
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFullerene derivative induced morphology of bulk heterojunction blends: PIPCP:PC61BM†
Tzu-Yen Huang a,
Hongping Yanab,
Maged Abdelsamiea,
Victoria Savikhinac,
Sebastian A. Schneiderad,
Niva A. Rane,
Thuc-Quyen Nguyene,
Guillermo C. Bazane and
Michael F. Toney*a
a,
Hongping Yanab,
Maged Abdelsamiea,
Victoria Savikhinac,
Sebastian A. Schneiderad,
Niva A. Rane,
Thuc-Quyen Nguyene,
Guillermo C. Bazane and
Michael F. Toney*a
aStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, CA, USA. E-mail: mftoney@slac.stanford.edu
bDepartment of Chemical Engineering, Stanford University, Stanford, CA, USA
cDepartment of Electrical Engineering, Stanford University, Stanford, CA, USA
dDepartment of Chemistry, Stanford University, Menlo Park, CA, USA
eCenter for Polymers and Organic Solids, Department of Chemistry and Biochemistry, University of California—Santa Barbara, Santa Barbara, CA, USA
First published on 30th January 2019
Abstract
The performance of organic solar cells (OSCs) depends crucially on the morphology in bulk heterojunctions (BHJs), including the degree of crystallinity of the polymer and the amount of each material phase: aggregated donor, aggregated acceptor, and molecular mixed donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor phase. In this paper, we report the BHJ morphology of as-cast blend films incorporating the polymer PIPCP as the donor and [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) as the acceptor. Tracking the scattering intensity of PC61BM as a function of PC61BM concentration shows that PC61BM aggregates into donor-rich domains and there is little to no phase where the PC61BM and PIPCP are intimately mixed. We further find that on blending the scattering peak due to PIPCP ordering along the backbone increases with decreasing PIPCP fraction, which is attributed to improved ordering of PIPCP due to the presence of PC61BM. Our results suggest that the improved ordering of PIPCP along the backbone (consistent with an increased conjugation length) with blending contributes to the observed low open-circuit voltage energy loss.
:acceptor phase. In this paper, we report the BHJ morphology of as-cast blend films incorporating the polymer PIPCP as the donor and [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) as the acceptor. Tracking the scattering intensity of PC61BM as a function of PC61BM concentration shows that PC61BM aggregates into donor-rich domains and there is little to no phase where the PC61BM and PIPCP are intimately mixed. We further find that on blending the scattering peak due to PIPCP ordering along the backbone increases with decreasing PIPCP fraction, which is attributed to improved ordering of PIPCP due to the presence of PC61BM. Our results suggest that the improved ordering of PIPCP along the backbone (consistent with an increased conjugation length) with blending contributes to the observed low open-circuit voltage energy loss.
1. Introduction
Solution-processed organic solar cells (OSCs) are attractive as inexpensive renewable energy harvesting materials because of their low manufacturing costs, roll-to-roll processing, and flexible large-area devices.1–4 Steady progress in the development of conjugated polymers with donor/acceptor structures has resulted in power conversion efficiencies (PCEs) reaching greater than 13% for single cells and up to 17% for tandem cells5,6 based on the bulk heterojunction (BHJ) architecture. The BHJ is a blend of donor and acceptor semiconductors with partial phase separation and provides large interfacial area for charge separation. The phase-separated morphology consists of relatively pure donor and acceptor domains, and a molecularly mixed donor:acceptor region (i.e., the mixed phase). The donor- and acceptor-rich phases in the film form an interpenetrating network to transport charge carriers after exciton dissociation; however, in addition to the pure phase, the interface between the donor and the acceptor phase may not be molecularly sharp, but gradual. This molecular mixed phase in the BHJ film is still not fully understood, although there is some evidence that the mixed phase enhances generation of charge carriers.7 To date, the relative amounts of pure and mixed phase only have been reported for some polymer:fullerene systems8,9 and mixed phases demonstrated for certain well-known polymers such as poly(3-hexylthiophene-2,5-diyl) (P3HT), poly[(4,8-bis-(2-ethylhexyloxy)-benzo(1,2-b:4,5-b′)dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-)-2-carboxylate-2-6-diyl)] (PTB7), and poly(di(2-ethylhexyloxy)benzo[1,2-b:4,5-b′]dithiophene-co-octylthieno[3,4-c]pyrrole-4,6-dione) (PBDTTPD).10–13 In the case of the PBDTTPD polymer, X-ray scattering was utilized to quantify the mixed phase by varying the fullerene concentration in BHJs and determining the PC61BM diffraction. No PC61BM scattering was observed until the content of fullerene reached 20 wt%; therefore, Bartelt et al. concluded that ∼20 wt% of fullerene is intimately mixed with PBDTTPD.12 On the other hand, no mixed phase was observed in the high-performing small molecule X2:PCBM blend in,14,15 using a similar method.
While PIPCP:PC61BM BHJ blends exhibit a PCE of 6.4%, they are relevant in the context of their high open-circuit voltage (VOC) of 0.86 V.16 Ran et al. studied the energy loss (Eloss = Eg − eVOC, where Eg is the optical bandgap) for a series of polymer donors with PCE > 5%. Compared with most polymer BHJs, PIPCP:PC61BM blends exhibit an exceptionally low energy loss of Eloss = 0.52 eV, while most fullerene-based BHJs have Eloss > 0.6 eV.17,18 Furthermore, the absorption edge of PIPCP is redshifted after blending with PC61BM and the Urbach energy of the neat PIPCP and the blend are both small at ∼27 meV. Transition electron microscopy (TEM) from PIPCP:PCBM blends showed well defined PIPCP lattice planes due to crystalline regions in the blends, while in the neat PIPCP such regions were not observed. These results were interpreted as showing that for the blend film, the optoelectronic behavior was due to the increase in PIPCP conjugation length with the PIPCP molecule becoming more planar.18 PIPCP and several derivative polymers have also been investigated with grazing-incidence wide-angle X-ray scattering (GIWAXS).19–21
Ran et al. recently investigated the blend morphology of PIPCP:PC61BM BHJ film using resonant soft X-ray scattering (RSoXS)17 and a number of optoelectronic measurements. They showed that the as-cast PIPCP:PC61BM blend film forms phase-separated donor and acceptor domains with a size of ≈30 nm but that annealed films were mixed and not phase separated. This was inferred to suggest that PIPCP and PC61BM were highly miscible, consistent with calculations of the Flory–Huggins chi parameter. Ran et al.17 further comprehensively investigated the recombination and loss mechanisms and concluded that the mixed morphology caused the observed non-geminate recombination and low fill factor (FF) in devices. It is important to note here that the much less pronounced phase separation was only observed in annealed films and that the length scale of the RSoXS measurements was limited to a minimum of about 4–6 nm in diameter (set by the maximum scattering vector of 0.8 nm−1). Under these conditions, the RSoXS will not resolve mixing below about 4–6 nm, corresponding to between 30 and 100 fullerenes within the volume of a pure PC61BM cluster. The grazing-incidence wide-angle X-ray scattering (GIWAXS) method we adopt14,15 is, in contrast, sensitive at a smaller length scale of polymers and fullerene mixing. Thus, the accurate characterization, and thus our understanding, of donor:acceptor mixing is still limited.
In this work, we report the blend morphology of the PIPCP:PC61BM BHJ system with a varying ratio of components. GIWAXS is employed to investigate the BHJ morphology, using the methodology that we previously reported to elucidate the mixed phase.15 We find that there is a negligible amount of molecularly mixed phase in as-cast PIPCP:PC61BM BHJs. However, the PIPCP phase may contain isolated PC61BM clusters that lead to high charge recombination and low FF. The presence of aggregated PC61BM improves the order of PIPCP polymers and surprisingly a scattering peak along the in-plane direction at qxy = 0.25 Å−1 significantly increases with increasing PC61BM content, showing a strengthening of the order along the backbone chain. These results provide insight into the morphology in BHJs and help developing a new class of high VOC materials for OSCs.
2. Experimental
2.1 Materials
The synthesis of PIPCP, used as the donor material, has been described previously.16 PC61BM was purchased from Nano-C. Chloroform (CF) and chlorobenzene (CB) were purchased from Sigma-Aldrich. The chemical structures of PIPCP and PC61BM are displayed in Fig. 1. All chemicals were received and used without further purification. | ||
| Fig. 1 The chemical structure of (a) PIPCP and (b) PC61BM. | ||
2.2 Preparation of PIPCP:PC61BM blend films
The solution of PIPCP:PC61BM blend was prepared in the CF:CB (3:2 v/v%) co-solvent. To investigate the effect of PC61BM concentration on the morphology, the total solids concentration of active layer solution was maintained at 20 mg ml−1 while varying the composition of PC61BM (0–100 wt%). Prior to sample preparation, the Si substrates were cleaned in detergent, DI water, acetone, and isopropyl alcohol. After routine solvent cleaning, the Si substrates were treated with UV ozone for 15 min. The active layer solution (PIPCP:PC61BM) was spin-coated in a nitrogen glovebox (2000 rpm, 60 s) onto the Si substrate at room temperature and the films were not annealed. A Bruker Dektak XT-S Stylus profilometer (stylus force: 1–15 mg with LIS 3 sensor and scan length range: 55 mm (2 in.); 200 mm (8 in.) with scan stitching capability) was used to measure the film thickness of the active layer.
2.3 Grazing-incidence wide-angle X-ray scattering (GIWAXS) measurement
GIWAXS was measured at the Stanford Synchrotron Radiation Lightsource (SSRL) beamline 11-3 in a helium-filled chamber with an X-ray wavelength of 0.9752 Å and sample-to-detector distance of 25 cm at an incident angle of 0.12°. For the in situ thermal measurement, a GIWAXS image was taken initially at room temperature before any thermal treatment. The samples were then heated to the annealing temperature (50, 100, 150, 200 and 225 °C) and allowed to equilibrate at each temperature for 20 min before the data collection (at these elevated temperatures). The spectra were recorded on a 2D X-ray detector (MX225, Rayonix, L.L.C.) with a pixel size of 73 μm (3072 × 3072 pixels) and analyzed using the Nika and WAXStools package for Igor software (WaveMetrics, Inc.).15,22 In this work, the polar angle (χ) is defined as the angle of the observed diffraction from the normal to the substrate, with χ = 0° defined as perpendicular to the substrate and χ = 90° parallel to the substrate. The out-of-plane sector is considered at 0–20° and an in-plane sector is 70–90°.3. Results & discussion
Fig. 1(a) and (b) display the chemical structure of PIPCP and PC61BM. PIPCP contains a backbone comprised of IDT-PT-CPDT-PT repeat subunits where IDT = indacenodithiophene, PT = pyridyl[2,1,3]thiadiazole and CPDT = cyclopentadithiophene.3.1 GIWAXS of PIPCP:PC61BM blend films
GIWAXS was employed to quantify the mixed phase in BHJ films by tracking the PC61BM scattering intensity as outlined above. Fig. 2 shows the GIWAXS pattern of PIPCP:PC61BM BHJ blends incorporating 0, 10, 30, 50, 70 and 100 wt% of PC61BM. The complete GIWAXS patterns incorporating the different PC61BM ratios are provided in Fig. s1.† The pristine PIPCP film scattering pattern has peaks from lamellar stacking at q ≈ 0.4 Å−1 (≈17 Å d-spacing) and from π–π stacking at q = 1.43 Å−1 (4.4 Å d-spacing),16 with a rather broad polar angle distribution. The scattering from π–π stacking appears generally isotropic with slight preference in the out-of-plane direction which shows some preferred face-on orientation with respect to the Si substrate. As the ratio of PC61BM increases to 30 and 50 wt%, the PIPCP lamellar stacking becomes more angularly isotropic and the π–π stacking begins to overlap with the scattering of PC61BM. Further increasing PC61BM to more than 70 wt%, the scattering from PC61BM were observed at q = 0.68 and 1.35 Å−1 and the scattering peaks attributed to PIPCP become weaker.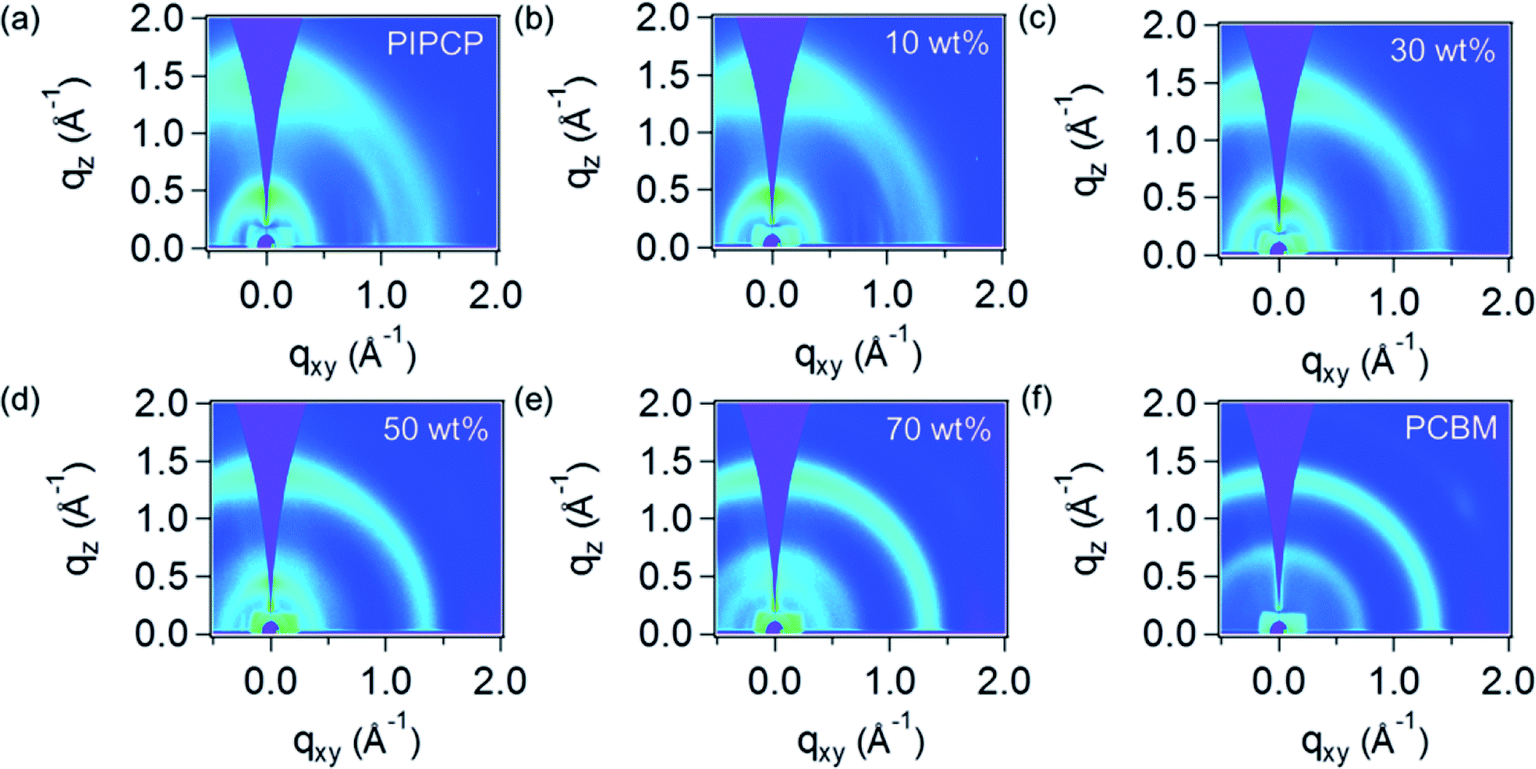 | ||
| Fig. 2 The GIWAXS pattern of PIPCP:PC61BM BHJ blends with various blend ratio. The labeled weight percentage corresponds to the content of PC61BM in the active layer: (a) 0% (or neat PIPCP), (b) 10%, (c) 30%, (d) 50%, (e) 70%, and (f) 100% (or neat PC61BM). | ||
By studying the PC61BM intensity as a function of PC61BM concentration, the behavior of PC61BM (aggregated vs. molecularly mixed with PIPCP) in the BHJ can be determined, similar to the method reported by Bartelt et al.12,14,15 The intensity vs. scattering vector profiles (integrated over all polar angles) of PIPCP:PC61BM blends are summarized in Fig. 3(a). These profiles are normalized at q = 1.0 Å−1, since scattering at this location is mostly due to diffuse background. The dependence of the PC61BM intensity at q = 0.68 Å−1 with concentration in this normalized plot shows that PC61BM contributes to the scattering even with as little as 10% PC61BM content. Fig. 3(b) shows a plot of the intensity at q = 0.68 Å−1 vs. PC61BM fraction and displays a linear dependence with no onset. The PC61BM scattering at q = 1.35 Å−1 displays a similar tendency as shown in Fig. s2.† These observations show that PC61BM is aggregated even at low weight fractions and little PC61BM exists in an intimately mixed phase (clusters less than about 2 nm) for the PIPCP:PC61BM BHJ system. The low FF for PIPCP:PC61BM BHJs may then be due to poorly connected donor or acceptor domains and/or shallow traps that lead to severe charge recombination.
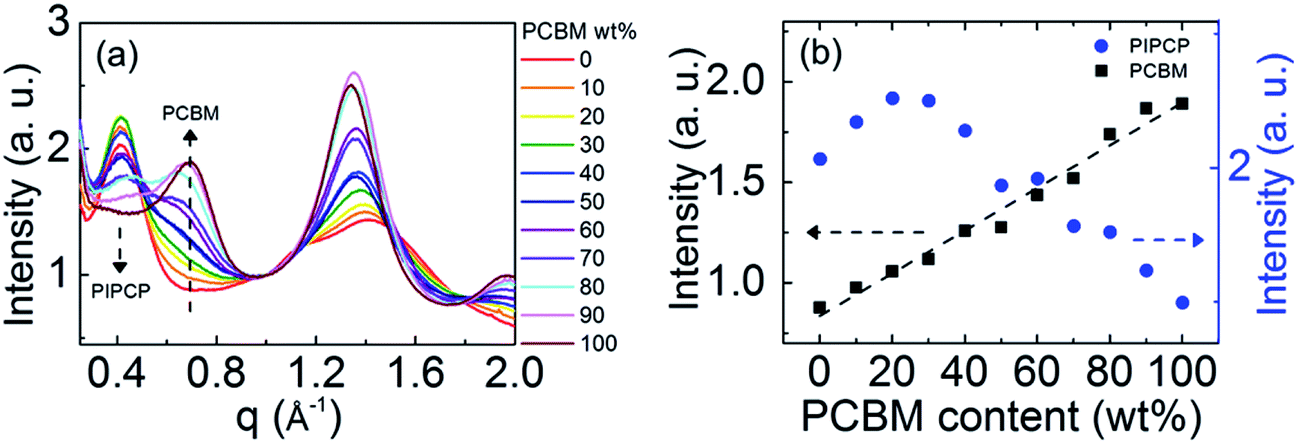 | ||
| Fig. 3 (a) Polar angle integrated intensity from GIWAXS pattern of PIPCP:PC61BM blends varying from 0 to 100 wt% of PC61BM. The data are normalized at q = 1.0 Å−1. (b) The intensity of PIPCP (q ≈ 0.4 Å−1) and PC61BM (q = 0.68 Å−1) scattering with different PC61BM content. The dashed line is a linear fit to the data. | ||
On the other hand, the scattering intensity of PIPCP at q ≈ 0.4 Å−1 increases as the PC61BM content increases (PIPCP fraction decreases) to 20%, implying that the extent of ordering of PIPCP crystal is enhanced by the introduction of PC61BM, then decreases with higher PC61BM content (>30%) in the film. This suggests that the ordering of PIPCP is improved in the presence of PC61BM in BHJs, consistent with TEM of PIPCP:PC61BM blends showing crystalline regions.18
Careful inspection of the GIWAXS for small q shows a scattering peak along the in plane direction at qxy = 0.25 Å−1 as can be seen by the enlarged GIWAXS pattern in Fig. 4(a). (Note that the asymmetry of the X-ray window only allows observation of the left side of the image.) This corresponds to a d-spacing of ≈25 Å, close to that estimated for a monomer unit. This, together with the in-plane (qxy) orientation of this peak, shows that the 0.25 Å−1 peak results from order along the PIPCP backbone. To gain more insight into the PIPCP morphological changes upon blending, we plot the GIWAXS in-plane line profiles of the left side of the images in Fig. 4(b). This shows how the PIPCP scattering varies on blending and remarkably illustrates that the backbone reflection at q = 0.25 Å−1 significantly increases with increasing PC61BM content. In addition, the lamellar stacking diffraction shifts to slightly higher q values with increasing PC61BM content, indicating a decrease in lamella d-spacing. To extract quantitative information of the lamella and backbone scattering, the line cuts in Fig. 4(b) are fit to a small background term (accounting for scattering from the substrate and sample cell) and three peaks due to the 0.25 Å−1 peak, the lamella peak and the PC61BM scattering, as shown in Fig. s3.†
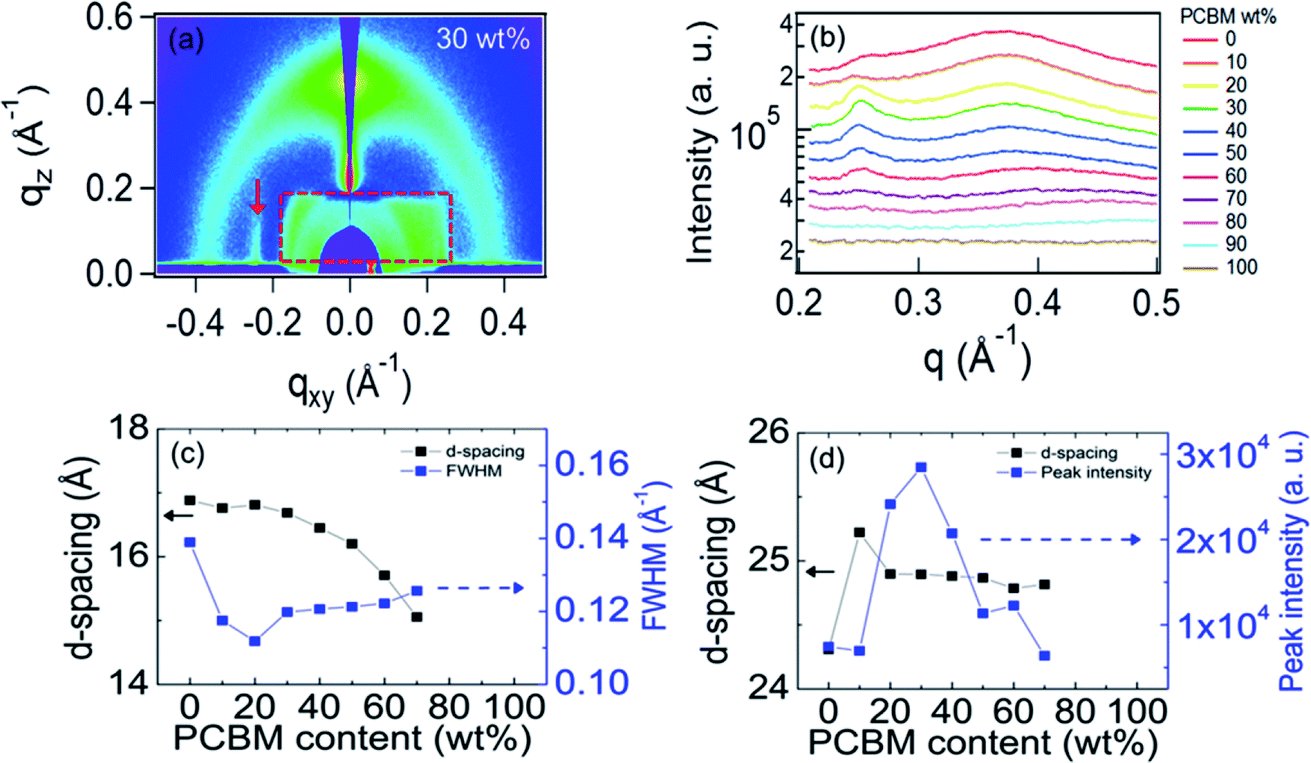 | ||
| Fig. 4 (a) The enlarged GIWAXS pattern of PIPCP:PC61BM BHJ blends incorporating 30 wt% PC61BM. Red rectangle (dashed line) is the X-ray window and arrow points to the backbone peak at 0.25 Å−1. (b) The in-plane line profiles from the GIWAXS pattern for different PIPCP:PC61BM blend ratio. (c) The lamellae d-spacing and FWHM at q ≈ 0.40 Å−1 with different PC61BM content. (d) The d-spacing along the polymer backbone and peak intensity at q = 0.25 Å−1 with different PC61BM content. (c and d) are taken from peak fits; peaks were not resolvable for PC61BM contents larger than 70%. | ||
Fig. 4(c) plots the PIPCP lamellar d-spacing and the peak full-width at half maximum (FWHM) upon increasing the PC61BM fraction. The d-spacing (16.8 Å) of PIPCP lamellar stacking remains constant for samples with PC61BM content up to 20 wt% but starts to decrease after incorporating ≥30 wt% of PC61BM. The FWHM of the lamellar peak here provides insight into the regularity of the lamellar packing. FWHM decreases slightly on initial blending, showing that the lamellar stacking becomes somewhat more ordered. Above about 30% PC61BM, the lamellae FWHM increases slightly with increasing PC61BM content. Importantly, as shown in Fig. 4(d), the backbone scattering peak intensity of PIPCP at 0.25 Å−1 increases significantly (more than 3×) upon increasing the PC61BM fraction from 0 to 30%, showing that the order along the PIPCP backbone is markedly increased. The similar tendency can be observed when the scattering intensity is normalized by the corresponding content of PIPCP (Fig. s4†). The improved backbone order of PIPCP that we observe is consistent with an increased effective conjugation length of polymer after blending of PC61BM and the observed red shift in the blend absorption.
3.2 Temperature dependence of PIPCP:PC61BM blend films
We used in situ GIWAXS measurements to monitor the thin film morphology evolution with thermal annealing, in order to understand any thermally induced changes and to verify our hypothesis that the q = 0.25 Å−1 peak is due to the ordering along the polymer backbone. We note that Wang et al. previously reported that no thermal transitions were observed for the neat PIPCP polymer up to a temperature of 280 °C.16 The PIPCP:PC61BM film incorporating 30 wt% PC61BM shows the highest intensity in PIPCP scattering peaks, and therefore, this blend ratio was selected for our annealing study. Fig. 5(a) and (b) display the GIWAXS pattern of the BHJ film with 30 wt% of PC61BM held at the annealing temperature of 200 and 225 °C. The remaining GIWAXS patterns with varying annealing temperature are provided in Fig. s5.† The GIWAXS of unannealed PIPCP:PC61BM film featured the backbone stacking, lamellar stacking, and π–π stacking peaks. In Fig. s5(a)–(d),† this pattern persisted with annealing to 150 °C. Further increasing the annealing temperature to 200 and 225 °C [Fig. 5(a) and (b), respectively] caused the lamellar stacking to shift to lower q values. The integrated in-plane line profiles with varying annealing temperature are shown in Fig. 5(c). The backbone scattering peak at about 0.25 Å−1 is unaffected with increasing temperature, corresponding to a nearly constant d-spacing of 25 Å (Fig. 5(d)). This behavior is expected, since the strong bonds within the backbone result in weak thermal expansion. This observation supports our assignment of this peak to backbone periodicity.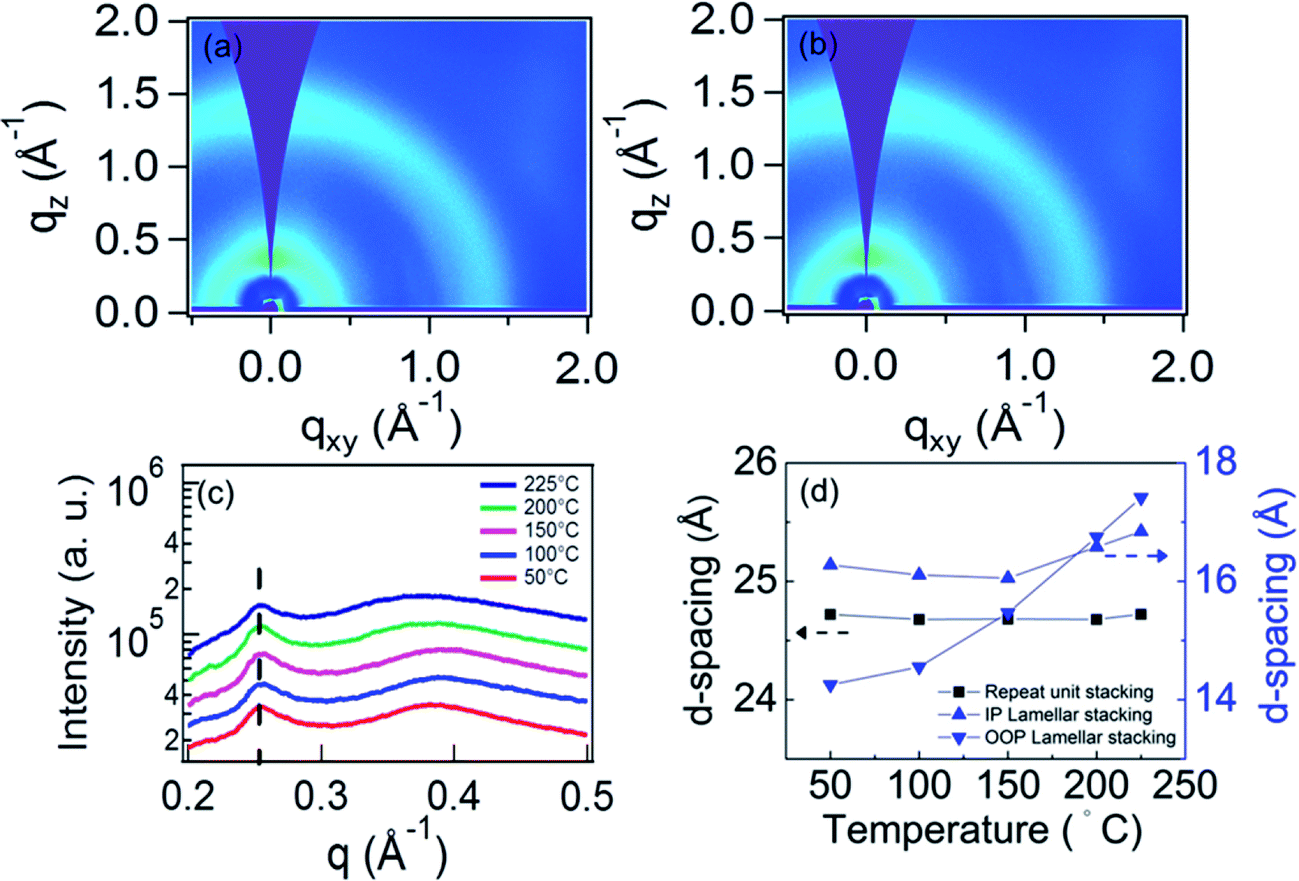 | ||
| Fig. 5 The GIWAXS pattern of PIPCP:PC61BM BHJ film incorporating 30 wt% of PC61BM with holding the annealing temperature at (a) 200 and (b) 225 °C. (c) The in-plane line profiles integrated from the GIWAXS pattern with holding the annealing temperature at 50, 100, 150, 200 and 225 °C. (d) The d-spacing of lamellar and repeat unit stacking with varying the annealing temperature. | ||
Fig. 5(d) also displays the d-spacing of the in-plane (IP) and out-of-plane (OOP) components of lamellar stacking with varying annealing temperature. Refraction of the X-rays can slightly increase the observed q position of scattering peaks in the out-of-plane direction.23 To account for refraction, we calculated the peak shift following the methodology in the literature,23 and found that refraction will result in a shift of 0.007 Å−1. Thus, we subtracted this from our results before calculating the d-spacing. When the annealing temperature is below 150 °C, the IP d-spacing is nearly constant but then slowly increases up to 225 °C. In contrast, the d-spacing of OOP lamellar stacking is smaller than for the IP and increases slightly non-linearly, with temperature. These observations suggest that the BHJ film is laterally constrained by the substrate below about 225 °C, but becomes unconstrained above this temperature. From the lamellar d-spacing in Fig. 5(d) we can estimate the linear coefficient of thermal expansion (CTE).24 The slightly nonlinear behavior in Fig. 5(d) complicates this calculation and hence we simply estimate the CTE of lamellar stacking by a line through the OOP d-spacing data and obtain (10 ± 5) × 10−4 °C−1. This is close, but somewhat larger than, the CTE of the P3HT lamella spacing (∼4.8 × 10−4 °C−1) and the CTE of the PTB7 π–π stacking (∼3.5 × 10−4 °C−1).24,25
The presence of a molecular mixed phase in the BHJ films has been reported in several polymer:fullerene systems. The mixed phase is observed for P3HT:PC61BM,7 poly(2,5-bis(3-tetradecylthiophen-2-yl)thieno[3,2-b]thiophene) (pBTTT):PC71BM,26 poly[[2,6′-4,8-di(5-ethylhexylthienyl)benzo[1,2-b;3,3-b]dithiophene]3-fluoro-2[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl] (PTB7-Th):PC71BM,27 and PBDTTPD:PC61BM.12 In contrast, the absence of the mixed phase is observed for poly[(5,6-difluoro-2,1,3-benzothiadiazol-4,7-diyl)-alt-(3,3′′′-di(2-octyldodecyl)-2,2′;5′,2′′;5′′,2′′′-quaterthiophen-5,5′′′-diyl)](PffBT4T-2OD or PCE11):PC61BM28 which is similar to our observation, as well as for some small molecule:fullerene OSCs.15 These results show that the mixed phase is varied in the reported systems, relying on the formation of preferable morphology that facilitates electron–hole separation. We also note that there are several methods to improve the order of polymers in BHJs such as solvent additives29 and thermal treatment.30,31 In the case of PIPCP, the ordering is improved by incorporating PC61BM. We speculate that the improved crystallinity of PIPCP:PC61BM blends is due to the interactions between the PIPCP branched sidechains and the PC61BM. Similar behavior was reported for a series of anthracene containing poly(p-arylene-ethynylene)-alt-poly(p-arylene-vinylene)s polymers, abbreviated AnE-PV. Here, the crystallinity of AnE-PVstat is enhanced after blending with PC61BM, and was ascribed interactions of the sidechains,32,33 consistent with recent work.34
4. Conclusion
We have investigated the blend morphology of PIPCP:PC61BM BHJs by varying the PC61BM composition within the BHJ. GIWAXS shows that PC61BM not only prefers to aggregate in the film, resulting in no mixed phase in the BHJ, but remarkably improves the order of the PIPCP polymer along the backbone, consistent with an increased conjugation length of the polymer. This behavior is unusual since most often, blending conjugated polymers with PCBM decreases the order in the polymer. We speculate that the improved backbone order (or increased conjugation length) due to PC61BM incorporation into the BHJ results to the low energy loss (Eloss) of the OSC device.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Work was supported by the Department of the Navy, Office of Naval Research Award No. N00014-14-1-0580, N00014-16-1-2520 and N00014-16-1-2678. Use of Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-76SF00515. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152. Tzu-Yen Huang acknowledges the support of Ministry of Science and Technology (MOST), Taiwan though the Overseas Project for Postdoctoral Research Abroad Program (PRAP) (105-2917-I-564-044). Victoria Savikhin acknowledges the support of the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship (NDSEG) program.References
- T. R. Andersen, H. F. Dam, M. Hosel, M. Helgesen, J. E. Carle, T. T. Larsen-Olsen, S. A. Gevorgyan, J. W. Andreasen, J. Adams, N. Li, F. Machui, G. D. Spyropoulos, T. Ameri, N. Lemaitre, M. Legros, A. Scheel, D. Gaiser, K. Kreul, S. Berny, O. R. Lozman, S. Nordman, M. Valimaki, M. Vilkman, R. R. Sondergaard, M. Jorgensen, C. J. Brabec and F. C. Krebs, Energy Environ. Sci., 2014, 7, 2925–2933 RSC.
- M. Kaltenbrunner, M. S. White, E. D. Głowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 CrossRef PubMed.
- L. H. Rossander, H. F. Dam, J. E. Carle, M. Helgesen, I. Rajkovic, M. Corazza, F. C. Krebs and J. W. Andreasen, Energy Environ. Sci., 2017, 10, 2411–2419 RSC.
- C. Lungenschmied, G. Dennler, H. Neugebauer, S. N. Sariciftci, M. Glatthaar, T. Meyer and A. Meyer, Sol. Energy Mater. Sol. Cells, 2007, 91, 379–384 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361(6407), 1094–1098 CrossRef CAS PubMed.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1800613 CrossRef PubMed.
- P. Westacott, J. R. Tumbleston, S. Shoaee, S. Fearn, J. H. Bannock, J. B. Gilchrist, S. Heutz, J. deMello, M. Heeney, H. Ade, J. Durrant, D. S. McPhail and N. Stingelin, Energy Environ. Sci., 2013, 6, 2756–2764 RSC.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- N. C. Miller, E. Cho, R. Gysel, C. Risko, V. Coropceanu, C. E. Miller, S. Sweetnam, A. Sellinger, M. Heeney, I. McCulloch, J.-L. Brédas, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2012, 2, 1208–1217 CrossRef CAS.
- B. A. Collins, J. R. Tumbleston and H. Ade, J. Phys. Chem. Lett., 2011, 2, 3135–3145 CrossRef CAS.
- W. Chen, T. Xu, F. He, W. Wang, C. Wang, J. Strzalka, Y. Liu, J. Wen, D. J. Miller, J. Chen, K. Hong, L. Yu and S. B. Darling, Nano Lett., 2011, 11, 3707–3713 CrossRef CAS PubMed.
- J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Fréchet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364–374 CrossRef CAS.
- C. Zhong, J. A. Bartelt, M. D. McGehee, D. Cao, F. Huang, Y. Cao and A. J. Heeger, J. Phys. Chem. C, 2015, 119, 26889–26894 CrossRef CAS.
- S. D. Oosterhout, V. Savikhin, M. A. Burgers, J. Zhang, Y. Zhang, S. R. Marder, G. C. Bazan and M. F. Toney, J. Phys. Chem. C, 2018, 122, 11136–11144 CrossRef CAS.
- S. D. Oosterhout, V. Savikhin, J. Zhang, Y. Zhang, M. A. Burgers, S. R. Marder, G. C. Bazan and M. F. Toney, Chem. Mater., 2017, 29, 3062–3069 CrossRef CAS.
- M. Wang, H. Wang, T. Yokoyama, X. Liu, Y. Huang, Y. Zhang, T.-Q. Nguyen, S. Aramaki and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 12576–12579 CrossRef CAS PubMed.
- N. A. Ran, J. A. Love, M. C. Heiber, X. Jiao, M. P. Hughes, A. Karki, M. Wang, V. V. Brus, H. Wang, D. Neher, H. Ade, G. C. Bazan and T.-Q. Nguyen, Adv. Energy Mater., 2018, 8, 1701073 CrossRef.
- N. A. Ran, J. A. Love, C. J. Takacs, A. Sadhanala, J. K. Beavers, S. D. Collins, Y. Huang, M. Wang, R. H. Friend, G. C. Bazan and T.-Q. Nguyen, Adv. Mater., 2016, 28, 1482–1488 CrossRef CAS PubMed.
- M. Wang, H. Wang, M. Ford, J. Yuan, C.-K. Mai, S. Fronk and G. C. Bazan, J. Mater. Chem. A, 2016, 4, 15232–15239 RSC.
- J. Yuan, M. J. Ford, W. Ma and G. C. Bazan, J. Mater. Chem. A, 2017, 5, 8903–8908 RSC.
- J. Yuan, N. A. Ran, M. J. Ford, M. Wang, M. K. Ravva, C.-K. Mai, X. Liu, J.-L. Brédas, T.-Q. Nguyen, W. Ma and G. C. Bazan, J. Mater. Chem. A, 2017, 5, 18618–18626 RSC.
- J. Ilavsky, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef CAS.
- M. F. Toney and S. Brennan, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 7963–7966 CrossRef.
- E. Verploegen, R. Mondal, C. J. Bettinger, S. Sok, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2010, 20, 3519–3529 CrossRef CAS.
- V. Savikhin, L. K. Jagadamma, L. J. Purvis, I. Robertson, S. D. Oosterhout, C. J. Douglas, I. D. W. Samuel and M. F. Toney, iScience, 2018, 2, 182–192 CrossRef CAS PubMed.
- A. C. Mayer, M. F. Toney, S. R. Scully, J. Rivnay, C. J. Brabec, M. Scharber, M. Koppe, M. Heeney, I. McCulloch and M. D. McGehee, Adv. Funct. Mater., 2009, 19, 1173–1179 CrossRef CAS.
- W. Huang, N. Chandrasekaran, S. K. K. Prasad, E. Gann, L. Thomsen, D. Kabra, J. M. Hodgkiss, Y.-B. Cheng and C. R. McNeill, ACS Appl. Mater. Interfaces, 2016, 8, 29608–29618 CrossRef CAS PubMed.
- N. Li, J. D. Perea, T. Kassar, M. Richter, T. Heumueller, G. J. Matt, Y. Hou, N. S. Güldal, H. Chen, S. Chen, S. Langner, M. Berlinghof, T. Unruh and C. J. Brabec, Nat. Commun., 2017, 8, 14541 CrossRef CAS PubMed.
- G. Jing, C. Wei, D. Letian, C. Chun-Chao, C. Wei-Hsuan, L. Yongsheng, L. Gang and Y. Yang, Adv. Mater., 2014, 26, 3142–3147 CrossRef PubMed.
- R. Heuvel, F. J. M. Colberts, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. C, 2018, 6, 3731–3742 RSC.
- T. Erb, U. Zhokhavets, G. Gobsch, S. Raleva, B. Stühn, P. Schilinsky, C. Waldauf and C. J. Brabec, Adv. Funct. Mater., 2005, 15, 1193–1196 CrossRef CAS.
- S. Rathgeber, J. Perlich, F. Kühnlenz, S. Türk, D. A. M. Egbe, H. Hoppe and R. Gehrke, Polymer, 2011, 52, 3819–3826 CrossRef CAS.
- C. Kästner, D. A. M. Egbe and H. Hoppe, J. Mater. Chem. A, 2015, 3, 395–403 RSC.
- V. Savikhin, M. Babics, M. Neophytou, S. Liu, S. D. Oosterhout, H. Yan, X. Gu, P. M. Beaujuge and M. F. Toney, Chem. Mater., 2018, 30, 7872–7884 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10488c |
| This journal is © The Royal Society of Chemistry 2019 |