
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies for the synthesis of fluorinated polyesters†
Xiaoman Zhaoab,
Jennifer Noroc,
Jiajia Fuab,
Carla Silva*c and
Artur Cavaco-Paulo *abc
*abc
aJiangsu Engineering Technology Research Center for Functional Textiles, Jiangnan University, Wuxi 214122, P. R. China
bInternational Joint Research Laboratory for Textile and Fiber Bioprocesses, Jiangnan University, Wuxi 214122, P. R. China
cCentre of Biological Engineering, University of Minho, Campus de Gualtar, Braga 4710-057, Portugal. E-mail: carla.silva@ceb.uminho.pt; artur@deb.uminho.pt
First published on 15th January 2019
Abstract
In this work we synthetized three fluorinated polyesters from dimethyl tetrafluorosuccinate (DMTFS), dimethyl hexafluoroglutarate (DMHFG), and dimethyl octafluoroadipate (DMOFA) and ethylene glycol. The influence of parameters like monomer's size, temperature, vacuum, ultrasound and catalyst, on the polyesters synthesis was evaluated. The conversion rates were assessed considering 1H NMR data and the results disclose the role of ultrasound (US) as crucial to attain high reaction conversion rates (≈20% of increase relatively to the reactions performed in absence of US). The effect of US was more relevant for the higher molecular weight monomers (DMHFG and DMOFA). The use of Candida antarctica lipase (immobilized CALB) marginally favors the synthesis reactions when fixing the other conditions. The size of the starting monomers influenced greatly the reaction conversion rates, as shorter monomers gave rise to high amount of product recovering. All the produced polyesters were isolated and fully characterized by NMR (1H and 19F), FTIR, TGA and MALDI-TOF.
Introduction
Aliphatic polyesters belong to a group of biodegradable polymers with extensive applications in biomaterials, surgical sutures, controlled release carriers and coatings.1,2 This type of polyesters are also widely used as thermoplastics and thermoset resins. Among them, poly(lactic acid) (PLA) is the most well-known aliphatic polyester, which can be used in fibres, food packaging materials and durable goods, with a global demand of around 360 kilo tons in 2013.2 Poly(butylene succinate) (PBS) is another important polyester applied in packaging films and disposable cutlery, with a global market of around 10–15 kilo tons per year.1,2Commercially available polyesters are mainly produced through different methodologies which include condensation polymerization of aliphatic dicarboxylic acids with diols, transesterification reaction of diesters with diols, polymerization of hydroxyl acids, and ring-opening polymerization of lactones.3 The chemical catalysis is traditionally applied for the synthesis of polyester commercial products however with some undesirable properties, already recognized, which include the need of high temperatures to conduct the experiments and the use of toxic and low selective metal catalysts.3 Unlike chemical catalysts, enzymatic catalysis has been often conducted to produce these polyesters and is processed under mild reaction conditions enabling structure retention when polymerizing unstable monomers and circumventing the introduction of metals. It also provides selectivity by avoiding the protection–deprotection steps leading to exceptional options of structural control.1–4 Lipase-catalyzed polycondensation reactions have been explored focusing on reactions between diols and activated diesters, such as 2,2,2-trichloroethyl and vinyl esters.4,5 Significant progresses related with the lipase-catalysed condensation polymerization of conventional (unactivated) diacids and diols have been also reported.6,7 A high number of works have described the use of Candida antarctica lipase B (CALB) on the lipase-catalysed synthesis of aliphatic polyesters. Together with the choice of the starting monomers and the use of enzymes, the reactors used for polyester synthesis have been optimized in order to increase operational stability, production yield and process simplification.8 We have recently studied the effect of ultrasound and vacuum on the lipase-catalysed synthesis of poly(ethylene glutarate)9 and on the synthesis of poly(ethylene malonate) and poly(ethylene phthalate).10 On both studies the effect of ultrasound and vacuum was relevant to achieve higher levels of conversion and reduce the amount of enzyme in the process. Other examples of synthesis optimization rely on the application of different batch reactors, variable-volume view reactors and packed bed reactors to perform the continuous lipase-catalysed ring-opening polymerization of ε-caprolactone.11,12
Despite all the important achievements so far, a challenge related to the synthesis of shorter-chain substrates is still under study. As mentioned by Gross et al.1 the production of polymer structures from 4-carbon building blocks by lipase catalysis would provide semi crystalline materials, chemically sensitive, third-component monomers that enhance polymer performance. Moreover, specific polyester applications, like coatings, might impose the use of monomer species with differentiated properties, namely resistance to erosion, solvent resistance, high thermal stability and good weatherability.13
The fluorinated species may satisfy these demands by improving the hydrophobicity and oleophobicity of powder coatings. They have been considered on the production of high performance coatings on different substrates, showing a good protection against corrosion, weathering, and environmental pollution.14,15
In this study our goal was to produce three polyesters, namely poly(ethylene tetrafluorosuccinate), poly(ethylene hexafluoroglutarate), poly(ethylene octafluoroadipate) from dimethyl tetrafluorosuccinate, dimethyl hexafluoroglutarate, and dimethyl octafluoroadipate, respectively, and ethylene glycol. We aim to reduce the reaction time, often considered long for these polycondensation reactions, and eliminate the use of harmful organic solvents. We envisage to optimize the reactional conditions to obtain the highest conversion rates and for this different reactional conditions were tested: (a) lipase + vacuum; (b) lipase + ultrasound + vacuum, (c) vacuum, (d) ultrasound + vacuum. The polyesters obtained were characterized by 1H and 19F NMR – nuclear magnetic resonance, FTIR – Fourier-transform infrared spectroscopy, TGA – thermogravimetric analysis and MALDI-TOF – matrix-assisted laser ionization – time of flight.
Experimental
Materials and reagents
Fermase CALB™ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, a commercial Candida antarctica lipase B (CALB) immobilized on glycidyl methacrylate-ter-divinylbenzene-ter-ethylene glycol dimethacrylate (particle size of 150–300 μm, pore volume of 1.32 cm3 g−1, bulk density of 0.54 g cm−3 and an activity of 8000 Upropyl laurate/g) was received as a gift sample from Fermenta Biotech Ltd., Mumbai, India. Dimethyl tetrafluorosuccinate (DMTFS) (purity >95%), dimethyl hexafluoroglutarate (DMHFG) (purity >96%), and dimethyl octafluoroadipate (DMOFA) (purity >97%), were purchased from Tokyo Chemical Industry Co. Ltd., Japan. Ethylene glycol (EG) (purity ≥99%) was obtained from Sigma-Aldrich. Acetone (HPLC grade) and tetrahydrofuran (HPLC grade) were purchased from Fisher Scientific UK. Whatman® Filter paper was obtained from Whatman International Ltd., England. All the chemicals and enzyme were used directly without any further modification. An ultrasonic bath (US) (USC600TH, VWR International Ltd., USA) with frequency of 45 kHz and power of 120 W was used for reactions pre-incubation. A rotary vacuum evaporator (Vac) (Hei-VAP Advantage, Heidolph Instruments GmbH & Co. Germany) equipped with water bath and temperature controller was applied to conduct the enzymatic synthesis under vacuum at 100 rpm.
000, a commercial Candida antarctica lipase B (CALB) immobilized on glycidyl methacrylate-ter-divinylbenzene-ter-ethylene glycol dimethacrylate (particle size of 150–300 μm, pore volume of 1.32 cm3 g−1, bulk density of 0.54 g cm−3 and an activity of 8000 Upropyl laurate/g) was received as a gift sample from Fermenta Biotech Ltd., Mumbai, India. Dimethyl tetrafluorosuccinate (DMTFS) (purity >95%), dimethyl hexafluoroglutarate (DMHFG) (purity >96%), and dimethyl octafluoroadipate (DMOFA) (purity >97%), were purchased from Tokyo Chemical Industry Co. Ltd., Japan. Ethylene glycol (EG) (purity ≥99%) was obtained from Sigma-Aldrich. Acetone (HPLC grade) and tetrahydrofuran (HPLC grade) were purchased from Fisher Scientific UK. Whatman® Filter paper was obtained from Whatman International Ltd., England. All the chemicals and enzyme were used directly without any further modification. An ultrasonic bath (US) (USC600TH, VWR International Ltd., USA) with frequency of 45 kHz and power of 120 W was used for reactions pre-incubation. A rotary vacuum evaporator (Vac) (Hei-VAP Advantage, Heidolph Instruments GmbH & Co. Germany) equipped with water bath and temperature controller was applied to conduct the enzymatic synthesis under vacuum at 100 rpm.
CALB-catalyzed synthesis of polyesters from ethylene glycol and fluorinated di-ester substrates
Three different fluorinated polyesters, namely poly(ethylene tetrafluorosuccinate), poly(ethylene hexafluoroglutarate) and poly(ethylene octafluoroadipate) were respectively synthesized using different approaches: (1) pre-incubation of reactants (1:1) (molar ratio) (EG: 83 mg; DMTFS: 292 mg; DMHFG: 358 mg; DMOFA: 426 mg) under ultrasound for 1 h at 40 °C followed by 6 hours of vacuum at 40 °C or 70 °C, using 1% (w/v) of CALB; (2) incubation of reactants under vacuum for 7 hours at 40 °C or 70 °C, using 1% (w/v) of CALB; (3) pre-incubation of reactants (1:1) under ultrasound for 1 h at 40 °C followed by 6 hours of vacuum at 40 °C or 70 °C; (4) incubation of reactants under vacuum for 7 hours at 40 °C or 70 °C. The vacuum stage was ensured by placing the solutions in a rotary evaporator coupled with a vacuum pump with an end vacuum of 2 mbar. The reaction schemes related with the synthesis of the three polyesters are presented in Table 1.
 |
 |
 |
Isolation of the synthesized fluorinated polyesters
Aiming to separate the enzyme from the final reaction mixtures, tetrahydrofuran was firstly added to the final reaction solution and, when applied, the enzyme was filter out from the reaction system. After evaporation of the tetrahydrofuran, the reaction solution was collected using acetone and kept in extractor hood overnight to completely evaporate the acetone. The obtained fluorinated polyester products were prepared for 1H NMR analysis. The lyophilized final products were obtained as brown oils and were kept in the fridge for further characterization.Nuclear magnetic resonance – NMR
The 1H and 19F NMR spectra were acquired after dissolving the final products in 750 μL of deuterated chloroform (CDCl3, Cortecnet, France). The spectra were recorded using a Bruker Avance III 400 spectrometer (400 MHz, Bruker Corporation, Germany). The peak solvent was used as internal reference.Fourier transform infrared – FTIR
Fourier transform infrared (FTIR) spectra of the lyophilized products was recorded on a FTIR Bomem MB using NaCl cells. All the absorbance spectra were acquired in the range of ν 4000–500 cm−1 with a spectral resolution of 8 cm−1.Thermogravimetric analysis – TGA
TGA of synthesized polyesters and the starting materials was performed in a thermogravimetric analyser (PerkinElmer TGA 4000). The calibration was performed with metals, such as nickel, alumel and perkalloy, based on their Curie point reference. The temperature range was 30–600 °C (heating rate 10 °C min−1, sample weight: 10–13 mg) and the nitrogen flow rate was 20 mL min−1 with a gas pressure of 3.0 bar.16Matrix-assisted laser ionization – time of flight – MALDI-TOF
The polyesters were analysed by Matrix-Assisted Laser desorption/ionization with time-of-flight (MALDI-TOF) using 2,5-dihydroxybenzoic acid (DHB) as the matrix (≥99.5%). The mass spectra were acquired on an Ultra-flex MALDI-TOF mass spectrophotometer (Bruker Daltonics GmbH) equipped with a 337 nm nitrogen laser. For this, the samples were dissolved in a TA30 (30% acetonitrile/70% TFA) solution and mixed with a 20 mg mL−1 solution of DHB (1:1). Then a volume of 2 μL was placed in the ground steel plate (Bruker part no 209519) until dry. The mass spectra were acquired in linear negative mode.
Results and discussion
Effect of reactional conditions on the synthesis of fluorinated polyesters
The synthesis of fluorinated polymers is governed by different parameters including starting monomer's type and size, temperature, type of reactor, time, and presence of catalyst, among others. To optimize the synthesis of the three polyesters, it was imperative to study differentiated reactional conditions. Table 1 evidences the general synthetic strategy for the synthesis of the fluorinated polyesters. The synthesis reactions were carried with a fluorinated di-ester and ethylene glycol in an equimolar ratio. The copolymerization reactions were performed at different temperatures under ultrasound and vacuum in order to infer the best operational temperature. Following several works reported in literature,2,12,17 we started to test a high temperature, 70 °C, followed by testing 40 °C. The molar ratio of fluorinated di-esters to ethylene glycol was 1:1, and the catalyst was physically immobilized Candida antarctica lipase B (1% w/v). Increasing the ratio of one of the reagents did not led to a higher amount of polymer (data not shown).
In this work, we used di-esters with differentiated chain length, namely dimethyl tetrafluorosuccinate (DMTFS), 4 carbons, dimethyl hexafluoroglutarate (DMHFG), 5 carbons and dimethyl octafluoroadipate (DMOFA), 6 carbons, aiming to evaluate the role of the monomer's size on the final conversion rates and, when applied, to test the selectivity of the enzyme for these different substrates. After varying different processing conditions, namely temperature, time, ultrasound and enzyme, the conversion rates (calculated by 1H NMR) obtained are presented in Table 2.
| Starting reagents | Enzyme | Ultrasound | Vacuum | % Conversion ratea |
|---|---|---|---|---|
| a The values are the mean of 2 independent experiments. | ||||
| DMTFS:EG | — | 1 h; 40 °C | 6 h; 40 °C | 91.7 ± 1.2 |
| CALB | 1 h; 40 °C | 6 h; 40 °C | 94.2 ± 1.1 | |
| — | — | 7 h; 40 °C | 72.0 ± 0.9 | |
| CALB | — | 7 h; 40 °C | 72.7 ± 0.8 | |
| — | 1 h; 70 °C | 6 h; 70 °C | 85.9 ± 0.8 | |
| CALB | 1 h; 70 °C | 6 h; 70 °C | 100 ± 0.8 | |
| — | — | 7 h; 70 °C | 85.7 ± 1.3 | |
| CALB | — | 7 h; 70 °C | 97.4 ± 1.5 | |
| DMHFG:EG | — | 1 h; 40 °C | 6 h; 40 °C | 95.3 ± 0.7 |
| CALB | 1 h; 40 °C | 6 h; 40 °C | 90.1 ± 0.6 | |
| — | — | 7 h; 40 °C | 59.7 ± 1.2 | |
| CALB | — | 7 h; 40 °C | 63.5 ± 1.3 | |
| — | 1 h; 70 °C | 6 h; 70 °C | 89.4 ± 1.5 | |
| CALB | 1 h; 70 °C | 6 h; 70 °C | 80.4 ± 1.6 | |
| — | — | 7 h; 70 °C | 78.5 ± 1.5 | |
| CALB | — | 7 h; 70 °C | 88.9 ± 1.5 | |
| DMOFA:EG | — | 1 h; 40 °C | 6 h; 40 °C | 81.3 ± 1.3 |
| CALB | 1 h; 40 °C | 6 h; 40 °C | 85.7 ± 1.3 | |
| — | — | 7 h; 40 °C | 43.1 ± 1.8 | |
| CALB | — | 7 h; 40 °C | 36.6 ± 1.9 | |
| — | 1 h; 70 °C | 6 h; 70 °C | 32.6 ± 1.9 | |
| CALB | 1 h; 70 °C | 6 h; 70 °C | 83.7 ± 1.9 | |
| — | — | 7 h; 70 °C | 28.5 ± 2.0 | |
| CALB | — | 7 h; 70 °C | 66.3 ± 2.0 | |
From the data obtained one can infer that the synthesis performed in the presence of CALB is not favoured for all the cases studied. Moreover, even when higher conversion rates are obtained, the differences displayed are not significant and do not justify the use of a catalysts on the reaction. Contrarily, ultrasound played a determinant role on the polyesters biosynthesis, by improving the conversion rates of about 20%, in comparison with the experiments performed only under vacuum. The effect of ultrasound on the materials conversion is directly proportional to the molecular weight of the starting fluorinated di-ester, as can be seen by the conversion difference obtained without and with US. Apparently, 1 hour of ultrasound and 6 hours of vacuum are suitable to achieve high levels of conversion of around 90%. It is unquestionable and documented that the increase of the mass transfer, inherent to ultrasound pre-treatment, is crucial for the polymerization to go further.18 Considering the immiscibility of the reactants, the application of ultrasonication to the reactional mixture allowed the collapse of the cavitation bubbles producing significant structural and mechanical changes leading to the formation of a one-phase emulsion. Herein, when considering the heterogeneous system with immobilized enzyme CALB and the immiscible starting reactants, the reaction between ethylene glycol and the fluorinated di-esters was favoured and the interexchange of chemical groups took place by mass transfer effects promoted by cavitation and proper mixing with agitation. Considering previous related works, we may assume that ultrasound plays dual role in creating higher interfacial area of synthesis as well as in facilitating the process of interfacial transport to form polyester chain by breaking of covalent bonds of the reagents. In addition, it is proved that ultrasound helps to substantial reduce the synthesis reaction time. In the conventional process the mass transfer is very slow as compared to ultrasonication requiring higher energy for activation, thus compromising the synthesis reaction rates.19 The increase of the surface contact created might have also accelerate the polymerization reaction.20–22 The vacuum effect on polyesters synthesis is of extreme importance since the residual alcohols and water can be further eliminated from the reaction under higher vacuum, which facilitates the chain growth of co-polyesters.
It is also noteworthy the conversion differences observed, for the same reactional conditions, between the three fluorinated di-esters starting reactants. Being the fluorinated di-esters the bulkier substrates, and considering the higher molecular weight DMOFA starting monomer, one may predict that as the synthesis occurs, the reaction is hindered to proceed due to the stereo-impediment of the high amount of fluorine atoms. This assumption is corroborated by the amount of polymer formed which, independently on the reactional conditions, presents always the lowest amount of products isolated (Table 2). Also, for the cases when the enzyme is applied, the reactants size might hinder its access to the enzyme's active site which is hampered by the immobilized nature of the catalyst. These assumptions are supported by the results obtained for the shorter di-ester starting reactant, DMTFS, which reveal the best overall results, as in absence or in presence of high-energy environment. A smaller monomer size is expected to facilitate the access to the enzyme's active site and higher amount of monomer is converted. Likewise, stereo-impediments are less evident, when in absence of a catalyst, comparing to the larger substrates used (glutarate and adipate), since only half of the fluorine atoms are present in the structure.
From the data obtained it might be questionable the high conversions obtained in the absence of enzyme on the system. This is easily justified by the high reactivity of the fluorinated starting materials against hydrolysis. Additionally, the presence of a nucleophile in the medium (ethylene glycol) makes the formation of the polymers inevitable. Furthermore, the use of methylene esters substrates, allows the formation of methanol as sub-product of the reaction. This compound is easily evaporated at the vacuum step, regarding its low boiling point (65 °C), which allows the propagation of the reaction. The temperature is a parameter described for greatly influence the biosynthesis of polyesters. Besides the optimization of polyester synthesis with the concomitant reduction of chemicals, we aimed herein to decrease the reactional temperature, and thus different temperatures were tested. Surprisingly, in absence or in presence of enzyme, lower temperatures (40 °C) allowed to achieve similar conversion levels as when higher temperatures (70 °C) were applied. We did not considered temperatures below 40 °C since previous experiments revealed this temperature as the minimal to successfully carry out the synthesis of polyesters.9,10,23 Temperatures above 70 °C were not studied since high conversion levels were reached when using this temperature and processing costs related with the use of elevated temperatures are therefore avoid.
Characterization of the fluorinated polyesters – NMR
All the reactions were followed by 1H NMR to confirm polyester synthesis and evaluate the material conversion rates. 19F NMR was also conducted to confirm polyester synthesis. Fig. 1 and 2 correspond to 1H NMR and 19F NMR, respectively, of polyesters synthesized in the absence of catalysts under ultrasound followed by vacuum for 6 h at 40 °C.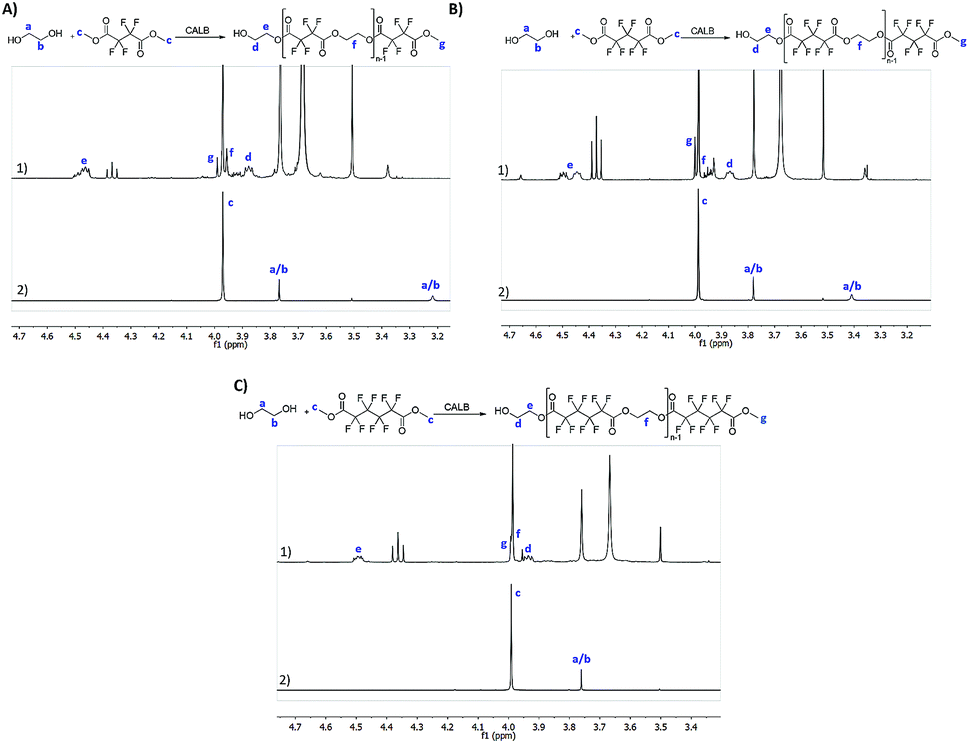 | ||
| Fig. 1 1H NMR (CDCl3) spectra of the synthesized polyesters and respective starting reactants: (A1) poly(ethylene tetrafluorosuccinate); (A2) dimethyl tetrafluorosuccinate; (B1) poly(ethylene hexafluoroglutarate); (B2) dimethyl hexafluoroglutarate; (C1) poly(ethylene octafluoroadipate) and (C2) dimethyl octafluoroadipate; the reactions were performed under the optimal conditions: US for 1 h at 40 °C followed by vacuum for 6 h at 40 °C. | ||
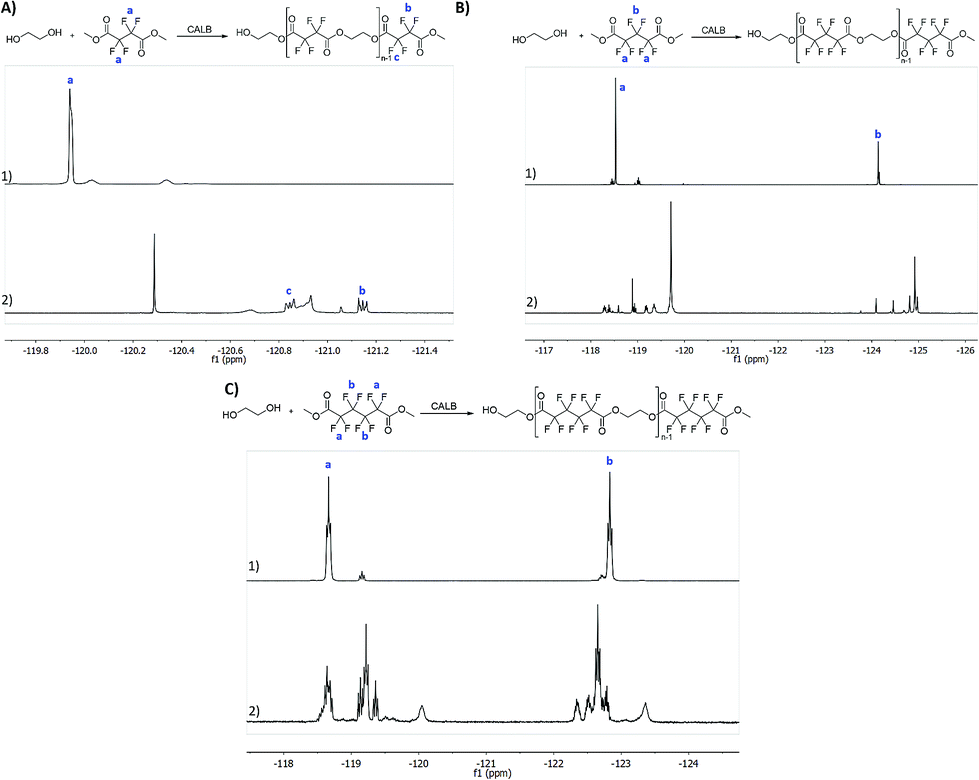 | ||
| Fig. 2 19F NMR (CDCl3) spectra of the synthesized polyesters and respective starting reactants: (A1) dimethyl tetrafluorosuccinate; (A2) poly(ethylene tetrafluorosuccinate); (B1) dimethyl hexafluoroglutarate; (B2) poly(ethylene hexafluoroglutarate); (C1) dimethyl octafluoroadipate and (C2) poly(ethylene octafluoroadipate); the reactions were performed under the optimal conditions: US for 1 h at 40 °C followed by vacuum for 6 h at 40 °C. | ||
1H NMR
1H NMR of the starting materials (mixed only in the NMR tube) and the fluorinated polyesters are shown in Fig. 1. The fluorinated monomers appear as a unique singlet, which correspond to the terminal CH3 (δH 4.0 ppm). The ethylene glycol, due to the presence of a fluorine compound in the NMR tube, appears as two distinct singlets for each CH2. One of the CH2 is observed near δH 3.75 ppm (in all fluorine compounds), while the other CH2 is observed at δH 3.22 ppm for the DMTFS, δH 3.41 ppm for DMHFG and δH 2.39 ppm for DMOFA. In the polymer's spectra two new peaks appear, corresponding to the ethylene glycol moiety at the polymer extremity. These two peaks are assigned as triplet or multiplet. The first peak is observed around δH 3.90 ppm, which correspond to proton d (CH2 near the terminal OH group); the other peak (proton e) is observed near δH 4.50 ppm, with a more complex peak in all cases, due to the proximity of the fluorinated ester group.19F NMR
From 19F NMR data different assumptions must be considered. DMTFS spectrum reveals only one peak at δF −119.9 ppm as a singlet, while in DMHFG spectrum two peaks, one at δF −124.1 ppm (fluors b) and the other at δF −118.5 ppm (fluors a), can be observed. The spectrum of DMOFA shows two triplets, one at δF −128.8 ppm (fluors b) ppm and the other at δF −118.7 ppm (fluors a). No starting material is detected in the spectra of all the polyesters produced. In all cases, the appearance of new peaks indicate a large variety of fluorine atoms at different chemical environments, consistent with the reaction synthesis. However, due to their complexity, the correct assignment of all peaks was not feasible.MALDI-TOF considering the high levels of material conversion, it was vital to characterize the new polyesters respecting to their molecular size. The maximum polymerization degree was evaluated by MALDI-TOF spectroscopy and is presented in Fig. 3 as the mass spectra of poly(ethylene tetrafluorosuccinate) after conversion using all the different reactional procedures. From the data achieved (Table 3) one can confirm that the combination of the ultrasound pre-treatment with CALB gave rise to longer polyesters (m/z = 859, DPavg = 4), as previously confirmed by NMR data. The reaction conditions involving US + vacuum gave rise to smaller oligomers (DPavg = 2), however confirming the previous findings related with NMR interpretation that ensure the feasibility of the polyesters synthesis in the absence of a biological catalyst. Moreover, despite the different average DP obtained, the maximum DP attained when using enzyme is only 1-fold higher, confirming the previous conversion findings. It is also noteworthy, that the presence of enzyme did not improve the size of the polyesters synthesized only under vacuum, which present similar polymerization degree (DPavg = 2).
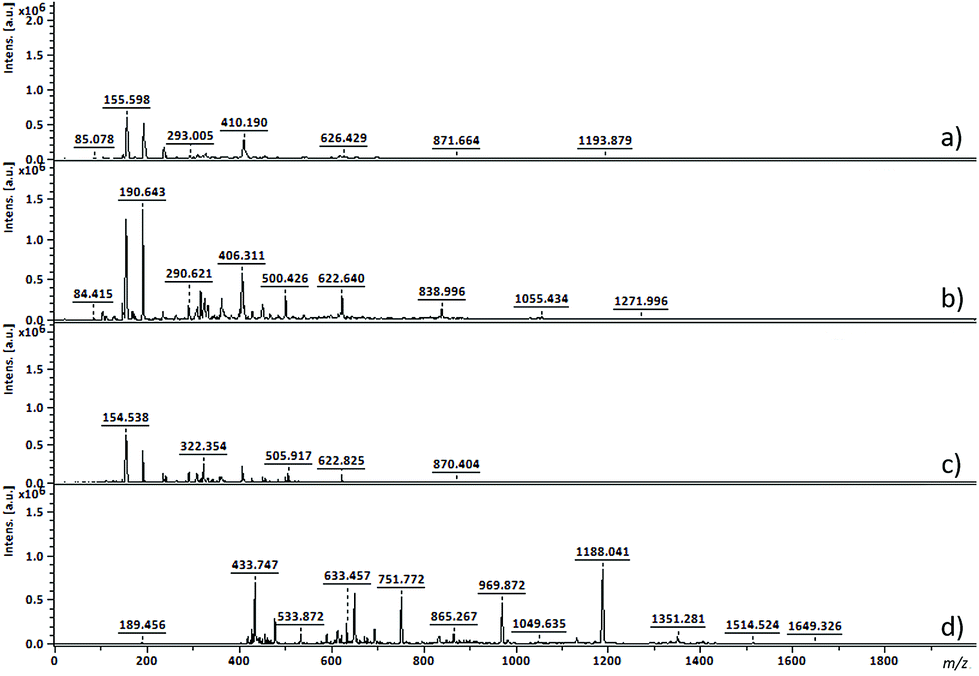 | ||
| Fig. 3 MALDI-TOF of synthesized poly(ethylene tetrafluorosuccinate) with equimolar ratio of reactants, using different conditions: (a) US 1 h 40 °C + Vac 6 h 40 °C without CALB; (b) Vac 7 h 40 °C without CALB; (c) Vac 7 h 40 °C, 1% (w/v) CALB and (d) US 1 h 40 °C + Vac 6 h 40 °C, 1% (w/v) CALB. | ||
| m/z | Mn | Mw | DPavg | PDI | |
|---|---|---|---|---|---|
| (a) US 1 h 40 °C + Vac 6 h 40 °C; without CALB | 1193 | 416 | 481 | 2 | 1.15 |
| (b) Vac 7 h 40 °C; without CALB | 1271 | 512 | 588 | 2 | 1.15 |
| (c) Vac 7 h 40 °C; 1% (w/v) CALB | 870 | 402 | 439 | 2 | 1.09 |
| (d) US 1 h 40 °C + Vac 6 h 40 °C; 1% (w/v) CALB | 1649 | 704 | 859 | 4 | 1.22 |
The maximum DP obtained for the other polyesters (DMHFG and DMOFA) was lower than the reported for DMTFS (data not shown), confirming the role of the starting reactants size on the synthesis performance.
FTIR
The different polyesters synthesized using the optimized conditions: 1 h US followed by 6 h vacuum at 40 °C, were characterized spectroscopically by FTIR. The spectra of the new polyesters obtained from fluorinated di-ester compounds (DMTFS/DMHFG/DMOFA) and ethylene glycol, under the previous optimal conditions presented, are depicted in Fig. S1.† The infrared absorption frequencies of the fluorinated compounds are listed in Table S1.† FTIR spectra of the fluorinated reactants reveal the presence of the carbonyl ester group at ν 1750 cm−1. In the ethylene glycol FTIR spectra is present the typical OH stretch at ν 3300 cm−1. In the polyester's spectra, the carbonyl group is still present (around ν 1780–1710 cm−1), while the OH stretch is less evident, considering that this group is only present in the extremity of the polymer. Other peaks, such as C–F stretch (∼ν 1150 cm−1) are also observed in the polyester's FTIR spectra. These results confirm the synthesis of the fluorinated polyesters by a incubation of the reactants under ultrasound followed by vacuum.TGA
The thermal stability, is a crucial parameter to evaluate the physical and chemical properties of the newly synthesized fluorinated polyesters by monitoring their weight loss as function of temperature. This property was evaluated using a thermogravimetric analyser under nitrogen atmosphere and the percentage of weight loss was plotted as a function of temperature. Fig. 4 shows the TGA curves and the decomposition temperatures at the maximum weight loss rate (Tdmax) for the starting reactants and for the polyesters synthetized under the optimized conditions. | ||
| Fig. 4 Thermogravimetric analysis of polyesters synthesized from fluorinated compounds: (a) dimethyl tetrafluorosuccinate; (b) dimethyl hexafluoroglutarate; (c) dimethyl octafluoroadipate and ethylene glycol, using: US for 1 h at 40 °C followed by 6 h of vacuum at 40 °C. | ||
As shown in Fig. 4, and in contrast to the starting materials that totally decompose at around 150 °C, three-step or four-step decomposition stages are detected for the fluorinated polyesters synthesized. It is evident a significant weight loss at around 109, 117 and 136 °C for poly(ethylene tetrafluorosuccinate), poly(ethylene hexafluoroglutarate) and poly(ethylene octafluoroadipate), respectively, which is inversely proportional to the starting reactants size. The weight losses of the first step observed for the three polyesters and EG were 52.857 wt% at 109 °C, 8.619 wt% at 50 °C, and 33.961 wt% at 68 °C, for DMTFS, DMHFG and DMOFA, respectively. Such a pre-major weight loss stage was mentioned previously after synthesis of poly(ethylene terephthalate), being attributed to the volatilization of small molecules, residual catalysts, 1,3-propanediol and carbon dioxide that devaluated from the chain end.24,25 In this study we found that the starting fluorinated reagents (DMTFS; DMHFG; DMOFA) were completely decomposed at around 150 °C while ethylene glycol (EG) evaporated at slightly higher temperature, nevertheless lower than 200 °C. These data corroborate the hypothesis that the first decomposition steps at temperatures lower than 200 °C is due to the oligomer degradation.
Above 200 °C, the decomposition of fluorinated polyesters reveal a much slower behavior (Table 4) reaching the total weight loss at around 400 °C. The thermal degradation temperature is affected by the structural parameters like molecular weight, crystallinity, orientation and chemical structure.26 From the data obtained the thermal stability of the three polyesters studied increased and is inversely proportional to the amount of fluorinated methylene groups. This is consistent with previous findings for the decomposition rates of PBT – polybutylene terephthalate or PPT – polypropylene terephthalate which were found to be are higher than that of PET, as the PBT or PPT contain one or two methylene groups less than PET, being more stable to decomposition.27
| Poly(ethylene tetrafluorosuccinate) | Poly(ethylene hexafluoroglutarate) | Poly(ethylene octafluoroadipate) | |||
|---|---|---|---|---|---|
| Δweight (%) | Tdmax (°C) | Δweight (%) | Tdmax (°C) | Δweight (%) | Tdmax (°C) |
| 52.86 | 109 | 8.62 | 50 | 33.96 | 68 |
| 9.08 | 280 | 46.90 | 117 | 35.33 | 136 |
| 32.57 | 394 | 7.91 | 278 | 8.11 | 258 |
| — | — | 32.38 | 398 | 18.62 | 382 |
The hydrophobic character of the new fluorinated polyesters produced was tested using cotton fabrics substrate as support. The preliminary results of time of water absorption demonstrate the promising ability of the fluorinated polyesters to confer hydrophobicity to the surfaces where they are applied. However deeper studies must be conducted, namely on the coating methodology, since low coating homogeneity was achieved (Fig. S2†).
Conclusions
In this work new fluorinated polyesters were synthetized by varying different parameters, namely size of monomer reactants, temperature, ultrasound, vacuum and presence of a catalyst, immobilized CALB. The best results in terms of synthesis conversion rate and polymerization degree were obtained when following the conditions: incubation under ultrasound for 1 h at 40 °C followed by incubation under vacuum (2 mbar) for 6 h at 40 °C. The presence of CALB reveal to be negligible since very small differences of conversion rates were obtained comparing with synthesis without enzyme. Moreover the maximum DP obtained does not justify the costs related with the usage of a catalysts during the reaction. We have found the role of ultrasound in the reactional procedure as critical, concerning the high conversion rates obtained when this pre-treatment was applied. The size of the fluorinated reactants played also an important role on the polyesters biosynthesis. The data obtained reveal that the reaction conversion rates and the DP are inversely proportional to the size of the starting reactants. The new fluorinated polyesters are presented herein as potential coating products for textile applications, namely when hydrophobic behaviour is required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Chinese government scholarship under the State Scholarship Fund (No. 201706790049), Training Fund for Excellent Doctoral Student in Jiangnan University, Key Projects of Governmental Cooperation in International Scientific and Technological Innovation (No. 2016 YFE0115700), Fundamental Research Funds for the Central Universities (No. JUSRP51622A). The study was also supported by the National Natural Science Foundation of China [No. 31470509], the 111 Project [No. B17021], Jiangsu Planned Projects for Postdoctoral Research Funds [No. 2018K018A]. This study was also supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of UID/BIO/04469/2013 unit and COMPETE 2020 (POCI-01-0145-FEDER-006684) and BioTecNorte operation (NORTE-01-0145-FEDER-000004) funded by European Regional Development Fund under the scope of Norte2020 – Programa Operacional Regional do Norte. Jennifer Noro and Carla Silva also thanks to FCT for funding (SFRH/BD/121673/2016 and SFRH/IF/00186/2015).References
- H. Azim, A. Dekhterman, Z. Jiang and R. A. Gross, Biomacromolecules, 2006, 7, 3093–3097 CrossRef CAS PubMed.
- Y. Jiang and K. Loos, Polymers, 2016, 8, 243 CrossRef.
- R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS PubMed.
- Y. Yu, D. Wu, C. Liu, Z. Zhao, Y. Yang and Q. Li, Process Biochem., 2012, 47, 1027–1036 CrossRef CAS.
- A. Mahapatro, A. Kumar, B. Kalra and R. A. Gross, Macromolecules, 2004, 37, 35–40 CrossRef CAS.
- K. R. Yoon, S.-P. Hong, B. Kong and I. S. Choi, Synth. Commun., 2012, 42, 3504–3512 CrossRef CAS.
- H. Uyama, K. Inada and S. Kobayashi, Polym. J., 2000, 32, 440 CrossRef CAS.
- J. Zhang, H. Shi, D. Wu, Z. Xing, A. Zhang, Y. Yang and Q. Li, Process Biochem., 2014, 49, 797–806 CrossRef CAS.
- X. Zhao, S. R. Bansode, A. Ribeiro, A. S. Abreu, C. Oliveira, P. Parpot, P. R. Gogate, V. K. Rathod and A. Cavaco-Paulo, Ultrason. Sonochem., 2016, 31, 506–511 CrossRef CAS PubMed.
- P. D. Tomke, X. Zhao, P. P. Chiplunkar, B. Xu, H. Wang, C. Silva, V. K. Rathod and A. Cavaco-Paulo, Ultrason. Sonochem., 2017, 38, 496–502 CrossRef CAS PubMed.
- S. R. Comim Rosso, E. Bianchin, D. de Oliveira, J. V. Oliveira and S. R. S. Ferreira, J. Supercrit. Fluids, 2013, 79, 133–141 CrossRef CAS.
- A. Mohammed Gumel, M. S. Annuar and Y. Chisti, Lipase catalyzed ultrasonic synthesis of poly-4-hydroxybutyrate-co-6-hydroxyhexanoate, 2012 Search PubMed.
- H. Miao, F. Bao, L. Cheng and W. Shi, J. Fluorine Chem., 2010, 131, 1356–1361 CrossRef CAS.
- L. Zhu, X. Hu, J. Xiong, B. Shentu and Z. Weng, Polym. Sci., Ser. B, 2015, 57, 677–686 CrossRef CAS.
- B. Pilch-Pitera, J. Appl. Polym. Sci., 2011, 124, 3302–3311 CrossRef.
- F. Signori, M.-B. Coltelli and S. Bronco, Polym. Degrad. Stab., 2009, 94, 74–82 CrossRef CAS.
- A. J. Mesiano, E. J. Beckman and A. J. Russell, Biotechnol. Prog., 2008, 16, 64–68 CrossRef PubMed.
- P. B. Subhedar and P. R. Gogate, Ultrason. Sonochem., 2016, 29, 67–75 CrossRef CAS PubMed.
- N. A. Methrath Liyakathali, P. D. Muley, G. Aita and D. Boldor, Bioresour. Technol., 2016, 200, 262–271 CrossRef CAS PubMed.
- K. Makino, M. M. Mossoba and P. Riesz, J. Phys. Chem., 1983, 87, 1369–1377 CrossRef CAS.
- F. M. D. Nora and C. D. Borges, Cienc. Rural, 2017, 47, 1–9 Search PubMed.
- R. B. N. Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559–1584 RSC.
- X. Zhao, J. Noro, J. Fu, H. Wang, C. Silva and A. Cavaco-Paulo, Process Biochem., 2018, 66, 82–88 CrossRef CAS.
- K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Thermochim. Acta, 2005, 435, 142–150 CrossRef CAS.
- X.-S. Wang, X.-G. Li and D. Yan, Polym. Test., 2001, 20, 491–502 CrossRef CAS.
- G. Bheemaneni, S. Saravana and R. Kandaswamy, Mater. Today, 2018, 5, 1807–1816 CrossRef.
- F. Samperi, C. Puglisi, R. Alicata and G. Montaudo, Polym. Degrad. Stab., 2004, 83, 3–10 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10341k |
| This journal is © The Royal Society of Chemistry 2019 |