has a significant effect on the activity of Co/CMC-A-1 for the catalytic conversion of CO2, and a higher temperature is preferred for facilitating the conversion of CO2. Overall, the performance of the activated CMC-based catalysts for the catalytic conversion of CO2 is comparable or superior to that of most other heterogeneous catalysts reported in Table S3† and the literature.20,42,43 Furthermore, the Co/CMC-A-1 catalyst showed good effect for the cycloaddition of epoxides and CO2 to cyclic carbonates, and the results can be found in the Table 2.
| Entry | Catalysts | Metal contentsb (mmol g−1) | Additive | Yields (%) | Selectivities | TOFs (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: propylene oxide (20 mmol), TBAB (7.2 mol%), catalyst (30 mg), CO2 (0.5 MPa), temperature (100 °C), time (1.5 h).b Determined by ICP.c Recycled for 5 times.d Reaction conditions: propylene oxide (20 mmol), TBAB (7.2 mol%), catalyst (30 mg), CO2 (1.0 MPa), temperature (100 °C), time (0.5 h).e Reaction conditions: propylene oxide (20 mmol), TBAB (7.2 mol%), catalyst (30 mg), CO2 (0.5 MPa), temperature (25 °C), time (48 h). | ||||||
| 1 | Co/CMC-A-1 | 0.60 | TBAB | 95 | ∼100 | 701 |
| 2 | Co/CMC-A-2 | 0.61 | TBAB | 95 | ∼100 | 695 |
| 3 | Zn/CMC-A-1 | 0.55 | TBAB | 95 | 9 | 761 |
| 4 | Co/CMC-A-1 | 0.60 | None | 26 | ∼100 | 195 |
| 5 | None | — | TBAB | 36 | ∼100 | — |
| 6 | CMC-A-1 | — | TBAB | 42 | ∼100 | — |
| 7 | Co/CMC-A-1c | 0.58 | TBAB | 95 | 99 | 719 |
| 8 | Co/CMC-A-1d | 0.60 | TBAB | 96 | 99 | 2103 |
| 9 | Co/CMC-A-1e | 0.60 | TBAB | 92 | 99 | 21 |
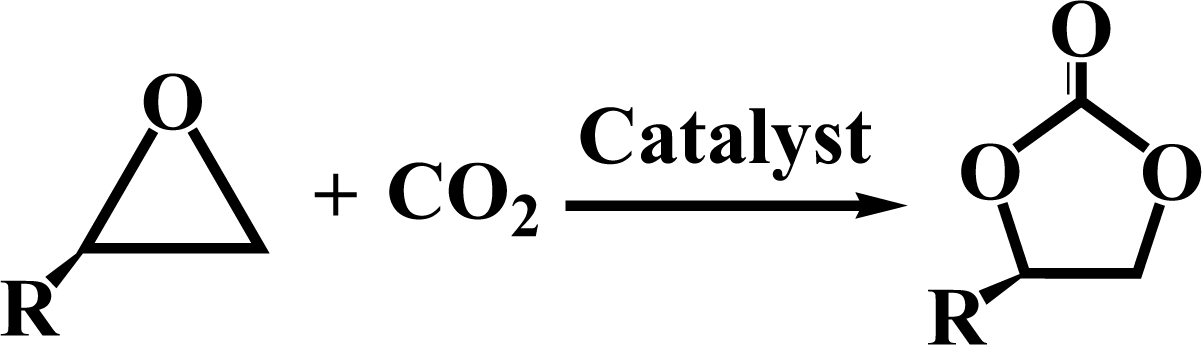












4. Conclusions
In summary, cow manure obtained from local farms was used as a precursor for the synthesis of porous carbon materials by direct carbonization followed by chemical activation. The chemical activation by KOH and NaNH2 significantly improved the porosity of cow manure-derived carbons. Electron microscopy and spectroscopic characterizations demonstrated the amorphous and disordered structure of the cow manure-derived carbons. The synthesized cow manure-derived carbons were used as heterogenous catalysts for CO2 conversion. It was found that the cow manure-derived carbons displayed competitive performance for the selective capture and catalytic conversion of CO2. The present study provides an alternative method for the preparation of heterogeneous catalysts for CO2 conversion from abundant bio-waste.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21573150).References
- S. Asefi-Najafabady, P. J. Rayner, K. R. Gurney, A. McRobert, Y. Song, K. Coltin, J. Huang, C. Elvidge and K. Baugh, J. Geophys. Res.: Atmos., 2014, 119, 10213–10231 Search PubMed.
- J. X. Mi, Y. N. Cao, J. C. Zhang, Y. D. Ma, C. Q. Chen, D. L. Li, X. Y. Lin and L. L. Jiang, Appl. Catal., A, 2018, 553, 36–42 CrossRef CAS.
- P. M. Cox, R. A. Betts, C. D. Jones, S. A. Spall and I. J. Totterdell, Nature, 2000, 408, 750 CrossRef CAS.
- T. L. Johnson and D. W. Keith, Energy Policy, 2004, 32, 367–382 CrossRef.
- F. J. Liu, K. Huang, S. M. Ding and S. Dai, J. Mater. Chem. A, 2016, 4, 14567–14571 RSC.
- Y. Zhang and D. S. Lim, ChemSusChem, 2015, 8, 2606–2608 CrossRef CAS.
- Q. W. Song, Z. H. Zhou and L. N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- F. J. Liu, K. Huang, Q. Wu and S. Dai, Adv. Mater., 2017, 29, 1700445 CrossRef PubMed.
- R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS PubMed.
- K. Chen, G. Shi, W. Zhang, H. Li and C. Wang, J. Am. Chem. Soc., 2016, 138, 14198–14201 CrossRef CAS.
- F. Huang, G. Lu, L. Zhao, H. Li and Z. X. Wang, J. Am. Chem. Soc., 2010, 132, 12388–12396 CrossRef CAS.
- Z. Z. Yang, L. N. He, Y. N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971–3975 RSC.
- C. M. Momming, E. Otten, G. Kehr, R. Frohlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed.
- X. Ma, B. Zou, M. Cao, S. L. Chen and C. Hu, J. Mater. Chem. A, 2014, 2, 18360–18366 RSC.
- Y. Li, B. Zou, C. Hu and M. Cao, Carbon, 2016, 99, 79–89 CrossRef CAS.
- K. Huang, J. Y. Zhang, F. J. Liu and S. Dai, ACS Catal., 2018, 8, 9079–9102 CrossRef CAS.
- S. W. Xu, Y. Lu, J. Li, Z. Y. Jiang and H. Wu, Ind. Eng. Chem. Res., 2006, 45, 4567–4573 CrossRef CAS.
- A. Zhu, T. Jiang, B. Han, J. Zhang, Y. Xie and X. Ma, Green Chem., 2007, 9, 169–172 RSC.
- L. Liang, C. Liu, F. Jiang, Q. Chen, L. Zhang, H. Xue, H. L. Jiang, J. Qian, D. Yuan and M. Hong, Nat. Commun., 2017, 8, 1233 CrossRef PubMed.
- G. Ji, Z. Z. Yang, H. Zhang, Y. Zhao, B. Yu, Z. Ma and Z. M. Liu, Angew. Chem., Int. Ed., 2016, 55, 9685–9689 CrossRef CAS PubMed.
- K. Huang, F. J. Liu, L. L. Jiang and S. Dai, ChemSusChem, 2017, 10, 4144–4149 CrossRef CAS PubMed.
- K. Huang, F. J. Liu and S. Dai, J. Mater. Chem. A, 2016, 4, 13063–13070 RSC.
- J. Liu, N. P. Wickramaratne, S. Z. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef CAS.
- H.-L. Peng, J.-B. Zhang, J.-Y. Zhang, F.-Y. Zhong, P.-K. Wu, K. Huang, J.-P. Fan and F. J. Liu, Chem. Eng. J., 2019, 359, 1159–1165 CrossRef CAS.
- M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- W. Z. Zhong, Z. Z. Zhang, Y. J. Luo, W. Qiao, M. Xiao and M. Zhang, Bioresour. Technol., 2012, 114, 281–286 CrossRef CAS.
- M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte and J. H. Clark, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- M. Biswal, A. Banerjee, M. Deo and S. Ogale, Energy Environ. Sci., 2013, 6, 1249–1259 RSC.
- C. D. do E. S. Barbosa, J. Corrêa, G. A. Medeiros, G. Barreto, K. G. Magalhaes, A. L. de Oliveira, J. Spencer, M. O. Rodrigues and B. A. D. Neto, Chem.–Eur. J., 2015, 21, 5055–5060 CrossRef CAS PubMed.
- I. Noshadi, B. Kanjilal and F. J. Liu, Appl. Catal., A, 2016, 513, 19–29 CrossRef CAS.
- K. Ramesh, K. S. Reddy, I. Rashmi, A. K. Biswas and K. R. Islam, Commun. Soil Sci. Plant Anal., 2016, 47, 1622–1629 CrossRef CAS.
- F. J. Liu, L. Wang, Q. Sun, L. F. Zhu, X. J. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948–16950 CrossRef CAS PubMed.
- Y. Y. Lv, F. Zhang, Y. Q. Dou, Y. P. Zhai, J. X. Wang, H. J. Liu, Y. Y. Xia, B. Tua and D. Y. Zhao, J. Mater. Chem., 2012, 22, 93–99 RSC.
- B. He, F. J. Liu and S. K. Yan, J. Mater. Chem. A, 2017, 5, 18064–18070 RSC.
- K. Huang, F. J. Liu, J.-P. Fan and S. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 36961–36968 CrossRef CAS.
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. Dou and L. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS.
- K.-Y. Park, J.-H. Jang, J.-E. Hong and Y.-U. Kwon, J. Phys. Chem. C, 2012, 116, 16848–16853 CrossRef CAS.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- J. W. F. To, J. J. He, J. G. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. C. Chen, W.-G. Bae, L. J. Pan, J. B.-H. Tok, J. Wilcox and Z. N. Bao, J. Am. Chem. Soc., 2016, 138, 1001–1009 CrossRef CAS PubMed.
- A. E. Creamer and B. Gao, Environ. Sci. Technol., 2016, 50, 7276–7289 CrossRef CAS PubMed.
- K. C. Bedin, A. C. Martins, A. L. Cazetta, O. Pezoti and V. C. Almeida, Chem. Eng. J., 2016, 286, 476–484 CrossRef CAS.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2011, 23, 1818–7289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10497b |
| This journal is © The Royal Society of Chemistry 2019 |