
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced phosphate sequestration by Fe(III) modified biochar derived from coconut shell†
Zhenxing Zhong a,
Guowen Yua,
Wenting Mob,
Chunjie Zhanga,
Hao Huangac,
Shengui Lib,
Meng Gaod,
Xiejuan Lu*a,
Beiping Zhanga and
Hongping Zhue
a,
Guowen Yua,
Wenting Mob,
Chunjie Zhanga,
Hao Huangac,
Shengui Lib,
Meng Gaod,
Xiejuan Lu*a,
Beiping Zhanga and
Hongping Zhue
aSchool of Environmental Science and Engineering, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: luxiejuanhust@163.com; Tel: +86-27-87792155
bDepartment of Urban Construction, Wuchang Shouyi University, Wuhan, 430064, China
cWuhan Planning and Design Company, Wuhan, 430014, China
dHuangshi Institute of Environmental Protection, Huangshi, 435000, China
eSchool of Civil Engineering & Mechanics, Huazhong University of Science and Technology, Wuhan 430074, China
First published on 3rd April 2019
Abstract
In this work, a novel Fe-modified coconut shell biochar (Fe-CSB) was synthesized and utilized to remove phosphate from aqueous solution. Characterization results confirmed that the iron in the Fe(III)-impregnated CSB existed mainly in the amorphous phase, as ferrihydrite and amorphous hydroxide, which substantially enhanced the phosphate adsorption. Batch experiments indicated that phosphate adsorption on the Fe-CSB was highly dependent on the pH, the humic acid, and temperature, while it was less affected by the nitrate. Phosphate adsorption by the CSB and Fe-CSB could be well described by the pseudo n-th order and Langmuir–Freundlich models. The fitting of the experimental data with the intra-particle diffusion model revealed that surface adsorption and inner-sphere diffusion were involved in the phosphate adsorption process, and that the latter was the rate-controlling step. Batch adsorption experiments and post-adsorption characterization results revealed that the phosphate adsorption by Fe-CSB was primarily governed by four mechanisms: ligand exchange, electrostatic attraction, chemical precipitation, and inner-sphere complexation. This work demonstrated that the modified Fe-CSB is an environmentally friendly and cost-effective bioretention medium and could open up new pathways for the removal of phosphorus from stormwater, as well as solve the problem of waste biomass pollution.
1. Introduction
Over the past four decades, China has witnessed huge changes in industrialization, urbanization, and intensive animal production. Consequently, numerous cities are facing urgent urban water environmental issues, such as non-point source pollution, contamination of surface and ground waters, as well as the functional changes of aquatic ecosystems.1,2 To improve the quality of the urban aquatic environment and reduce the adverse impacts of contaminated water bodies on the environment and public health, numerous wastewater treatment plants have been constructed and are being operated for the removal of nutrients (e.g. carbon, nitrogen and phosphorus) from most wastewaters. However, the nutrients deriving from non-point sources, such as stormwater runoff, are still not successfully controlled. Consequently, the ever-increasing of nutrient loading in urban water bodies has posed a serious threat to the urban aquatic ecosystem.In recent years, stormwater has been identified as an important source of phosphorus (P) and nitrogen (N) pollution, and the loading of nutrients (N and P) into urban water bodies has markedly intensified.3 Due to widespread urbanization, the retention functions of pollutants by urban watersheds had declined greatly,1 whereas the nutrient influxes via stormwater runoff continue to increase, thus leading to the deterioration of water quality and the decline in biodiversity in the aquatic environment.4–6 Phosphorus is a limiting nutrient for the growth and metabolism of aquatic animals and plants and is also an important indicator of water quality and of the health of aquatic ecosystems.7 One of the main issues concerning water quality is represented by eutrophication with dense algal blooms, which causes high turbidity and low dissolved oxygen. The reduction of human-induced P input is the key to counteract eutrophication in polluted water bodies. Consequently, it is becoming essential to explore effective technologies to eliminate phosphorus from stormwater runoff prior to its discharge into urban surface water.
A bioretention system is an extensively applied stormwater best management practice to control the runoff quantity and improve water quality in urban areas.8 However, P removal by bioretention system is highly variable and unstable, mainly due to their low capacity and the weak stability of P adsorbed on the filter media of the bioretention tanks, which results in high phosphorus leaching in the bioretention effluent.9,10 Therefore, enhanced phosphate removal via novel filter media within bioretention has been widely investigated and the application of effective, low-cost materials for phosphate removal has received great attention. Examples of the different materials used to remove P from water include: fly ash,11 red mud,12 waste-tailings,13 and metal oxides,14 however, many of these materials exhibit poor sorption, capacity and selectivity, as well as potential toxicity. Therefore, the investigation of low-cost, eco-friendly materials with high specific surface area, open pore structure and high adsorption capacity may offer significant advances for urban runoff control.
In recent years, biochar and modified biochar have been widely recognized as environmentally friendly and low-cost adsorbents for the removal of various contaminants (heavy metals, organic compounds and nutrients) from aqueous solutions.15–17 Nevertheless, the improvement of P adsorption in bioretention system that employ biochar, especially Fe(III) decorated biochar, has been seldom investigated, and the sorption mechanisms need to be further addressed.
In this work, a promising adsorbent for phosphate adsorption was synthesized by introducing Fe(III) hydroxide into coconut shell biochar (CSB). The main objectives were to: (1) prepare the Fe(III) hydroxide impregnated porous biochars and evaluate the characteristics of phosphate uptake by the CSBs using adsorption kinetics and isotherms; (2) investigate the effects of temperature, pH values and coexisting compounds on phosphate adsorption capability; (3) explore the underlying mechanism of phosphate adsorption by the CSBs with a combination of scanning electron microscopy (SEM), X-ray diffraction (XRD), and Fourier transform infrared spectroscopy (FTIR), etc.
2. Materials and methods
2.1. Chemicals and materials
The coconut shell biochar (CSB) used in the study was purchased from Guangdong, China. After washing with deionized water (DI) to remove impurities (mainly residual ashes), the biochar was oven-dried (70 °C) and passed through a 1 mm sieve and sealed in a container prior to use. The characterization of the CSB used is listed in Table 1. Phosphate solutions were prepared by dissolving potassium phosphate monobasic (KH2PO4, ≥99.5%) in DI water. Ferric chloride (FeCl3, ≥99.0%) and all the other reagents were above analytical grade, and were purchased from Sinopharm Chemical Reagent Co. Ltd.| Item | Fe | Al | Ca | Mg | TP | C (%) | H (%) | O (%) | N (%) | H/C | O/C | (O + N)/C |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (mg g−1) | ||||||||||||
| CSB | 1.61 | 2.36 | 5.1 | 2.65 | 0.13 | 84.31 | 1.42 | 13.54 | 0.48 | 0.02 | 0.16 | 0.17 |
| Fe-CSB | 106.6 | 0.78 | 3.44 | 0.86 | 0.11 | 28.89 | 5.28 | 32.18 | 0.42 | 0.18 | 1.11 | 1.13 |
2.2. CSB modification
One portion of pretreated CSB particles was immersed in 0.5 mol L−1 FeCl3 solutions at 80 °C for 6 h, and the initial pH of solution was adjusted to 7.0 using 0.1 mol L−1 HCl and NaOH. After washed with DI water to no precipitation (using 1 mol L−1 NaOH solution to detect the filtrate), the modified CSB particles were dried at 105 °C for 12 h and stored for use. The resulting biochar sample was henceforth referred as Fe-CSB. The physicochemical properties of the Fe-CSB are summarized in Tables 1 and 2.| Property | CSB | Fe-CSB |
|---|---|---|
| pH | 6.3 | 7.8 |
| BET surface area (m2 g−1) | 760.5 | 547.0 |
| Surface area of micropore (m2 g−1) | 576.9 | 369.6 |
| Surface area of mesopore (m2 g−1) | 183.6 | 177.4 |
| Average pore diameter (nm) | 2.1 | 2.3 |
| Total pore volume (cm3 g−1) | 0.40 | 0.32 |
| Zeta potential (mV) | −23.3 | 35.5 |
2.3. Adsorption kinetics and isotherms
For the sorption kinetics experiments, 0.05 g adsorbent (CSB or Fe-CSB) was added in 20 mL phosphate solution, and the initial concentration was 20 mg L−1. 0.02 mol L−1 KCl was used as a background electrolyte. The pH was adjusted to 7.0 using 0.1 mol L−1 HCl or NaOH. The sealed vials were placed in a mechanical shaker bath at 25 ± 1 °C, shaken at 150 rpm. Samples were drawn periodically for phosphate concentration analysis.The phosphate sorption isotherms on the CSBs were studied by adding 0.05 g CSB or Fe-CSB to 20 mL phosphate solutions with a series of initial concentrations (2–100 mg L−1) in the sealed vials. The sealed vials were then shaken in an incubator shaker with thermostat at 25 ± 1 °C under 150 rpm for 24 h. The samples were periodically drawn and filtered through a 0.45 μm membrane. Besides, the pH of mixtures was maintained at 7.0. Control experiments without CSB and Fe-CSB result in less than 5% loss of P.
2.4. Effects of initial pH, temperature and coexisting matters
In a test procedure similar to the above, different initial pH values (3.0–12.0) of solutions were used to investigate the effects on P adsorption by the CSB and Fe-CSB. The effects of co-existing compounds (NO3− and humic acid (HA)) on phosphate adsorption were evaluated in batch test with an initial phosphate concentration of 20 mg L−1. The concentration ranges of NO3− and HA were imposed at 0–50 mg L−1 and 0–20 mg L−1, respectively. Moreover, the influence of various temperatures (298, 308 and 318 K) on the uptake of phosphate was also studied. The same procedures were used to analyze for phosphate concentration in suspensions.2.5. Desorption kinetics
To evaluate the desorption performance of phosphate from the CSB and Fe-CSB, 20 mL of centrifuged supernatant was removed from an adsorption test, and the vials were refilled to the original volume by adding 0.02 mol L−1 KCl solution without phosphate. The mixtures were re-equilibrated for 48 h under the same conditions (pH, temperature, rotating velocity). After 0.5, 1, 2, 4, 8, 24, and 48 h, the samples were taken out for the analysis of phosphate and desorbed phosphate was calculated. NaOH solution was used to regenerate the Fe-CSB after the adsorption process, and four adsorption–desorption cycles were carried out to evaluate the reusability of Fe-CSB.2.6. Analysis and characterization
The concentrations of P in the filtrates were determined by molybdenum blue spectrophotometric method with a UV-2600 UV/vise spectrometer (Shimadzu, Japan). Portions of CSB and Fe-CSB were digested with HNO3–HF–HClO4, and the contents of total Fe, Al, Mg, Ca and TP of the CSB and Fe-CSB were analyzed by ICP-AES (PerkinElmer, USA). The textural characteristics of the CSB and Fe-CSB were determined using a N2 adsorption–desorption test at 77 K (ASAP-3000, Micromeritics). Scanning electron microscope (Quanta 200, Holland) was applied to analyze the surface morphology and probe the surface elements of the CSB and Fe-CSB. X-ray diffraction (XRD) of randomly oriented powders of the samples was performed on a Phillips PW 3050/60 diffractometer using monochromated Cu Kα radiation, operating at 40 kV and 30 mA, from 10° to 80° (2θ), with a scanning step of 0.02° at 1° per minute. The analyses of four primary elements (C, H, O and N) in materials were conducted using an elemental analyzer (ThermoFinnigan, EA112 CHN, USA). The changes between the surface groups before and after phosphate were recorded by FTIR spectroscopy (Bruker, Vector 22, Germany). A B1-ZetaPlus (Brookhaven Instruments, USA) apparatus was used to measure the zeta potential of the CSBs before and after phosphate adsorption.3. Results and discussion
3.1. Properties of the CSB and Fe-CSB
The quantitative elemental compositions of the CSB and Fe-CSB are summarized in Table 1. Compared with the original CSB, the content of Fe in the Fe-CSB remarkably increased after modification, while the contents of Al, Ca, and Mg moderately decreased. The modified Fe-CSB contained mainly metallic elements, as was also validated by the Energy Dispersive Spectrometer (EDS) results. As exhibited in Fig. 1(a) and (b), the EDS spectrum of the SEM image focusing area exhibited a pronounced peak relative to Fe. The XPS spectrum also confirmed that Fe was present in the modified Fe-CSB (Fig. 2(a)). The peaks relative to Si, Al, and Ca were also visible in the EDS spectra of the CSBs. Besides, the XRD spectra indicated that the raw CSB was dominated with a broad peak at 23.0° (Fig. 2(b)), which is accounted for the cellulose crystal plane.18,19 However, no sharp peaks were obtained in the XRD patterns, indicating that the iron present in the Fe-CSB sample mainly existed in the amorphous state (e.g., 2-line ferrihydrite), which exhibits low crystallinity.20,21 From the SEM spectra in Fig. 1(a), it is quite clear that the surface of the original CSB was relatively smooth, and a part of the intrinsic characteristics of the pristine biochar were manifested, while the surface morphology of the Fe-CSB was rough and porous. Compared with the pristine CSB, numerous microsized and nanosized pores were irregularly distributed on the surface of the Fe-CSB. The observations were also verified by the increased value of the average pore diameter in the Fe-CSB.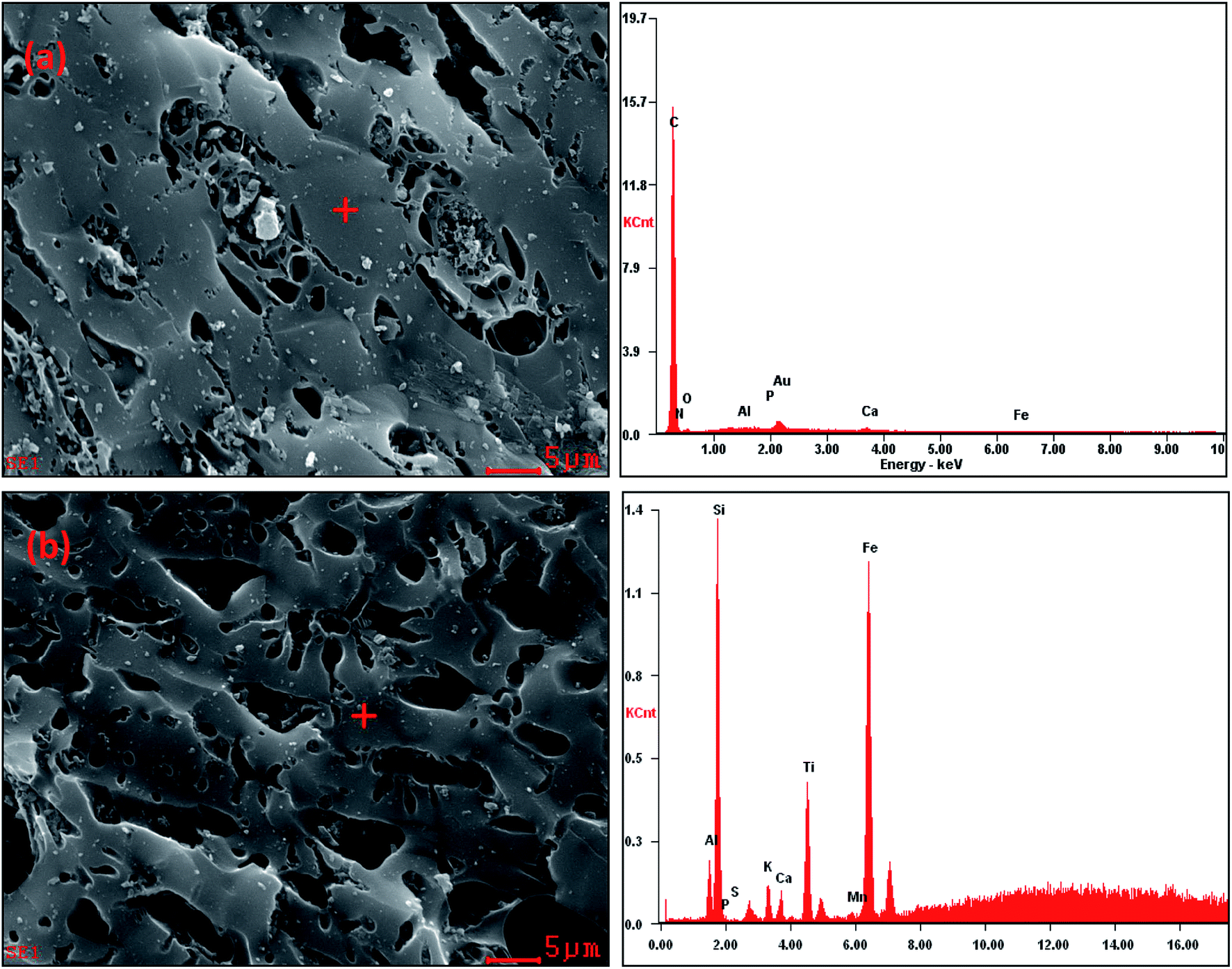 | ||
| Fig. 1 SEM images and the corresponding EDS spectra of the CSB (a) and Fe-CSB (b). | ||
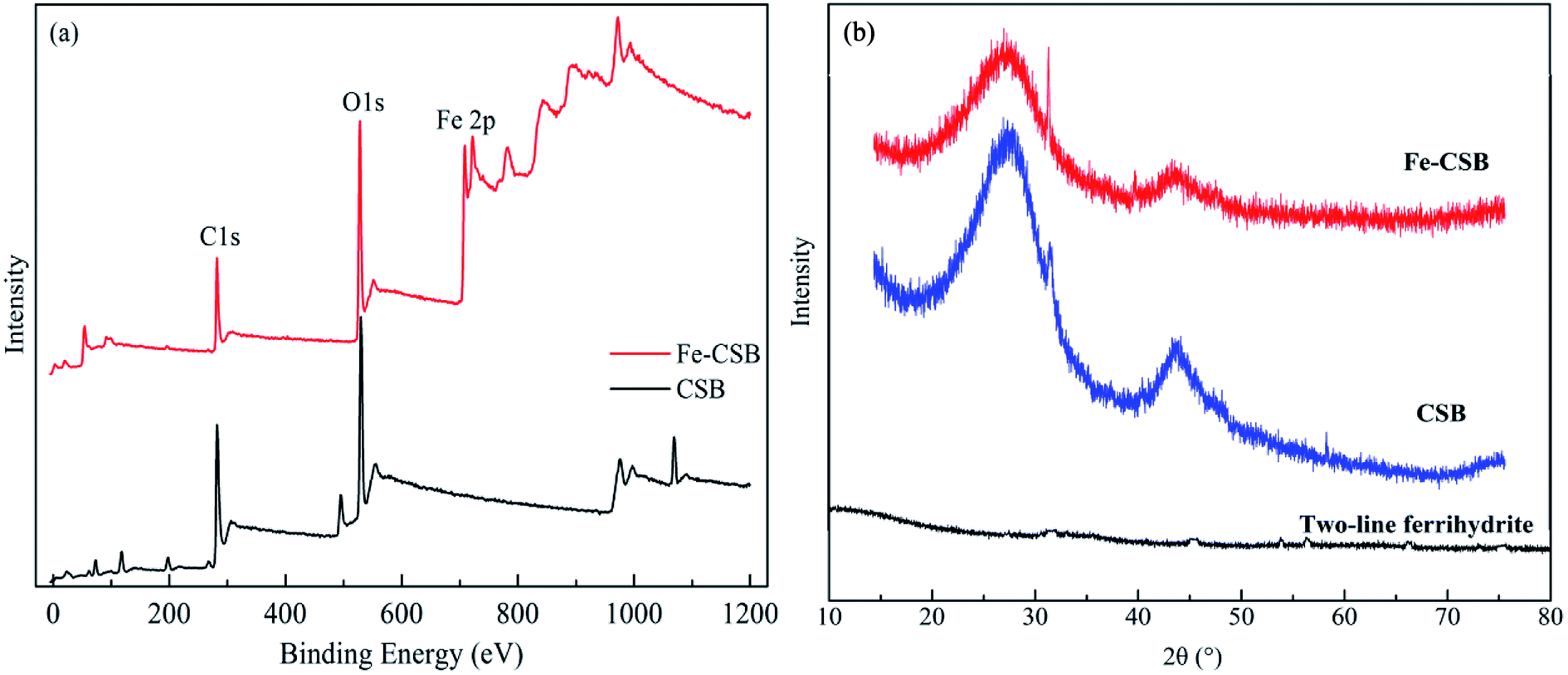 | ||
| Fig. 2 XPS (a) and XRD (b) spectra of the CSB and Fe-CSB. | ||
From Table 1, it can be noted that the Fe-CSB had a lower C content and higher O content than the CSB, which was associated with its lower hydrophobicity. The ratios of H/C, O/C, and (O + N)/C in the Fe-CSB sample were much higher than those of the CSB, implying that there were more oxygen-containing functional groups (e.g., –COOH and –OH) on the surface of the former.19,22 The results were confirmed by the more numerous and stronger absorption peaks appearing in the FTIR spectrum of the Fe-CSB compared to that of the pristine CSB.23 As shown in Table 2, the BET surface areas of the CSB and Fe-CSB were 760.5 and 547.0 m2 g−1, respectively, implying that the homogenous deposition of the Fe(III) hydroxide over the entire CSB matrix caused the partial blocking of the micropore and mesopore channels of Fe-CSB. Although the raw CSB sample exhibited a higher specific surface area and total pore volume than the Fe-CSB, the average pore diameter of the former was lower. Importantly, the zeta potentials of the CSB and Fe-CSB were −23.32 mV and 35.45 mV, respectively. This was attributed to the change in content of the iron hydroxide and oxygen-containing groups on the surface of the Fe-CSB, which occurred during the modification process. Based on the above results, the surface properties of the CSB were ameliorated substantially following the Fe(III) modification process.
3.2. Sorption kinetics and intraparticle diffusion model
The kinetic profiles of phosphate adsorption by the CSB and Fe-CSB are displayed in Fig. 3(a). It is quite obvious that the initial adsorption of phosphate by the two adsorbents was fast and was followed by a comparatively slow adsorption state. Equilibrium conditions were reached after 5 and 24 h, respectively. This result was agreement with the findings of Zeng et al.24 With regards to the CSB, the phosphate adsorption sharply increased with reaction time, and over 97% phosphate was eliminated after 3 h. The initial fast adsorption may be attributed to the electrostatic interactions between the positively charged metal oxides (Al2O3, CaO, MgO) on the surface of the pristine CSB and the negatively charged P species (PO43−, HPO42−, H2PO4−).19,25 Compared with the CSB, the P adsorption on the Fe-CSB was slightly slower, but the adsorbed amount increased steadily with the reaction time, resulting in a significantly higher adsorption capacity. The equilibrium adsorption capacity of the Fe-CSB was 4.2 mg g−1, that of the CSB was 2.2 mg g−1, suggesting that there was a stronger driving force for P adsorption onto the Fe-CSB. Similar results for the adsorption of P onto iron-decorated natural and engineered sorbents were also reported in the other works.26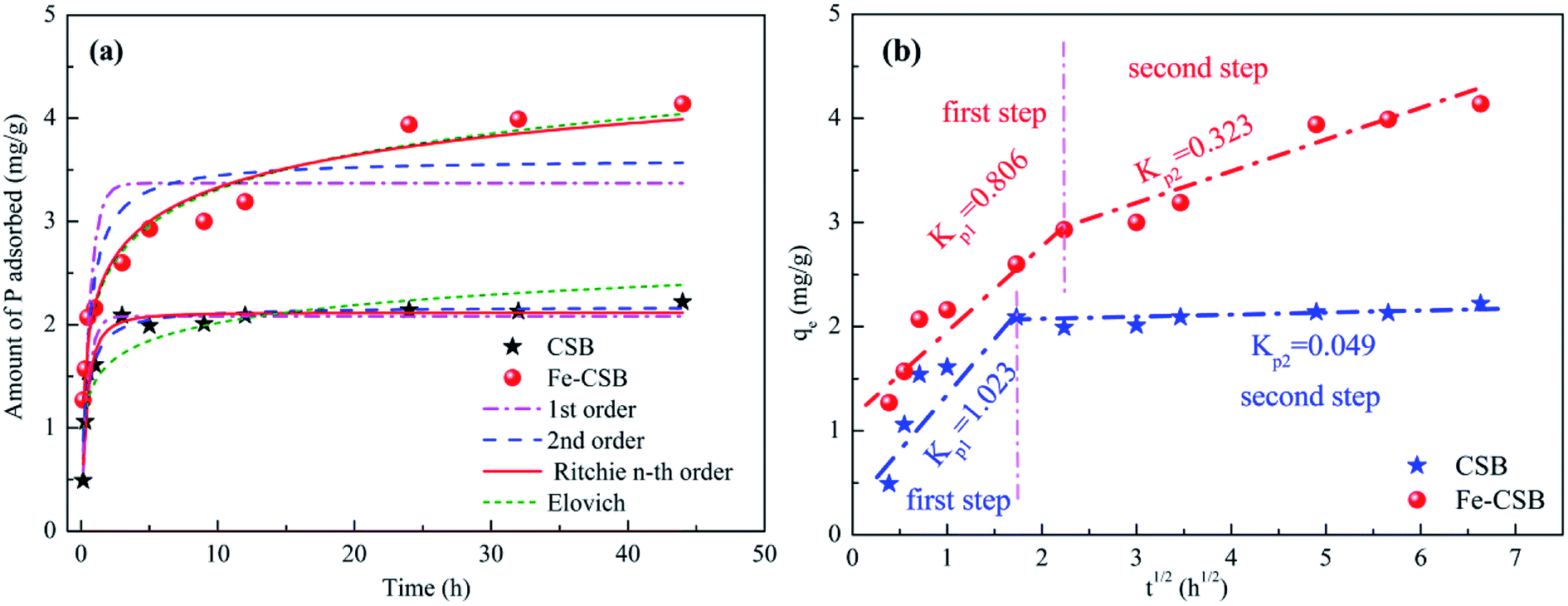 | ||
| Fig. 3 Sorption kinetic data and modeling for phosphate on the CSB and Fe-CSB (a); and the intra-particle diffusion model (b). | ||
To explore the sorption mechanism, the kinetic data were fitted with pseudo 1st order, pseudo 2nd order, n-th-order, and Elovich models (Table S1†),5 and the obtained parameters, along with the correlation coefficient (R2) and the root mean squared error (RMSE) values, were reported in Table 3. Lower RMSE values indicated a better fitting of the experimental data. Taking both the R2 and RMSE into consideration, the n-th-order model (R2 = 0.959 & 0.967, RMSE = 0.098 & 0.247) was found to match the data slightly better than the other three models proposed. Consequently, it was concluded that the uptake of phosphate by the CSB and Fe-CSB may be governed by two or more sorption mechanisms.23 Earlier research had demonstrated that intraparticle diffusion may be involved in the removal of pollutants by biochar materials.27
| Adsorbent | Models | Parameter 1 (gn−1 (mgn−1 h)−1) | Parameter 2 (mg g−1) | Parameter 3 | R2 | RMSE |
|---|---|---|---|---|---|---|
| CSB | 1st-order | k1 = 2.177 | qe = 2.082 | 0.951 | 0.109 | |
| 2nd-order | k2 = 1.453 | qe = 2.175 | 0.955 | 0.105 | ||
| n-th-order | kn = 1.819 | qe = 2.117 | n = 1.523 | 0.959 | 0.098 | |
| Elovich | b = 4.014 (g mg−1) | a = 81.681 (mg g−1 h−1) | 0.788 | 0.228 | ||
| Fe-CSB | 1st-order | k1 = 1.724 | qe = 3.369 | 0.662 | 0.514 | |
| 2nd-order | k2 = 0.592 | qe = 3.606 | 0.807 | 0.388 | ||
| n-th-order | kn = 0.090 | qe = 4.128 | n = 2.652 | 0.967 | 0.247 | |
| Elovich | b = 2.018 (g mg−1) | a = 39.20 (mg g−1 h−1) | 0.962 | 0.276 |
On the basis of these observations, the intraparticle diffusion model was applied to investigate the rate-determining steps and mechanisms of phosphate adsorption by the CSB and Fe-CSB (Table S2†).28 As seen in Fig. 3(b), a multi-linearity relation existed between qt and t1/2 over a wide range of reaction time, suggesting the presence of an external mass transfer resistance during P adsorption. Two separate regions were observed for phosphate adsorption by the CSB and Fe-CSB. During the first steep-sloped period, most of the phosphate anions (about 94% and 71%, respectively) were rapidly adsorbed on the exterior surfaces of the CSB (Kp1 = 1.023) and Fe-CSB (Kp1 = 0.806), which was ascribed to the mass transfer taking place in the diffusion boundary layer.24 During the second period, the intraparticle diffusion rate of the CSB (Kp2 = 0.049) slowed down sharply due to the achievement of adsorption equilibrium. As to the Fe-CSB, although the exterior surface reached adsorption saturation, P anions were impregnated into the inner-sphere of the Fe-CSB, where they were firmly adsorbed. Since the phosphate slowly penetrated into the inner-sphere, the diffusion resistance gradually increased, leading to the decrease of the diffusion rate (Kp2 = 0.323). The lower diffusion rate in the Fe-CSB may have also been related to the smaller pores, stronger electrostatic repulsion, and lower phosphate concentration in the solution.28 Additionally, the C values of Fe-CSB (1.212 and 2.092) were obviously larger than those of CSB (0.463 and 1.886), which indicated that the boundary layer in the Fe-CSB was thicker than that of the CSB (Table S2†). The result further confirmed that the Fe(III) hydroxide was loaded on the surface of the Fe-CSB and enhanced the phosphate removal. Thus, it could be postulated the phosphate adsorption on the exterior surface was the rate-limiting step of P adsorption on the CSB, while intra-particle diffusion was rate-limiting for P adsorption on the Fe-CSB.29
3.3. Sorption isotherms and thermodynamics
Phosphate sorption isotherms on the CSB and Fe-CSB fitted by three models were investigated in the temperature range of 298–318 K (Table S1† and Fig. 4), and the corresponding parameters were reported in Table 4. From Fig. 4, it can be seen that the amount of P adsorbed by the CSB and Fe-CSB sharply increased when the reaction temperatures increased from 298 K to 308 K, while the adsorption amount increased only slightly when the temperature was further increased to 318 K. It has been reported that the relatively higher temperatures enhance ion diffusion in solution and create strong interaction forces between the phosphate anions and the biochars. Consequently, the phosphate can be more easily adsorbed by the CSB and Fe-CSB.30,31 The above results implied that the primary mechanism for phosphate adsorption by the CSBs was an endothermic process. In addition, the positive ΔH0 values of the phosphate adsorption by the CSBs also confirmed the endothermic process of the P adsorption for temperatures of 298–318 K (Table S3†).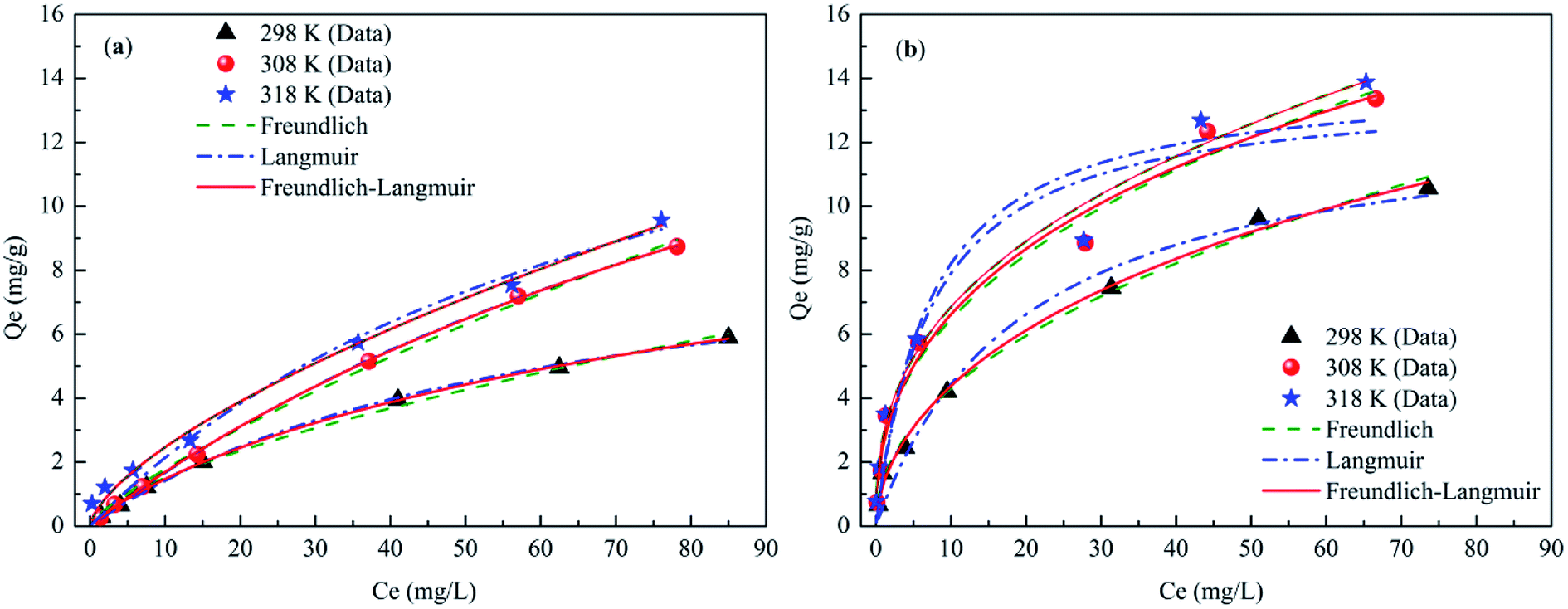 | ||
| Fig. 4 Sorption isotherms data and modeling for phosphate on the CSB (a) and Fe-CSB (b) at different temperatures. | ||
| Models & parameters | CSB | Fe-CSB | ||||
|---|---|---|---|---|---|---|
| 298 K | 308 K | 318 K | 298 K | 308 K | 318K | |
| Freundlich | ||||||
| KF | 1.541 | 0.291 | 0.542 | 1.471 | 2.618 | 2.873 |
| n | 0.336 | 1.273 | 1.518 | 2.144 | 2.548 | 2.650 |
| R2 | 0.995 | 0.997 | 0.991 | 0.991 | 0.980 | 0.980 |
| RSME | 0.126 | 0.147 | 0.268 | 0.278 | 0.591 | 0.572 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Langmuir | ||||||
| qm | 9.832 | 18.75 | 23.31 | 13.05 | 23.70 | 34.06 |
| KL | 0.017 | 0.008 | 0.013 | 0.051 | 0.136 | 0.140 |
| R2 | 0.998 | 0.996 | 0.976 | 0.976 | 0.939 | 0.9225 |
| RMSE | 0.075 | 0.060 | 0.446 | 0.519 | 1.050 | 1.222 |
|
||||||
| Langmuir–Freundlich | ||||||
| qm | 13.23 | 25.46 | 26.84 | 36.0 | 60.88 | 65.32 |
| KLF | 0.017 | 0.008 | 0.011 | 0.038 | 0.044 | 0.0391 |
| n | 0.872 | 0.974 | 0.891 | 0.563 | 0.445 | 0.422 |
| R2 | 0.999 | 0.999 | 0.994 | 0.993 | 0.983 | 0.983 |
| RMSE | 0.037 | 0.057 | 0.268 | 0.244 | 0.580 | 0.547 |
Three isotherm models (Freundlich, Langmuir, and Langmuir–Freundlich), described in Table S1,† were used to fit the experimental data relative to the CSB and Fe-CSB.5,24 The obtained parameters, along with the R2 and the RMSE values for the three models, were also summarized in Table 4. Although all the isotherm models closely reproduced the data with R2 values exceeding 0.92, the Langmuir–Freundlich model fitted the experimental data better than the other two models, presenting the highest R2 values (0.983–0.999) and the lowest RMSE values (0.0365–0.5802) for all three temperatures. These results implied that phosphate adsorption onto the CSB and Fe-CSB could be more accurately described by the Langmuir–Freundlich model and the interactions between phosphate and the surfaces of the CSBs were mainly influenced by both the Langmuir and Freundlich processes. This observation coincided with the results of the kinetic research, which had revealed that the adsorption of phosphate by the CSB and Fe-CSB could be controlled by two or more mechanisms.12,32
The maximum phosphate adsorption capacities (qm) of the Fe-CSB obtained from the Langmuir–Freundlich model were approximately 36.0, 60.88, and 65.32 mg g−1 at 298 K, 308 K, and 318 K, respectively. Compared with other Fe-based materials reported for phosphate adsorption, the Fe-CSB displayed a relatively superior capacity (Table S4†). For instance, the maximum capacities of phosphate adsorption on Fe(III)-doped activated carbon, magnetic Fe–Zr binary oxide, and ferrihydrite were about 8.13, 13.6 and 22.17 mg g−1, respectively. The higher phosphate adsorption capacity could be attributed to the relatively high Fe content and more numerous active sites on the Fe-CSB.33,34 Moreover, when taking into consideration the cost of raw materials and the preparation processes, the Fe(III) modified CSB can be considered as a promising and low-cost medium to be employed in bioretention systems for P removal, since the feedstock used for its preparation is a cheap and natural biomass, widely available in the country.
3.4. Effect of pH on P adsorption
The pH value is one of the most crucial variables affecting the adsorption process of compounds as it determined the charge state. As depicted in Fig. 5(a), the adsorption profile of P by the CSB and the Fe-CSB was highly pH-dependent, and the acidic or weak acidic conditions favored P adsorption on the CSB and Fe-CSB. | ||
| Fig. 5 Effect of initial pH on P adsorption on the CSBs and final pH (a), the variations of zeta potential of Fe-CSB before and after of P adsorption (b). | ||
As for the CSB, the P adsorption amount only decreased slightly when the initial pH increased from 3 to 7. With a further increase of pH from 7 to 12, the phosphate adsorption decreased sharply. Similarly, the P adsorption capacity by the Fe-CSB decreased mildly from 6.53 to 3.8 mg g−1 when the initial pH increased from 3 to 9, but it decreased dramatically when then initial pH was increased from 9 to 12. Similar results were also reported during the P adsorption by other Fe-based and metal-containing sorbents.19,24 The main phosphate anions formed were monovalent H2PO4− and divalent HPO42− in the pH ranges of 3–7 and 7–9, respectively. Meanwhile, the surfaces of the CSB and Fe-CSB (pHpzc = 7.6) were protonated and carried positive charges at low pH. As a result, the positively charged surface of the CSB and Fe-CSB were more likely to adsorb the negatively charged phosphate anions (HPO42− and H2PO4−). The higher adsorption capacity of phosphate at lower pH values was attributed to the stronger electrostatic attraction.11 Conversely, a higher pH caused the surfaces of the CSB and Fe-CSB to hold more negative charges, which would strongly repulse the main phosphate species (HPO42− and PO43−), which also carried negative charges in the pH range of 9–12. Accordingly, the lower adsorption amount in weak alkaline and alkaline solutions was accounted for in terms of the increased repulsion.24
On the other hand, some iron in the Fe-CSB was easily dissolved and existed as Fe3+ at low pH values. Therefore, the chemical precipitation of the Fe3+ and P ions as FePO4 may also be have contributed significantly to the P removal, since the FePO4 has a low Ksp value (7.9 × 1021).24,35 Furthermore, the Fe-CSB also contained low-solubility metal oxides, such as CaO, MgO and Al2O3, etc., whose dissolution could have been facilitated by the presence of phosphate anions, which could have led to the formation of phosphate precipitates, such as AlPO4, Ca3(PO4)2, and Mg3(PO4)2 (Table S5†).5,11
3.5. Effect of coexisting compounds on P adsorption
Urban stormwater usually contains coexisting compounds (e.g., inorganic and organic matters),36 that could interfere with the phosphate removal through various pathways. In this study, the influence of nitrate (NO3−) and humic acid (HA) in six concentration levels on the phosphate uptake by the CSBs were evaluated at the initial pH of 7.0 (Fig. 6).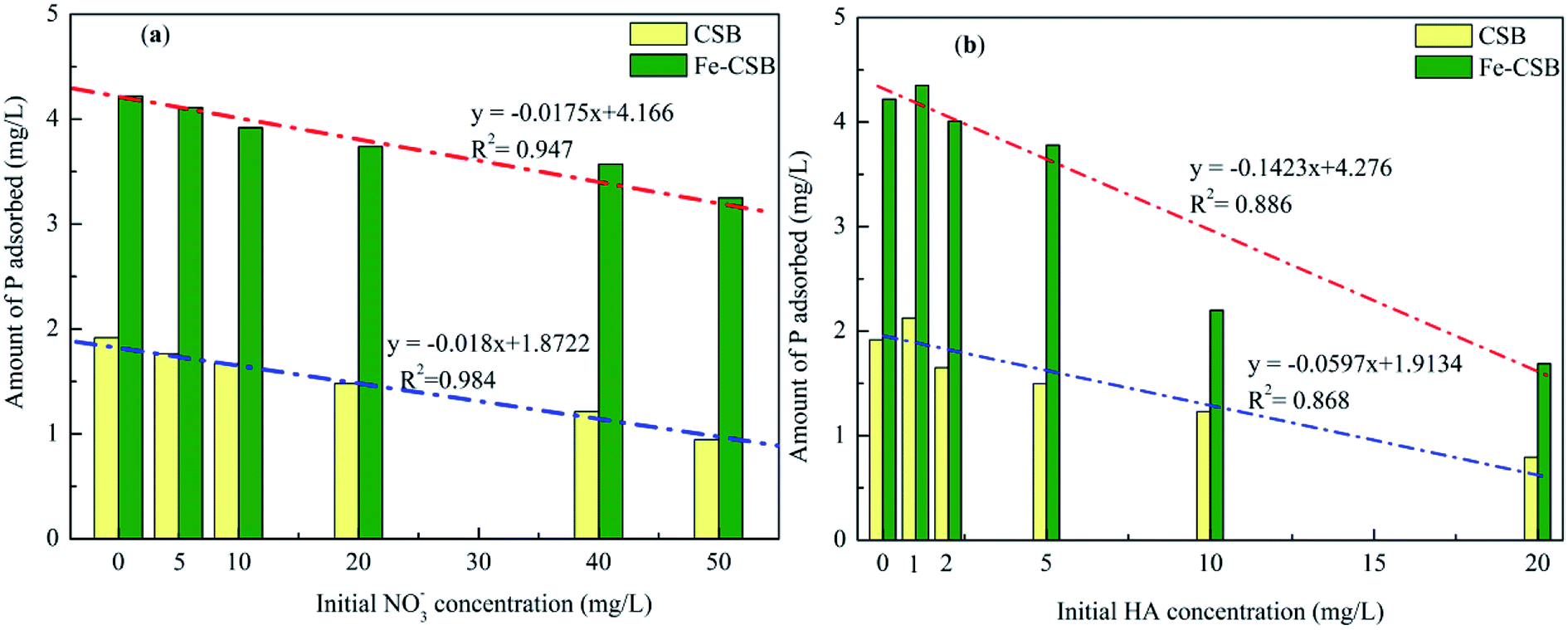 | ||
| Fig. 6 Effects of NO3− (a) and HA (b) on P adsorption on the CSB and Fe-CSB. | ||
As shown in Fig. 6(a), when the NO3− concentrations increased from 0 to 50 mg L−1, the adsorption amount of phosphate by the CSB and Fe-CSB was decreased by 51.0% and 23.0%, respectively. Obviously, the presence of the NO3− had a more severe effect on the phosphate adsorption by the CSB. This may be explained by the competitive adsorption of NO3−. Previous research revealed that anions adsorbed by the outer-sphere interactions of the adsorbent were substantially sensitive to the ionic strength of the solution in which they were immersed and that the adsorption process was clearly restrained by the competition with weakly adsorbing anions, such as NO3−, since electrolytes could also form outer-sphere complexes via electrostatic forces. Conversely, anions adsorbed by inner-sphere interactions of the adsorbent exhibited little sensitivity to the ionic strength of the solution.37 The observation was in line with the results from the analysis of the intraparticle diffusion model and indicated that inner-sphere diffusion played a key role in phosphate adsorption by Fe-CSB.
On the basis of the above results, it was concluded that a large amount of P (about 51%) was adsorbed by the exterior surface of the CSB, which could be easily influenced by the coexisting NO3−. On the other hand, on the Fe-CSB, most of the phosphate (about 77%) was adsorbed by the interior surface and could not be replaced by the NO3− coexisting in the solution. Meanwhile, although the adsorption capacity of Fe-CSB was considerably higher than that of the CSB, the lower amount of P adsorbed on the CSB (0.98 mg g−1) and Fe-CSB (0.97 mg g−1) was nearly equal when the NO3− concentrations increased from 0 to 50 mg L−1. The finding also confirmed that electrostatic attraction was the main mechanism of P adsorption on the CSB, but not on the Fe-CSB. Additionally, based on the high R2 values (0.984 and 0.947), the strong negative correlation between the amount of P adsorbed by the CSBs and the NO3− concentration was demonstrated (Fig. 6(a)). The results further confirmed that the adsorption of P by the CSB was largely governed by electrostatic attraction.
As shown in the Fig. 6(b), humic acid (HA) had a noticeable effect on the adsorption of phosphate. When the HA concentrations increased from 0 to 20 mg L−1, the amount of P adsorbed by the CSB and Fe-CSB decreased by 58.9% and 60.0%, respectively. The results may be ascribed to the competition or blocking effect of the surface sites of the CSBs by the HA.5,38 Antelo et al. revealed that HA and phosphate could compete for the goethite surface, thus explaining the adsorption of HA and the decrease of in phosphate adsorption in the presence of HA.39 Hiemstra et al. found that HA was the most effective competitor, and the higher number of functional groups increased oxyanion competition.40 Additionally, previous researchers have verified that ligand exchange (–COOH and –OH of humic acid versus –OH on the surface of materials) is the primary mechanism for HA adsorption on the surfaces of carbon and clay.41
Finally, for the above bi-solute adsorption conditions, a linear correlation was found between the amount of P adsorbed by the CSBs and the HA concentrations. The R2 values for the correlation describing P adsorption on the CSB and Fe-CSB in the HA/P bi-solute system were 0.886 and 0.868, respectively, which were clearly worse than those relative to the NO3−/P bi-solute system (R2 = 0.984 and 0.947). These results implied that the presence of HA could considerably influence the P adsorption by multiple reaction mechanisms. Violante et al. demonstrated that organic ligands could co-precipitate with OH–Al or OH–Fe groups on the surface of adsorbents and form organo–mineral complexes, the regenerative complexes differed in chemical composition, surface features, and reactivity toward phosphate.42
3.6. Desorption kinetics
To further evaluate the availability of the CSB and Fe-CSB for P adsorption, desorption studies were performed. The desorption of P from the CSB and Fe-CSB gradually increased with time 0.5 to 48 h, and the percentages of released P varied between 10.9–27.7% and 7.4–10.0%, respectively (Fig. S2†). Compared with the CSB, the amount of P desorbed from Fe-CSB was considerably lower, which proved that the Fe-CSB possessed higher P adsorption and immobilization capacities than the CSB. Since desorption is more likely to occur for physically adsorbed compounds, the above result also suggested that physical adsorption played a more important role in the phosphate uptake by the raw CSB, while chemical adsorption may have been the primary process on the Fe-CSB.43 Meanwhile, it is quite clear that phosphate adsorption by the Fe-CSB was not entirely reversible and the bonding between the Fe-CSB granules and phosphate anions was probably stable due to the high content of Fe in the Fe-CSB.24 This observation is in agreement with the reports of Zhao et al.43 and Zhang et al.,37 who noted that the phosphate adsorbed by iron oxide was relatively stable. Finally, NaOH solution was used to regenerate the Fe-CSB after the adsorption processes, and four adsorption–desorption cycles were carried out and the corresponding results are obtained in Fig. S3.† In the previous two cycles, the adsorbed phosphate could be effectively extracted by the NaOH solution, and 85.1% and 75.7% of phosphate could be adsorbed again on the Fe-CSB. After two cycles, the amount of adsorbed phosphate sharply decreased to 65.3% and 63.1%, indicating that part of active sites on the surface of Fe-CSB were lost. The implication of these results is that the Fe-CSB has the potential to be used as an effective bioretention medium for phosphate elimination from stormwater owing to its low cost and excellent adsorption capacity.3.7. Sorption mechanisms of P on the CSB and Fe-CSB
To achieve insight into the sorption mechanisms of P by the Fe-CSB, a series of additional analyses were conducted. Firstly, as presented in Table 1, the Fe-CSB contained a high content of Fe and some Al, Mg and Ca. These metal oxides may release metal ions into the solution and exist as Fe3+, Ca2+, Mg2+, and Al3+ under low initial pH conditions. Subsequently, surface precipitation probably enhanced the removal of phosphate through the formation of insoluble precipitates between the phosphate anions (H2PO4− and HPO42−, PO43−) and these metal ions, due to the low Ksp values of the precipitated (Table S5†).11,24,44Secondly, it was reported that the Fe hydroxide formed on the surfaces of Fe-based materials could easily be protonated in acid solutions (Fe–OH + H+ = Fe–OH2+), leading to the formation of positively-charged surfaces, while it was deprotonated in alkaline solutions (Fe–OH–H+ = Fe–O−), forming negatively-charged surfaces.45 As displayed in Fig. 5(b), the point of zero charge (pHpzc) value for the surface layer of Fe-CSB before phosphate adsorption was 7.6. Hence, the surface of the Fe-CSB was protonated and possessed a positive charge in the pH ranges of 3–7. As a result, the main phosphate anions (HPO42−, H2PO4−) could be strongly adsorbed by the Fe-CSB through electrostatic interaction.44 A further increase of the pH value of the solution, caused the gradual deprotonation of the Fe-CSB surface, which therefore carried a more negative charge. Consequently, the amount of phosphate adsorbed decreased dramatically (Fig. 5(a)). Besides, the zeta potential of Fe-CSB after phosphate adsorption clearly decreased from 7.6 to 5.1, owing to the collection of the negatively-charged phosphate anions on the surface of the Fe-CSB.24 In addition, it has been found that a strong negative correlation existed between the initial pH values and the amount of phosphate adsorbed by the Fe-CSB (R2 = 0.951, Fig. S2†). These results demonstrated that the electrostatic attraction between the phosphate anions and the positively charged surfaces may be an important mechanism for phosphate uptake by the Fe-CSB.
Thirdly, as discussed above (Section 3.4), the phosphate uptake by the Fe-CSB was notably influenced by the pH values of the solution. By comparing the initial and final pH values during the adsorption process, it was quite clear that the final pH values were obviously higher than the initial ones (Fig. 5(a)), suggesting that the hydroxyl groups on the surface of the Fe-CSB were replaced by the phosphate anions and were then released into the solution. Hence, the final pH values of the aqueous solutions strongly increased in the pH range of 3–7. The high adsorption capacity for phosphate also coincided with the significant increase of pH values under acidic condition. However, the final pH values increased only slightly when the initial pH was in the range of 9–12. The corresponding low phosphate adsorption capacity confirmed that only a few of the hydroxyl groups were exchanged with phosphate anions and released into the solution under alkaline conditions.33,41 These findings implied that the ligand exchange was a primary mechanism for phosphate removal by the Fe-CSB.
On the other hand, as mentioned above, phosphate adsorption by the Fe-CSB was slightly affected by the different concentrations of nitrate (0–50 mg L−1), suggesting that the inner-sphere complexation may have been involved in the phosphate uptake by the Fe-CSB.46,47 Furthermore, according to the results of the intraparticle diffusion model, the phosphate uptake process consisted of two steps. Firstly, the phosphate anions (H2PO4−, HPO42− and PO43−) were quickly adsorbed on the exterior surface of the Fe-CSB, being transferred from the liquid phase to the liquid–solid surface through ligand exchange and electrostatic interactions. Secondly, the P anions slowly penetrated into the inner-sphere of the Fe-CSB and inner-sphere complexes were formed by bonding with the hydroxide groups, abundantly present in the amorphous regions of the Fe-CSB. Besides, the increase in average pore diameter of the Fe-CSB after modification also favored the penetration of phosphate into the internal area.5,48 The FTIR spectra of the CSB and Fe-CSB before and after phosphate uptake were plotted in Fig. 7. After P adsorption, the FTIR spectrum of the Fe-CSB-P exhibited important differences compared to that of the Fe-CSB and CSB. With regards to the Fe-CSB-P, the appearance of three peaks at 618, 528 and 456 cm−1 was ascribed to the stretching vibration of Fe–O, which validated the involvement of the Fe–O bond in phosphate adsorption.49,50 The emergence of three asymmetric vibration peaks, relative to the P–O bonds, at 970, 1066, and 1167 cm−1 revealed that phosphate was firmly adsorbed on the surface of the metal oxide (Fe–O and Al–O) through the formation of monodentate and bidentate complexes on the inner-sphere surface of Fe-CSB.14,25,45,51,52 The absorption peak located at 3446 cm−1 may have been related to the –OH stretching vibration of hydrogen-bonded groups (H⋯H) and water molecules (H2O).45 The bands at 2920, 2850, and 1379 cm−1 were attributed to the CH2 group in biochars.53 The peak at 1735 cm−1 was ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the ester bond.53 The benzene ring CC stretching peaks at 1457 and 1542 cm−1 proved that aromatization occurred during the synthesis of the Fe-CSB.25 This finding was also consistent with the higher value of (O + N)/C in the Fe-CSB.
O stretching vibration of the ester bond.53 The benzene ring CC stretching peaks at 1457 and 1542 cm−1 proved that aromatization occurred during the synthesis of the Fe-CSB.25 This finding was also consistent with the higher value of (O + N)/C in the Fe-CSB.
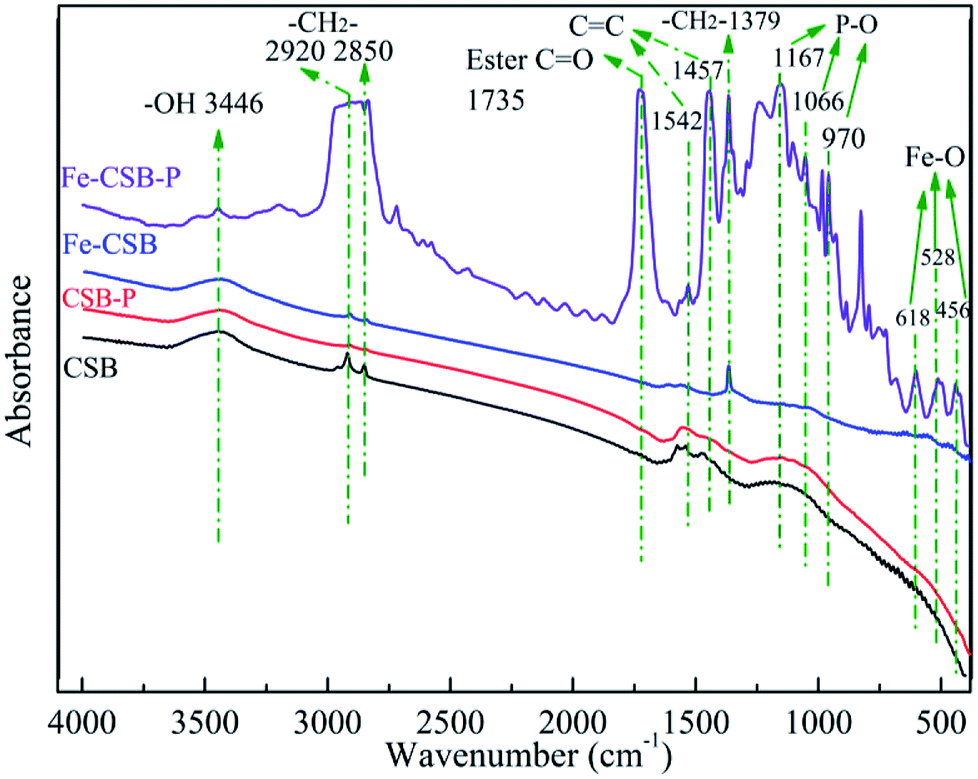 | ||
| Fig. 7 FTIR spectrum of the CSB and Fe-CSB before and after adsorption. | ||
The XPS spectra of Fe-CSB before and after adsorption of phosphate were analyzed, and the corresponding results were exhibited in Fig. 8. Observably, the O 1 s spectra showed that the peak at ∼532.2 eV shifted to lower binding energy (∼531.3 eV) after phosphate adsorption, and the relatively intensity increased from 65.5% to 74.3% (Fig. 8(a)). This shift agreed well with the formation of Fe–O–P bonds.54,55 The binding energy of the Fe 2p peak at ∼712.8 eV after phosphate adsorption was apparently higher than that of the Fe-CSB while similar to the reference samples of FePO4, which further confirmed that the Fe–O–P bonds were formed.56 Meanwhile, after phosphate adsorption, the percentages of –OH (∼530.3 eV) and –COOH (∼289.1 eV) decreased from 34.5% and 32.4% to 25.7% and 13%, respectively, which further confirmed that the hydroxyl groups on the surface of Fe-CSB were replaced by the phosphate ions. Based on the aforementioned evidences, four possible mechanisms that govern the adsorption process of phosphate from aqueous solutions by the novel Fe-CSB could be proposed and were illustrated in Fig. 9.
 | ||
| Fig. 8 XPS survey of Fe-CSB and Fe-CSB-P. High resolution XPS spectra of O 1s (a), Fe 2p (b), P 2p (c) and C 1s (d) for the samples. | ||
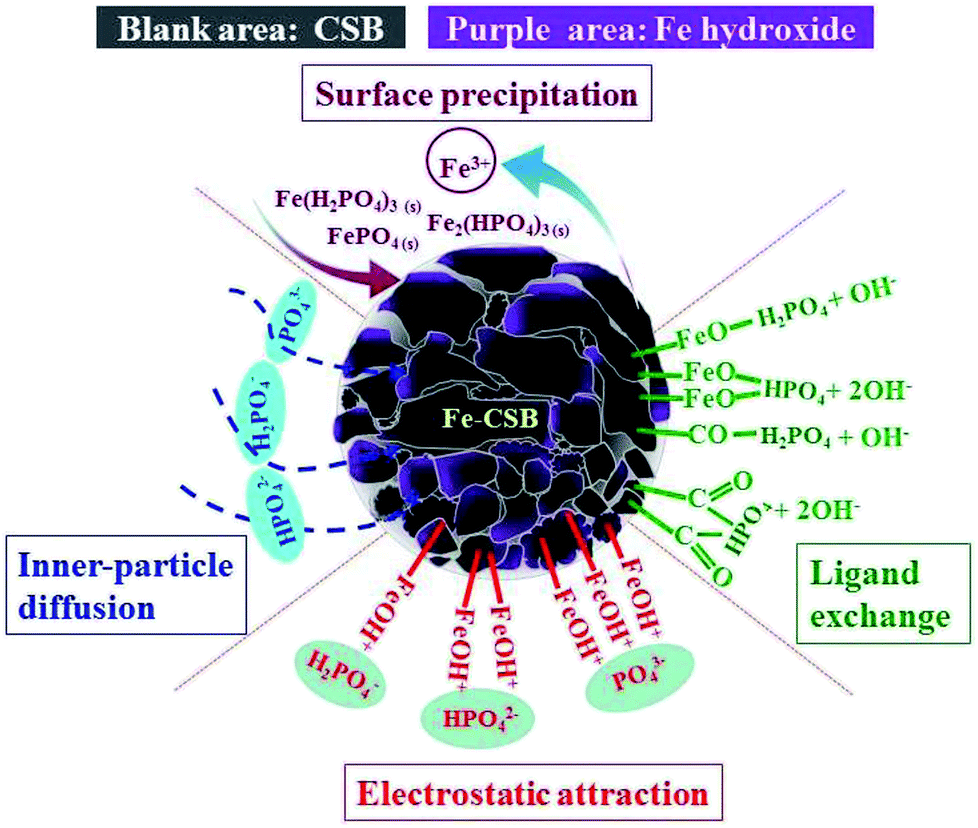 | ||
| Fig. 9 Possible mechanisms of phosphate adsorption onto Fe-CSB. | ||
4. Conclusions
This study revealed that the Fe-CSB deriving from coconut shell has excellent phosphate adsorption properties. The Fe-CSB showed high adsorption capacity up to 36.0 mg g−1, which was 2.73 times that of the pristine CSB. Adsorption isotherms were well fitted by Langmuir–Freundlich model and kinetics were represented by pseudo n-th order. Phosphate adsorption by the CSBs was an endothermic process, and the adsorption capacity was highly affected by the pH, the humic acid, and temperature. The results from mathematical modeling and characterization demonstrated that the phosphate adsorption by the Fe-CSB was primarily governed by four mechanisms: ligand exchange, electrostatic attraction, chemical precipitation and inner-sphere complexation. Thus, the Fe-CSB could be considered a promising alternative bioretention medium or an environmentally friendly material for phosphate removal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key Research and Development Program of China (2016YFC0400704) and the Logy Pillar Program of China (No. 2012BAC05B02). We are grateful to the anonymous reviewers for their valuable suggestions.References
- B. Pernet-Coudrier, W. Qi, H. Liu, B. Muller and M. Berg, Environ. Sci. Technol., 2012, 46, 5294–5301 CrossRef CAS PubMed.
- C. J. Vorosmarty, P. B. McIntyre, M. O. Gessner, D. Dudgeon, A. Prusevich, P. Green, S. Glidden, S. E. Bunn, C. A. Sullivan, C. R. Liermann and P. M. Davies, Nature, 2010, 467, 555–561 CrossRef CAS PubMed.
- N. B. Grimm, S. H. Faeth, N. E. Golubiewski, C. L. Redman, J. Wu, X. Bai and J. M. Briggs, Science, 2008, 319, 756–760 CrossRef CAS PubMed.
- A. J. Erickson, J. S. Gulliver and P. T. Weiss, Water Res., 2012, 46, 3032–3042 CrossRef CAS.
- Y. Yao, B. Gao, J. Chen and L. Yang, Environ. Sci. Technol., 2013, 47, 8700–8708 CrossRef CAS.
- Y. Tong, W. Zhang, X. Wang, R.-M. Couture, T. Larssen, Y. Zhao, J. Li, H. Liang, X. Liu, X. Bu, W. He, Q. Zhang and Y. Lin, Nat. Geosci., 2017, 10, 507–511 CrossRef CAS.
- W. W. Li, H. Q. Yu and B. E. Rittmann, Nature, 2015, 528, 29–31 CrossRef CAS PubMed.
- C. Hsieh and A. P. Davis, J. Environ. Eng., 2005, 131, 1521–1531 CrossRef CAS.
- J. Li and A. P. Davis, Water Res., 2016, 90, 141–155 CrossRef CAS PubMed.
- S. W. O'Neill and A. P. Davis, J. Environ. Eng., 2012, 138, 328–336 CrossRef.
- S. G. Lu, S. Q. Bai, L. Zhu and H. D. Shan, J. Hazard. Mater., 2009, 161, 95–101 CrossRef CAS PubMed.
- W. Huang, S. Wang, Z. Zhu, L. Li, X. Yao, V. Rudolph and F. Haghseresht, J. Hazard. Mater., 2008, 158, 35–42 CrossRef CAS PubMed.
- P. L. Sibrell, G. A. Montgomery, K. L. Ritenour and T. W. Tucker, Water Res., 2009, 43, 2240–2250 CrossRef CAS.
- H. Liu, X. Sun, C. Yin and C. Hu, J. Hazard. Mater., 2008, 151, 616–622 CrossRef CAS.
- E. Hiller, V. Tatarkova, A. Simonovicova and M. Bartal, Chemosphere, 2012, 87, 437–444 CrossRef CAS PubMed.
- O. S. Amuda, A. A. Giwa and I. A. Bello, Biochem. Eng. J., 2007, 36, 174–181 CrossRef CAS.
- J. Zhang, M. Lu, J. Wan, Y. Sun, H. Lan and X. Deng, Biochem. Eng. J., 2018, 130, 104–112 CrossRef CAS.
- W. J. Liu, H. Jiang, K. Tian, Y. W. Ding and H. Q. Yu, Environ. Sci. Technol., 2013, 47, 9397–9403 CrossRef CAS PubMed.
- R. Li, J. J. Wang, B. Zhou, Z. Zhang, S. Liu, S. Lei and R. Xiao, J. Cleaner Prod., 2017, 147, 96–107 CrossRef CAS.
- M. Arias, J. Da Silva-Carballal, L. Garcia-Rio, J. Mejuto and A. Nunez, J. Colloid Interface Sci., 2006, 295, 65–70 CrossRef CAS PubMed.
- J. Yan, T. Jiang, Y. Yao, S. Lu, Q. Wang and S. Wei, J. Environ. Sci., 2016, 42, 152–162 CrossRef PubMed.
- Y. Wu, Y. Fan, M. Zhang, Z. Ming, S. Yang, A. Arkin and P. Fang, Biochem. Eng. J., 2016, 105, 27–35 CrossRef CAS.
- L. Chen, X. L. Chen, C. H. Zhou, H. M. Yang, S. F. Ji, D. S. Tong, Z. K. Zhong, W. H. Yu and M. Q. Chu, J. Cleaner Prod., 2017, 156, 648–659 CrossRef CAS.
- L. Zeng, X. Li and J. Liu, Water Res., 2004, 38, 1318–1326 CrossRef CAS.
- R. Li, J. J. Wang, B. Zhou, M. K. Awasthi, A. Ali, Z. Zhang, L. A. Gaston, A. H. Lahori and A. Mahar, Sci. Total Environ., 2016, 559, 121–129 CrossRef CAS PubMed.
- N. Boujelben, J. Bouzid, Z. Elouear, M. Feki, F. Jamoussi and A. Montiel, J. Hazard. Mater., 2008, 151, 103–110 CrossRef CAS PubMed.
- R. Weerasooriya, H. J. Tobschall, W. Seneviratne and A. Bandara, J. Hazard. Mater., 2007, 147, 971–978 CrossRef CAS PubMed.
- Q. Sun and L. Yang, Water Res., 2003, 37, 1535–1544 CrossRef CAS PubMed.
- J. Lalley, C. Han, X. Li, D. D. Dionysiou and M. N. Nadagouda, Chem. Eng. J., 2016, 284, 1386–1396 CrossRef CAS.
- Z. Wang, X. Yu, B. Pan and B. Xing, Environ. Sci. Technol., 2010, 44, 978–984 CrossRef CAS PubMed.
- M. Alkan, M. Doğan, Y. Turhan, Ö. Demirbaş and P. Turan, Chem. Eng. J., 2008, 139, 213–223 CrossRef CAS.
- Q. Yue, Y. Zhao, Q. Li, W. Li, B. Gao, S. Han, Y. Qi and H. Yu, J. Hazard. Mater., 2010, 176, 741–748 CrossRef CAS PubMed.
- K. W. Jung, T. U. Jeong, H. J. Kang and K. H. Ahn, Bioresour. Technol., 2016, 211, 108–116 CrossRef CAS PubMed.
- P. Liao, S. Yuan, W. Xie, W. Zhang, M. Tong and K. Wang, J. Colloid Interface Sci., 2013, 390, 189–195 CrossRef CAS PubMed.
- G. Yu, B. Zhang, X. Lu, J. Li, J. Chen and J. Zuo, Arabian J. Geosci., 2015, 8, 3491–3499 CrossRef CAS.
- E. Risch, J. Gasperi, M. C. Gromaire, G. Chebbo, S. Azimi, V. Rocher, P. Roux, R. K. Rosenbaum and C. Sinfort, Water Res., 2018, 128, 412–423 CrossRef CAS PubMed.
- G. Zhang, H. Liu, R. Liu and J. Qu, J. Colloid Interface Sci., 2009, 335, 168–174 CrossRef CAS.
- I. Safarik, K. Pospiskova, E. Baldikova and M. Safarikova, Biochem. Eng. J., 2016, 116, 17–26 CrossRef CAS.
- J. Antelo, F. Arce, M. Avena, S. Fiol, R. López and F. Macías, Geoderma, 2007, 138, 12–19 CrossRef CAS.
- T. Hiemstra, S. Mia, P. B. Duhaut and B. Molleman, Environ. Sci. Technol., 2013, 47, 9182–9189 CrossRef CAS PubMed.
- X. Wang, L. Shu, Y. Wang, B. Xu, Y. Bai, S. Tao and B. Xing, Environ. Sci. Technol., 2011, 45, 9276–9283 CrossRef CAS PubMed.
- A. Violante, M. Pigna, M. Ricciardella and L. Gianfreda, in Developments in Soil Science, ed. A. Violante, P. M. Huang, J. M. Bollag and L. Gianfreda, Elsevier, 2002, vol. 28, pp. 279–295 Search PubMed.
- Y. Zhao, Q. Yue, Q. Li, B. Gao, S. Han and H. Yu, J. Hazard. Mater., 2010, 182, 309–316 CrossRef CAS PubMed.
- M. Mallet, K. Barthelemy, C. Ruby, A. Renard and S. Naille, J. Colloid Interface Sci., 2013, 407, 95–101 CrossRef CAS PubMed.
- Q. Yin, R. Wang and Z. Zhao, J. Cleaner Prod., 2018, 176, 230–240 CrossRef CAS.
- F. Gérard, Geoderma, 2016, 262, 213–226 CrossRef.
- J. Liu, R. Zhu, T. Xu, Y. Xu, F. Ge, Y. Xi, J. Zhu and H. He, Chemosphere, 2016, 144, 1148–1155 CrossRef CAS PubMed.
- C. C. Chusuei, D. W. Goodman, M. J. V. Stipdonk, D. R. Justes, K. H. Loh and E. A. Schweikert, Langmuir, 1999, 15, 7355–7360 CrossRef CAS.
- S. F. Lim, Y. M. Zheng and J. P. Chen, Langmuir, 2009, 25, 4973–4978 CrossRef CAS PubMed.
- C. Gong, D. Chen, X. Jiao and Q. Wang, J. Mater. Chem., 2002, 12, 1844–1847 RSC.
- F. Long, J.-L. Gong, G.-M. Zeng, L. Chen, X.-Y. Wang, J.-H. Deng, Q.-Y. Niu, H.-Y. Zhang and X.-R. Zhang, Chem. Eng. J., 2011, 171, 448–455 CrossRef CAS.
- K. A. Karimaian, A. Amrane, H. Kazemian, R. Panahi and M. Zarrabi, Appl. Surf. Sci., 2013, 284, 419–431 CrossRef CAS.
- B. Chen, D. Zhou and L. Zhu, Environ. Sci. Technol., 2008, 42, 5137–5143 CrossRef CAS PubMed.
- H. Fu, Y. Yang, R. Zhu, J. Liu, M. Usman, Q. Chen and H. He, J. Colloid Interface Sci., 2018, 530, 704–713 CrossRef CAS PubMed.
- L. Fang, R. Liu, J. Li, C. Xu, L. Z. Huang and D. Wang, Water Res., 2018, 130, 243–254 CrossRef CAS PubMed.
- A. Pacella, M. Fantauzzi, F. Turci, C. Cremisini, M. R. Montereali, E. Nardi, D. Atzei, A. Rossi and G. B. Andreozzi, Geochim. Cosmochim. Acta, 2014, 127, 221–232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10400j |
| This journal is © The Royal Society of Chemistry 2019 |