
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: TMEM88 inhibits fibrosis in renal proximal tubular epithelial cells by suppressing the transforming growth factor-β1/Smad signaling pathway
Huicong Li†
 *,
Yunqian Wang†,
Baoping Chen and
Jun Shi
*,
Yunqian Wang†,
Baoping Chen and
Jun Shi
Department of Nephrology, Huaihe Hospital of Henan University, No. 115 Ximen Street, Kaifeng 475000, Henan Province, China. E-mail: huiconcon@126.com; Fax: +86-0371-23906686; Tel: +86-0371-23906686
First published on 28th February 2019
Abstract
Transmembrane protein 88 (TMEM88) belongs to a member of the TMEM family, and was reported to be involved in fibrogenesis. However, the biological role of TMEM88 in renal fibrosis has not been elucidated. Therefore, the objective of this study was to investigate the effect of TMEM88 on cell proliferation and extracellular matrix (ECM) accumulation in a TGF-β1-induced human renal proximal tubular epithelial cell line (HK2). Our results showed that TMEM88 was downregulated in renal fibrotic tissues and TGF-β1-treated HK2 cells. In addition, TMEM88 overexpression inhibited TGF-β1-induced cell proliferation and migration in HK2 cells. Furthermore, TMEM88 overexpression reduced the production of α-SMA, collagen I, and collagen III in TGF-β1-stimulated HK2 cells. Mechanistically, TMEM88 overexpression suppressed the phosphorylation status of Smad2 and Smad3 in TGF-β1-stimulated HK2 cells. In conclusion, data from our experiments demonstrate that TMEM88 plays a pivotal role in the pathological process of renal fibrosis. TMEM88 inhibited fibrosis in renal proximal tubular epithelial cells by suppressing the TGF-β1/Smad signaling pathway.
1. Introduction
Renal fibrosis is a main pathological process of chronic kidney diseases progressing to end-stage renal disease.1 It is characterized by tubular atrophy, fibroblast/myofibroblast activation and the deposition of extracellular matrix (ECM).2 Until now, the efficacy of current strategies available for inhibiting or reversing renal fibrosis remains unsatisfactory.3,4 Transforming growth factor β1 (TGF-β1) is known as a key pro-fibrotic factor in renal fibrosis.5,6 During development of renal fibrosis, TGF-β1 can induce myofibroblastic activation and abundant deposition of collagen and fibronectin in renal tubulointerstitium.7 Thus, developing effective approaches to suppress TGF-β1-induced ECM accumulation is of significant importance for prevention of renal fibrosis.The transmembrane (TMEM) family of proteins plays important roles in regulating cell proliferation, inflammatory response, olfaction, neuronal excitability, smooth muscle contraction and nociception.8–11 Transmembrane protein 88 (TMEM88) belongs to a member of TMEMs family. Previous studies showed that it is expressed abnormally in several types of cancer and implicated in the development and progression of tumor.12–14 Besides, TMEM88 was reported to be involved in fibrogenesis. One study reported that the expression of TMEM88 was greatly reduced in human liver fibrotic tissues, and TMEM88 significantly repress the activation and proliferation of hepatic stellate cells (HSCs).15 However, the biological role of TMEM88 in renal fibrosis has not been elucidated. Therefore, the objective of this study was to investigate the effect of TMEM88 on cell proliferation and ECM accumulation in TGF-β1-induced human renal proximal tubular epithelial cells.
2. Materials and methods
2.1. Tissue sample collection
Fresh renal biopsy samples were obtained from ten healthy individuals (3 males and 7 females; mean age of 43.1 ± 4.7 years) and ten patients with renal fibrosis (4 males and 6 females; mean age of 45.8 ± 3.4 years) at the Department of Nephrology, Huaihe Hospital of Henan University (China). The stage of all patients was confirmed by means of clinical diagnosis as well as a series of examinations, and the assessment was blind to renal pathologists. All specimens were immediately frozen in liquid nitrogen and stored at −80 °C until analysis. All experiments were performed in accordance with the Guidelines of Huaihe Hospital of Henan University, and experiments were approved by the ethics committee at Huaihe Hospital of Henan University. Informed consents were obtained from human participants of this study.2.2. Cell culture
Human renal proximal tubular epithelial cell line (HK2) was obtained from the American Type Cell Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Sigma, St. Louis, MO, USA). All cells were grown at 37 °C under 5% CO2.2.3. Plasmid constructs and cell transfection
The full length cDNA-encoding sequence of TMEM88 was sub-cloned into pcDNA3.1 vectors, and pcDNA3.1 empty vector was used as a negative control. HK2 cells (1 × 105 cells per well) were seeded into 96-well plates, grown for 24 h to reach 80% confluence, and transfected with pcDNA3.1-TMEM88 or vector using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 24 h, according to the manufacturer's instructions.2.4. RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from tissues and cells using Trizol reagent (Invitrogen). 5 μg total RNA for each sample were reversed transcribed into first strand cDNA the First-Strand cDNA Synthesis Kit (Invitrogen) according to the manufacturer's instruction. qRT-PCR was performed on the ABI prism 7000 sequence detection system (Applied Biosystems, Eugene, OR, USA) using the SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's instructions. The specific primers for TMEM88 were forward, 5′-GAAGCTTATGCGCGAGTACAAAG-3′ and reverse, 5′-GGAATTCCTATTGTATGTTACATG-3′; and β-actin forward, 5′-GAGGCA CTCTTCCAGCCTTC-3′ and reverse, 5′-GGATGTCCACGTCACACTTC-3′. Relative quantification was evaluated using the method of 2−ΔΔCt.2.5. Western blot analysis
Tissues or cells were homogenized and lysed using RIPA lysis buffer (Beyotime, Nantong, China). Equal amount of protein was separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to a nitrocellulose membrane (Millipore). The membranes were blocked with 5% defatted milk in TBST buffer at room temperature for 1 h, and then incubated with primary antibodies overnight at 4 °C. The primary antibodies were anti-TMEM88, anti-p-Smad2, anti-Smad2, anti-p-Smad3, anti-Smad3 and anti-GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h. Finally, the antigen–antibody complexes were determined using an enhanced chemiluminescence (Gibco, Rockville, MD). The absorbance values of the target proteins were performed through Gel-Pro Analyzer version 4.0 software (Media Cybernetics, Silver Spring, MD, USA).2.6. Cell proliferation assay
Cell proliferation was evaluated using the MTT assay. Briefly, HK2 cells (1 × 104 cells per well) were seeded in 96-well plates and then transfected with TMEM88 or pcDNA3.1 for 24 h, respectively, followed by stimulation with TGF-β1 (10 ng mL−1) for 24 h. Next, 20 μL MTT solution (5 mg mL−1) was added into each well, and samples were incubated for 4 h at 37 °C. The medium was then removed from each well, and the resulting MTT formazan product was solubilized in 150 μL dimethylsulfoxide (DMSO). The absorbance was read at 490 nm using EL800 microplate reader (BioTek Instruments, Inc., Winooski, VT).2.7. Cell migration assay
Cell migration was performed using the Transwell chamber (Corning Costar Corp., Cambridge, MA, USA) assay. Briefly, the treated HK2 cells (1 × 104 cells per well) were suspended in serum-free medium and plated on the upper chamber, and the lower chamber was filled with 10% serum-containing DMEM. After 24 h incubation, cells on the top chamber were removed with a cotton swab. Migrated cells on the lower surface were fixed in 95% ethanol, stained with 0.1% crystal violet for 15 min and counted under a light microscope (Olympus, Tokyo, Japan).2.8. Enzyme-linked immunosorbent assay (ELISA)
HK2 cells were incubated with TMEM88 or vector for 24 h, and then treated with TGF-β1 (10 ng mL−1) for additional 24 h. Afterwards, the cell culture medium was centrifuged, and the supernatant was collected. The productions of type I collagen, α-SMA and fibronectin in cell supernatant were evaluated using commercially available ELISA kits, respectively, according to the manufacturer's instructions (Sigma).2.9. Statistical analysis
Statistical analyses were performed using the SPSS 17.0 (SPSS, Inc., Chicago, USA). All data are presented as the mean![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ±standard deviation (S.D.) from at least three independent experiments. Data were analyzed with a t-test, where the means between two groups were compared, or with one-way ANOVA plus Tukey's post hoc multiple-comparison test to compare mean values across multiple groups. p < 0.05 was considered significant.
±standard deviation (S.D.) from at least three independent experiments. Data were analyzed with a t-test, where the means between two groups were compared, or with one-way ANOVA plus Tukey's post hoc multiple-comparison test to compare mean values across multiple groups. p < 0.05 was considered significant.
3. Results
3.1. TMEM88 was downregulated in renal fibrotic tissues and TGF-β1-treated HK2 cells
We first examined the expression of TMEM88 in tissues of renal fibrosis. As indicated in Fig. 1A, TMEM88 mRNA was significantly lower in renal fibrosis tissues than that of in normal renal tissues. We also detected the protein expression of TMEM88 and found that TMEM88 protein level was also reduced in renal fibrosis tissues (Fig. 1B). We then performed qRT-PCR to detect TMEM88 mRNA in TGF-β1-treated HK2 cells. We found that stimulation with TGF-β1 in HK2 cells decreased TMEM88 mRNA levels (Fig. 1C). These findings were further supported by western blot results (Fig. 1D). | ||
| Fig. 1 TMEM88 was downregulated in renal fibrotic tissues and TGF-β1-treated HK2 cells. (A) TMEM88 mRNA was detected in renal fibrosis tissues and normal renal tissues. *P < 0.05 vs. Normal group. (B) TMEM88 protein was detected in renal fibrosis tissues and normal renal tissues. *P < 0.05 vs. Normal group. N, represents normal renal tissues; R, represents renal fibrosis tissues. HK2 cells were incubated with TGF-β1 for 24 h. (C and D) The expression of TMEM88 at both mRNA and protein was evaluated by qRT-PCR and western blot. *P < 0.05 vs. control group. | ||
3.2. TMEM88 inhibited the proliferation and migration in TGF-β1-treated HK2 cells
To further examine the role of TMEM88 in renal fibrosis, HK2 cells with overexpression of TMEM88 were established. After transfection, TMEM88 mRNA and protein levels were evaluated by qRT-PCR and western blot, respectively. TMEM88 mRNA and protein levels were markedly increased in HK2 cells transfected with overexpression of TMEM88, compared with the plasmid group (Fig. 2A and B). Then, we performed the MTT assay to evaluate the potential role of TMEM88 in cell proliferation. The results demonstrated that TGF-β1 treatment significantly induced the proliferation of HK2 cells, which was reversed by TMEM88 overexpression (Fig. 2C). Additionally, HK2 cells subjected to TGF-β1 displayed increased cell migration, as indicated by the Transwell migration assay, compared with the un-treated cells. However, overexpression of TMEM88 significantly inhibited TGF-β1-induced cell migration (Fig. 2D).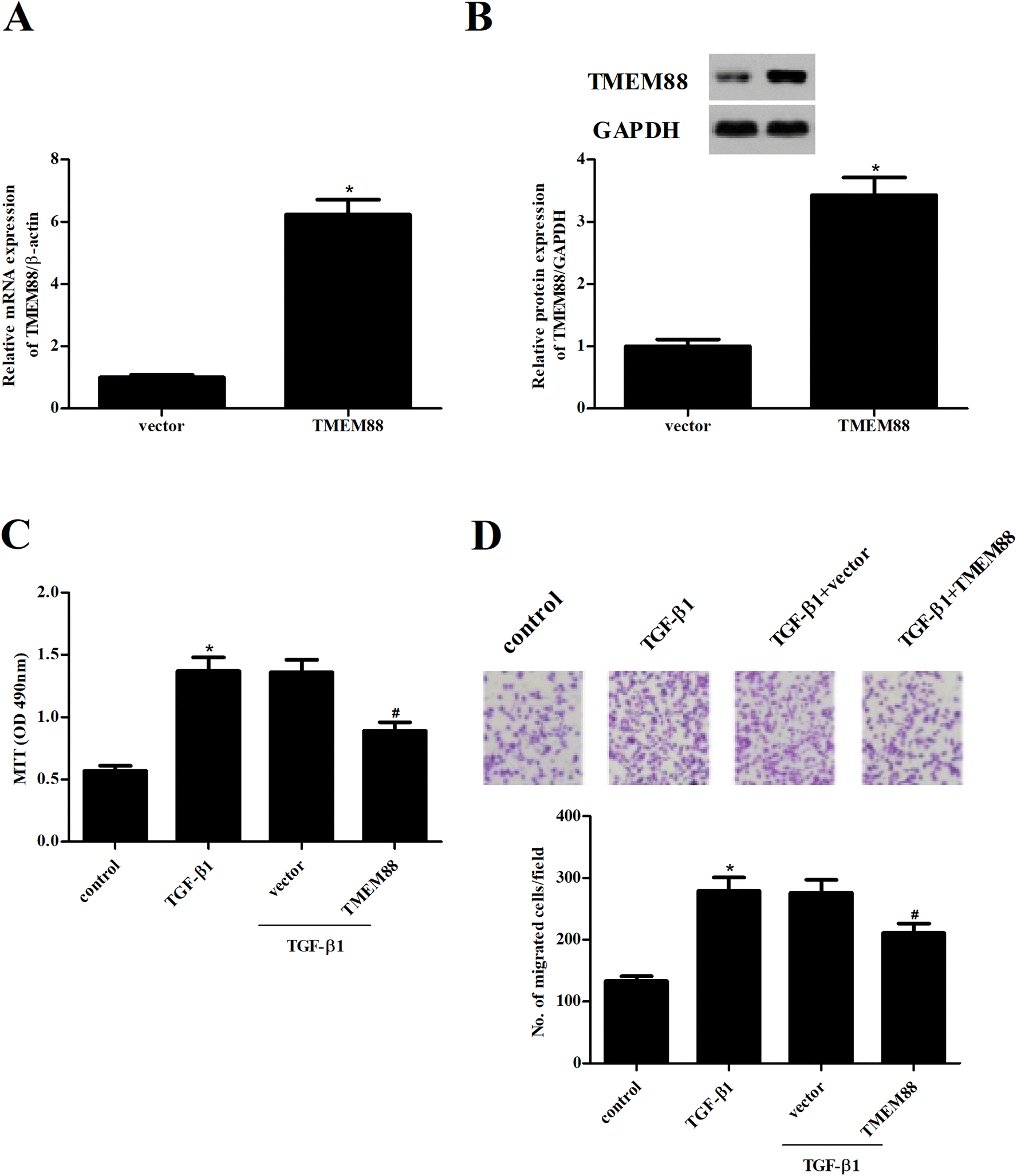 | ||
| Fig. 2 TMEM88 inhibited the proliferation and migration in TGF-β1-treated HK2 cells. HK2 cells were transfected with TMEM88 or vector for 24 h, respectively. (A and B) The transfection efficiency was evaluated by qRT-PCR and western blot. HK2 cells were incubated with TMEM88 or vector for 24 h, and then treated with TGF-β1 (10 ng mL−1) for additional 24 h. (C) Cell proliferation was detected using the MTT assay. (D) Cell migration was measured using Transwell migration assay. *P < 0.05 vs. control group, #P < 0.05 vs. TGF-β1 group. | ||
3.3. TMEM88 inhibited the production of ECM in TGF-β1-treated HK2 cells
Renal fibrosis is characterized by the excessive production and accumulation of ECM proteins, such as collagen and fibronectin. Thus, we examined the effect of TMEM88 on ECM expression in TGF-β1-stimulated HK2 cells using ELISA. As shown in Fig. 3A–C, the production of type I collagen, α-SMA and fibronectin was markedly increased in HK2 cells exposed to TGF-β1 stimulation. However, TMEM88 overexpression efficiently attenuated TGF-β1-mediated upregulation of ECM in HK2 cells. | ||
| Fig. 3 TMEM88 inhibited the production of ECM in TGF-β1-treated HK2 cells. HK2 cells were incubated with TMEM88 or vector for 24 h, and then treated with TGF-β1 (10 ng mL−1) for additional 24 h. (A–C) The production of type I collagen, α-SMA and fibronectin was evaluated using ELISA kits. *P < 0.05 vs. control group, #P < 0.05 vs. TGF-β1 group. | ||
3.4. TMEM88 inhibited the activation of Smad signaling in TGF-β1-treated HK2 cells
Finally, we sought to explore the molecular mechanism of how TMEM88 inhibits fibrogenesis caused by TGF-β1 in HK2 cells. The results of western blot analysis indicated that as compared with the un-treated group, TGF-β1 treatment significantly up-regulated phosphorylation of Smad2 and Smad3, but not the levels of Smad2, Smad3 in HK2 cells. However, TMEM88 significantly reduced the phosphorylation status of Smad2 and Smad3 in TGF-β1-stimulated HK2 cells (Fig. 4). | ||
| Fig. 4 TMEM88 inhibited the activation of Smad signaling in TGF-β1-treated HK2 cells. HK2 cells were incubated with TMEM88 or vector for 24 h, and then treated with TGF-β1 (10 ng mL−1) for additional 1 h. (A) The phosphorylation levels of Smad2 and Smad3 were detected using western blot. (B) Quantitative analysis of p-Smad2/Smad2. (C) Quantitative analysis of p-Smad3/Smad3. *P < 0.05 vs. control group, #P < 0.05 vs. TGF-β1 group. | ||
4. Discussion
In the present study, we showed that TMEM88 was downregulated in renal fibrotic tissues and TGF-β1-treated HK2 cells. In addition, TMEM88 overexpression inhibited TGF-β1-induced cell proliferation and migration in HK2 cells. Furthermore, TMEM88 overexpression reduced the production of α-SMA, collagen I, collagen III in TGF-β1-stimulated HK2 cells. Mechanistically, TMEM88 overexpression suppressed phosphorylation status of Smad2 and Smad3 in TGF-β1-stimulated HK2 cells.TGF-β1 is the most potent pro-fibrotic factor in the development of renal fibrosis.16 Herein, to establish an in vitro model of renal fibrosis, HK2 cells, a human renal proximal tubular epithelial cell line, were treated with TGF-β1. Our data showed that TGF-β1 treatment induced the proliferation/migration and ECM production in HK2 cells, suggesting that in vitro model of renal fibrosis was successful. Then, we used TGF-β1-stimulated HK2 cells to examine the role of TMEM88 in renal fibrosis, and results showed that TMEM88 was lowly expressed in renal fibrosis tissues and TGF-β1-treated HK2 cells. Our findings are consistent with previous studies showing that TMEM88 expression was down-regulated in the human liver fibrotic tissues, primary HSCs from fibrotic mice and activated HSC-T6 cells.15 In addition, proliferation and migration of renal proximal tubular epithelial cells play important roles in the pathological process of renal fibrosis.17 Our data showed TMEM88 overexpression inhibited the proliferation and migration of HK2 cells exposed to TGF-β1. These data implying that TMEM88 attenuated renal fibrosis in vitro through suppressing HK2 cell proliferation and migration.
Excessive deposition of ECM proteins is a hallmark of renal fibrosis.18 TGF-β1 is reported to induce renal fibrosis via the synthesis of ECM components.19,20 In addition, it was reported that TMEM88 effectively inhibits TGF-β1-inducing fibrosis-related gene expression in skin fibroblasts.21 Consistent with the results of previous studies, herein, we observed that TMEM88 overexpression reduced the production of α-SMA, collagen I, collagen III in TGF-β1-stimulated HK2 cells.
A growing body of evidence indicates that the TGF-β1/Smad pathway plays a critical role in progression of renal fibrosis.22–24 After binding to its receptor, Smad 2/3 is phosphorylated and binds with Smad4 to form multimers, then activated R-Smads translocate to the nucleus and induce the expression of target genes, such as collagen and fibronectin.25 In addition, Smad3 may be the most prominent downstream mediator in TGF-β-induced renal fibrosis.26 For example, a study showed that Smad3 inhibition could improve renal fibrosis in type 1 diabetic kidney disease.27 Thus, the block of TGF-β1/Smad pathway is an effective approach for preventing the progression of renal fibrosis. In the present study, we found that TMEM88 overexpression suppressed phosphorylation status of Smad2 and Smad3 in TGF-β1-stimulated HK2 cells. These observations suggest that the TGF-β1/Smad pathway was involved in TMEM88-regulated renal fibrosis in vitro.
5. Conclusion
In conclusion, data from our experiments demonstrate that TMEM88 plays a pivotal role in the pathological process of renal fibrosis. TMEM88 inhibited fibrosis in renal proximal tubular epithelial cells, at least in part, via suppressing the TGF-β1/Smad signaling pathway. This study only evaluated the effect of TMEM88 on an in vitro model of renal fibrosis, an in vivo study will be considered in the following studies.Conflicts of interest
The authors declare no conflicts of interest.References
- H. W. Schnaper, Front. Biosci., 2003, 8, e68 CrossRef CAS.
- Y. Liu, Kidney Int., 2006, 69, 213–217 CrossRef CAS PubMed.
- P. Boor, K. Šebeková, T. Ostendorf and J. Floege, Nephrol., Dial., Transplant., 2007, 22, 3391–3407 CrossRef CAS PubMed.
- B. M. Klinkhammer, R. Goldschmeding, J. Floege and P. Boor, Adv. Chronic. Kidney Dis., 2017, 24, 117–129 CrossRef PubMed.
- M. Iwano, Front. Biosci., 2010, 15, 229–238 CrossRef.
- Y. S. Kanwar, Am. J. Pathol., 2012, 1147–1150 CrossRef CAS PubMed.
- X. M. Meng, M. K. Tang, J. Li and H. Y. Lan, Front. Physiol., 2015, 6, 82 Search PubMed.
- J. Guo, L. Chen, N. Luo, W. Yang, X. Qu and Z. Cheng, Oncol. Rep., 2015, 33, 3124–3130 CrossRef CAS PubMed.
- T. Xu, L. X. Pan, Y. X. Ge, P. Li, X. M. Meng, C. Huang and J. Li, Inflammopharmacology, 2017, 1–10 Search PubMed.
- F. Huang, X. Wang, E. M. Ostertag, T. Nuwal, B. Huang, Y. N. Jan, A. I. Basbaum and L. Y. Jan, Nat. Neurosci., 2013, 16, 1284 CrossRef CAS PubMed.
- B. Manoury, A. Tamuleviciute and P. Tammaro, J. Physiol., 2010, 588, 2305–2314 CrossRef CAS PubMed.
- L. M. De, H. Cardenas, E. Vieth, R. Emerson, M. Segar, Y. Liu, K. Nephew and D. Matei, Gynecol. Oncol., 2016, 142, 539–547 CrossRef PubMed.
- X. Yu, X. Zhang, Y. Zhang, G. Jiang, X. Mao and F. Jin, OncoTargets Ther., 2015, 6, 25034–25045 Search PubMed.
- Y. X. Ge, C. H. Wang, F. Y. Hu, L. X. Pan, J. Min, K. Y. Niu, L. Zhang, J. Li and T. Xu, J. Cell. Physiol., 2017, 233, 79–87 CrossRef PubMed.
- S. P. Cai, X. Y. Cheng, P. J. Chen, X. Y. Pan, T. Xu, C. Huang, X. M. Meng and J. Li, Mol. Immunol., 2016, 80, 58–67 CrossRef CAS PubMed.
- W. Qin, A. C. K. Chung, X. R. Huang, X. Meng, D. S. C. Hui, C. Yu, J. J. Y. Sung and H. Y. Lan, J. Am. Soc. Nephrol., 2011, 22, 1462–1474 CrossRef CAS PubMed.
- M. Pang, L. Ma, N. Liu, M. Ponnusamy, T. C. Zhao, H. Yan and S. Zhuang, J. Cell. Biochem., 2011, 112, 2138–2148 CrossRef CAS PubMed.
- Y. Liu, Nat. Rev. Nephrol., 2011, 7, 684–696 CrossRef CAS PubMed.
- Z. Yao, S. Yang, W. He, L. Li, R. Xu, X. Zhang, H. Li, R. Zhan, W. Sun and J. Tan, Sci. Rep., 2015, 5, 17032 CrossRef CAS PubMed.
- B. Sutariya, D. Jhonsa and M. N. Saraf, Immunopharmacol. Immunotoxicol., 2016, 38, 39 CrossRef CAS PubMed.
- H. Zhao, F. Lu, S. Cui, X. Zhang, W. Wang, E. Si and Z. Yuan, Biomed. Pharmacother., 2017, 95, 1436–1440 CrossRef CAS PubMed.
- M. Sato, Y. Muragaki, S. Saika, A. B. Roberts and A. Ooshima, J. Clin. Invest., 2003, 112, 1486–1494 CrossRef CAS PubMed.
- Y. Zhang, S. Wang, S. Liu, C. Li and J. Wang, Int. Urol. Nephrol., 2015, 47, 1965–1975 CrossRef CAS PubMed.
- H. Y. Lan and C. K. Chung, Semin. Nephrol., 2012, 32, 236–243 CrossRef CAS PubMed.
- B. A. Hocevar, T. L. Brown and P. H. Howe, EMBO J., 2014, 18, 1345–1356 CrossRef PubMed.
- X. Qu, X. Li, Y. Zheng, Y. Ren, V. G. Puelles, G. Caruana, D. J. Nikolic-Paterson and J. Li, Am. J. Pathol., 2014, 184, 944–952 CrossRef CAS PubMed.
- J. Li, X. Qu, J. Yao, G. Caruana, S. D. Ricardo, Y. Yamamoto, H. Yamamoto and J. F. Bertram, Diabetes, 2010, 59, 2612–2624 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |