
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of 2,2-dimethylbenzopyran derivatives as potent neuroprotection agents†
Fangyu
Du
a,
Qifan
Zhou
a,
Xiaoxiao
Fu
b,
Yajie
Shi
a,
Yuanguang
Chen
a,
Wuhong
Fang
a,
Jingyu
Yang
*b and
Guoliang
Chen
 *a
*a
aKey Laboratory of Structure-Based Drug Design & Discovery of Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, P. R. China. E-mail: spucgl@163.com
bDepartment of Pharmacology, Shenyang Pharmaceutical University, Shenyang 110016, P. R. China. E-mail: yangjingyu2006@gmail.com
First published on 21st January 2019
Abstract
The development of novel neuroprotection agents is of great significance for the treatment of ischemic stroke. In this study, a series of compounds comprising 2,2-dimethylbenzopyran groups and cinnamic acid groups have been synthesized. Preferential combination principles and bioisostere that improved the neuroprotective effect of the compounds were identified for this series via biological activity assay in vitro. Meanwhile, a functional reversal group of the acrylamide amide resulted in the most active compounds. Among them, BN-07 significantly improved the morphology of neurons and obviously increased cell survival rate of primary neurons induced by oxygen glucose deprivation (OGD), superior to clinically used anti-ischemic stroke drug edaravone (Eda). Overall, our findings may provide an alternative strategy for the design of novel anti-ischemic stroke agents with more potency than Eda.
1 Introduction
Ischemic stroke is one of most common cerebrovascular diseases that seriously endangers human health and features high incidence, high recurrence, high disability and high fatality.1,2 Ischemic stroke causes extensive cellular loss that impairs brain functions, resulting in severe disabilities. Despite being a major public health concern, effective disease management strategies for the treatment of ischemic stroke are limited. The need to develop effective therapeutic approaches for treating stroke is compelling. Currently, anti-ischemic stroke drugs that are in clinical trials or launched mainly include thrombolytic drugs, antiplatelet aggregation drugs, neuroprotective drugs, and drugs promoting angiogenetic effects.3,4Among multiple treatment strategies implicated in the pathogenesis of ischemia, neuroprotection is the most attractive protocol. Clinical drugs mainly play a role in different pathophysiological stress processes that mediate brain tissue damage after ischemic stroke, including vascular dilation and increasing cerebral blood flow,5,6 antioxidative stress,7 anti-apoptosis,8 inhibiting excitotoxicity9 and inhibiting inflammation.10
Ischemia causes a shortage of glucose and oxygen and thus a consumption of ATP, which results in the failure of the sodium potassium pump and leads to membrane depolarization. Then, the membrane depolarization conversely induces multiple mechanisms, involving increasing intracellular calcium, triggering cell death signalling pathways such as apoptosis, autophagy, and necroptosis.11 The increase of intracellular calcium triggers the production of reactive oxygen species (ROS), which initiates cell death cascades and activates inflammatory responses, consequently resulting in ischemic neuronal death.12 Therefore, increased oxidative stress has been considered as the primary cause. Although ROS can be cleaned by a defense system in normal tissues, superabundant ROS can result in neuronal damage or death under an ischemic condition because the antioxidant defense system is interrupted.13–16 Therefore, ROS scavengers may represent potential candidates for the treatment of brain ischemia injury. Many approaches have been indicated to reduce the generation of ROS and to block the ROS-mediated neuronal cell death, involving utilization of antioxidants or inhibitors of cell death signalling molecules.17–19
Zang et al. discovered that pyrano[3,2-a]carbazole alkaloid 1 was an effective agent against ischemic stroke (Fig. 1) exhibited potent activity to scavenge free radical.20 Meanwhile, a previous study reported that a kind of benzopyran derivatives 2 (Fig. 1) could protect against ischemia on the stroke model of mice, the symptoms and physiological functions were obviously improved after administration.21 Edaravone (5-methyl-2-phenyl-1,2-dihydropyrazol-3-one, Fig. 1), a free radical scavenger, has been approved by the PMDA of Japan as a new drug mainly for the treatment of ischemic stroke since 2001.22 In a multicenter, randomized, placebo-controlled, double-blind study on acute ischemic stroke patients, edaravone represents a neuroprotective agent which is potentially useful for the treatment of acute ischemic stroke, because it can exert significant effects on outcome as compared with placebo.23
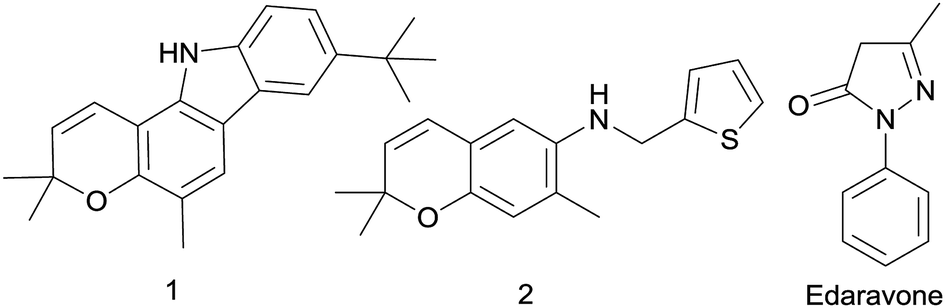 | ||
| Fig. 1 Structures of 2,2-dimethylbenzopyran derivatives and edaravone. | ||
Previous studies have demonstrated that 2,2-dimethylbenzopyran derivatives and cinnamic acid analogues are important natural active compounds with broad pharmacological activities, such as antibacterial, anti-tumor and neuroprotection.24–28 Luo and Agon et al. disclosed that cinnamic acid analogues such as caffeic acid, ferulic acid, cinnamic acid could enhance the activity of lactate dehydrogenase (LDH), superoxide dismutase (SOD) and lower the malondialdehyde (MDA) contents, which were used to protect cardiovascular system.29,30 Inspired by the neuroprotective activity and the outstanding structure of 2,2-dimethylbenzopyran derivatives and cinnamic acid analogues, we therefore designed and synthesized a novel of benzopyran compounds using molecular hybridization approach, affording compounds BA-01–BA-07. Meanwhile, we replaced benzene ring with following aromatic cycles, including pyridine, thiophene, furan and naphthalene according to bioisostere, giving compounds BA-08–BA-11. Meanwhile, functional group reversal was induced in many cases to obtain compounds BN-01–BN-05 and further modifications were carried out to obtain compounds BN-06–BN-10. Excitingly, experimental results of oxygen glucose deprivation (OGD) model displayed that some novel compounds exhibited better activity of neuroprotection than edaravone.
2 Results and discussion
2.1 Chemistry
The synthetic routes for the designed compounds are shown in Schemes 1 and 2. In general, 3-chloro-3-methylbut-1-yne (3) was easily prepared from commercially available 2-methylbut-3-yn-2-ol (2). (E)-3-Argioacrylic acid derivatives (Scheme 1) were either purchased or synthesized in one step from commercially available aromatic aldehyde (4a–4k) condensed with malonic acid via Knoevenagel condensation to give 5a–5k. Commercially available 4-acetamidophenol (6) reacted with 3-chloro-3-methylbut-1-yne (3) by Williamson ether synthesis in the presence of CuCl to give 7. The subsequent high temperature facilitated [3,3]-σ rearrangement of 7 afforded N-(2,2-dimethyl-2H-chromen-6-yl)acetamide (8) using diphenyl ether as a solvent. Treatment of 8 with 50% NaOH aqueous deprotected the acetyl group from the amine moiety, and heating in the presence of ethanol, and then generated the key intermediate 2,2-dimethyl-2H-chromen-6-amine (9). The synthesis of various substituted 2,2-dimethylbenzopyran derivatives BA-01–BA-11 (Scheme 1) was accomplished using a common intermediate 9 which coupled to the cinnamic acid analogues 5a–5k under standard amide coupling conditions (EDCI, HOBt, THF) as shown in Scheme 1.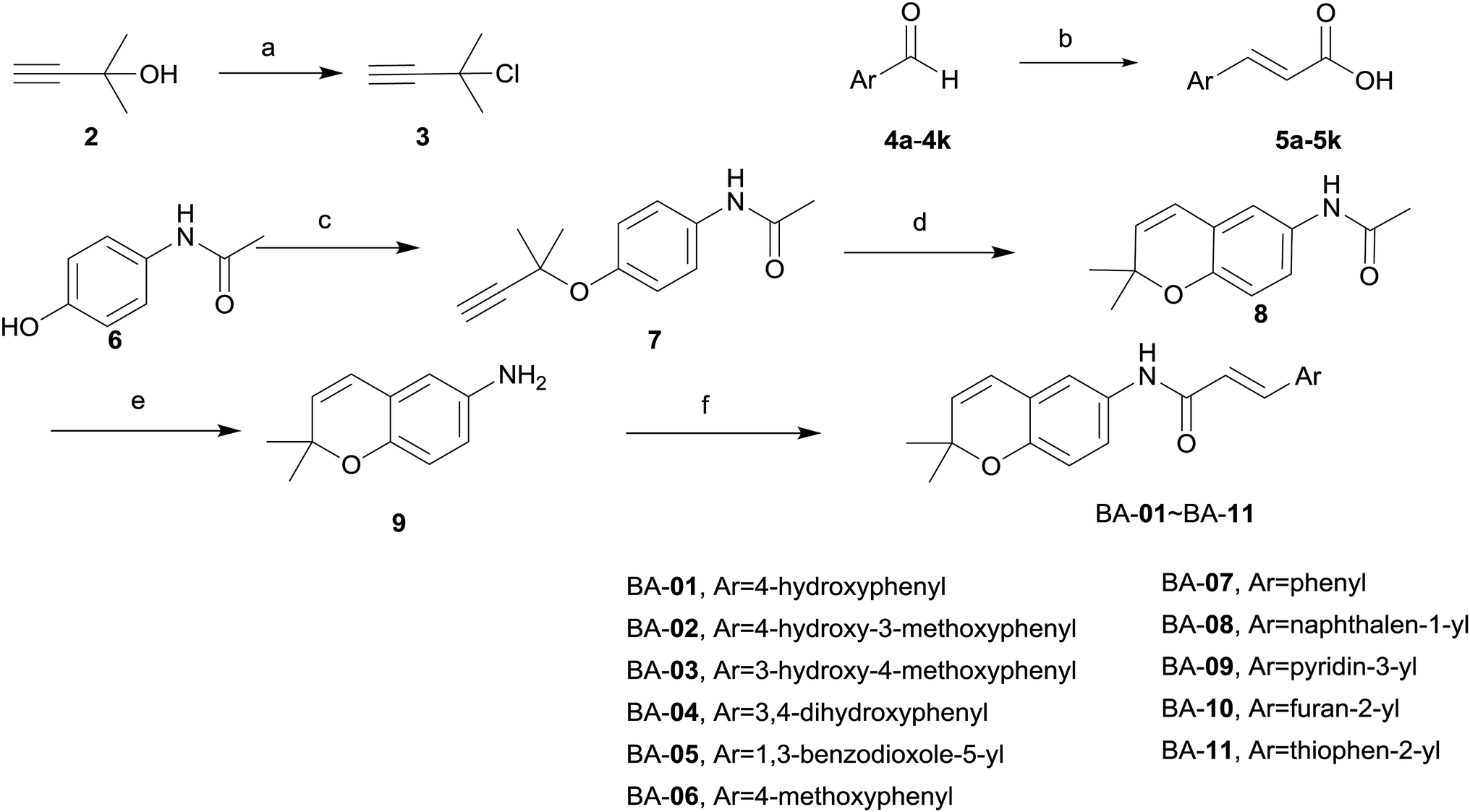 | ||
| Scheme 1 Synthesis of BA compounds. Reaction and conditions: (a) CuCl, CaCl2, conc. HCl, r.t., 1 h; (b) CH2(COOH)2, pyridine, piperidine, toluene, reflux, 12 h; (c) K2CO3, KI, PEG 600, acetone, reflux, 72 h; (d) diphenyl ether, reflux, 2 h; (e) 50% NaOH, ethanol, reflux, 12 h; (f) EDCI, HOBt, THF, r.t., 14 h. | ||
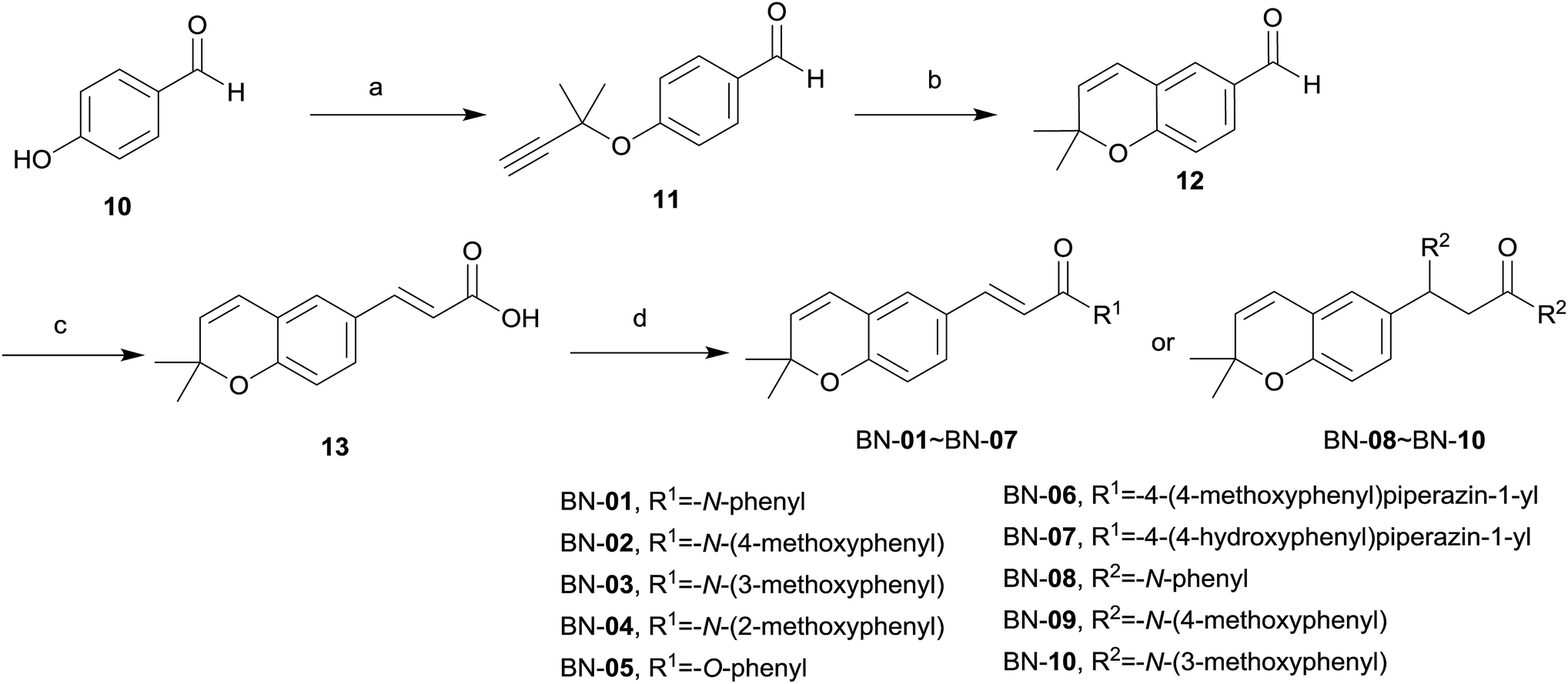 | ||
| Scheme 2 Synthesis of BN compounds. Reaction and conditions: (a) DBU, CuCl, acetonitrile, r.t., 4 h; (b) PhNMe2, reflux, 2 h; (c) CH2(COOH)2, pyridine, piperidine, toluene, reflux, 12 h; (d) HX-Ar, EDCI, HOBt, THF, r.t., 14 h. | ||
The synthesis of BN-01–BN-10 (Scheme 2) began with the use of Williamson ether synthesis to transform the phenolic hydroxyl group present in 4-hydroxybenzaldehyde 10 to ether to provide 11. The alkyne group in 11 was then rearranged by treatment with high temperature in N,N-dimethylaniline to afford the corresponding aldehyde 12. This entity was condensed with malonic acid to afford the key intermediate carboxylic acid 13via Knoevenagel condensation. Target compounds BN-01–BN-10 were made in a similar fashion of amide coupling starting from commercially available substituted aniline and substituted phenol. Interestingly, addition reaction was occurred simultaneously at the position of α,β-unsaturated double bond in the process of synthesis for compounds BN-01–BN-03, resulting in three novel compounds BN-08–BN-10.
2.2 In vitro biological activity assay
To explore the therapeutic potential of BA-01–BA-10 and BN-01–BN-11, in this study, we applied an in vitro oxygen glucose deprivation (OGD) model of primary cortical neurons cultures to investigate whether the series of compounds protected neurons against ischemic stroke.β-Tubulin is the skeletal protein of neuron which can represent location and shape of neuron. As shown in Fig. 2, neurons were evenly distributed in the field of view and only a small amount of nucleus didn't show the maker of green fluorescence (Fig. 2). β-tubulin III and DAPI double fluorescent staining showed that the isolated neurons were successfully cultured in vitro with high purity (>90%), which can be used for the subsequent experiments.31
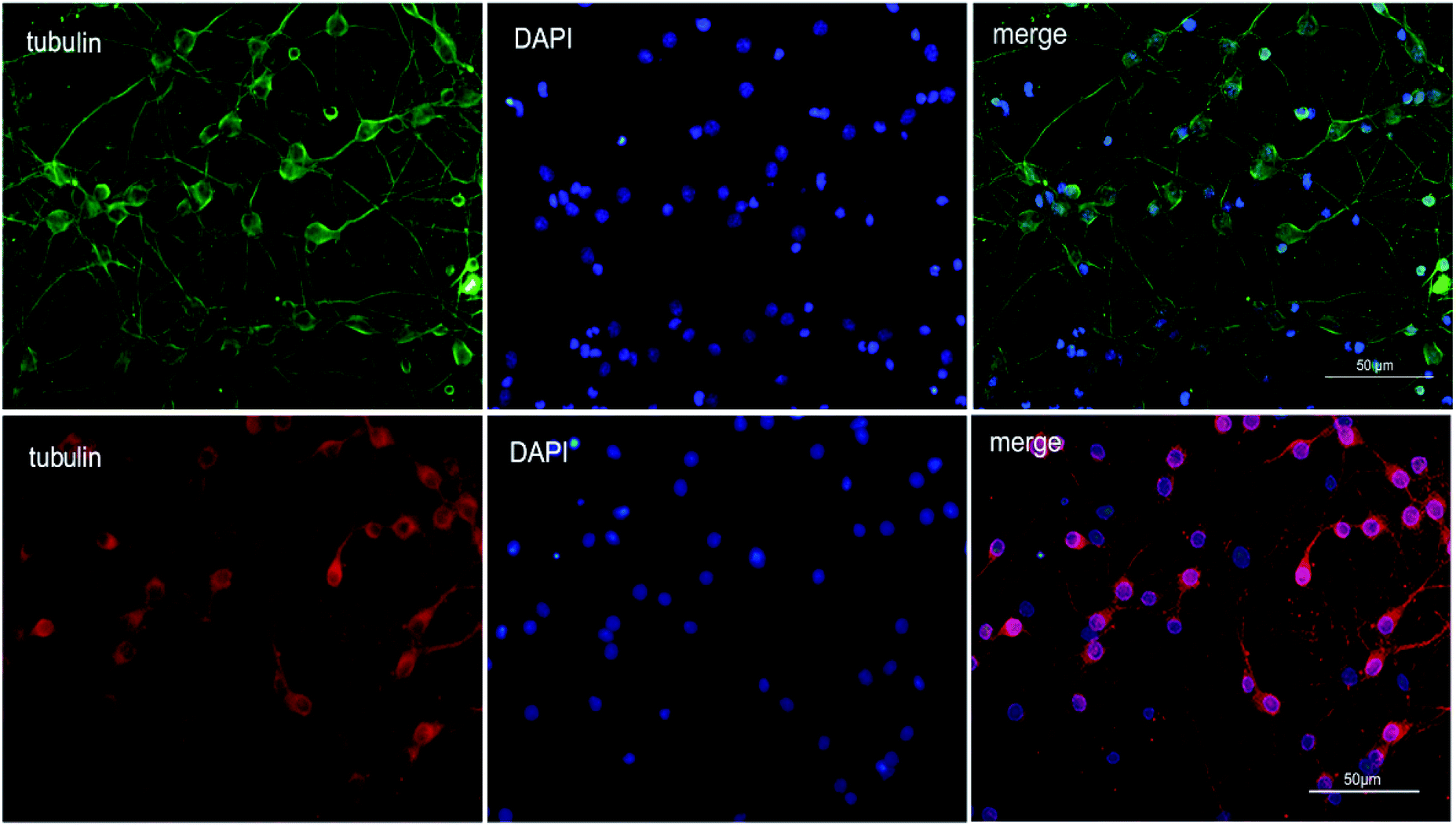 | ||
| Fig. 2 Purity identification of rat primary cortical neurons. | ||
| Sample | Control | 100 μM | Sample | Control | 100 μM |
|---|---|---|---|---|---|
| a Significance: *P < 0.05, **P < 0.01, ***P < 0.001 vs. control group. b The dose of administration was 100 μM and data were expressed as mean ± standard error (SE). | |||||
| BA-01 | 1.00 ± 0.015 | 0.75 ± 0.021*** | BN-01 | 1.00 ± 0.011 | 0.85 ± 0.038** |
| BA-02 | 1.00 ± 0.015 | 0.76 ± 0.025*** | BN-02 | 1.00 ± 0.011 | 0.93 ± 0.0080 |
| BA-03 | 1.00 ± 0.015 | 0.77 ± 0.012*** | BN-03 | 1.00 ± 0.011 | 0.89 ± 0.02 |
| BA-04 | 1.00 ± 0.015 | 0.56 ± 0.014*** | BN-04 | 1.00 ± 0.011 | 0.88 ± 0.038 |
| BA-05 | 1.00 ± 0.011 | 0.85 ± 0.037** | BN-05 | 1.00 ± 0.011 | 0.86 ± 0.023* |
| BA-06 | 1.00 ± 0.011 | 0.86 ± 0.018* | BN-06 | 1.00 ± 0.011 | 0.91 ± 0.028 |
| BA-07 | 1.00 ± 0.011 | 0.78 ± 0.046*** | BN-07 | 1.00 ± 0.011 | 0.96 ± 0.0063 |
| BA-08 | 1.00 ± 0.011 | 0.73 ± 0.025*** | BN-08 | 1.00 ± 0.011 | 0.99 ± 0.019 |
| BA-09 | 1.00 ± 0.011 | 0.49 ± 0.047*** | BN-09 | 1.00 ± 0.011 | 0.80 ± 0.047*** |
| BA-10 | 1.00 ± 0.011 | 0.84 ± 0.026** | BN-10 | 1.00 ± 0.011 | 0.81 ± 0.058*** |
| BA-11 | 1.00 ± 0.011 | 0.89 ± 0.054 | |||
To further investigate the neuroprotective effects exerted by the interesting compounds, we next attempted to assess cell viability against OGD model after administration by MTT method using edaravone as a positive drug. Cultured cortical neuron of SD rats were randomly divided into control group, model group, positive control group, and test group (1 μM, 30 μM, 100 μM). As shown in Table 2, cell viability was significantly decreased (p < 0.001) after OGD treatment for 12 h compared with the control, which indicated that OGD model was succeed. For BA compounds, the results showed that compounds BA-(03, 05, 07, 08, 09, 11) could significantly improve the survival rate of neuron (OGD 12 h) at a dose of 1 μM (P < 0.05).
| Sample | Control | Model | Sample in 1 μM | Sample in 30 μM | Sample in 100 μM | Edaravoned |
|---|---|---|---|---|---|---|
| a Significance: ###P < 0.001 vs. control group. Significance: *P < 0.05, **P < 0.01, ***P < 0.001 vs. model group. b Data were expressed as mean ± standard error (SE). c At this concentration, the compound was cytotoxic. d The concentration was 100 μM. | ||||||
| BA-01 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.52 ± 0.023 | 0.28 ± 0.040*** | —c | 0.54 ± 0.0069* |
| BA-02 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.42 ± 0.024** | 0.30 ± 0.046*** | —c | 0.54 ± 0.0069* |
| BA-03 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.59 ± 0.038* | 0.31 ± 0.035*** | —c | 0.54 ± 0.0069* |
| BA-04 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.42 ± 0.054* | 0.23 ± 0.019*** | —c | 0.54 ± 0.0069* |
| BA-05 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.56 ± 0.027* | 0.40 ± 0.012*** | 0.29 ± 0.013*** | 0.54 ± 0.0069* |
| BA-06 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.50 ± 0.040 | 0.33 ± 0.032*** | 0.37 ± 0.068** | 0.54 ± 0.0069* |
| BA-07 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.53 ± 0.028* | 0.40 ± 0.074*** | —c | 0.54 ± 0.0069* |
| BA-08 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.57 ± 0.044* | 0.51 ± 0.064 | —c | 0.54 ± 0.0069* |
| BA-09 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.54 ± 0.028* | 0.29 ± 0.011*** | —c | 0.54 ± 0.0069* |
| BA-10 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.46 ± 0.025 | 0.25 ± 0.0017*** | 0.19 ± 0.0017*** | 0.54 ± 0.0069* |
| BA-11 | 1.00 ± 0.012 | 0.49 ± 0.094### | 0.56 ± 0.022* | 0.25 ± 0.021*** | 0.21 ± 0.0023*** | 0.54 ± 0.0069* |
Another dose of compounds could decrease the survival rate of neurons or reveal no significant improvement (Table 2, and ESI, Fig. S2†). According to the results, the ring of cinnamic acid analogues had a better activity when substituted with dialkoxy groups on the benzene ring. When the substituents were replaced with hydroxyl groups, the activity was decreased or disappeared, such as BA-01, BA-04. Interestingly, various aromatic cycles, including pyridine, thiophene, furan and naphthalene, showed different efficacy, and BA-09, replaced with naphthalene, exhibited a better activity which increased survival rate of neuron (ODG model) from 49% to 57%. BA-09, BA-11, respectively substituted with pyridine, thiophene, maintained the same biological activity. On the contrary, 3-(furan-2-yl)acrylamide (BA-10) lost neuroprotective effect. Considering that series compounds of BA didn't significantly improve the survival rate of neurons, and we, therefore, designed and synthesized a novel series of 2,2-dimethylbenzopyran derivatives using functional reversal group of α,β-unsaturated amide group to afford BN compounds.
To our excitement, the cell viability results suggested that series compounds of BN can obviously increase the survival rate of neurons (OGD 12 h) at dose of 1 μM, 30 μM and 100 μM which were much superior to positive drug of edaravone (56%), compared with model group. (P < 0.001) (Table 3, and ESI, Fig. S3†). Experimental data demonstrated that activity was greatly increased when aniline substituted with methoxy such as BN-02, 03, 04, and the survival rate of neurons were 87%, 87%, 83% respectively, compared with BN-01 (73%). Moreover, the meta and para substituted compounds were more potent than the ortho. Attempt to replace aniline with phenol proved successful which displayed 81% survival rate of neuron (BN-05). Inspired by neuroprotective effects of cinnamamide derivatives,32,33 we synthesized amide with N-substituted piperazine showing superior biological activity. Among them, BN-07, substituted with 4-(piperazin-1-yl)phenol, increased the survival rate of neuron from 41% to 98%. Correspondingly, BN-06, substituted with 1-(4-methoxyphenyl)piperazine, maintained the similar activity (75%), which indicated that hydrophile group of 1-phenylpiperazine was contributed to neuroprotective effect. Interestingly, three compounds without double bonds (BN-08, 09, 10) maintained good biological activity which increased the survival rates of neuron to more than 80%. The results demonstrated that the α,β-unsaturated double bond isn't essential for the activity. Based on the neuroprotective activities in vitro shown in Tables 2 and 3, it was observed that series of compounds BN revealed better pharmaceutical effects of neuroprotection which were suitable for further research of anti-ischemic stroke.
| Sample | Control | Model | Sample in 1 μM | Sample in 30 μM | Sample in 100 μM | Edaravoned |
|---|---|---|---|---|---|---|
| a Significance: ###P < 0.001 vs. control group. Significance: *P < 0.05, **P < 0.01, ***P < 0.001 vs. model group. b Data were expressed as mean ± standard error (SE). c At this concentration, the compound was cytotoxic. d The concentration was 100 μM. | ||||||
| BN-01 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.73 ± 0.067*** | 0.67 ± 0.044*** | 0.60 ± 0.012*** | 0.56 ± 0.015*** |
| BN-02 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.87 ± 0.013*** | 0.74 ± 0.012*** | 0.50 ± 0.025*** | 0.56 ± 0.015*** |
| BN-03 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.87 ± 0.0084*** | 0.77 ± 0.0016*** | 0.58 ± 0.0090*** | 0.56 ± 0.015*** |
| BN-04 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.83 ± 0.012*** | 0.73 ± 0.0066*** | 0.41 ± 0.064 | 0.56 ± 0.015*** |
| BN-05 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.81 ± 0.010*** | 0.73 ± 0.0091*** | 0.69 ± 0.017*** | 0.56 ± 0.015*** |
| BN-06 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.75 ± 0.014*** | 0.77 ± 0.018*** | 0.70 ± 0.035*** | 0.56 ± 0.015*** |
| BN-07 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.98 ± 0.029*** | 0.83 ± 0.013*** | 0.66 ± 0.032*** | 0.56 ± 0.015*** |
| BN-08 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.89 ± 0.014*** | 0.79 ± 0.017*** | 0.69 ± 0.015*** | 0.56 ± 0.015*** |
| BN-09 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.85 ± 0.015*** | 0.77 ± 0.010*** | —c | 0.56 ± 0.015*** |
| BN-10 | 1.00 ± 0.0051 | 0.41 ± 0.014### | 0.81 ± 0.0090*** | 0.77 ± 0.0035*** | —c | 0.56 ± 0.015*** |
 | ||
| Fig. 3 Morphology of OGD neurons after administration of BN-07. | ||
2.3 Prediction of ADMET properties
BBB penetrations of all compounds were predicted by Discovery Studio 3.0 (see ESI, Fig. S5† and Table 4). The ADMET plot figure displayed that most target compounds had potent permeability of blood brain barrier (BBB) and appropriate values of clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P which were conducive to the development of neuroprotective effect. Among them, BN compounds revealed higher permeability of blood brain barrier than BA compounds. Therefore, predicted data manifested that BN compounds have the potential to exhibit better activity of neuroprotective in vivo.
P properties
P which were conducive to the development of neuroprotective effect. Among them, BN compounds revealed higher permeability of blood brain barrier than BA compounds. Therefore, predicted data manifested that BN compounds have the potential to exhibit better activity of neuroprotective in vivo.
P properties
| Sample | BBBa | clogP |
Sample | BBBa | clogP |
|---|---|---|---|---|---|
| a BBB ≥ 0.7, very high permeability; 0 ≤ BBB < 0.7, high permeability; −0.52 < BBB < 0, medium permeability; BBB ≤ −0.52, low permeability. | |||||
| BA-01 | 0.020 | 3.627 | BN-01 | 0.424 | 3.869 |
| BA-02 | −0.126 | 3.611 | BN-02 | 0.278 | 3.853 |
| BA-03 | −0.126 | 3.611 | BN-03 | 0.278 | 3.853 |
| BA-04 | −0.384 | 3.385 | BN-04 | 0.278 | 3.853 |
| BA-05 | 0.070 | 3.638 | BN-05 | 0.686 | 4.517 |
| BA-06 | 0.278 | 3.853 | BN-06 | 0.454 | 4.112 |
| BA-07 | 0.424 | 3.869 | BN-07 | 0.197 | 3.886 |
| BA-08 | 0.705 | 4.778 | BN-08 | 0.547 | 4.992 |
| BA-09 | −0.110 | 2.719 | BN-09 | 0.677 | 5.600 |
| BA-10 | 0.039 | 3.265 | BN-10 | 0.254 | 4.89 |
| BA-11 | 0.340 | 3.595 | |||
3 Conclusion
A novel series of 2,2-dimethylbenzopyran derivatives was created from an initial 2,2-dimethyl-2H-chromen-6-amine lead using the molecular hybridization approach, bioisostere and functional group reversal strategies. Subsequently, their activity of neuroprotection was evaluated in oxygen glucose deprivation (OGD) model. Experimental results revealed that some novel compounds exhibit better activity of neuroprotection than edaravone. The efficacy improvement was achieved via lead transformation of cinnamamide to phenylacrylamide and by the incorporation of substituent at the amide position. These efforts culminated with the identification of an optimized molecule, compound BN-07, which displayed good efficacy in neurons of OGD model and will be further optimized as a lead compound. Efforts to determine the neuroprotection mechanism of these compounds are currently under way in our laboratory.4 Experimental section
4.1 Chemistry
All reagents used were commercially available. All the 1H-NMR and 13C-NMR spectra were recorded on a Bruker 400 MHz and 600 MHz spectrometer in DMSO-d6. Chemical shifts (δ) were expressed in parts per million using tetramethylsilane as an internal reference. ESI-MS data were obtained using an Agilent 1100 instrument. LC-MS-ESI was recorded by Agilent 1100 Series MSD Trap (SL). GC-MS-ESI was recorded by Agilent 6890-5975 GC-MS. Melting points were measured on X-4 digital melting point instrument. The purity was determined by Shimadzu LC-2010AHT high performance liquid chromatography. Reactions were monitored by thin-layer chromatography (TLC) and visualized by UV-light. Column chromatography was performed on silica gel (160–200 mesh).:1), which was recrystallized from absolute ethanol to give the light yellow solid (10.3 g, 47.3%), mp 116–119 °C. ESI-MS m/z: 218.1 [M + H]+, 240.1 [M + Na]+, 435.2 [2M + H]+, 457.2 [2M + Na]+.
(E)-3-(2,2-Dimethyl-2H-chromen-6-yl)acrylic acid 13 was obtained as a light yellow solid (7.41 g, 50.1%), mp 181–184 °C. 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 12.24 (s, 1H), 7.48 (d, J = 15.6 Hz, 1H), 7.44–7.41 (m, 2H), 6.76 (d, J = 8.0 Hz, 1H), 6.42 (d, J = 9.9 Hz, 1H), 6.34 (d, J = 15.6 Hz, 1H), 5.81 (d, J = 9.9 Hz, 1H), 1.39 (s, 6H).
(E)-3-(4-Hydroxyphenyl)acrylic acid (5a). 5a was obtained as a light yellow solid in 30.4% yield, mp 212–214 °C.
(E)-3-(3-Hydroxy-4-methoxyphenyl)acrylic acid (5c). 5c was obtained as a light yellow solid in 78.4% yield, mp 123–125 °C.
(E)-3-(Benzo[d][1,3]dioxol-5-yl)acrylic acid (5e). 5e was obtained as a light yellow solid in 80.7% yield, mp 242–244 °C.
(E)-3-(4-Methoxyphenyl)acrylic acid (5f). 5f was obtained as a light yellow solid in 73.1% yield, mp 171–173 °C.
(E)-3-(Naphthalen-1-yl)acrylic acid (5h). 5h was obtained as a light yellow solid in 50.3% yield, mp 210–212 °C.
(E)-3-(Pyridin-3-yl)acrylic acid (5i). 5i was obtained as a white solid in 56.6% yield, mp 235–237 °C.
(E)-3-(Thiophen-2-yl)acrylic acid (5k). 5k was obtained as a light yellow solid in 76.4% yield, mp 144–146 °C.
:EtOAc = 4:1 as eluent.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(4-hydroxyphenyl)acrylamide (BA-01). BA-01 was isolated as a light yellow solid (0.21 g, 23.0%), mp 230–233 °C. 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 9.93–9.92 (m, 2H), 7.46–7.42 (m, 4H), 7.35 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 6.81 (d, J = 8.5 Hz, 2H), 6.70 (d, J = 8.8 Hz, 1H), 6.57 (d, J = 15.6 Hz, 1H), 6.40 (d, J = 9.8 Hz, 1H), 5.77 (d, J = 9.8 Hz, 1H), 1.36 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 164.1, 159.6, 148.7, 140.3, 132.2, 132.1, 129.9, 126.3, 122.4, 121.3, 120.7, 119.2, 117.9, 116.3, 76.3, 27.7. ESI-MS m/z: 322.1 [M + H]+, 643.1 [2M + H]+, 665.2 [2M + Na]+.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(4-hydroxy-3-methoxyphenyl)acrylamide (BA-02). BA-02 was isolated as a light yellow solid (0.30 g, 29.4%), mp 88–91 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.65 (d, J = 15.6 Hz, 1H), 7.42 (s, 1H), 7.16–7.09 (m, 3H), 7.02 (s, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 6.37 (d, J = 15.6 Hz, 1H), 6.31 (d, J = 10.0 Hz, 1H), 5.84 (s, 1H), 5.64 (d, J = 10.0 Hz, 1H), 3.93 (s, 3H), 1.43 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 164.1, 149.7, 148.3, 146.7, 142.0, 131.5, 131.2, 127.3, 123.5, 122.3, 121.6, 120.9, 118.6, 118.5, 116.5, 114.8, 109.8, 76.3, 56.0, 27.9. ESI-MS m/z: 352.2 [M + H]+, 374.2 [M + Na]+, 350.4 [M − H]−.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(3-hydroxy-4-methoxyphenyl)acrylamide (BA-03). BA-03 was isolated as a light yellow solid (0.20 g, 19.7%), mp 185–188 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.62 (d, J = 15.4 Hz, 1H), 7.45 (s, 1H), 7.42 (s, 1H), 7.19 (d, J = 8.0 Hz, 1H), 7.13 (s, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.73 (d, J = 8.4 Hz, 1H), 6.37 (d, J = 15.4 Hz, 1H), 6.28 (d, J = 9.8 Hz, 1H), 5.62 (d, J = 9.8 Hz, 1H), 3.90 (s, 3H), 1.41 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 164.1, 149.7, 148.2, 145.8, 141.7, 131.5, 131.3, 128.3, 2, 122.3, 121.9, 121.5, 120.9, 119.0, 118.6, 116.5, 120.6, 110.5, 76.3, 56.0, 27.9. ESI-MS m/z: 352.3 [M + H]+, 350.4 [M − H]−.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(3,4-dihydroxyphenyl)acrylamide (BA-04). BA-04 was isolated as a gray solid (0.33 g, 33.9%), mp 220–222 °C. 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 9.91 (s, 1H), 9.45 (s, 1H), 9.19 (s, 1H), 7.43 (d, J = 2.3 Hz, 1H), 7.37–7.33 (m, 2H), 6.99 (d, J = 1.8 Hz, 1H), 6.89 (dd, J = 8.2 Hz, 1.8 Hz, 1H), 6.77 (d, J = 8.2 Hz, 1H), 6.70 (d, J = 8.5 Hz, 1H), 6.49 (d, J = 15.6 Hz, 1H), 6.39 (d, J = 9.8 Hz, 1H), 5.76 (d, J = 9.8 Hz, 1H), 1.36 (s, 6H). 13C NMR (150 MHz, DMSO-d6) δ 164.1, 148.7, 148.1, 146.1, 140.7, 133.2, 132.2, 126.7, 122.4, 121.3, 121.2, 120.6, 119.0, 117.9, 116.3, 116.2, 114.3, 76.3, 60.2, 28.0. ESI-MS m/z: 338.1 [M + H]+, 360.1 [M + Na]+, 675.3 [2M + H]+, 697.2 [2M + Na]+.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(1,3-benzodioxole-5-yl)acrylamide (BA-05). BA-05 was isolated as a light yellow solid (0.23 g, 22.9%), mp 151–153 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.64 (d, J = 15.3 Hz, 1H), 7.42 (s, 1H), 7.16–7.15 (m, 2H), 7.03–7.02 (m, 2H), 6.81 (d, J = 7.88 Hz, 1H), 6.74 (d, J = 8.52 Hz, 1H), 6.34 (d, J = 15.3 Hz, 1H), 6.31 (d, J = 9.72 Hz, 1H), 6.01 (s, 2H), 5.64 (d, J = 9.72 Hz, 1H), 1.43 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 164.1, 149.7, 149.2, 148.2, 141.6, 131.5, 131.3, 129.1, 124.1, 122.2, 121.5, 121.0, 119.0, 118.6, 116.5, 108.5, 106.4, 101.5, 76.3, 27.9. ESI-MS m/z: 350.1 [M + H]+, 372.1 [M + Na]+, 699.2 [2M + H]+, 721.2 [2M + Na]+.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(naphthalen-1-yl)acrylamide (BA-08). BA-08 was isolated as a white solid (0.08 g, 7.8%), mp 186–189 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 8.60 (d, J = 15.1 Hz, 1H), 8.27 (d, J = 8.4 Hz, 1H), 7.91–7.89 (m, 2H), 7.77–7.75 (m, 1H), 7.60–7.55 (m, 2H), 7.53–7.50 (m, 2H), 7.24–7.22 (m, 2H), 6.79 (d, J = 8.4 Hz, 1H), 6.61 (d, J = 15.1 Hz, 1H), 6.36 (d, J = 10 Hz, 1H), 5.68 (d, J = 10.0 Hz, 1H), 1.46 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 163.7, 149.8, 143.8, 139.2, 133.7, 131.5, 130.9, 130.1, 128.8, 128.6, 127.0, 126.8, 126.2, 125.4, 123.6, 122.2, 121.6, 121.0, 119.9, 118.6, 116.5, 76.3, 27.9. ESI-MS m/z: 356.1 [M + H]+, 378.1 [M + Na]+, 711.3 [2M + H]+, 733.3 [2M + Na]+.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(pyridin-3-yl)acrylamide (BA-09). BA-09 was isolated as a light yellow solid (0.25 g, 29.4%), mp 105–107 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.50 (d, J = 15.0 Hz, 1H),7.46 (s, 1H), 7.42 (s, 1H), 7.16–7.10 (m, 2H), 6.75–6.73 (m, 1H), 6.59–6.58 (m, 1H), 6.47 (s, 1H), 6.40 (d, J = 15.0 Hz, 1H), 6.31 (d, J = 9.8 Hz, 1H), 5.64 (d, J = 9.8 Hz, 1H), 1.43 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 163.0, 150.2, 150.1, 148.8, 138.0, 134.9, 131.6, 131.0, 130.8, 123.7, 123.4, 122.1, 121.6, 121.0, 118.6, 116.5, 76.4, 27.9. ESI-MS m/z: 296.1 [M + H]+, 591.2 [2M + H]+, 613.2 [2M + Na]+.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(furan-2-yl)acrylamide (BA-10). BA-10 was isolated as a light yellow solid (0.25 g, 29.4%), mp 105–107 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.50 (d, J = 15.0 Hz, 1H), 7.46 (s, 1H), 7.42 (s, 1H), 7.16–7.10 (m, 2H), 6.75–6.73 (m, 1H), 6.59–6.58 (m, 1H), 6.47 (s, 1H), 6.40 (d, J = 15.0 Hz, 1H), 6.31 (d, J = 9.8 Hz, 1H), 5.64 (d, J = 9.8 Hz, 1H), 1.43 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 163.7, 151.3, 149.7, 144.2, 131.5, 131.2, 128.7, 122.2, 121.6, 120.9, 118.6, 118.5, 116.5, 114.2, 112.3, 76.3, 27.9. ESI-MS m/z: 296.1 [M + H]+, 591.2 [2M + H]+, 613.2 [2M + Na]+.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(thiophen-2-yl)acrylamide (BA-11). BA-11 was isolated as a yellow solid (0.40 g, 46.6%), mp 125–127 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.84 (d, J = 15.1 Hz, 1H), 7.41 (s, 1H), 7.34 (d, J = 5.0 Hz, 1H), 7.24–7.22 (m, 2H), 7.15 (d, J = 7.4 Hz, 1H), 7.05–7.03 (m, 1H), 6.74 (d, J = 8.6 Hz, 1H), 6.34–6.29 (m, 1H), 6.30 (d, J = 9.8 Hz, 1H), 1.42 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 163.6, 149.8, 139.9, 134.5, 131.5, 131.1, 130.6, 128.1, 127.6, 122.2, 121.5, 121.0, 119.8, 118.6, 116.5, 76.3, 27.9. ESI-MS m/z: 312.1 [M + H]+, 334.2 [M + Na]+, 622.9 [2M + H]+, 645.1 [2M + Na]+, 310.0 [M − H]−.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-(4-methoxyphenyl)acrylamide (BA-06). BA-06 was isolated as a white solid (0.51 g, 43.5%), mp 173–175 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.68 (d, J = 15.4 Hz, 1H), 7.47–7.43 (m, 3H), 7.33–7.30 (m, 1H), 7.17 (d, J = 8.0 Hz, 1H), 6.88 (d, J = 8.4 Hz, 2H), 6.74 (d, J = 8.6 Hz, 1H), 6.39 (d, J = 15.4 Hz, 1H), 6.20 (d, J = 9.6 Hz, 1H), 5.63 (d, J = 9.6 Hz, 1H), 3.83 (s, 3H), 1.42 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 164.2, 161.0, 149.7, 141.6, 131.5, 131.3, 129.5, 127.4, 122.3, 121.6, 120.9, 118.6, 118.5, 116.5, 114.3, 76.3, 55.4, 27.7. ESI-MS m/z: 336.1 [M + H]+, 671.3 [2M + H]+, 693.2 [2M + Na]+.
(E)-N-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-3-phenylacrylamide (BA-07). BA-07 was isolated as a light yellow solid (0.40 g, 32.6%), mp 154–157 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.71–7.76 (d, J = 15.5 Hz, 1H), 7.54–7.52 (m, 2H), 7.43 (s, 1H), 7.39–7.31 (m, 3H), 7.20–7.16 (m, 2H), 6.75 (d, J = 8.6 Hz, 1H), 6.41 (d, J = 15.5 Hz, 1H), 5.96 (d, J = 9.8 Hz, 1H), 5.64 (d, J = 9.8 Hz, 1H), 1.43 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 163.8, 149.8, 142.0, 134.7, 131.5, 131.1, 129.9, 128.9, 127.9, 122.2, 121.6, 121.0, 120.9, 118.6, 116.5, 76.3, 27.9. ESI-MS m/z: 306.1 [M + H]+, 328.1 [M + Na]+, 611.3 [2M + H]+, 633.2 [2M + Na]+.
(E)-3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-N-(4-methoxyphenyl)acrylamide (BN-02). BN-02 was isolated as a yellow solid (0.22 g, 30.2%), mp 140-143 °C. 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 10.00 (s, 1H), 7.61 (d, J = 9.0 Hz, 2H), 7.45 (d, J = 15.6 Hz, 1H), 7.36 (dd, J = 8.3 Hz, 1.8 Hz, 1H), 7.33 (d, J = 1.8 Hz, 1H), 6.90 (d, J = 9.0 Hz, 2H), 6.80 (d, J = 8.3 Hz, 1H), 6.63 (d, J = 15.6 Hz, 1H), 6.45 (d, J = 9.8 Hz, 1H), 5.82 (d, J = 9.8 Hz, 1H), 3.73 (s, 3H), 1.39 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 164.3, 156.4, 154.8, 141.7, 131.4, 131.3, 129.1, 127.4, 126.1, 121.8, 121.7, 121.3, 118.3, 116.7, 114.2, 77.2, 55.5, 28.2. ESI-MS m/z: 671.3 [2M + H]+, 693.2 [2M + Na]+.
3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-N-(4-methoxyphenyl)-3-((4-methoxyphenyl)amino)propanamide (BN-09). BN-09 was isolated as a white solid (0.08 g, 8.4%), mp 130–133 °C.1H-NMR (400 MHz, DMSO-d6): δ (ppm) 9.70 (s, 1H), 7.43 (d, J = 9.0 Hz, 2H), 7.12 (dd, J = 8.2, 1.7 Hz, 1H), 7.06 (d, J = 1.5 Hz, 1H), 6.84 (d, J = 9.0 Hz, 2H), 6.64–6.62 (m, 3H), 6.49 (d, J = 8.9 Hz, 2H), 6.33 (d, J = 9.8 Hz, 1H), 5.74 (d, J = 7.9 Hz, 1H), 5.59 (d, J = 9.8 Hz, 1H), 4.67 (q, J = 14.4, 7.3 Hz, 1H), 3.70 (s, 3H), 3.58 (s, 3H), 2.72 (dd, J = 14.2, 7.8 Hz, 1H), 2.58 (dd, J = 14.2, 6.4 Hz, 1H), 1.32 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 169.0, 156.5, 153.0, 152.3, 140.3, 134.3, 131.0, 130.6, 126.8, 124.2, 122.2, 122.1, 121.4, 116.6, 116.2, 114.8, 114.1, 76.3, 56.6, 55.6, 55.5, 45.2, 28.1, 28.0. ESI-MS m/z: 459.2 [M + H]+.
(E)-3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-N-(2-methoxyphenyl)acrylamide (BN-04). BN-04 was isolated as a light yellow solid (0.27 g, 37.1%), mp 115–116 °C. 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 10.00 (s, 1H), 7.61 (d, J = 9.0 Hz, 2H), 7.45 (d, J = 15.6 Hz, 1H), 7.37 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.33 (d, J = 1.6 Hz, 1H), 6.90 (d, J = 9.0 Hz, 2H), 6.80 (d, J = 8.4 Hz, 1H), 6.63 (d, J = 15.6 Hz, 1H), 6.45 (d, J = 9.8 Hz, 1H), 5.82 (d, J = 9.8 Hz, 1H), 3.73 (s, 3H), 1.40 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 164.1, 154.8, 147.8, 141.7, 131.3, 129.2, 128.0, 127.5, 126.0, 123.6, 121.8, 121.3, 121.2, 119.9, 118.7, 116.7, 109.8, 77.0, 55.7, 28.2. ESI-MS m/z: 336.1 [M + H]+, 358.1 [M + Na]+, 671.3 [2M + H]+, 693.3 [2M + Na]+.
(E)-3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-acrylate-phenyl ester (BN-05). BN-05 was isolated as a white solid (0.25 g, 37.6%), mp 93–94 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.77 (d, J = 15.9 Hz, 1H), 7.42–7.38 (m, 2H), 7.36 (dd, J = 8.4 Hz, 2.1 Hz, 1H), 7.24–7.21 (m, 2H), 7.17–7.15 (m, 2H), 6.80 (d, J = 8.4 Hz, 1H), 6.47 (d, J = 15.9 Hz, 1H), 6.34 (d, J = 9.9 Hz, 1H), 5.67 (d, J = 9.9 Hz, 1H), 1.46 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 165.7, 155.5, 150.9, 146.3, 131.4, 129.7, 129.4, 126.9, 126.4, 125.6, 121.7, 121.6, 121.4, 116.9, 114.4, 77.2, 28.3. ESI-MS m/z: 307.0 [M + H]+, 345.0 [M + Na]+, 635.7 [M + Na]+.
(E)-3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-1-(4-(4-hydroxyphenyl)piperazin-1-yl)prop-2-en-1-one (BN-07). BN-07 was isolated as a red brown solid (0.04 g, 4.7%), mp 193–196 °C. 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 8.90 (s, 1H), 7.49–7.46 (m, 2H), 7.42 (d, J = 15.3 Hz, 1H), 7.13 (d, J = 15.3 Hz, 1H), 6.84–6.81 (m, 2H), 6.76 (d, J = 8.0 Hz, 1H), 6.67–6.65 (m, 2H), 6.42 (d, J = 10.0 Hz, 1H), 5.81 (d, J = 10.0 Hz, 1H), 3.81 (s, 2H), 3.68 (s, 2H), 2.95 (s, 4H), 1.38 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 165.9, 155.5, 154.7, 146.3, 143.2, 131.3, 129.7, 129.0, 127.9, 125.9, 122.3, 121.8, 121.3, 118.2, 116.7, 114.4, 113.9, 77.0, 51.0, 49.9, 45.6, 40.0, 28.2. ESI-MS m/z: 391.2 [M + H]+, 781.3 [2M + H]+.
3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-N-phenyl-3-(phenylamino)propanamide (BN-08). BN-08 was isolated as a white solid (0.14 g, 16.2%), mp 143–146 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.57 (m, 1H), 7.33 (d, J = 7.6 Hz, 2H), 7.28 (d, J = 7.6 Hz, 2H), 7.14–7.06 (m, 4H), 6.97 (d, J = 1.9 Hz, 1H), 6.73–6.69 (m, 2H), 6.62 (d, J = 7.8 Hz, 2H), 6.23 (d, J = 9.8 Hz, 1H), 5.58 (d, J = 9.8 Hz, 1H), 4.85 (s, 1H), 4.76 (t, J = 6.1 Hz, 1H), 2.8 (d, J = 6.2 Hz, 2H), 1.40 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 169.0, 152.4, 146.4, 137.4, 134.1, 131.1, 129.2, 129.0, 126.8, 124.6, 124.2, 122.2, 121.5, 120.3, 118.5, 116.7, 114.4, 76.4, 55.4, 45.5, 28.1, 28.0. ESI-MS m/z: 399.2 [M + H]+, 437.1 [M + K]+, 797.3 [2M + H]+.
3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-N-(3-methoxyphenyl)-3-((3-methoxyphenyl)amino)propenamide (BN-10). BN-10 was isolated as a white solid (0.22 g, 22.1%), mp 145–146 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.50 (1H, s), 7.18 (t, J = 8.2 Hz, 1H), 7.09–7.07 (m, 2H), 7.04 (t, J = 8.2 Hz, 1H), 6.97 (d, J = 2.0 Hz, 1H), 6.80 (d, J = 7.9 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.63 (dd, J = 8.2 Hz, 2.0 Hz, 1H), 6.29–6.22 (m, 3H), 6.18 (s, 1H), 5.58 (d, J = 9.8 Hz, 1H), 4.74 (t, J = 6.1 Hz, 1H), 3.72 (s, 3H), 3.69 (s, 3H), 2.81 (d, J = 6.1 Hz, 2H), 1.40 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 168.8, 160.6, 160.0, 152.5, 146.7, 138.5, 133.2, 131.1, 130.0, 129.6, 127.0, 124.4, 122.1, 121.5, 116.7, 112.4, 110.5, 108.0, 105.9, 104.7, 101.1, 76.6, 56.2, 55.3, 55.1, 45.0, 28.1, 28.0. ESI-MS m/z: 459.1 [M + H]+, 481.2 [M + Na]+, 497.1 [M + K]+, 457.0 [M − H]−.
(E)-3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-N-(3-methoxyphenyl)acrylamide (BN-03). BN-03 was isolated as a yellow brown solid (0.11 g, 15.1%), mp 145–148 °C. 1H-NMR (400 MHz, CDCl3): δ (ppm) 7.66 (d, J = 15.4 Hz, 1H), 7.45 (s, 2H), 7.28 (d, J = 8.4 Hz, 1H), 7.24 (t, J = 8.1 Hz, 1H), 7.13 (s, 1H), 7.05–7.03 (m, 1H), 6.75 (d, J = 8.4 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.39 (d, J = 15.4 Hz, 1H), 6.29 (d, J = 10.0 Hz, 1H), 5.65 (d, J = 10.0 Hz, 1H), 3.81 (s, 3H), 1.44 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 164.5, 160.2, 154.9, 142.3, 139.5, 131.3, 129.7, 129.1, 127.3, 126.2, 121.7, 121.3, 118.1, 116.8, 112.0, 110.3, 105.4, 77.0, 55.3, 28.2. ESI-MS m/z: 336.1 [M + H]+, 358.1 [M + Na]+, 671.3 [2M + H]+, 693.3 [2M + Na]+.
(E)-3-(2,2-Dimethyl-2H-1-benzopyran-6-yl)-1-(4-(4-methoxyphenyl)piperazin-1-yl)prop-2-en-1-one (BN-06). BN-06 was isolated as a light yellow solid (0.27 g, 30.8%), mp 165–166 °C. 1H-NMR (400 MHz, DMSO-d6): δ (ppm) 7.49–7.46 (m, 2H), 7.42 (d, J = 15.3 Hz, 1H), 7.13 (d, J = 15.3 Hz, 1H), 6.84–6.81 (m, 2H), 6.85–6.82 (m, 2H), 6.76 (d, J = 8.0 Hz, 1H), 6.41 (d, J = 10.0 Hz, 1H), 5.81 (d, J = 10.0 Hz, 1H), 3.83 (s, 2H), 3.72–3.68 (m, 5H), 3.01 (s, 4H), 1.39 (s, 6H). 13C NMR (150 MHz, CDCl3) δ 165.8, 154.6, 154.4, 145.2, 142.9, 131.3, 129.0, 128.7, 128.0, 125.9, 121.8, 121.3, 120.8, 119.0, 116.7, 114.5, 114.1, 77.0, 55.6, 51.5, 51.0, 45.9, 42.2, 28.2. ESI-MS m/z: 405.2 [M + H]+, 427.2 [M + Na]+.
4.2 In vitro neuroprotection activity
Edaravone injection (30 mg/20 mL) were purchased from simcere Co., Ltd (Jiangsu, China). DMEM/F12 medium, neurobasal medium, B27 supplement, DMEM glucose-free culture medium, penicillin–streptomycin, FBS and Fetal bovine serum were purchased from Gibco Company (USA). L-Lysine homopolymer hydrobromide, thiazolyl blue (MTT), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). DAPI was purchased from Dojindo Laboratories (Japan). β-Tubulin antibody was purchased from Abcam Company (UK).
After 24 h of pretreatment with compounds of interest, the control group was cultured in neurobasal/B27 medium at 37 °C. The model group, positive control group, and test group were conducted with glucose-free DMEM medium before placing them into an anaerobic chamber loaded with a gas mixture containing 5% CO2 and 95% N2 (v/v) for 5 min at 20 L min−1. The cell cultures within the anaerobic chamber were kept in a humidified incubator at 37 °C for 12 h. The morphology of primary cortical neurons and cell viability were measured respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project 201602707 Natural Science Foundation of Liaoning Province.Notes and references
- A. S. Go, D. Mozaffarian, V. L. Roger, E. J. Benjamin, J. D. Berry, W. B. Borden, D. M. Bravata, S. Dai, E. S. Ford, C. S. Fox, S. Franco, H. J. Fullerton, C. Gillespie, S. M. Hailpern, J. A. Heit, V. J. Howard, M. D. Huffman, B. M. Kissela, S. J. Kittner, D. T. Lackland, J. H. Lichtman, L. D. Lisabeth, D. Magid, G. M. Marcus, A. Marelli, D. B. Matchar, D. K. McGuire, E. R. Mohler, C. S. Moy, M. E. Mussolino, G. Nichol, N. P. Paynter, P. J. Schreiner, P. D. Sorlie, J. Stein, T. N. Turan, S. S. Virani, N. D. Wong, D. Woo and M. B. Turner, Circulation, 2013, 127, e6–e245 Search PubMed.
- J. D. Pandian, V. Padma, P. Vijaya, P. N. Sylaja and J. M. Murthy, Int. J. Stroke., 2007, 2, 17–26 CrossRef.
- X. Chen and K. Wang, Acta Pharm. Sin. B, 2016, 6, 522–530 CrossRef.
- I. D. Kilic, A. Hakeem, K. Marmagkiolis, A. Paixao, I. Grunwald, D. Mutlu, S. Abou-Sherif, B. Gundogdu, S. Kulaksizoglu, I. Ates, M. Wholey, O. Goktekin and M. Cilingiroglu, Cardiovasc. Revasc. Med., 2018 DOI:10.1016/j.carrev.2018.07.010.
- Z. H. Mu, Z. Jiang, X. J. Lin, L. P. Wang, Y. Xi, Z. J. Zhang, Y. T. Wang and G. Y. Yang, CNS Neurosci. Ther., 2016, 22, 316–324 CrossRef CAS PubMed.
- T. Zhang, W. Yan, Q. Li, J. Fu, K. Liu, W. Jia, X. Sun and X. Liu, Neurol. Res., 2011, 33, 396–404 CrossRef CAS.
- A. M. Rao, J. F. Hatcher and R. J. Dempsey, J. Neurosci. Res., 1999, 58, 697–705 CrossRef CAS.
- H. Guo, I. Singh, Y. Wang, R. Deane, T. Barrett, J. A. Fernández, N. Chow, J. H. Griffin and B. V. Zlokovic, Eur. J. Neurosci., 2009, 29, 1119–1130 CrossRef.
- X. Zhao, R. Strong, P. Piriyawat, R. Palusinski, J. C. Grotta and J. Aronowski, Stroke, 2010, 41, 363–367 CrossRef CAS.
- D. M. Abdallah, N. N. Nassar and R. M. Abd-El-Salam, Brain Res., 2011, 1385, 257–262 CrossRef CAS.
- Z. H. Zhou, J. F. Lu, W. W. Liu, A. Manaenko, X. H. Hou, Q. Y. Mei, J. L. Huang, J. P. Tang, J. H. Zhang, H. G. Yao and Q. Hu, Pharmacol. Ther., 2018, 191, 23–42 CrossRef CAS.
- (a) K. Szydlowska and M. Tymianski, Calcium, Cell Calcium, 2010, 47, 122–129 CrossRef CAS; (b) E. Candelario-Jalil, Curr. Opin. Invest. Drugs, 2009, 10, 644–654 CAS.
- J. Wu, J. Ling, X. Wang, T. Li, J. Liu, Y. Lai, H. Ji, S. Peng, J. Tian and Y. Zhang, J. Med. Chem., 2012, 55, 7173–7181 CrossRef CAS.
- S. Panagiotou and S. Saha, Front. Neurosci., 2015, 9, 182 Search PubMed.
- H. Chen, H. Yoshioka, G. S. Kim, J. E. Jung, N. Okami, H. Sakata, C. M. Maier, P. Narasimhan, C. E. Goeders and P. H. Chan, Antioxid. Redox Signaling, 2011, 14, 1505–1517 CrossRef CAS.
- M. Cini and A. Moretti, Neurobiol. Aging, 1995, 16, 53–57 CrossRef CAS.
- P. H. Chan, C. J. Epstein, H. Kinouchi, H. Kamii, S. F. Chen, E. Carlson, J. Gafni, G. Yang and L. Reola, Adv. Neurol., 1996, 71, 271–280 CAS.
- K. Murakami, T. Kondo, M. Kawase, Y. Li, S. Sato, S. F. Chen and P. H. Chan, J. Neurosci., 1998, 18, 205–213 CrossRef CAS.
- K. Suzuki, Adv. Drug Delivery Rev., 2009, 61, 287–289 CrossRef CAS.
- Y. Zang, X. Song, C. Li, J. Ma, S. Chu, D. Liu, Q. Ren, Y. Li, N. Chen and D. Zhang, Eur. J. Med. Chem., 2018, 143, 438–448 CrossRef CAS.
- K. Y. Dae, K. K. Sun, S. H. Young, J. M. Kook, Y. S. Eun, P. S. Sup, S. H. Woo and Y. S. Wan, KR100769732B[P], 2007-10-23.
- https://integrity.thomson-pharma.com/integrity/xmlxsl/pk_prod_list.xml_prod_list_card_pr?p_id=129588%26p_tsearch=A .
- C. Reis, O. Akyol, W. M. Ho, C. Araujo, L. Huang, R. Applegate II and J. H. Zhang, BioMed Res. Int., 2017, 2017, 4863079 Search PubMed.
- F. M. C. Barros, B. Pippi, R. R. Dresch, D. Betina, S. C. Luciano, M. A. Apel, A. M. Fuentefria and G. L. von Poser, Ind. Crops Prod., 2013, 44, 294–299 CrossRef CAS.
- L. Wang, G. Chen, X. Lu, S. Wang, S. Han, Y. Li, G. Ping, X. Jiang, H. Li, J. Yang and C. Wu, Eur. J. Med. Chem., 2015, 89, 88–97 CrossRef CAS.
- Q. Hong, Z. C. Ma, H. Huang, Y. G. Wang, H. L. Tan, C. R. Xiao, Q. D. Liang, H. T. Zhang and Y. Gao, Eur. J. Pharmacol., 2016, 777, 1–8 CrossRef CAS.
- Y. Deng, N. Wang, Q. Zhu, X. M. Xie, J. A. Duan and A. W. Ding, Pharmacol. Clin. Chin. Mater. Med., 2008, 32–34 CAS.
- M. N. Liao, Y. P. Wang, Y. P. Zhang, R. D. Ma, X. Y. Zhang, L. J. Yu and T. X. Yu, Chin. J. Cell Biol., 2010, 32, 840–849 CAS.
- L. Luo and Y. Sun, J. Chin. Med. Mater., 2011, 34, 1750–1753 CAS.
- P. Agon, P. Goethals, D. V. Haver and J. M. Kaufman, J. Pharm. Pharmacol., 1991, 43, 597–600 CrossRef CAS.
- S. Y. Xu, Y. F. Hu, W. P. Li, Y. M. Wu, Z. Ji, S. N. Wang, K. Li and S. Y. Pan, Int. J. Biol. Sci., 2014, 10, 873–881 CrossRef CAS.
- C. G. Zhao, L. W. Zhang, H. Wang and D. Y. Huang, Food Sci., 2005, 26, 215–222 Search PubMed.
- B. Wu, L. Zhou and H. H. Cai, Chin. Chem. Lett., 2008, 19, 1163–1166 CrossRef CAS.
- T. Wu, W. Wang, X. S. Ding, M. J. Feng, Y. Gao and W. B. Yin, Pharmaceutical and Clinical Research, 2007, 15, 347–351 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10424g |
| This journal is © The Royal Society of Chemistry 2019 |