DOI:
10.1039/C8RA10365H
(Paper)
RSC Adv., 2019,
9, 13787-13796
Enhancing the photocatalytic activity of Cu0.25Zn0.75S nanodisks by metallic Ag loading in the visible-light region
Received
18th December 2018
, Accepted 5th April 2019
First published on 3rd May 2019
Abstract
Ag-loaded Cu0.25Zn0.75S (Ag/Cu0.25Zn0.75S) photocatalysts were synthesized for the photodegradation of organic pollutants such as rhodamine B (RhB), methyl violet (MV) and ciprofloxacin hydrochloride (CIP) under visible-light irradiation. Metallic Ag facilitated the enhancement of the photocatalytic activity of Cu0.25Zn0.75S nanodisks, and a Ag loading content of 11% exhibited great degradation efficiency for the degradation of RhB with the assistance of H2O2 under acidic conditions. This sample presented slight deactivation for the visible-light-driven degradation of RhB after five cycles. In addition, the excellent photocatalytic activity of Ag/Cu0.25Zn0.75S was obtained for the removal of MV and CIP. ·O2− is mainly responsible for the efficient activity of the photocatalytic process.
1. Introduction
Transition metal sulfides have attracted extensive attention in solar energy utilization and environmental protection.1–8 Among the transition metal sulfide semiconductors, ZnS and CuS are considered to be promising photocatalysts due to their low cost and nonexistent secondary pollution.4,9,10 Some studies have shown that the combination of ZnS with a wide band gap (3.72 eV for the cubic phase and 3.77 eV for the hexagonal phase) and CuS with a narrow band gap (2.2 eV) can make up for the defects of a single compound and significantly enhance photoelectric response performance and the visible-light photocatalytic activity.2,11–13 Pouretedal et al. reported that the visible-light-driven photoactivity of Zn1−xCuxS (Zn0.90Cu0.10S) nanocomposites was higher than that of Zn1−xNixS (Zn0.94Ni0.06S) nanocomposites.14 ZnS/CuS composites with a ZnS core and CuS shell prepared by the ion-exchange route exhibited inferior durability due to photocorrosion.5,15,16 A CuS/ZnS core/shell structure was developed to passivate the trap states of CuS and restrain the photocorrosion, which can efficiently decompose methylene blue and rhodamine B under visible-light irradiation.4,17,18 CuS–ZnS-based materials such as CuxZnyS films were prepared by pulsed laser deposition,19 spray pyrolysis,20 electrochemical deposition and photochemical deposition12,21–25 to enhance the visible-light photocatalytic capacity. Besides, Cu–Zn–S-sensitized TiO2 nanotube arrays (Cu–Zn–S/TiO2 NTAs) exhibited excellent photocatalytic activity compared with TiO2 NTAs.12
However, the wide band gap of CuxZnyS has been confirmed by some studies.24,26,27 The band gap can be tuned by elemental doping. Doping can facilitate the transmission of photo-generated electrons and prevent the photocorrosion of the ZnS–CuS composites. Shifu C. et al. prepared Ce2S3–ZnS–CuS by the ball milling method and found that the photocatalytic activity of Ce2S3–ZnS–CuS was much higher than that of ZnS–CuS.28 In addition, noble metals (such as Ag, Pt, Au) were frequently used as electron traps to promote the interfacial electron transfer processes.29–31 Among them, Ag was the most suitable in industrial applications for its high efficiency, low cost and corrosion resistance. The doping of Ag at an optimum amount can improve the photocatalytic activity of ZnS by increasing visible light absorption and enhancing the charge separation by trapping electrons.32–34 In addition to the doping, the loading of noble metals also showed better photocatalytic efficiency, such as in the Pt–BiOI nanoplates,29 Ag/Au-doped CeO2 nanoparticles,35 and AgAg2S–TiO2 nanocomposites.36
In our recent study, CuxZn1−xS nanodisks were prepared by a hydrothermal method and presented the enhanced photocatalytic activity of RhB compared with those of pure ZnS and CuS.37 Though CuxZn1−xS nanodisks had a narrow band gap, the catalytic activity and durability were affected by photocorrosion during the cycle testing process. Therefore, Ag/CuxZn1−xS composites were further designed to improve photocatalytic efficiency and stability. Different amounts of Ag were deposited on the surface of the Cu0.25Zn0.75S nanodisks by a photocatalytic deposition method, and the photocatalytic performance of the as-obtained Ag/Cu0.25Zn0.75S for the degradation of organic contaminants was investigated under visible-light irradiation.
2. Experimental section
2.1 Synthesis of the Cu0.25Zn0.75S nanodisk
The Cu0.25Zn0.75S nanodisk was synthesized by a one-step hydrothermal method, as described in our previous work.37 In a typical procedure, the total mass of Cu(NO3)2·3H2O and Zn(NO3)2·6H2O of 2, 4, 6 and 8 mmol with a Cu/Zn molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 15 mmol of thiourea and 1 g of PVP were dissolved in 160 mL acetonitrile solution (50% v/v) and stirred at room temperature for 2 h. The resultant solution was transferred into a 200 mL Teflon-lined autoclave, which was then maintained at different temperatures (140–200 °C) for different time periods (3–12 h) and was left to cool to room temperature. The product was directly centrifuged and the resulting supernatant was discarded. The product was washed with water and ethanol three times and vacuum-dried at 50 °C for 10 h, providing the sample in the powder form. The effects of the total mass of cupric nitrate and zinc nitrate, the reaction temperature and the reaction time on the structure of the Cu0.25Zn0.75S nanodisk were investigated.
:3, 15 mmol of thiourea and 1 g of PVP were dissolved in 160 mL acetonitrile solution (50% v/v) and stirred at room temperature for 2 h. The resultant solution was transferred into a 200 mL Teflon-lined autoclave, which was then maintained at different temperatures (140–200 °C) for different time periods (3–12 h) and was left to cool to room temperature. The product was directly centrifuged and the resulting supernatant was discarded. The product was washed with water and ethanol three times and vacuum-dried at 50 °C for 10 h, providing the sample in the powder form. The effects of the total mass of cupric nitrate and zinc nitrate, the reaction temperature and the reaction time on the structure of the Cu0.25Zn0.75S nanodisk were investigated.
2.2 Synthesis of the Ag/Cu0.25Zn0.75S catalyst
Ag/Cu0.25Zn0.75S was prepared by a photocatalytic deposition method.29 Brief descriptions are as follows. 0.1 g of Cu0.25Zn0.75S was dispersed in different concentrations of AgNO3 solution (the mass of Ag was 5%, 7%, 9%, 11% and 13% of Cu0.25Zn0.75S) by a sonicator and stirred at room temperature for 1 h. The resultant solution was irradiated by a 300 W metal halide lamp for 9 hours. The product was centrifuged and washed with water and ethanol three times. Finally, the sample was vacuum-dried at 50 °C for 10 h. These obtained samples were defined as 5-Ag/Cu0.25Zn0.75S, 7-Ag/Cu0.25Zn0.75S, 9-Ag/Cu0.25Zn0.75S, 11-Ag/Cu0.25Zn0.75S, and 13-Ag/Cu0.25Zn0.75S.
2.3 Characterization of the catalysts
Scanning electron microscopy (SEM) images were obtained using a JSM-5610LV/INCA scanning electron microscope operating at 20 kV. Powder X-ray diffraction (XRD) patterns were conducted with a Shimadzu LabX XRD-6000 X-ray diffractometer with Cu Kα radiation (1.54 Å) and a scanning speed of 5° min−1. The accelerating voltage and emission current were 40 kV and 100 mA, respectively. X-ray photoelectron spectra (XPS) were recorded by using an ESCALAB 250Xi (Thermo Fisher) with an operating voltage of 15 kV and a spot size of 400 μm. The Brunauer–Emmett–Teller (BET) specific surface areas were carried out using a Quantachrome NOVA-2020 analyzer. UV-vis diffuse reflectance spectra (DRS) were performed using a Shimadzu UV-2101 PC spectrophotometer in the range of 200–800 nm. The photoluminescence (PL) spectra were recorded by using a Hitachi F-4500 spectrometer. The actual contents of Cu, Zn and Ag in the catalysts were detected by an inductively coupled plasma emission spectrometer (ICP).
2.4 Photocatalytic reaction procedure
To measure the photocatalytic activity of the obtained samples, the photodegradation of RhB aqueous solutions were performed under visible-light irradiation using a 300 W xenon lamp with a UV cut-off filter (the output spectrum of 320–780 nm). 20 mg of the Ag/Cu0.25Zn0.75S samples were dispersed into 50 mL of 50 mg L−1 RhB solution. The suspensions were stirred for 1 h in the dark prior to irradiation to achieve the absorption–desorption equilibrium. 5 mL of the samples were taken out every 10 min and centrifuged for analysis using UV-vis spectroscopy (Unico UV-4802) during illumination. The impact of solution pH and hydrogen peroxide on the photodegradation of RhB were investigated. The stability of Ag/Cu0.25Zn0.75S with optimal performance was performed for five cycles according to the above reaction conditions. The used 11-Ag/Cu0.25Zn0.75S sample was washed with deionized water and ethanol and vacuum-dried at 60 °C for 6 h; it was then reused for the photodegradation of RhB. Moreover, the degradation efficiency of MV and CIP by 11-Ag/Cu0.25Zn0.75S with an optimal performance was compared with that of RhB under the same conditions. In addition, the active species (superoxide radical (·O2−), hydroxyl radical (·OH) and hole (h+)) were evaluated by adding to the free radical captures (p-BQ, t-BuOH, and EDTA-2Na), respectively.
3. Results and discussion
3.1 Characterization of the photocatalysts
The morphology and structure of the as-synthesized samples in different conditions were characterized by SEM, as shown in Fig. 1–3. The obtained Cu0.25Zn0.75S samples were uniform nanodisks. Temperature had a slight influence on the shape and structure of the products (Fig. 1). The crystallinity of the products increased with temperature (Fig. 1). Therefore, the reaction temperature in subsequent experiments was 200 °C. Upon further prolonging the reaction time, the diameter of the nanodisk increased, and extra crystal particles were deposited on the nanodisks (Fig. 2). The optimal reaction time was 6 h. The concentrations of Cu2+ and Zn2+ also influenced the morphology and scale of the nanodisks. At low concentrations, the formed CuS and ZnS nanoparticles were limited, which resulted in the incomplete growth of the Cu0.25Zn0.75S nanodisks (Fig. 3A–C). When the total mass of cupric nitrate and zinc nitrate was 4 mmol, the CuS and ZnS nanoparticles grew in an orientation to form perfect nanodisks (Fig. 3D–F). However, as the amount of Cu2+ and Zn2+ continued to increase, the excess nanoparticles were deposited onto the surface of the nanodisks (Fig. 3G–L). Thus, the optimal amount of Cu2+ and Zn2+ was 4 mmol. As shown in Fig. 4, the SEM results suggested that the Ag nanoparticles were dispersed on the surface of the Cu0.25Zn0.75S nanodisks.
 |
| | Fig. 1 SEM images of Cu0.25Zn0.75S samples prepared at different temperatures: 140 °C (A–C), 160 °C (D–F), 180 °C (G–I), 200 °C (J–L). | |
 |
| | Fig. 2 SEM images of Cu0.25Zn0.75S prepared with varying reaction times of 3 h (A–C), 6 h (D–F), 8 h (G–I), 12 h (J–L). | |
 |
| | Fig. 3 SEM images of Cu0.25Zn0.75S prepared with varying Cu and Zn total contents of 2 mmol (A–C), 4 mmol (D–F), 6 mmol (G–I), 8 mmol (J–L). | |
 |
| | Fig. 4 SEM images of 11-Ag/Cu0.25Zn0.75S. | |
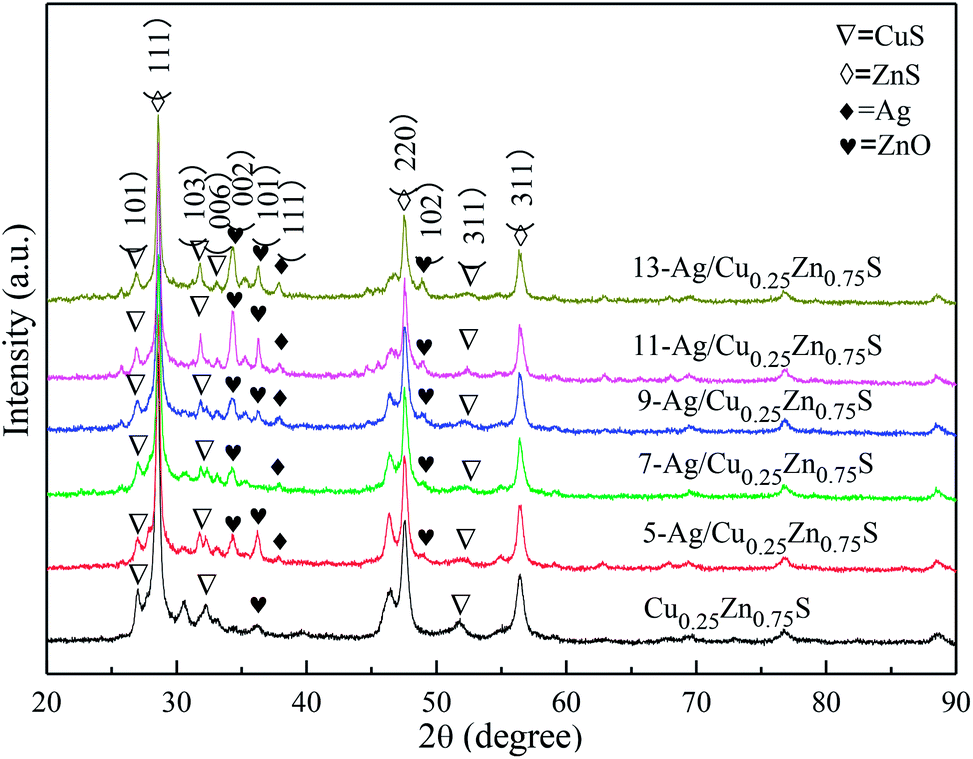
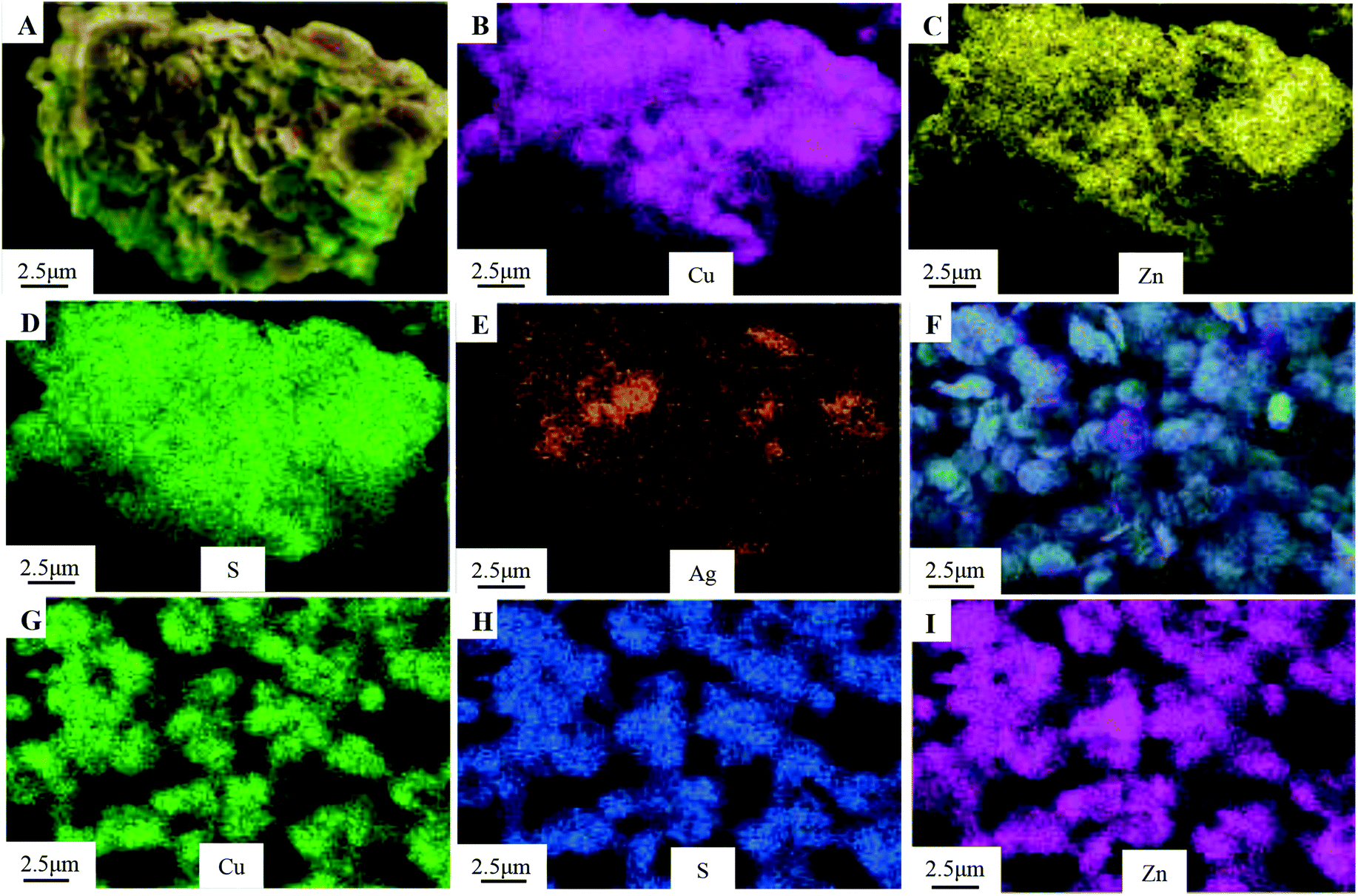
The XRD patterns of the as-obtained Cu0.25Zn0.75S and Ag/Cu0.25Zn0.75S samples loaded with 5–13% silver are shown in Fig. 5. The Cu0.25Zn0.75S composite consisted of CuS and ZnS crystals and a small amount of ZnO. The actual contents of Cu and Zn in Cu0.25Zn0.75S detected by ICP showed that the content of Cu and Zn was similar, and their content accounted for about 70 wt% of Cu0.25Zn0.75S (Table 2). This result also implied the existence of zinc oxide. The peaks at 2θ = 38.1° and 64.4° were indexed to the (111) and (220) planes of Ag with a cubic structure, and the intensity of the peaks was enhanced with increasing Ag amount. At low Ag content, the diffraction peaks of Ag were not apparent. The mapping images also indicated that Ag, Cu, Zn and S elements were observed in the Ag-loaded Cu0.25Zn0.75S samples (Fig. 6). The actual content of Ag loading was 7.48 wt% (Table 2). It is worth noting that the characteristic peak of ZnO was more obvious after loading Ag, especially in the 11-Ag/Cu0.25Zn0.75S sample. This suggests that the loading of Ag promotes the formation of ZnO. The reason may be that oxygen generated during the decomposition of silver nitrate contributes to the formation of ZnO. The actual contents of Cu and Zn in 11-Ag/Cu0.25Zn0.75S also showed that though the contents of Cu and Zn slightly decreased, the proportion in Cu0.25Zn0.75S increased. This also implied that part of the ZnS may be converted into ZnO.
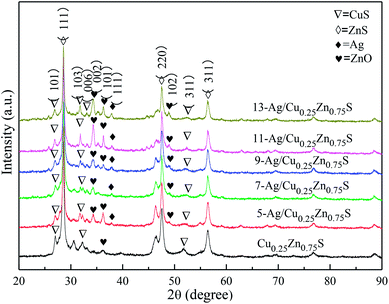 |
| | Fig. 5 XRD patterns of Ag/Cu0.25Zn0.75S and Cu0.25Zn0.75S. | |
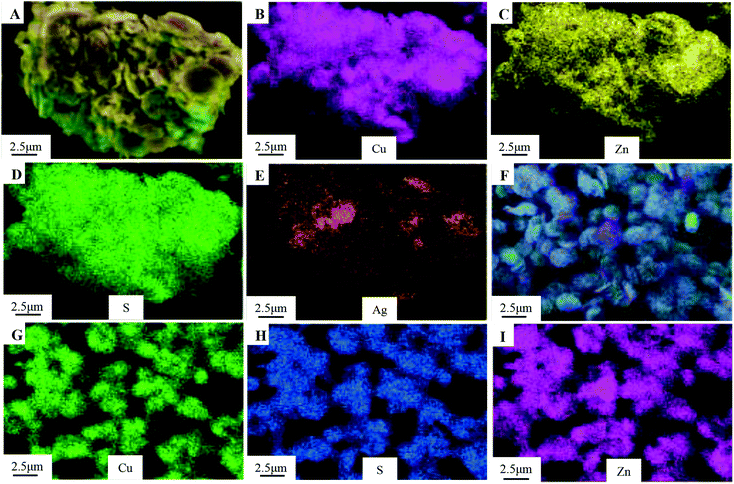 |
| | Fig. 6 Element mapping images of 11-Ag/Cu0.25Zn0.75S (A–E) and Cu0.25Zn0.75S (F–I). | |
Fig. 7 showed that the various Ag amounts of Ag/Cu0.25Zn0.75S catalysts displayed the type II hysteresis loop of the nitrogen adsorption–desorption curve. The BET surface areas of the Ag/Cu0.25Zn0.75S catalysts decreased with the increase in the Ag amount (Table 1). Fig. 8 showed the UV-visible absorption spectra of the Cu0.25Zn0.75S and Ag/Cu0.25Zn0.75S composites. In the UV-visible absorption spectra of Cu0.25Zn0.75S, a significant increase in the absorption at wavelengths shorter than 350 nm can be assigned to the intrinsic band-gap absorption of ZnS.5,38,39 The absorption between 500 nm and 800 nm can be attributed to the d–d transition of Cu2+, displaying the substitution of Zn2+ to form CuS.4 The loading of Ag enhanced the absorption of visible light. All Ag/Cu0.25Zn0.75S samples had a wide adsorption range from 250 to 800 nm, with especially strong absorption in the range of 700–800 nm, implying that these samples had good visible-light photocatalytic activity. The band gap energy values of all the samples were calculated using the Kubelka–Munk equation (Table 1). The estimated band gap energy value for 11-Ag/Cu0.25Zn0.75S was the lowest, which was 1.69 eV. This means that 11-Ag/Cu0.25Zn0.75S can more efficiently utilize visible light to drive photochemical reactions.6,40
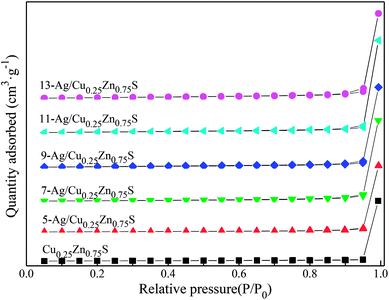 |
| | Fig. 7 N2 adsorption–desorption isotherms of Ag/Cu0.25Zn0.75S and Cu0.25Zn0.75S. | |
Table 1 Texture parameters of Cu0.25Zn0.75S and Ag/Cu0.25Zn0.75S
| Samples |
Specific surface area (m2 g−1) |
Pore size (nm) |
Pore volume (cm3 g−1) |
Band gap energya (eV) |
| The band gap energy of the Cu0.25Zn0.75S and Ag/Cu0.25Zn0.75S samples were calculated using the Kubelka–Munk equation. |
| Cu0.25Zn0.75S |
35.26 |
1.54 |
0.082 |
2.25 |
| 5-Cu0.25Zn0.75S |
31.34 |
1.47 |
0.071 |
1.93 |
| 7-Cu0.25Zn0.75S |
29.67 |
1.34 |
0.064 |
1.79 |
| 9-Cu0.25Zn0.75S |
27.32 |
1.44 |
0.059 |
1.75 |
| 11-Cu0.25Zn0.75S |
25.49 |
1.35 |
0.053 |
1.69 |
| 13-Cu0.25Zn0.75S |
24.32 |
1.31 |
0.051 |
1.83 |
Table 2 The actual contents of Cu, Zn and Ag in the Cu0.25Zn0.75S and 11% Ag/Cu0.25Zn0.75S samples detected by ICP
| sample |
Cu |
Zn |
Ag |
| Cu0.25Zn0.75S |
32.31% |
37.86% |
|
| 11% Ag/Cu0.25Zn0.75S |
31.44% |
35.90% |
7.48% |
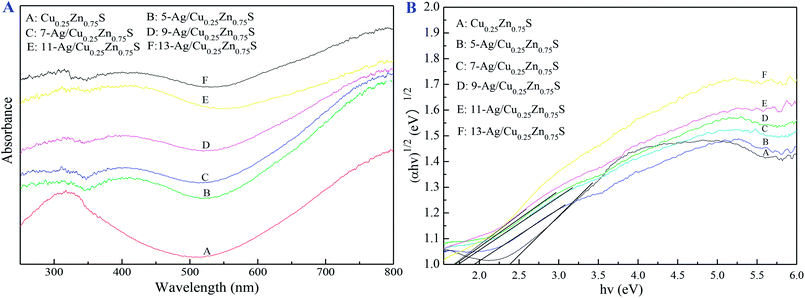 |
| | Fig. 8 UV-vis spectra of Cu0.25Zn0.75S and Ag/Cu0.25Zn0.75S. | |
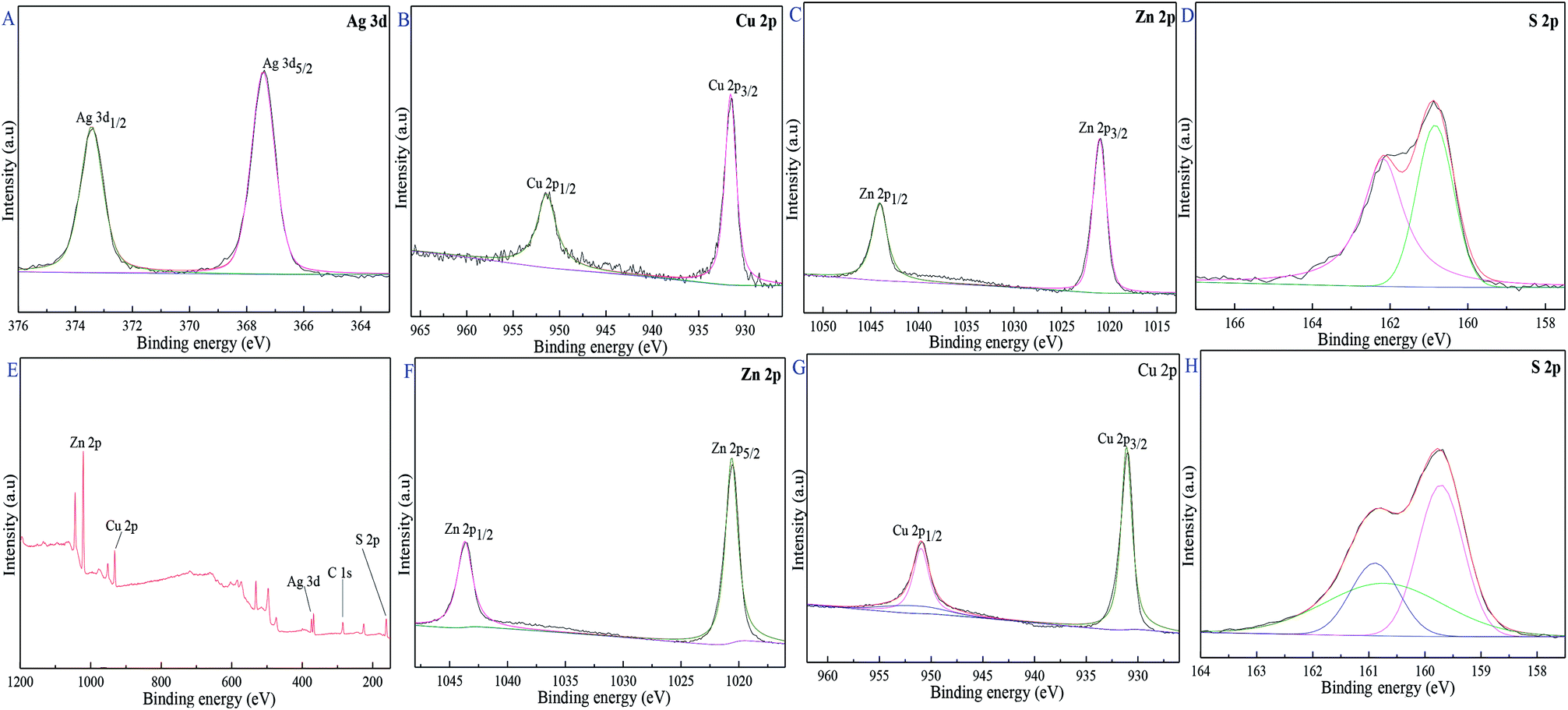
XPS was used to illuminate the surface composition and electronic state of Cu0.25Zn0.75S and 11-Ag/Cu0.25Zn0.75S samples. Fig. 9C and F showed that the two typical binding energy peaks of Zn 2p at 1020.9 8 eV and 1043.98 eV were ascribed to the binding energies of Zn 2p3/2 and Zn 2p1/2, respectively, indicating the presence of pure Zn2+ ions.7 The binding energy peaks of Cu 2p3/2 and Cu 2p1/2 at 931.48 eV and 951.08 eV corresponded to the typical values of Cu2+ in CuS (Fig. 9B and H).41 Fig. 9D and I showed that the peaks at 160.99 eV and 162.28 eV were attributed to S 2p3/2 and S 2p1/2, respectively, matching well with that of S2− ions.42–45 The characteristic binding energies of Ag 3d5/2 and 3d3/2 were 367.38 eV and 373.28 eV (Fig. 9A), respectively, implying the existence of zero valent Ag.46–48 These results indicated that the surface of 11-Ag/Cu0.25Zn0.75S sample mainly contained Zn2+, Cu2+, Ag and S2−.
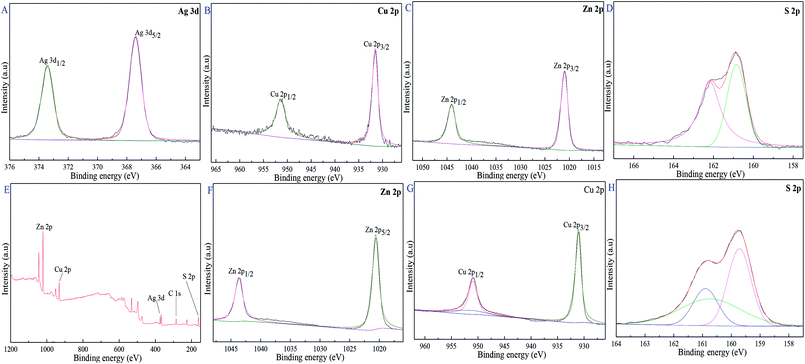 |
| | Fig. 9 XPS spectra of 11-Ag/Cu0.25Zn0.75S and Cu0.25Zn0.75S, 11-Ag/Cu0.25Zn0.75S (A–E), Cu0.25Zn0.75S (F–H). | |
3.2 Photocatalytic capacity
Ag/Cu0.25Zn0.75S was performed for the degradation of organic contaminants under visible-light irradiation, such as RhB, MV and CIP. In contrast with Cu0.25Zn0.75S, loading Ag could improve the photocatalytic activity of Cu0.25Zn0.75S due to its surface plasmon resonance effect. The photodegradation efficiency of RhB increased first and then decreased with the increase in the Ag loading content, of which 11-Ag/Cu0.25Zn0.75S exhibited the best photocatalyst activity for the removal of RhB (Fig. 10A). Ag nanoparticles, serving as the electron sink, were responsible for the efficient separation of photoexcited electron–hole pairs. However, high Ag content led to the overlapping agglomerates and reduced surface area.29 This negative effect was unsuitable for visible-light-driven photocatalytic performance. 11-Ag/Cu0.25Zn0.75S had better photocatalytic activity and efficiency compared with some similar catalysts, such as Cu2ZnSnS4–Pt and Cu2ZnSnS4–Au heterostructured nanoparticles,39 CuS/ZnS core/shell nanocrystals,18 Zn1−xCuxS and Zn1−xNixS nanoparticles,14 Ce2S3–ZnS–CuS nanoparticles,28 graphene oxide/Ag/Ag2S–TiO2 nanocomposites36 and ZnS–Ag2S ternary composites.7
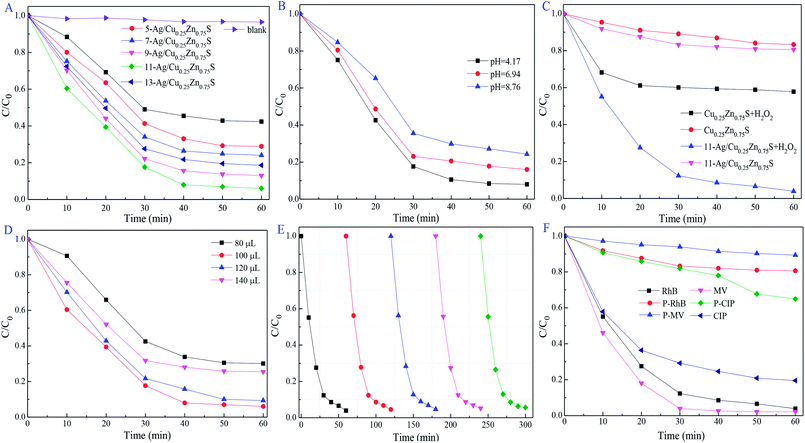 |
| | Fig. 10 Influence of Ag loading concentration in Ag/Cu0.25Zn0.75S samples (A), pH value (B), effects of H2O2 dosage (C and D), the cycling degradation (E) and different organic matter (F) on photodegradation activity. | |
pH plays an important role in the photocatalytic reactions for the removal of the pollutants.49 11-Ag/Cu0.25Zn0.75S had higher photocatalytic activity under acidic conditions (pH = 4.17) due to the modified surface charge of the photocatalyst and the maximized active radicals (Fig. 10B). The optimized pH condition may be suitable for the formation of ·OH and ·O2− radicals with the consumed e−/h+ pairs, inducing the promising photocatalytic reaction. In the photocatalytic system, ·O2− radicals generated from O2 molecules can react with H+ ions to form the ·OOH species, which were further reduced to H2O2 molecules. Subsequently, H2O2 can react with an e− to generate OH− ions and ·OH radicals, of which OH− ions are consumed by h+ to generate ·OH radicals. As the electron acceptor, moderate amounts of H2O2 promote the separation of electrons and holes and produces ·OH radicals. As shown in Fig. 10C and D, the best photodegradation activity for RhB removal was obtained by the presence of 100 μL of H2O2. The photocatalytic activity of 11-Ag/Cu0.25Zn0.75S increased first and then decreased with an increase in the H2O2 content due to the viable ·OH radicals scavenged by excess H2O2.50 The durability of 11-Ag/Cu0.25Zn0.75S for the visible-light-driven photocatalytic degradation of RhB changed little after five cycles, as shown in Fig. 10E. The morphology and structure of the used 11-Ag/Cu0.25Zn0.75S sample were not influenced during the cycle testing experiment (Fig. 11), indicating the superior stability of 11-Ag/Cu0.25Zn0.75S.
 |
| | Fig. 11 SEM images of 11-Ag/Cu0.25Zn0.75S after the photodegradation reaction. | |
To further investigate the photodegradation activity of 11-Ag/Cu0.25Zn0.75S on organic pollutants, MV and CIP were also carried out under the same conditions. As shown in Fig. 10F, MV was completely removed within 30 min, and the degradation efficiency of CIP was about 80% after 60 min under visible-light irradiation. It was concluded that 11-Ag/Cu0.25Zn0.75S was a potential photocatalyst for the removal of organic pollutants.
The trapping experiments were conducted to investigate the active radicals in the photocatalytic process of RhB removal, of which ethylenediamine tetraacetic acid disodium salt (EDTA-2Na), p-benzoquinone (p-BQ) and tert-butanol (t-BuOH) served as the trapping agents for h+, ·O2− and ·OH radicals, respectively.8,51–53 As shown in Fig. 12A, EDTA-2Na slightly suppressed the photodegradation activity, implying that the h+ radical was not crucial in the photocatalysis process. In contrast, only about 40% of RhB was decomposed by 11-Ag/Cu0.25Zn0.75S after 60 min in the presence of p-BQ. It was suggested that the ·O2− radical is the dominant oxidative species. The reduced degradation rate was also detected by the addition of t-BuOH, signifying the important role of the ·OH radical.
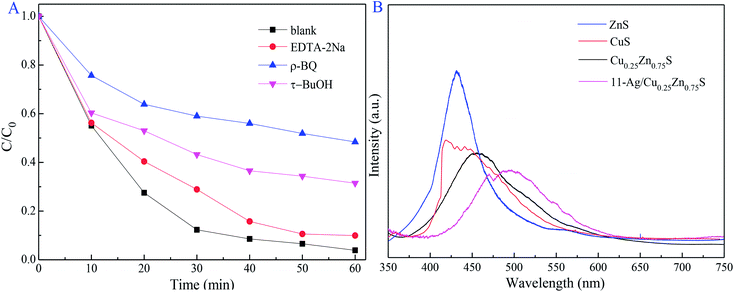 |
| | Fig. 12 Influence of the radical scavenger on photodegradation activity and PL spectra (A) of ZnS, CuS, Cu0.25Zn0.75S and (B) 11-Ag/Cu0.25Zn0.75S. | |
PL emission spectrum was widely used for illustrating the photo-generated electron–hole pair recombination process.54 Compared with CuS, ZnS and Cu0.25Zn0.75S, 11-Ag/Cu0.25Zn0.75S exhibited a lower photoluminescence intensity at room temperature (Fig. 12B). It was concluded that the low recombination of photoexcited electron–hole pairs led to the superior photocatalytic activity of 11-Ag/Cu0.25Zn0.75S.
4. Conclusion
Cu0.25Zn0.75S and Ag/Cu0.25Zn0.75S photocatalysts were prepared by the hydrothermal method and the photocatalytic deposition method, respectively. Ag loading enhanced the photocatalytic activity of the Cu0.25Zn0.75S nanodisks, and the optimal amount of Ag loading was 11 wt% (the actual content of Ag loading was 7.48 wt%). 11-Ag/Cu0.25Zn0.75S had great photocatalytic activity and stability for RhB, in addition to its excellent photodegradation effect on MV and CIP. ·O2− is mainly responsible for the efficient activity of the photocatalytic process. Ag loading can efficiently separate the photogenerated electrons and holes and promote the interfacial electron transfer processes to remarkably enhance the photocatalytic activity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support of this work by National Natural Science Foundation of China (Grant No: 21506103 and 51608512), Sichuan Science and Technology Program (Grant No: 2018JY0451), Science Foundation of the Education Department of Sichuan Province (Grant No: 17ZA0221), and Neijiang Science-technology Support Plan Projects.
References
- J. Zhang, J. Yu, Y. Zhang, Q. Li and J. R. Gong, Nano Lett., 2011, 11, 4774–4779 CrossRef CAS PubMed.
- P. Wang, Y. Gao, P. Li, X. Zhang, H. Niu and Z. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 15820–15827 CrossRef CAS PubMed.
- X. Xu, J. Bullock, L. T. Schelhas, E. Z. Stutz, J. J. Fonseca, M. Hettick, V. L. Pool, F. T. Kong, M. F. Toney and X. Fang, Nano Lett., 2016, 16, 1925–1932 CrossRef CAS PubMed.
- C. Feng, X. Meng, X. Song, X. Feng, Y. Zhao and G. Liu, RSC Adv., 2016, 6, 110266–110273 RSC.
- Y. B. Shao, L. H. Wang and J. H. Huang, RSC Adv., 2016, 6, 84493–84499 RSC.
- V. Selvaraj, RSC Adv., 2016, 6, 4227–4236 RSC.
- D. A. Reddy, R. Ma, M. Y. Choi and T. K. Kim, Appl. Surf. Sci., 2015, 324, 725–735 CrossRef.
- H. Wei, W. A. Mcmaster, J. Z. Y. Tan, L. Cao, D. Chen and R. A. Caruso, J. Phys. Chem. C, 2017, 121, 22114–22122 CrossRef CAS.
- D. P. Leonard, H. Pan and M. D. Heagy, ACS Appl. Mater. Interfaces, 2015, 7, 24543–24549 CrossRef CAS PubMed.
- M. Saranya, R. Ramachandran, E. J. J. Samuel, S. K. Jeong and A. N. Grace, Powder Technol., 2015, 279, 209–220 CrossRef CAS.
- M. Li, J. Su and L. Guo, Int. J. Hydrogen Energy, 2008, 33, 2891–2896 CrossRef CAS.
- T. T. Thanhthuy, P. Sheng, C. A. Huang, J. Li, L. Chen, L. Yuan, C. A. Grimes and Q. Cai, Chem. Eng. J., 2012, 210, 425–431 CrossRef.
- J. Kaur, M. Sharma and O. P. Pandey, Superlattices Microstruct., 2015, 77, 35–53 CrossRef CAS.
- H. R. Pouretedal and M. H. Keshavarz, J. Alloys Compd., 2010, 501, 130–135 CrossRef CAS.
- B. Zeng, X. Chen, C. Chen, X. Ning and W. Deng, J. Alloys Compd., 2014, 582, 774–779 CrossRef CAS.
- J. Yu, J. Zhang and S. Liu, J. Phys. Chem. C, 2010, 114, 13642–13649 CrossRef CAS.
- S. Harish, J. Archana, M. Navaneethan, S. Ponnusamy, A. Singh, V. Gupta, D. K. Aswal, H. Ikeda and Y. Hayakawa, RSC Adv., 2017, 7, 34366–34375 RSC.
- U. T. D. Thuy, N. Q. Liem, C. M. A. Parlett, G. M. Lalev and K. Wilson, Catal. Commun., 2014, 44, 62–67 CrossRef CAS.
- M. Feng, H. Zhou, W. Guo, D. Zhang, L. Ye, W. Li, J. Ma, G. Wang and S. Chen, J. Alloys Compd., 2018, 750, 750–756 CrossRef CAS.
- X. Xu, Q. Chen, H. Yuan, N. Yi, J. Chen, X. Zhu, Y. Yamaguchi and X. Dou, J. Mater. Sci.: Mater. Electron., 2017, 28, 18085–18089 CrossRef CAS.
- K. Yang and M. Ichimura, Jpn. J. Appl. Phys., 2011, 50, 584–587 Search PubMed.
- K. Yang, Y. Nakashima and M. Ichimura, J. Electrochem. Soc., 2012, 159, H250–H254 CrossRef CAS.
- A. Giaccherini, G. Montegrossi, F. D. Benedetto and M. Innocenti, Electrochim. Acta, 2018, 262, 135–143 CrossRef CAS.
- B. Tong and M. Ichimura, Jpn. J. Appl. Phys., 2016, 55, 098004 CrossRef.
- D. Man, K. Yang and M. Ichimura, Semicond. Sci. Technol., 2012, 27, 125007 CrossRef.
- A. M. Diamond, L. Corbellini, K. R. Balasubramaniam, S. Chen, S. Wang, T. S. Matthews, L. W. Wang, R. Ramesh and J. W. Ager, Phys. Status Solidi, 2012, 209, 2101–2107 CrossRef CAS.
- D. E. Ortíz-Ramos, L. A. González and R. Ramirez-Bon, Mater. Lett., 2014, 124, 267–270 CrossRef.
- C. Shifu, J. Mingsong and Y. Yunguang, J. Nanosci. Nanotechnol., 2012, 12, 4898–4904 CrossRef CAS PubMed.
- C. Yu, J. C. Yu, C. Fan, H. Wen and S. Hu, Mater. Sci. Eng., B, 2010, 166, 213–219 CrossRef CAS.
- L. Sun, S. Zhao, Z. Gao and Z. Cheng, Chem. J. Chin. Univ., 2017, 38, 907–914 CAS.
- P. Rai, S. M. Majhi, Y. T. Yu and J. H. Lee, RSC Adv., 2015, 5, 76229–76248 RSC.
- M. Kimi, L. Yuliati and M. Shamsuddin, Adv. Mater. Res., 2014, 1024, 368–371 Search PubMed.
- G. Murugadoss, R. Jayavel, M. R. Kumar and R. Thangamuthu, Appl. Nanosci., 2016, 6, 503–510 CrossRef CAS.
- J. Kaur, A. Gupta and O. P. Pandey, Sol. Energy, 2018, 176, 678–687 CrossRef CAS.
- M. Mittal, A. Gupta and O. P. Pandey, Sol. Energy, 2018, 165, 206–216 CrossRef CAS.
- S. Shuang, L. Ruitao, C. Xiaoyang, X. Zheng, Z. Jian and Z. Zhengjun, RSC Adv., 2018, 8, 5784–5791 RSC.
- X. Zheng, W. Fu, H. Peng and J. Wen, J. Environ. Chem. Eng., 2018, 6, 9–18 CrossRef CAS.
- S. Zu, Z. Wang, B. Liu, X. Fan and G. Qian, J. Alloys Compd., 2009, 476, 689–692 CrossRef CAS.
- X. Yu, A. Shavel, X. An, Z. Luo, M. Ibánez and A. Cabot, J. Am. Chem. Soc., 2014, 136, 9236–9239 CrossRef CAS PubMed.
- L. Wang, B. Cao, K. Wei, M. Hybertsen, K. Maeda, K. Domen and P. G. Khalifah, Inorg. Chem., 2013, 44, 9192–9205 CrossRef PubMed.
- C. Feng, L. Zhang, M. Yang, X. Song, H. Zhao, Z. Jia, K. Sun and G. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 15726 CrossRef CAS PubMed.
- X. J. Zhang, G. S. Wang, Y. Z. Wei, L. Guo and M. S. Cao, J. Mater. Chem. A, 2013, 1, 12115–12122 RSC.
- D. A. Reddy, D. H. Kim, S. J. Rhee, C. U. Jung, B. W. Lee and C. Liu, J. Alloys Compd., 2014, 588, 596–604 CrossRef.
- C. Chen, Y. Xie, G. Ali, S. H. Yoo and S. O. Cho, Nanoscale Res. Lett., 2011, 6, 462 CrossRef PubMed.
- W. Zhang, L. Zhang, Z. Hui, X. Zhang and Y. Qian, Solid State Ionics, 2000, 130, 111–114 CrossRef CAS.
- H. Yu, H. Yang, R. Yao and X. Z. Guo, Acta Phys.-Chim. Sin., 2014, 30, 1384–1390 CAS.
- H. Yang, Y. Liu, Q. Shen, L. Chen, W. You, X. Wang and J. Sheng, J. Mater. Chem., 2012, 22, 24132–24138 RSC.
- K. Nischala, T. N. Rao and N. Hebalkar, Colloids Surf., B, 2011, 82, 203–208 CrossRef CAS PubMed.
- M. López-Muñoz, J. Aguado, A. Arencibia and R. Pascual, Appl. Catal., B, 2011, 104, 220–228 CrossRef.
- L. Wei, C. Shifu, Z. Wei and Z. Sujuan, J. Hazard. Mater., 2009, 164, 154 CrossRef CAS PubMed.
- L. Yin, Y. Shi, L. Lu, R. Fang, X. Wan and H. Shi, Catalysts, 2016, 6, 69 CrossRef.
- Z. Chen, W. Wang, Z. Zhang and X. Fang, J. Phys. Chem. C, 2013, 117, 19346–19352 CrossRef CAS.
- F. T. Li, X. J. Wang, Y. Zhao, J. X. Liu, Y. J. Hao, R. H. Liu and D. S. Zhao, Appl. Catal., B, 2014, 144, 442–453 CrossRef CAS.
- H. Yamashita, A. Yuichi Ichihashi, M. Anpo, M. Hashimoto, C. Louis and M. Che, ChemInform, 1997, 28, 16041–16044 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Fuyan Kanga,
Hao Peng
a,
Fuyan Kanga,
Hao Peng