
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe preparation of MgO nanopowders synthesized via an improved polyacrylamide gel method
Xiaojun Zhaoa,
Haitang Yang *bc,
Pengfei Wubc,
Xiaozhong Huangbc and
Xiaofeng Wanga
*bc,
Pengfei Wubc,
Xiaozhong Huangbc and
Xiaofeng Wanga
aSchool of Materials Science and Engineering, Central South University, Changsha 410083, China
bSchool of Aeronautics and Astronautics, Central South University, Changsha 410083, China. E-mail: hai.tang.ouyang@hotmail.com; Tel: +86-18207480255
cHunan Key Laboratory of Advanced Fibers and Composites, Central South University, Changsha 410083, China
First published on 14th May 2019
Abstract
In order to address the issue of metal ion incorporation during polymerization, citric acid was used as a chelating agent to improve the polyacrylamide gel route. In the present work, MgO nanoparticles were synthesized via this improved method. The calcination temperature of the gel precursor containing magnesium nitrate was determined by thermogravimetry and differential scanning calorimetry (TG-DSC). The phases and microstructures of MgO nanopowders were identified via X-ray diffraction (XRD), transmission electron microscopy (TEM) and specific surface area measurements (BET). The results showed that the nanoparticles synthesized under 600 °C were pure, globular and about 5–20 nm in size with a narrow distribution. Furthermore, the coalescence and growth of the MgO nanograins were amazingly observed with increasing calcination temperatures and calcination time. The influence of calcination temperature on the morphology and growth behavior is greater than that of the calcination duration. The activation energy for grain growth was estimated to be 31.43 kJ mol−1, and the dominant growth mechanism was predicted to be related to the grain-rotation-induced grain coalescence (GRIGC) mechanism.
1. Introduction
The polyacrylamide gel technique is a simple, convenient and inexpensive process for synthesizing nanoparticles in a time-saving manner from salt solutions containing the polymer; thus it has been widely used to synthesize nanoparticles of a variety of oxide materials and SOFCs in recent years.1–3 Specifically, BeO,4 2SiO2–3Al2O3,5 α-Al2O3,6,7 Zr2O3,8 BiO2,9 YVO4:Eu,10 SnO2:Eu,11 Zr0.84Y0.16O1.92,12 La0.85Sr0.15Ga0.85Mg0.15O2.85 (ref. 13) and other oxide systems14–18 are typical examples. For this method, organic acrylamide (AM) has always been used as the precursor to form a polyacrylamide hydrogel in a salt solution.19 Then, steric entrapment of the stoichiometric cation solution occurs in nanocavities formed inside the gel; in other words, a homogeneous micro-solution with cations in the desired stoichiometry is produced. Therefore, nanopowders could be obtained by inhibiting gel aggregation during the calcination of the gel and salt. However, this method is not suitable for synthesizing some oxide nanoparticles that contain Mg2+, Ca2+ and other divalent ions. This is due to the fact that Ca2+ and Mg2+ ions easily capture free radicals from the initiator and inhibit the polymerization of acrylamide.Highly active MgO nanoparticles with large specific surface areas are extensively investigated in different applications as catalysts, catalyst supports, and destructive adsorbents for a large number of pollutants.20 In order to reduce or eliminate the effect of metal ions on polymerization, the polyacrylamide gel route was improved by the introduction of citric acid as a chelating agent. The aim of this work was to synthesize MgO nanoparticles via the improved polyacrylamide gel route, evaluate the effect of the calcination process on the properties of MgO powders, and identify the underlying synthesis mechanisms. Additionally, the growth of the MgO nanograins was investigated in this work.
2. Experimental
2.1 Raw materials
Magnesium nitrate hexahydrate (Mg(NO3)2·6H2O), acrylamide (AM), 2-hydroxypropane-1,2,3-tricarboxylic acid (citric acid), N,N′-methylenebis(acrylamide) (MBAM), and ammonium persulfate (APS) were used as the raw materials in the present work. All of the A. R. grade reagents are produced by Sinopharm Group Co. Ltd.2.2 Synthesis procedure
The experimental procedure for the polyacrylamide gel route for preparing MgO nanoparticles is reported in our previous work.14 Initially, Mg(NO3)2·6H2O, AM and MBAM were dissolved in distilled water to prepare a transparent 2 M Mg(NO3)2 solution with magnetic stirring, where the molar ratio of AM and MBAM monomers was 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the concentration of organic agents was 5 wt%. To chelate the metal ions of Mg2+, citric acid (5 wt%) was also added into the solution until a clear solution was observed. A small amount of ammonium persulfate was added in the form of a solution (APS: 10 wt%). Thereafter, the temperature of the solution was increased slowly to 60 °C in a water bath. The solution turned gradually from liquid to hydrogel in 1 h. Afterwards, the gel was dried at 80 °C for 48 h in an oven under vacuum. The xerogel thus formed contained homogenous magnesium ions locked by citric acid. Then the xerogel was calcined at various temperatures (500 °C, 600 °C, 700 °C and 800 °C) and times (0.5 h, 1 h, 1.5 h, and 2 h) in an oxygen atmosphere to determine the grain-growth kinetics of the MgO nanograins, with a heating rate of 5 °C min−1. Finally, the prepared MgO nanoparticles were furnace-cooled to room temperature, as shown in Table 1.
:1 and the concentration of organic agents was 5 wt%. To chelate the metal ions of Mg2+, citric acid (5 wt%) was also added into the solution until a clear solution was observed. A small amount of ammonium persulfate was added in the form of a solution (APS: 10 wt%). Thereafter, the temperature of the solution was increased slowly to 60 °C in a water bath. The solution turned gradually from liquid to hydrogel in 1 h. Afterwards, the gel was dried at 80 °C for 48 h in an oven under vacuum. The xerogel thus formed contained homogenous magnesium ions locked by citric acid. Then the xerogel was calcined at various temperatures (500 °C, 600 °C, 700 °C and 800 °C) and times (0.5 h, 1 h, 1.5 h, and 2 h) in an oxygen atmosphere to determine the grain-growth kinetics of the MgO nanograins, with a heating rate of 5 °C min−1. Finally, the prepared MgO nanoparticles were furnace-cooled to room temperature, as shown in Table 1.
| Duration time/h | Calcination temperature/°C | Particle sizes from specific surface area/nm | Particle sizes from XRD patterns/nm |
|---|---|---|---|
| 0.5 | 600 | 18.3 | 10 ± 3 |
| 1 | 600 | 19.7 | 11 ± 2 |
| 1.5 | 600 | 23.8 | 12 ± 3 |
| 5 | 600 | 32.7 | 17 ± 2 |
| 2 | 600 | 27.4 | 15 ± 2 |
| 2 | 500 | 20.1 | 10 ± 4 |
| 2 | 700 | 33.4 | 18 ± 2 |
| 2 | 800 | 46.3 | 33 ± 1 |
2.3 Characterization
To define the precise calcination temperatures, the magnesium nitrate salt and precursor gels (xerogels) were evaluated by thermo-gravimetric analysis (TGA-DSC/TGA, STA 449C, Netzsch) with a heating rate of 5 °C min−1 from room temperature to 800 °C and an air flow of 30 cm3 min−1. The phase and crystallinity of synthesized powders were analyzed via X-ray diffraction (XRD), using a Cu-Kα radiation (D/Max2550VB+, Rigaku) source in the range of 2θ = 30–90°. The mean diameter of crystallites was determined from X-ray diffraction line broadening using the Scherrer formula. The particle size, distribution and morphology of the prepared powders were examined by field-emission scanning electron microscopy (SEM, Sirion-2000, Philips) and transmission electron microscopy (TEM, CM-300, Philips). Specimens for transmission electron microscopy were prepared by suspending the fine powder in alcohol on carbon grids. Specific surface areas of synthesized powders were measured by the Brunauer–Emmett–Teller (BET) isotherm technique with nitrogen adsorption using a micromeritics ASAP 2000 surface area analyzer.3. Results and discussion
3.1 Effect of citric acid
The polyacrylamide gel route has been widely used to synthesize a variety of oxide nanoparticles. In the polyacrylamide gel method, the polymerization is a typical free radical process, which is initiated by an initiator. In the xerogel, nanosized salts were captured in the nanocaves of the polymer network, which was formed via the polymerization of monomer, acrylamide, and the crosslinker, N,N′-methylenebisacrylamide, in the solution. In a typical process of this method, organic acrylamide is used as the precursor to form polyacrylamide hydrogel in solution. Then, a steric entrapment of the stoichiometric cation solution occurs in the nanocavities formed inside the gel, that is, a homogeneous micro-solution with cations in the desired stoichiometry is produced. The gel with salt is calcined, and nanosize powders are obtained by inhibiting their aggregation via the network of gel. However, some elements (Mg, Ca, Ni, Mn) can react with the amino group of acrylamide.21 Since the solution of initiator (APS) is alkaline, these elements also react with OH− ions and form hydroxide sediments. Thus, the presence of these elements can inhibit polymerization.It is difficult to control the reaction mechanisms for the synthesis of oxides due to the different chemical behavior of each cation. Thus, a rational route to form a network has been proposed with the aid of organic polymers or chelates. In the present work, citric acid (HO–C(CH2–COOH)2–COOH) was chosen as the chelating agent to chelate Mg2+ ions. Fig. 1 shows the structures of the formed compounds. In the present work, the transparent Mg(NO3)2 solution containing citric acid was converted to a gel following the addition of the polymerization initiator, APS. Agrawal et al.22 also reported that citric acid (HO–C(CH2–COOH)2–COOH) is an excellent chelator for Mg2+ ions because they form stable compounds in solution.
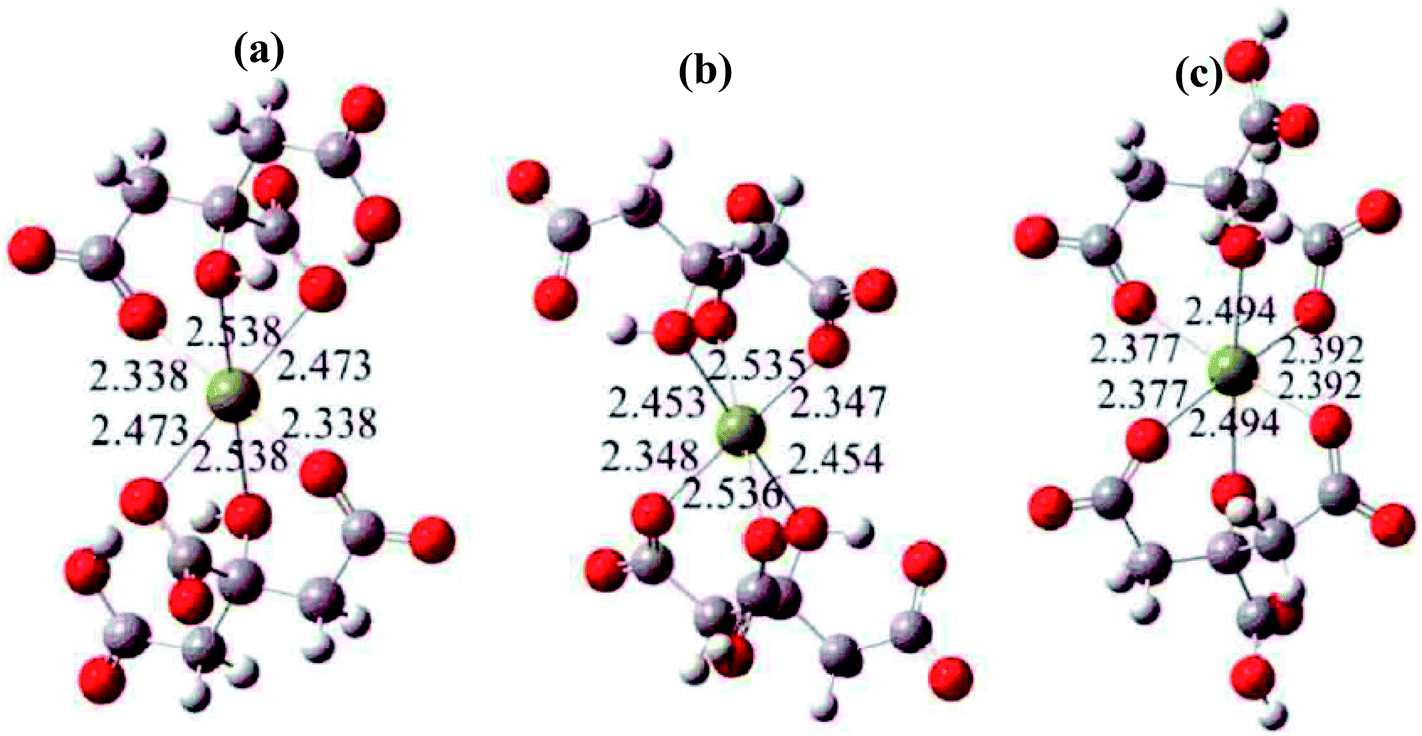 | ||
| Fig. 1 The most likely chelate structures between citric acid and Mg2+ ions, where the yellow balls represent Mg2+ ions. | ||
In this investigation, acrylamide gel, consisting of long polymeric chains that are crosslinked to create a tangled network and are soaked in an aqueous liquid, was used as the raw material. Due to the presence of tangled network, cations that are entrapped into a solution in nanocavities form inside the gel. A steric entrapment of a stoichiometric cation solution occurs, that is, a homogeneous micro-solution with cations in the desired stoichiometry is formed. Depending on the chemical composition and other factors, the gels varied in consistency from soft to a rigid solid. Polymerization of the gel proceeds through a chain reaction, the first step of which is the combination of an initiator with the acrylamide, which is thereby activated.5 As the chain of polyacrylamide grows, the active site shifts to its free end. Bisacrylamide (i.e., N,N-methylenebisacrylamide), which consists of two acrylamide units joined through their –CONH2 groups via a methylene group, can link two growing chains. Hence, bisacrylamide enables the formation of crosslinked chains, resulting in a complex topology with loops, branches, and interconnections.6,7
Since a gel would react with the acrylamide monomers and form complexes in which the element is bound to their amino groups, the formation of the gel as an intermediate form of matter between solid and liquid should be avoided. Thus, in the present work, transition metals (Cu, Ni, Mn), rare-earth elements (La, Y) and metalloid elements of the p-group (Bi) were used to impede the formation of the gel. For this reason, the formation of gels containing copper and yttrium, for example, in the Y-123 phase (YBa2–Cu3O7),6 was only possible when a low concentration of cations was used.
Citric acid is used to chelate with various cationic precursors by forming a polybasic acid. It is the most widely used chelating agent for the polyacrylamide gel process and forms relatively stable complexes with a variety of metal cations in its ionized form. Moreover, the introduction of citric acid also played a positive role on the drying and calcination processes of the precursor gel. As reported in our previous work,19 during these processes without citric acid, the precursor gels were able to expand and even weakly explode due to the intense decomposition of the nitrate group. However, the presence of citric acid suppresses the decomposition of the nitrate group, leading to a decrease in the reaction rate.
3.2 Thermal decomposition of precursors
Fig. 2 shows the thermal analysis (TG-DSC) of the precursor xerogel containing magnesium nitrate salt. It can be seen from the DSC results that three peaks were present at 192 °C, 393 °C and 533 °C, respectively. These peaks were assigned to the decomposition of the gel through the dehydration of magnesium nitrate, the decomposition of anhydrous magnesium nitrate, and the thermal decomposition of the gel, respectively. Also, it is interesting to observe a three-stage weight loss in the TG curve. The first stage with a significant weight loss below 250 °C is due to the removal of residual water in the dried gel4 and the dehydration of magnesium nitrate.23 Following this stage, the anhydrous magnesium nitrate and side-chain of the organic gel decompose, which is consistent with the results reported by Wu et al.24 In the third stage, the temperatures ranged between about 450 °C and 600 °C, where an exothermic peak appeared in the curve of DSC due to the decomposition of the backbone of the organic gel.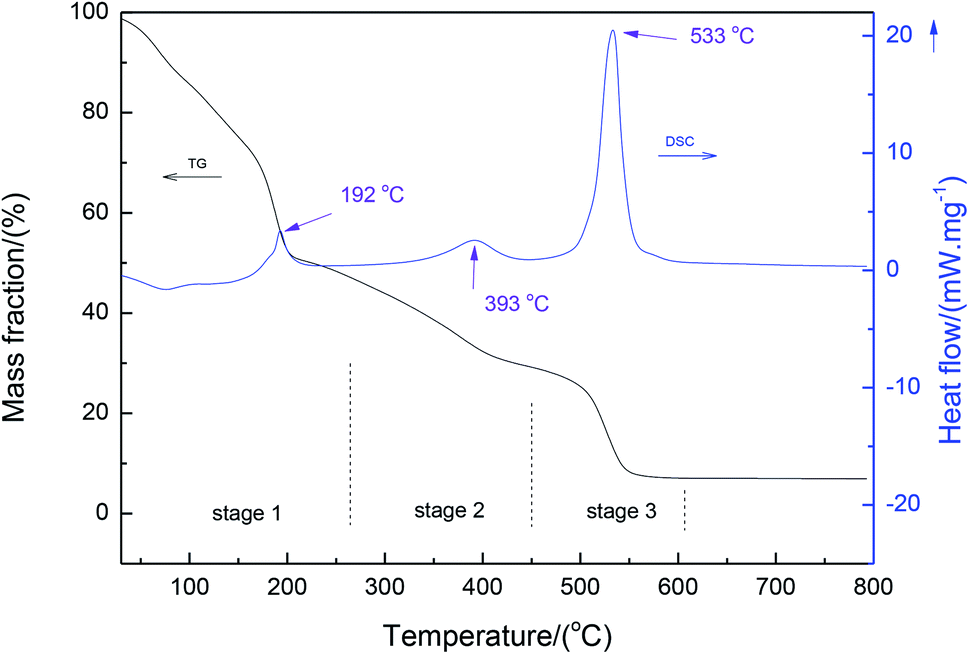 | ||
| Fig. 2 DSC and TG curves of the precursor xerogel containing magnesium nitrate salt. | ||
3.3 Growth of MgO nanopowders
Fig. 3 shows the X-ray diffraction patterns from the samples calcined at 500–800 °C (according to Fig. 2) in air for 0.5–5 h.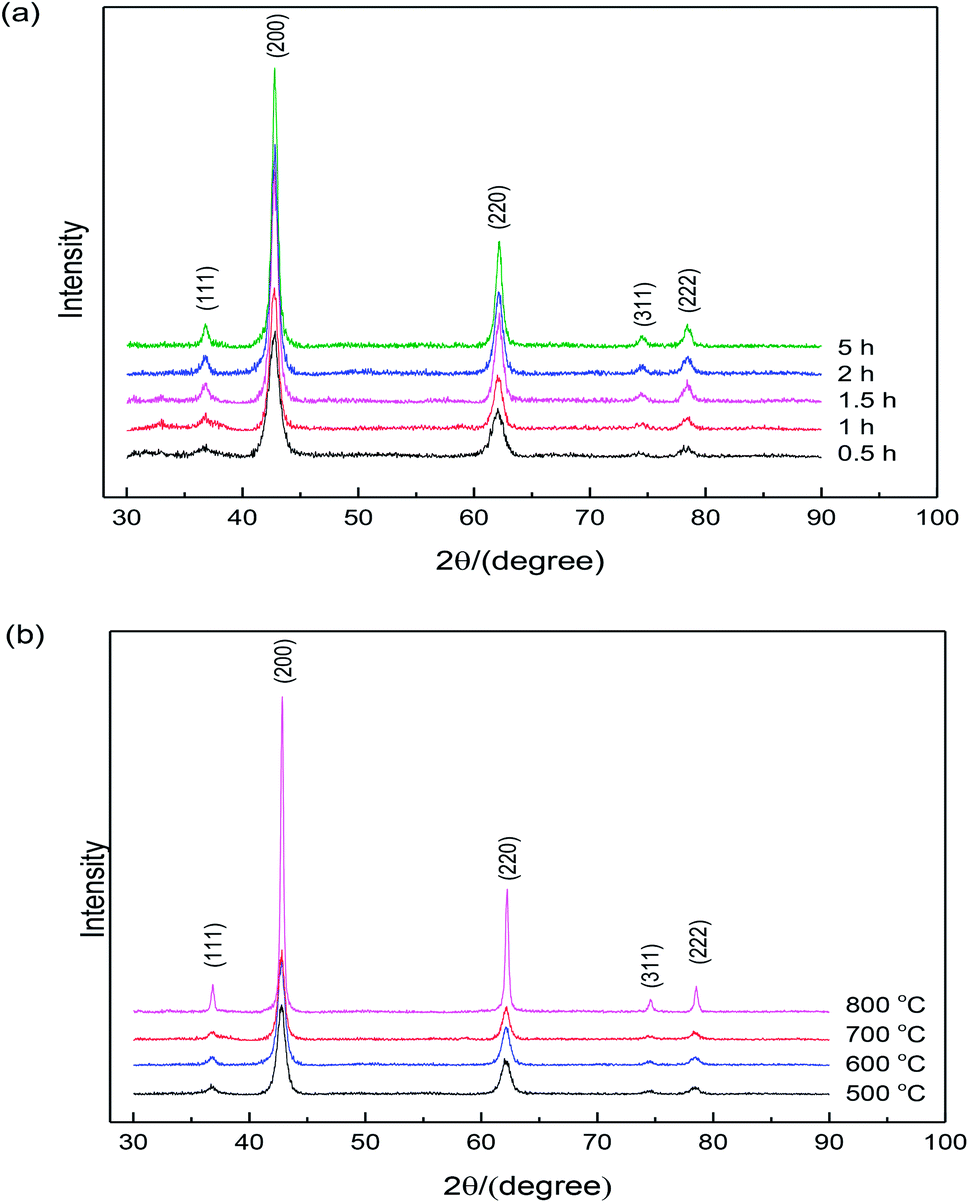 | ||
| Fig. 3 X-ray diffraction patterns of MgO nanopowders: (a) calcination temperature is 600 °C and (b) calcination time is 2 h. | ||
The clear diffraction patterns of MgO (based on the JPCDS reference code 35-0818) were detected in all cases, confirming the formation of pure MgO after calcination at 600 °C for different times. Note that the powders heated under 500 °C for 2 h were pure MgO, although this temperature is lower than the decomposition temperature of the sample according to the DSC curve shown in Fig. 2. It might be due to the duration time of 2 h, which was much longer than the time during thermal analysis. Additionally, the broadened diffraction peaks indicate that the crystallite size of the sample was very small, and the diffraction intensity increased with the increase in the calcination time, indicating the growth of nano-sized MgO particles. Based on the XRD patterns, the average crystallite sizes of the specimens could be calculated according to the Scherrer formula. Table 1 shows the crystallite sizes determined by XRD and BET. It is clearly seen that the MgO particles were nano-sized and the results calculated from specific surface area match well with the results determined from XRD. In addition, an increase in particle size was observed with the increase in the calcination time.
Fig. 4 shows the TEM micrographs and histograms for the size distribution of the specimens calcined at 600 °C for different durations. It is obvious that calcination at 600 °C for up to 5 h caused no significant change in the shape, and the average size of the particles increased from 10–20 nm to 10–40 nm with a narrow size distribution, as shown in the histograms for size distribution, which is in agreement with the results of XRD and specific surface area measurements. The calcinated MgO nanoparticles with large specific surface areas are attractive candidates for catalysts, catalyst supports, and destructive adsorbents for a large number of pollutant-related applications.
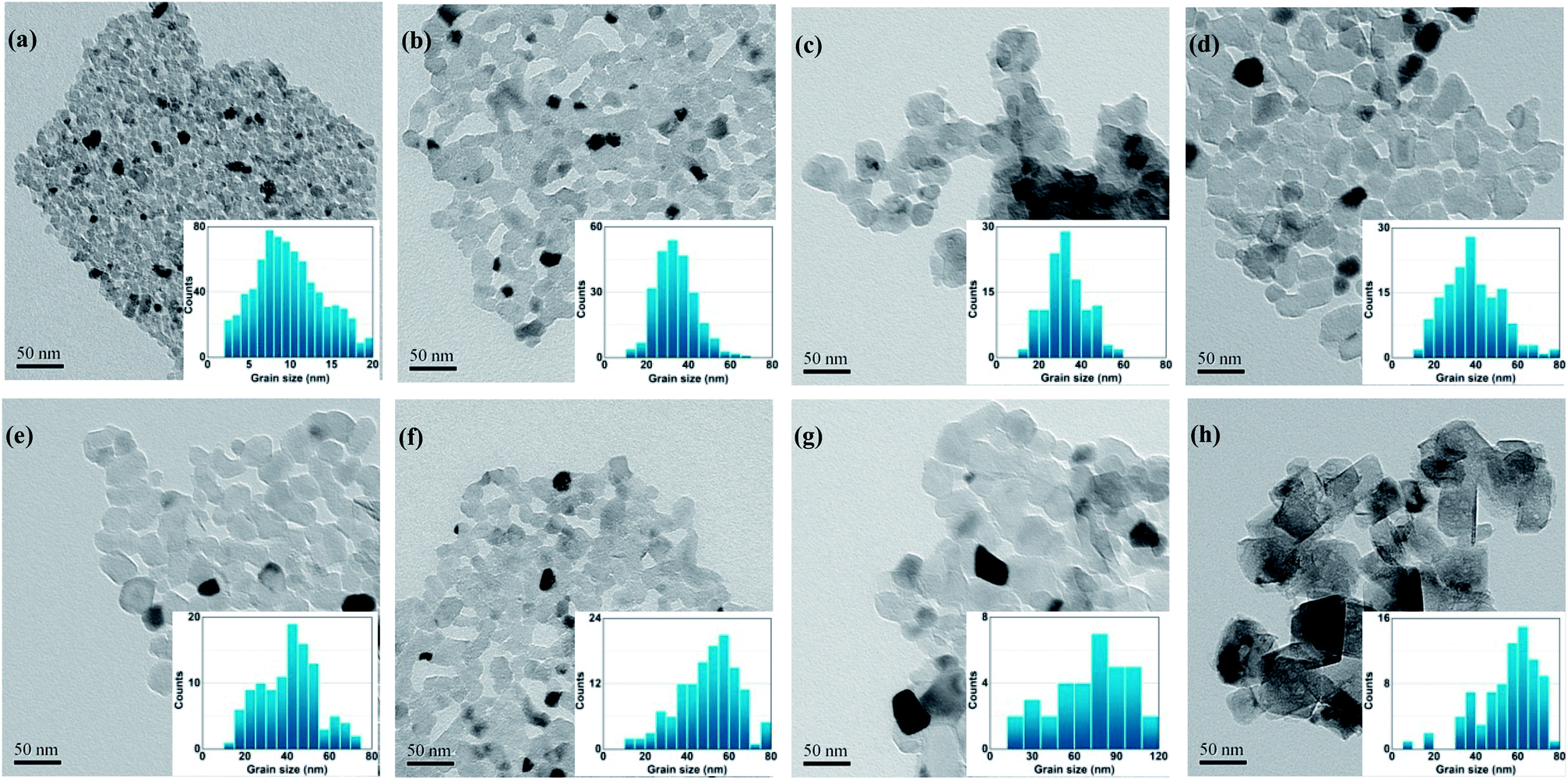 | ||
| Fig. 4 TEM images and histograms for the size distribution of MgO nanopowders calcined under different conditions: (a) 600 °C, 0.5 h; (b) 600 °C, 1 h; (c) 600 °C, 1.5 h; (d) 600 °C, 2 h; (e) 600 °C, 5 h; (f) 500 °C, 2 h; (g) 600 °C, 2 h and (h) 700 °C, 2 h. | ||
Grain-growth describes the increase in grain size of a single-phase solid. The phenomenological equation for grain-growth at a fixed temperature can be described as follows:
| Dtm − D0 = Kt | (1) |
log(K) + 1/mlog(t). The slope in the log(Dt) vs. log(t) plot can give the exponent m to deduce the grain-growth mechanism. Fig. 5 shows the plot of ln(Dt) as a function of log(t) and 1/T. It can be found from Fig. 5(a) that the grain-growth exponent is 3.70. According to the grain-rotation-induced grain coalescence (GRIGC) grain-growth mechanism,25 the rotation of MgO grains among neighboring grains results in a coherent grain–grain interface (the grains assume the same crystallographic orientation), which leads to the coalescence of neighboring grains via the elimination of common grain boundaries, thus forming a single larger MgO grain.
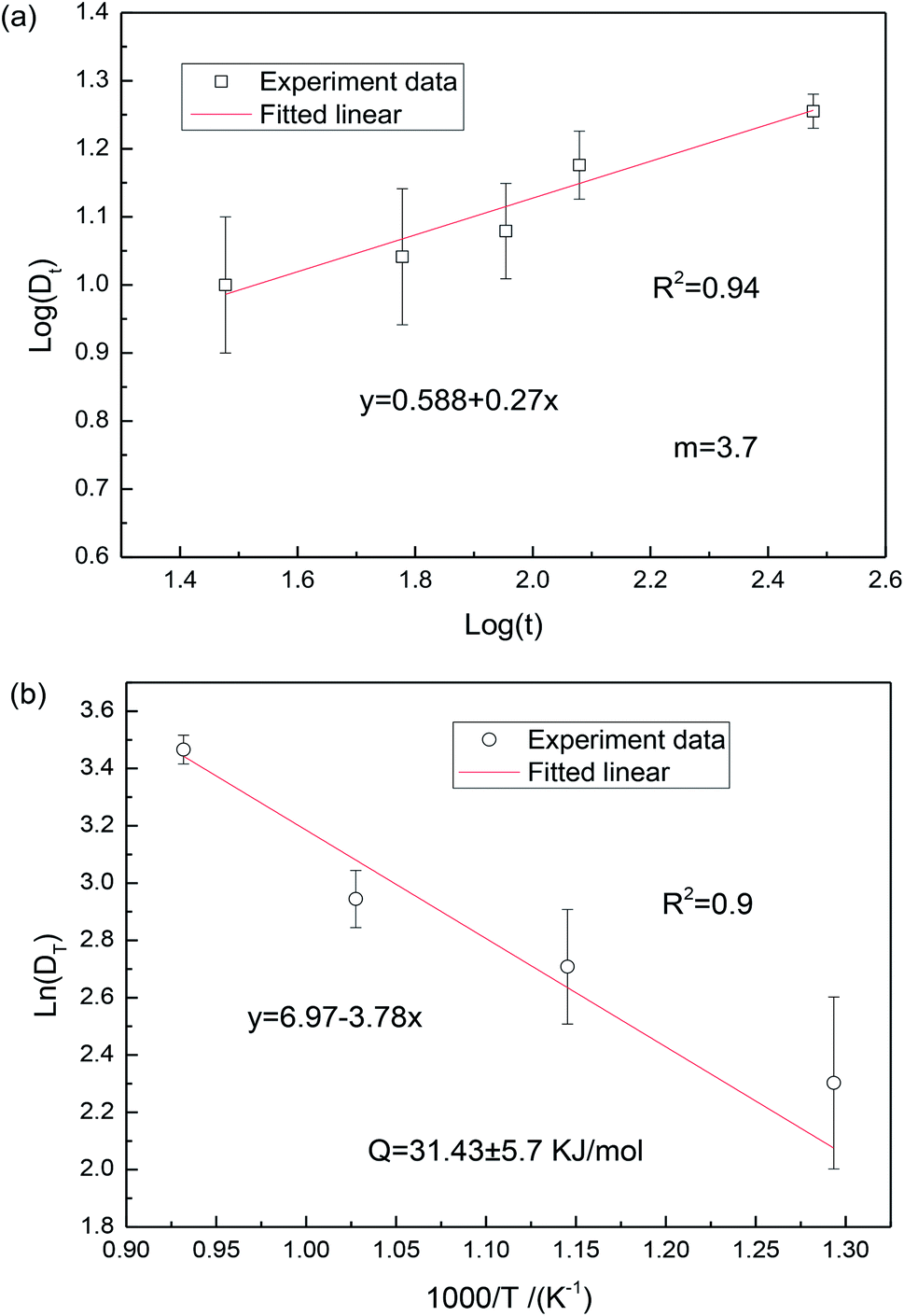 | ||
| Fig. 5 Grain growth of MgO nanopowders. (a) Grain sizes versus calcination time of MgO nanoparticles at 600 °C, and (b) the relationship between grain sizes and calcination temperature reflects the activation energy for grain growth. | ||
It should also be noted that the MgO grains would grow at a calcination temperature above 600 °C. Therefore, grain-growth based on grain-rotation-induced grain coalescence mechanism is a thermal activation process, satisfying the well-known Arrhenius equation,24
|
DT = D0e(−Q/RT)
| (2) |
| ln(DT) − ln(D0) = Q/RT | (3) |
The grain size DT was measured from the TEM micrographs (Fig. 4), where more than 50 nanograin diameters were evaluated on each sample and the average values were reported. According to eqn (3), the activation energy Q was estimated to be 31.43 kJ mol−1, which is significantly lower than the value of 161 kJ mol−1 reported by Chaim et al.26 This could be mainly attributed to the different fabrication methods. In the present work, the polyacrylamide gel method was used to synthesize the MgO nanoparticles, where the presence of a tangled polymeric network would constrain the aggregation and growth of MgO grains during the synthesis, leading to the reduction of activation energy. Additionally, the activation energy for surface diffusion in MgO, which changed from the adsorption and segregation effects at the surface, is sensitive to the impurity type and content.27 Similar low activation energies in MgO are related to surface diffusion by the GRIGC mechanism.24 In general, based on the Ostwald ripening theory, due to the fact that the chemical potential of the atoms in nanosized grains is very high, the MgO grains with low activation energy are active in growth.
4. Conclusions
In the present work, MgO nanopowders have been successfully synthesized via an improved polyacrylamide gel method. It was found that the calcination temperature of organic precursors with sulfate is around 550 °C, which is 30 °C lower than the typical calcination temperature. Due to the thermal stability of the polymeric network, powder aggregation was inhibited at high calcination temperatures, resulting in MgO particle sizes of about 10–40 nm with a narrow distribution. With the increase in the calcination temperature and calcination time, the coalescence and growth of MgO nanograins could be observed. The influence of calcination temperature on the morphology and growth behavior is greater than its duration and the growth becomes anisotropic. The activation energy for grain growth was estimated to be 31.43 kJ mol−1, and the dominant growth mechanism was most likely to be related to the grain-rotation-induced grain coalescence (GRIGC) mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (U1507110). In addition, I would also like to acknowledge the support and helpful discussion from the people of Hunan Key Laboratory of Advanced Fibers and Composites.References
- S. A. Hassanzadeh-Tabrizi and E. Taheri-Nassaj, Ceram. Int., 2013, 39(6), 6313–6317 CrossRef CAS.
- X. H. Su, G. Bai, J. Zhang, J. Zhou and Y. J. Jia, Appl. Surf. Sci., 2018, 442, 12–19 CrossRef CAS.
- H. Bessaies-Bey, R. Baumann, M. Schmitz, M. Radler and N. Roussel, Cem. Concr. Res., 2015, 76, 98–106 CrossRef CAS.
- X. F. Wang, R. C. Wang, C. Q. Peng, T. T. Li and B. Liu, J. Mater. Sci. Technol., 2011, 27(2), 147–152 CrossRef CAS.
- A. L. Castro, M. R. Nunes, A. P. Carvalho, F. M. Costa and M. H. Florêncio, Solid State Sci., 2008, 10(5), 602–606 CrossRef CAS.
- H. Z. Wang, L. Gao, W. Q. Li and Q. Li, Nanostruct. Mater., 1999, 11(8), 1263–1267 CrossRef CAS.
- M. Tahmasebpour, A. A. Babaluo, S. Shafiei and E. Pipelzadeh, Powder Technol., 2009, 191(1–2), 91–97 CrossRef CAS.
- M. Tahmasebpour, A. A. Babaluo and A. M. K. Razavi, J. Eur. Ceram. Soc., 2008, 28(4), 773–778 CrossRef CAS.
- C. Pan, X. Li, F. Wang and L. Wang, Ceram. Int., 2008, 34(2), 439–441 CrossRef CAS.
- H. Zhang, X. Fu, S. Niu, G. Sun and Q. Xin, J. Solid State Chem., 2004, 177(8), 2649–2654 CrossRef CAS.
- X. Fu, H. Zhang, S. Niu and Q. Xin, J. Solid State Chem., 2005, 178(3), 603–607 CrossRef CAS.
- A. Tarancón, G. Dezanneau, J. Arbiol, F. Peiro and J. R. Morante, J. Power Sources, 2003, 118(1–2), 256–264 CrossRef.
- N. Liu, Y. Yuan, P. Majewski and F. Aldinger, Mater. Res. Bull., 2006, 41(3), 461–468 CrossRef CAS.
- S. Q. Wu, L. N. He, F. P. Wang and T. Q. Lei, Trans. Nonferrous Met. Soc. China, 2003, 13(S1), 80–83 CAS.
- S. Q. Wu, Y. Y. Liu, F. P. Wang and L. Zhao, Trans. Nonferrous Met. Soc. China, 2005, 15(S2), 280–283 CAS.
- Z. Y. Wang, C. Q. Peng, R. C. Wang, X. F. Wang and B. Liu, Chin. J. of Nonferrous Met., 2013, 23(2), 480–486 CAS.
- C. J. Liu, R. M. Yu, Z. W. Xu, J. Cai, X. H. Yan and X. T. Luo, Trans. Nonferrous Met. Soc. China, 2007, 17(5), 1093–1099 CrossRef CAS.
- X. H. Jia, X. D. Zeng, L. P. Zhu and J. Y. Yang, Chin. J. of Nonferrous Met., 2015, 25(4), 1032–1038 CAS.
- X. Wang, R. Wang, C. Peng, T. Li and B. Liu, J. Sol-Gel Sci. Technol., 2011, 57(2), 115–127 CrossRef CAS.
- E. Esmaeili, A. Khodadadi and Y. Mortazavi, J. Eur. Ceram. Soc., 2009, 29(6), 1061–1068 CrossRef CAS.
- A. Sin and P. Odier, Adv. Mater., 2000, 12(9), 649–652 CrossRef CAS.
- S. Agrawal, A. Parveen and A. Azam, Mater. Lett., 2016, 168, 125–128 CrossRef CAS.
- J. G. Speight, Lange's handbook of chemistry, McGraw-Hill Inc, New York, 16th edn, 2004 Search PubMed.
- S. Q. Wu, Y. Y. Liu, L. N. He and F. P. Wang, Mater. Lett., 2004, 58(22–23), 2772–2775 CrossRef CAS.
- D. Moldovan, V. Yamakov, D. Wolf and S. R. Phillpot, Phys. Rev. Lett., 2002, 89(20), 206101 CrossRef PubMed.
- S. Kleiman and R. Chaim, Mater. Lett., 2007, 61(23–24), 4489–4491 CrossRef CAS.
- J. M. Vieira and R. J. Brook, Structure and Properties of MgO and Al2O3 Ceramics, in Advances in Ceramics, ed. W. D. Kingery, The American Ceramics Society Inc, Columbus, 1984, vol. 10 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |