DOI:
10.1039/C8RA10190F
(Paper)
RSC Adv., 2019,
9, 4635-4643
Fabrication of interlayer β-CD/g-C3N4@MoS2 for highly enhanced photodegradation of glyphosate under simulated sunlight irradiation
Received
12th December 2018
, Accepted 24th January 2019
First published on 6th February 2019
Abstract
Graphitic carbon nitride (g-C3N4) has been considered to be a promising metal-free photocatalyst, although the high recombination rate of charge carriers and poor absorption of visible light have limited its applications. In order to overcome these problems, an interlayer composite photocatalyst that comprised β-cyclodextrin (β-CD), oxygen-doped C3N4 (O-C3N4) and molybdenum disulfide (MoS2) was successfully constructed for the highly enhanced photodegradation of glyphosate in this study. The structure and morphology, optical properties, and photoelectrochemical properties of the prepared photocatalyst were characterized via a series of characterization techniques. The average fluorescence lifetime of the composite photocatalyst was extended from 6.67 ns to 7.30 ns in comparison with that of g-C3N4, which indicated that the composite photocatalyst enhanced the absorption of visible light and also inhibited the recombination of electron–hole pairs. The mass ratio of MoS2 that corresponded to O-C3N4/MoS2-5 enabled the highest removal rate under simulated sunlight irradiation, which was almost twice that achieved using pure g-C3N4. Relative species scavenging experiments revealed that ·O2− was the main species during the process of photodegradation. Besides, a toxicity test indicated that glyphosate became less toxic or non-toxic after photodegradation. This study provided an effective, feasible and stable photocatalyst driven by simulated sunlight irradiation for the highly enhanced photodegradation of glyphosate.
1. Introduction
Currently, increasing amounts of herbicides are used worldwide to improve crop yields. Glyphosate, which is a non-selective herbicidally active compound, is used on a large scale in the field of agriculture, and the annual amount of this herbicide applied reached 125![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons in 2014 and is growing continuously.1,2 However, glyphosate possesses the characteristics of stability, toxicity and durability, which lead to a great deal of environmental pollution. Hence, it is imperative to develop an efficient and eco-friendly method to solve this problem. Thus far, photocatalytic technology has been considered to be the most promising technology for solving this problem owing to its advantages of cost-effectiveness, energy efficiency, and the removal of recalcitrant compounds and their intermediates.3,4
000 tons in 2014 and is growing continuously.1,2 However, glyphosate possesses the characteristics of stability, toxicity and durability, which lead to a great deal of environmental pollution. Hence, it is imperative to develop an efficient and eco-friendly method to solve this problem. Thus far, photocatalytic technology has been considered to be the most promising technology for solving this problem owing to its advantages of cost-effectiveness, energy efficiency, and the removal of recalcitrant compounds and their intermediates.3,4
In recent years, owing to its low toxicity, favorable physicochemical stability, low-priced raw materials and suitable band gap (2.7 eV), g-C3N4 has been considered to be a promising metal-free catalyst for use in photochemistry and photocatalysis.5 However, the high recombination rate of charge carriers, low electrical conductivity and poor absorption of visible light (<460 nm) limit the applications of pristine g-C3N4.6 Therefore, the development of a kind of photocatalyst with a narrower band gap that enables better separation of electron–hole pairs is conducive to the enhancement of photocatalytic activity.
Doping with heteroatoms, controlling the morphology and copolymerization are effective approaches for modifying the electronic structure and energy band configuration.7–9 In comparison with metal atom dopants, non-metal atoms can optimize the electronic structure, broaden the light absorption range and improve effective charge separation, as well as having the advantages of eco-friendliness, economy and sustainable value.10,11 In recent years, Zhang et al. synthesized oxygen-doped g-C3N4 with a three-dimensional pore architecture by hydrothermal treatment and calcination to be used in a photocatalytic hydrogen evolution reaction, and the hydrogen evolution rate achieved using oxygen-doped g-C3N4 was higher by a factor of 11.3 than that achieved using bulk g-C3N4 under visible-light irradiation.11 However, the complex preparation and expensive raw material led us to employ an easier synthesis strategy and a more available material to obtain oxygen-doped g-C3N4. β-CD, which is a common inexpensive cyclic oligosaccharide, successfully attracted our attention as an oxygen-containing precursor because it can rapidly decompose via the cleavage of chemical bonds at approximately 350 °C, and the small chemical groups of β-CD and melamine may undergo self-assembly during the synthesis of graphitic carbon nitride. The presence of heteroatoms can adjust the band gap of g-C3N4 and hence improve the reducing power of electrons and inhibit the recombination of electrons and holes.12,13 However, there still exist some drawbacks in that the utilization of visible light is not high enough and the photocatalytic removal rate of organic contaminants still cannot reach a high level.
2D molybdenum disulfide (MoS2) has been intensively investigated as a prospective photocatalyst owing to its important properties, such as the large distance between stacked layers, atomic thickness, flexibility, and stability in both alkaline and acidic media.14,15 Ren et al. designed 3D CoFe2O4 nanorod/flower-like MoS2 nanosheet heterojunctions as recyclable visible-light-driven photocatalysts for the degradation of organic dyes, and the degradation efficiency for Congo red was 94.9%, which was about 12 times that achieved using pure MoS2.16 Liu et al. also developed 0D MoS2/2D g-C3N4 heterojunctions for enhanced photocatalytic and electrochemical hydrogen evolution.17 They all improved the photocatalytic activity by adjusting the transport paths for electrons and holes between different interfaces of the photocatalysts. Hence, this can provide an opportunity for us to search for available materials with a conduction band (CB) and valence band (VB) and adjust the transport paths for electrons and holes after composite formation, which can enhance the photocatalytic capacity for contaminants. It is a coincidence that MoS2 and O-C3N4 both possess the ability to prolong electron lifetimes, improve the efficiency of charge separation and widen the light absorption edge.
Inspired by this understanding, we report a novel hybrid photocatalyst based on oxygen-doped g-C3N4 and MoS2 for efficiently photodegrading glyphosate. The synthetic process comprised three steps: (i) O-C3N4 was prepared by stirring β-CD and melamine; (ii) MoS2 was synthesized via a facile one-pot hydrothermal process; and (iii) O-C3N4/MoS2 composites were prepared. Besides, the chemical structure, morphology, and optical properties of the composite materials that were obtained were characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), UV-vis diffuse reflectance spectroscopy (DRS), and photoluminescence (PL), and electron spin resonance (ESR) spectroscopy was used to determine the predominant radicals in the photodegradation system. The photocatalytic activity of the as-prepared photocatalyst was evaluated via the photodegradation of glyphosate under simulated sunlight irradiation. Finally, a pot experiment was conducted to investigate the toxicity of the degradation products. This study can provide an accessible route for the synthesis of an efficient and eco-friendly composite photocatalyst for removing water pollution by glyphosate or its analogues.
2. Experimental
2.1 Materials and chemicals
β-CD was purchased from Sichuan Kelong Chemical Reagent, China. Sodium molybdate dihydrate (Na2MoO4·2H2O) and N,N-dimethylformamide (DMF, 99.8%) were obtained from Adams Reagent Co., Ltd. Melamine and thiourea were purchased from Aladdin Industrial Corporation and Tianjin Beilian Chemical Development Co., Ltd, respectively. Analytically pure ethyl alcohol was purchased from Tianjin Yongsheng Chemical Reagent, China. Glyphosate was obtained from Jiangsu Fengshan Co., Ltd. ITO glass with a resistivity of 6 Ω was purchased from South China Xiangcheng Technology. All reagents that were employed for the preparation of the catalyst were of analytical grade and were used without further purification.
2.2 Preparation of photocatalysts
2.2.1 Preparation of oxygen-doped g-C3N4. Oxygen-doped g-C3N4 was synthesized from melamine and β-CD.10 In brief, melamine (10.00 g) and β-CD (0.15 g) were dispersed into 60 mL deionized water under stirring at 85 °C in a water bath to remove the water completely. Then, the solid mixture that was obtained was calcined in a tube furnace at 600 °C for 2 h at a ramp rate of 5 °C min−1 and was denoted as O-C3N4. Pure g-C3N4 was obtained under the same conditions without using β-CD.
2.2.2 Preparation of MoS2. In this study, 10 mmol Na2MoO4·2H2O and 50 mmol thiourea were dissolved in 30 mL deionized water under intensive stirring for 1 h, and then the mixture was transferred into a 50 mL Teflon-lined stainless steel autoclave, which was sealed tightly and heated in an oven at 200 °C for 24 h.18 Then the sample that was obtained was centrifuged at 7000 rpm for 10 min and washed with ethyl alcohol and deionized water alternately several times. The material that was obtained was dried in a vacuum oven at 80 °C for 12 h before use.
2.2.3 Synthesis of oxygen-doped g-C3N4@MoS2. In a typical process, different mass ratios (1%, 3%, 5%, and 7%) of MoS2 were initially dispersed into 50 mL DMF and then treated with ultrasound for 2 h in order to obtain exfoliated MoS2 layers. After that, 1 g as-prepared O-C3N4 was dispersed into the above solution and treated with ultrasound for a further 1 h, followed by intensive stirring for 12 h to enable a better interaction between the exfoliated MoS2 layers and O-C3N4. Then the solvent was removed by vacuum distillation to form the composite photocatalysts, which were denoted as O-C3N4/MoS2-1, O-C3N4/MoS2-3, O-C3N4/MoS2-5, and O-C3N4/MoS2-7, respectively. Moreover, a composite of g-C3N4 and MoS2 in a mass ratio of 5% was obtained by the same method.
2.3 Characterization
XRD was employed to determine the structural properties of the photocatalysts using a Rigaku Geigerflex D/Max B diffractometer with Cu Kα radiation at a scanning rate of 0.02° s−1 in the 2θ range from 10° to 80° at room temperature. The morphologies and microstructures of the samples were observed with a transmission electron microscope (Tecnai G2 F20, USA) at 100 kV. The physical surface properties of the samples were determined using the nitrogen adsorption–desorption method (Micromeritics Instruments 3Flex), and the adsorption isotherm was generated using N2 at 77 K. XPS measurements were conducted using a Thermo ESCALAB 250XI system to identify the chemical states of the samples with an Mg Kα X-ray source (1253.6 eV) under a vacuum pressure of <10−6 Pa, and the spectra were calibrated to the C 1s peak at 284.8 eV. The optical band gaps of the photocatalysts were determined with a UV-visible spectrophotometer (Hitachi U4100, Japan). The PL spectra of the photocatalysts were recorded with a fluorescence spectrophotometer (FLSP 920, UK) at room temperature using an Xe lamp as the excitation light source with an excitation wavelength of 330 nm. Electron spin resonance (ESR) signals were obtained using a JES FA200 spectrometer equipped with a 300 W Xe lamp as a visible light source.
2.4 Photocatalytic performance tests
To determine the photodegradation efficiency of the as-prepared catalysts, a 300 W Xe lamp (CEL-HXF300, China) with AM1.5 cut-off filters was used as the light source. A water circulation system was used to maintain the temperature at 25 °C. A photodegradation efficiency test under solar light was carried out in Shihezi, Xinjiang on 11 July 2018 from 12:00 to 15:00, and the temperature was about 37 °C. In each test, 20 mg of the catalyst was dispersed into 50 mL of a 9 g L−1 aqueous solution of glyphosate. In order to achieve adsorption–desorption equilibrium on the surface of the catalyst, the mixture was initially stirred in the dark for 30 min and then exposed to light irradiation for a constant time. Finally, the mixture was filtered through a 0.45 μm Millipore filter (Taitan Company, China) to remove the catalyst. The residual aqueous solution of glyphosate was analyzed using a UV-vis spectrophotometer (UV-5100, China) at a wavelength of 292 nm. The removal efficiency (η) of the photocatalyst was calculated by eqn (1), and the apparent rate constants (k) for the samples were calculated by eqn (2):| | |
η = (C0 − Ct)/C0 × 100%
| (1) |
where C0 and Ct are the initial concentration of glyphosate and the concentration after an irradiation time of t, respectively.
Besides, the presence of active species was detected by trapping experiments to reveal the mechanism of O-C3N4/MoS2. Ethylenediaminetetraacetic acid disodium (EDTA-2Na), isopropyl alcohol (IPA) and 1,4-benzoquinone (BQ) were used as a hole (h+) scavenger, hydroxyl radical (·OH) scavenger and superoxide radical (·O2−) scavenger, respectively. In brief, 10 mM solutions of the radical scavengers were placed into 50 mL of a 9 mg L−1 solution of glyphosate, and then the suspensions were irradiated with simulated sunlight for the same time. Finally, the removal rate (η) of glyphosate was calculated to determine the main role of the active species.
2.5 Photoelectrochemical measurements
Photoelectrochemical measurements on the as-prepared catalysts were performed using a CHI-760 electrochemical system (Shanghai, China) equipped with a conventional three-electrode electrochemical cell. The as-prepared photocatalysts were coated on ITO substrates (ca. 2 cm2) and used as the working electrode. A Pt electrode and a saturated calomel electrode were employed as the counter electrode and reference electrode, respectively. A 0.2 M solution of Na2SO4 was used as the electrolyte, and a 300 W Xe lamp with AM1.5 cut-off filters was used as the light source in this system.
3. Results and discussion
3.1 Structure and morphology characterization
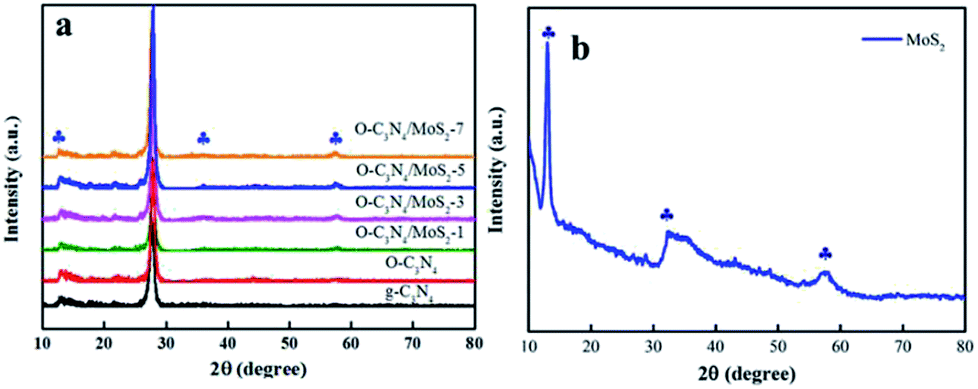
3.1.1 XRD analysis. The XRD patterns of pure g-C3N4, MoS2, and O-C3N4/MoS2 are displayed in Fig. 1. In the case of as-prepared g-C3N4, there exist two significant diffraction peaks at 2θ = 12.7° and 27.4°, which originated from in-plane repeating motifs of the continuous heptazine framework and the interlayer stacking structure of the conjugated aromatic sheets, respectively, and can be perfectly indexed to the (100) and (002) crystal planes, respectively.19 O-C3N4 exhibited similar diffraction peaks to those of pristine g-C3N4 and possessed high crystallinity, which indicated that the oxygen atoms may have bonded to the adjacent C or N atoms in g-C3N4. Moreover, all the typical peaks of MoS2, as shown in Fig. 1b, are located at 13.1°, 32.9° and 58.6°, which correspond to the (002), (100) and (110) planes, respectively. Hence, it can be distinctly identified as hexagonal 2H-MoS2.20 Furthermore, the peaks of MoS2 could be detected in the spectra of O-C3N4/MoS2, and we can hence conclude that MoS2 and O-C3N4 successfully combined with each other, which can change the charge transport paths and be beneficial for the photodegradation of contaminants.
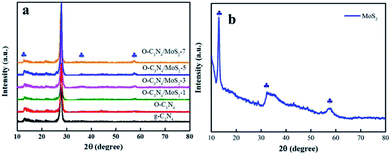 |
| | Fig. 1 X-ray diffraction patterns of (a) pure g-C3N4 and O-C3N4/MoS2 and (b) MoS2. | |
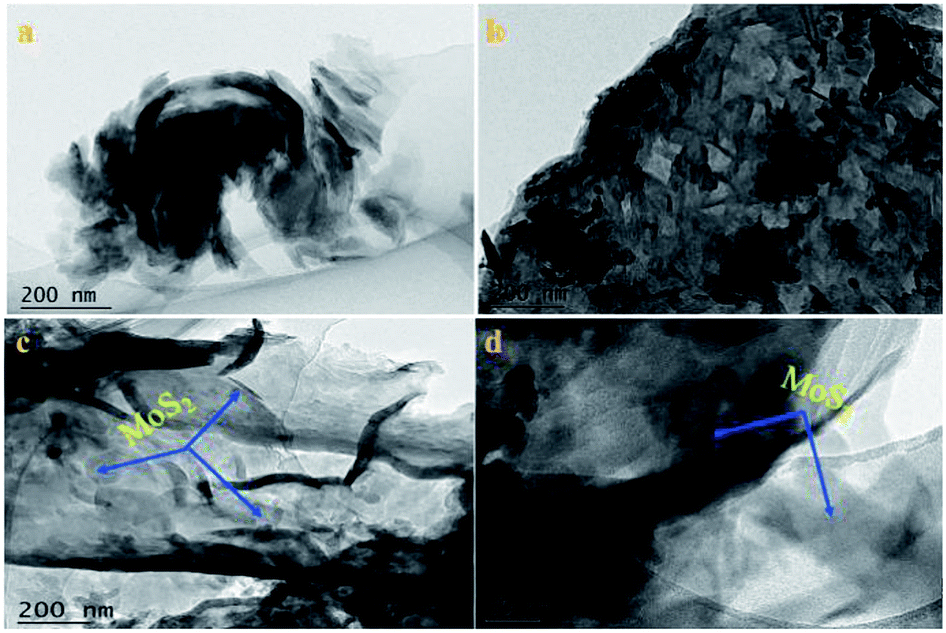
3.1.2 TEM analysis. The microstructure and morphology of the samples were revealed by TEM, as shown in Fig. 2. As shown in Fig. 2a, a TEM image of a light yellow powder of g-C3N4 revealed a typical stacked sheet structure, which demonstrated that the as-prepared g-C3N4 consisted of numerous sheets. Moreover, the morphology of oxygen-containing graphitized carbon did not destroy the microstructure of pristine g-C3N4, as seen in Fig. 2b, which shows a similar sheet morphology to that of pristine g-C3N4. In Fig. 2c and d, the images of the O-C3N4/MoS2-5 hybrid illustrate that thin and sheet-like O-C3N4 was decorated by MoS2 nanosheets with a random distribution and exhibited an irregular interlayer morphology after ultrasonic exfoliation and stirring during the synthesis of the composite photocatalyst. The thin layer can contribute to the acceleration of fast electron transfer between the surface of O-C3N4 and MoS2 and inhibit the recombination of electrons and holes, which will enhance the photocatalytic degradation of glyphosate.
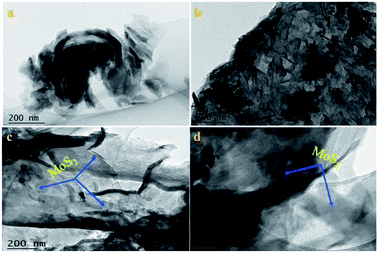 |
| | Fig. 2 TEM images of (a) g-C3N4, (b) O-C3N4 and (c and d) O-C3N4/MoS2-5. | |
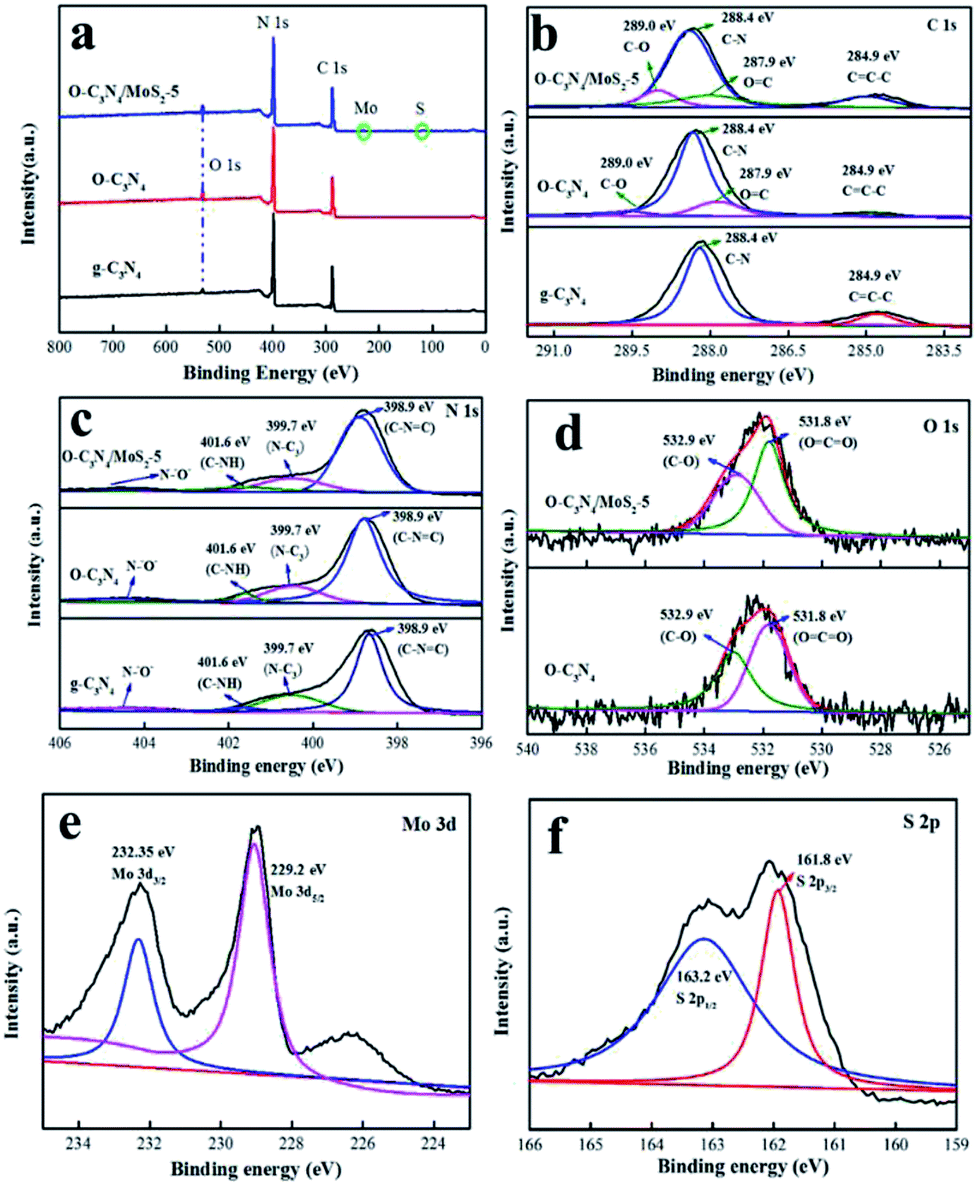
3.1.3 XPS analysis. The valence states and surface chemical compositions of pure g-C3N4, O-C3N4 and O-C3N4/MoS2-5 were characterized by XPS analysis. As shown in Fig. 3a, the elements C, N and O were all present in g-C3N4, O-C3N4 and O-C3N4/MoS2-5. Moreover, two weak peaks due to Mo and S elements were detected in the spectrum of O-C3N4/MoS2-5 owing to the low content of MoS2. Fig. 3b–f show the high-resolution C 1s, N 1s, O 1s, Mo 3d and S 2p spectra, respectively, of g-C3N4, O-C3N4 and O-C3N4/MoS2-5. The high-resolution C 1s spectrum displays two peaks at 288.4 and 284.9 eV, which were assigned to surface adventitious carbon or sp2 C–C bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C) formed during pyrolysis.22 In comparison with the spectrum of g-C3N4, two new peaks located at 289.0 and 287.9 eV appeared in the spectra of O-C3N4 and O-C3N4/MoS2-5, which were ascribed to C–O and OC–O bonds, respectively.10,11,21 As shown in Fig. 3c, deconvolution of the N 1s spectrum revealed four peaks at 404.2, 401.6, 399.7 and 398.8 eV, which correspond to charging effects and C–NH, N–(C)3 and C–NC bonds, respectively, which is consistent with reports by Zou et al. and Ran et al.22,23 In particular, two peaks centered at 532.9 and 531.8 eV were detected in the spectra of O-C3N4 and O-C3N4/MoS2-5, which were due to O–CO and C–O groups. Moreover, the C/N ratio increased from 0.460 in g-C3N4 to 0.503 in O-C3N4/MoS2-5, which led to nitrogen vacancies.24 We can conclude that oxygen atoms from β-CD were doped into the framework of g-C3N4 during pyrolysis, and some oxygen atoms may have replaced nitrogen atoms in the framework.10,25 Furthermore, O–CO and C–O groups are beneficial for photodegradation.10 The corresponding high-resolution Mo 3d spectrum can be deconvolved into two peaks centered at 232.35 and 229.2 eV, which correspond to Mo 3d3/2 and Mo 3d5/2, respectively,18,26 and demonstrate the primary presence of Mo4+ species in O-C3N4/MoS2-5. Besides, there exist two peaks located at 163.2 and 161.8 eV, which were attributed to S 2p1/2 and S 2p3/2, respectively, and represent the typical binding energies of S2−.26 Therefore, the XPS results illustrate that oxygen atoms were doped into the framework of g-C3N4 and replaced some of the N atoms in the crystal lattice to form O–CO and C–O groups, whereas MoS2 was successfully loaded onto the surface of O-C3N4, which can lead to the transfer of photoinduced electrons from one interface to another and prolong the lifetime of electrons, which provides more opportunities for the generation of ·O2− and hence improves the photocatalytic activity.
C–C) formed during pyrolysis.22 In comparison with the spectrum of g-C3N4, two new peaks located at 289.0 and 287.9 eV appeared in the spectra of O-C3N4 and O-C3N4/MoS2-5, which were ascribed to C–O and OC–O bonds, respectively.10,11,21 As shown in Fig. 3c, deconvolution of the N 1s spectrum revealed four peaks at 404.2, 401.6, 399.7 and 398.8 eV, which correspond to charging effects and C–NH, N–(C)3 and C–NC bonds, respectively, which is consistent with reports by Zou et al. and Ran et al.22,23 In particular, two peaks centered at 532.9 and 531.8 eV were detected in the spectra of O-C3N4 and O-C3N4/MoS2-5, which were due to O–CO and C–O groups. Moreover, the C/N ratio increased from 0.460 in g-C3N4 to 0.503 in O-C3N4/MoS2-5, which led to nitrogen vacancies.24 We can conclude that oxygen atoms from β-CD were doped into the framework of g-C3N4 during pyrolysis, and some oxygen atoms may have replaced nitrogen atoms in the framework.10,25 Furthermore, O–CO and C–O groups are beneficial for photodegradation.10 The corresponding high-resolution Mo 3d spectrum can be deconvolved into two peaks centered at 232.35 and 229.2 eV, which correspond to Mo 3d3/2 and Mo 3d5/2, respectively,18,26 and demonstrate the primary presence of Mo4+ species in O-C3N4/MoS2-5. Besides, there exist two peaks located at 163.2 and 161.8 eV, which were attributed to S 2p1/2 and S 2p3/2, respectively, and represent the typical binding energies of S2−.26 Therefore, the XPS results illustrate that oxygen atoms were doped into the framework of g-C3N4 and replaced some of the N atoms in the crystal lattice to form O–CO and C–O groups, whereas MoS2 was successfully loaded onto the surface of O-C3N4, which can lead to the transfer of photoinduced electrons from one interface to another and prolong the lifetime of electrons, which provides more opportunities for the generation of ·O2− and hence improves the photocatalytic activity.
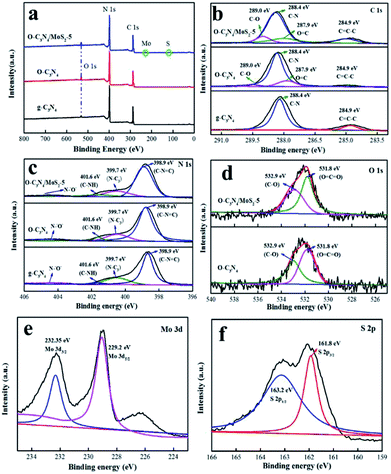 |
| | Fig. 3 XPS spectra of g-C3N4, O-C3N4 and O-C3N4/MoS2-5: (a) survey spectra, (b and c) high-resolution C 1s and N 1s spectra of g-C3N4, O-C3N4 and O-C3N4/MoS2-5, (d) high-resolution O 1s spectra of O-C3N4 and O-C3N4/MoS2-5, and (e and f) high-resolution Mo 3d and S 2p spectra of O-C3N4/MoS2-5. | |
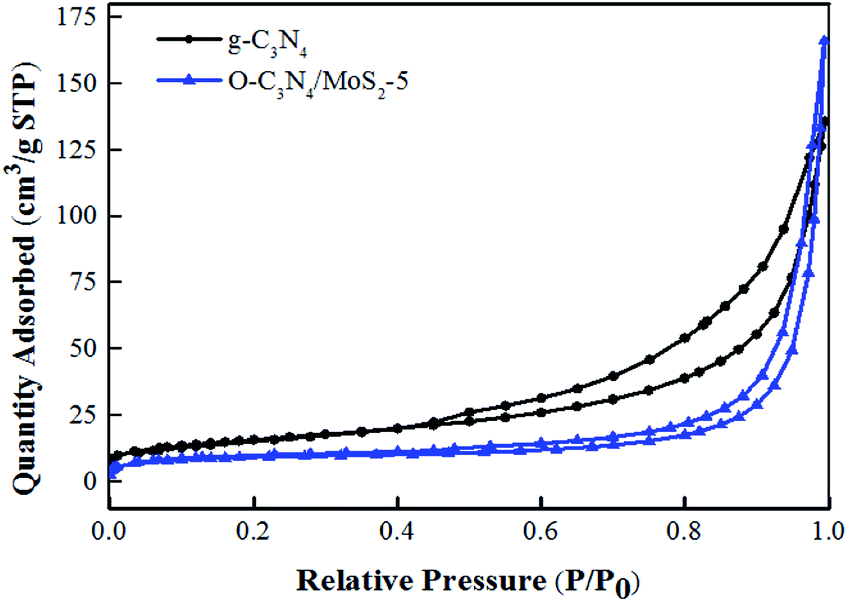
3.1.4 BET analysis. As shown in Fig. 4, the N2 adsorption–desorption isotherm curves of g-C3N4 and O-C3N4/MoS2-5 are classed as type IV isotherms with an H3 hysteresis loop according to the IUPAC classification system, which suggests slit-shaped pores produced by the layered structure, which was in close accordance with the results of SEM characterization.18,27 Moreover, some surface characteristics of as-prepared g-C3N4 and O-C3N4/MoS2-5 are listed in Table 1. The samples were all mesoporous materials, and the BET specific surface area of O-C3N4/MoS2-5 decreased in comparison with that of pure g-C3N4. However, the pore volume and average pore size increased. It is clear that the surface of pristine g-C3N4 was rough and many nanosheets were stacked together and formed some pores, whereas the surface of O-C3N4 was also rough and some pores were present, but the surface of O-C3N4/MoS2-5 was relatively smooth, as seen from the TEM images. This may be accounted for by the fact that O-C3N4 was exfoliated to form a sheet-like material and MoS2 was successfully loaded onto the surface of O-C3N4, which was accompanied by covering the pores and gaps between stacks, as well as the formation of the interlayer structure. Hence, O-C3N4/MoS2-5 possessed a smaller BET specific surface area and larger pore volume and average pore size, which was similar to the results that Cao et al. reported.28
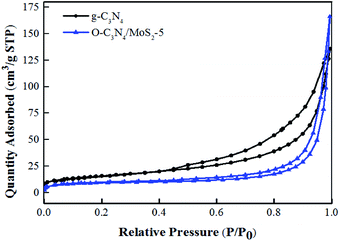 |
| | Fig. 4 N2 adsorption–desorption isotherm curves of g-C3N4 and O-C3N4/MoS2-5. | |
Table 1 Surface characteristics of as-prepared g-C3N4 and O-C3N4/MoS2-5
| Sample |
SBET (m2 g−1) |
Pore volume (cm3 g−1) |
Average pore size (Å) |
| g-C3N4 |
56.83 |
0.21 |
165.40 |
| O-C3N4/MoS2-5 |
33.89 |
0.25 |
459.13 |
3.2 Optical properties and photoelectrochemical properties
3.2.1 UV-vis diffuse reflectance spectra (DRS). The DRS spectra were employed to investigate the optical absorption properties of g-C3N4, O-C3N4, MoS2 and O-C3N4/MoS2-5. The band gap (Eg) of g-C3N4 and MoS2 was estimated according to Tauc's equation: (αhv) = A(hv − Eg)n/2. As shown in Fig. 5, the Eg values of pure g-C3N4 and MoS2 were approximately 2.7 and 1.84 eV, respectively, which was in accordance with previous reports.7,29–31 To act as a photocatalyst, a material should have conduction and valence bands, which can be calculated using the following empirical equations:where the values of X for MoS2 and g-C3N4 are 5.323 and 4.72, respectively, E0 is the energy of free electrons on the hydrogen scale (∼4.5 eV), and Eg is the band gap.32 On the basis of the above equations, the calculated positions of the CB and VB of MoS2 are −0.097 eV and 1.743 eV, respectively, and the calculated positions of the CB and VB of g-C3N4 are −1.13 eV and 1.57 eV, respectively.
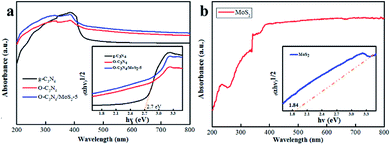 |
| | Fig. 5 UV-vis diffuse reflectance spectra of (a) g-C3N4, O-C3N4 and O-C3N4/MoS2-5 and (b) MoS2; the insets show plots of (αhv)1/2 vs. photon energy (hv). | |
After doping with oxygen atoms and the formation of a composite with MoS2, the absorption edge of O-C3N4/MoS2-5 underwent a red shift, which led to a wider absorption edge that reached about 537 nm. This may have occurred because MoS2 has a narrow band gap and has the potential to absorb infrared light. Fortunately, MoS2 was successfully loaded onto the surface of O-C3N4 and the photoinduced electrons would be transferred between the surface of O-C3N4 and MoS2, which indicated higher visible-light absorption efficiency and higher efficiency in the separation of electrons and holes.33 Ultimately, the narrow band gap and wide absorption edge extended the spectral response and thus enhanced the ability to photodegrade contaminants.16,34
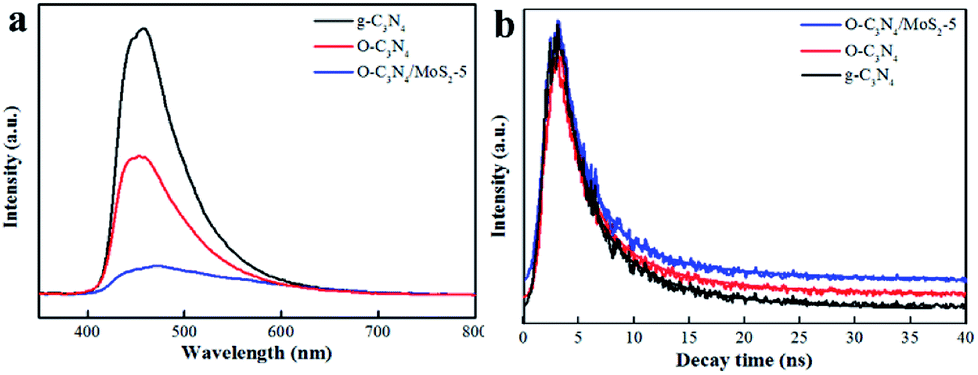
3.2.2 Photoluminescence (PL) spectra. PL was measured to study the ability of the photogenerated carriers to recombine. The results are displayed in Fig. 6a. The emission peak of O-C3N4/MoS2-5 underwent a red shift and the intensity of this peak decreased in comparison with that of pure g-C3N4, which suggested that the recombination of electrons and holes was effectively inhibited.11,21 In order to further understand the process of charge separation and transfer in the samples, the fluorescence decay time was measured, as shown in Fig. 6b,11,21 and the tendency of the decay time to change was determined. As shown in Table 2, the average fluorescence lifetime (τ) of O-C3N4/MoS2-5 was calculated to be 7.30 ns, which was longer than those of pristine g-C3N4 and O-C3N4, which suggested a longer electron lifetime and higher separation efficiency, which may improve the ability to photodegrade contaminants. Moreover, it can be clearly seen that O-C3N4/MoS2-5 exhibited a longer fast decay lifetime (τ1, 2.35 ns) and slow decay lifetime (τ2, 8.36 ns) than g-C3N4 (Table 2), which indicated that the separation of photoinduced electrons and holes in O-C3N4/MoS2-5 was more effective owing to the interlayer structures that were present and hence the photoinduced electrons had more opportunities to reconvert O2 into ·O2− and enhance the ability to photodegrade contaminants.27,35
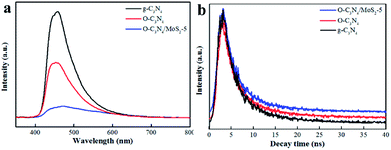 |
| | Fig. 6 (a) PL spectra and (b) time-resolved photoluminescence spectra of samples with an excitation wavelength of 330 nm. | |
Table 2 Dynamic analysis of emission decay for the different photocatalysts
| Sample |
A1 (%) |
τ1 (ns) |
A2 (%) |
τ2 (ns) |
τ (ns) |
| O-C3N4/MoS2-5 |
43.35 |
2.35 |
56.65 |
8.36 |
7.30 |
| O-C3N4 |
46.42 |
2.34 |
53.58 |
8.12 |
6.97 |
| g-C3N4 |
47.69 |
2.32 |
52.31 |
7.84 |
6.67 |
3.2.3 Transient photocurrent response and cyclic voltammogram (CV). In order to further investigate the photoresponse ability and the efficiency of the transfer and separation of charge carriers, the transient photocurrent responses and cyclic voltammograms of pure g-C3N4 and O-C3N4/MoS2-5 were recorded. From Fig. 7a, it can be seen that both pure g-C3N4 and O-C3N4/MoS2-5 exhibited fast and reproducible photocurrent responses, whereas O-C3N4/MoS2-5 exhibited a higher transient photocurrent response, which indicated that the lifetimes of the photoinduced charge carriers in O-C3N4/MoS2-5 were effectively prolonged and the ability to separate the photogenerated electrons and holes was higher, which was in accordance with the analysis of the PL and time-resolved photoluminescence spectra.21,26 The photocatalysts were dispersed in ethyl alcohol, doped onto the surface of ITO glass and fixed with naphthol. However, the fixed materials gradually detached, since a certain amount of cyclic voltage existed, which led to a decrease in the current. Moreover, CVs were recorded to determine the oxidation capacity of the samples. In Fig. 7b, it is clearly seen that O-C3N4/MoS2-5 exhibited a higher anodic peak and higher electric current at the same voltage in comparison with pure g-C3N4, which suggested that more photoinduced holes were present, which can provide evidence that O-C3N4/MoS2-5 displayed higher efficiency in the separation of photoinduced electrons and holes, which is consistent with the above characterization results, and exhibited a longer electron lifetime and higher separation efficiency, which will lead to a higher photodegradation efficiency, which is in close accordance with the above characterization results.36
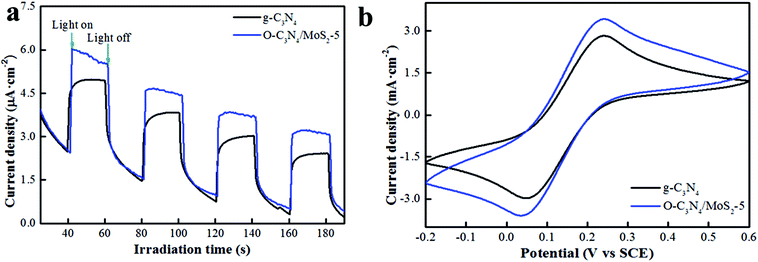 |
| | Fig. 7 (a) Transient photocurrent responses and (b) cyclic voltammograms of pure g-C3N4 and O-C3N4/MoS2-5. | |
3.3 Photocatalytic activity and photocatalyst stability
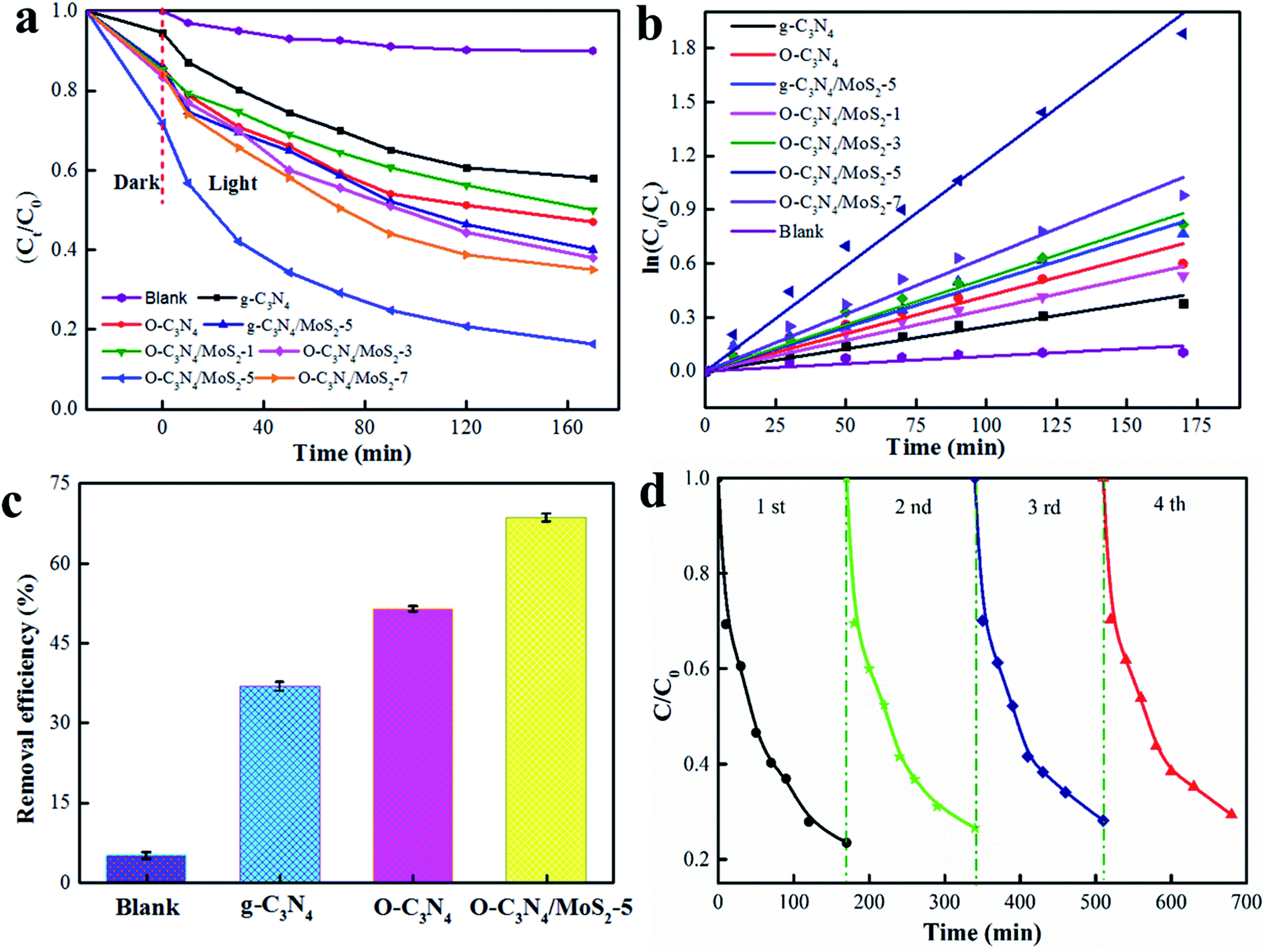
The photodegradation activities are shown in Fig. 8a. The photodegradation rate of pristine g-C3N4 was about 40%, whereas the O-C3N4/MoS2 composite in which the mass ratio of MoS2 accounted for 5% exhibited the highest photocatalytic activity, which reached about 84% after 170 min. This may have occurred because O-C3N4/MoS2-5 has a wider light absorption edge, a longer electron lifetime and a higher efficiency in the separation of electrons and holes in comparison with pure g-C3N4, which are in accordance with the characterization results and are all beneficial for the photodegradation of glyphosate. However, when the amount of MoS2 in the composite photocatalysts increased or decreased, the photodegradation rate for glyphosate decreased in comparison with that achieved using O-C3N4/MoS2-5, which may have occurred because a larger amount of MoS2 may limit the absorption of light, whereas a smaller amount of MoS2 could hinder the transfer of electrons between MoS2 and O-C3N4. Because O-C3N4 has a more negative CB in comparison with MoS2 (about −0.097 eV),10,37 photogenerated electrons in O-C3N4 after light irradiation can be effectively trapped by MoS2 for the degradation of glyphosate at the appropriate ratio of MoS2 to O-C3N4 because the interlayer structure can shorten the transfer distance of the electrons between the surface of MoS2 and O-C3N4 and inhibit the recombination of electrons and holes effectively.22,37 Fitted kinetic curves for the photodegradation of glyphosate are presented in Fig. 8b, and the results indicate that the photodegradation of glyphosate fitted a first-order kinetic model well. O-C3N4/MoS2-5 exhibited the highest rate constant, which reached 0.0518 min−1.
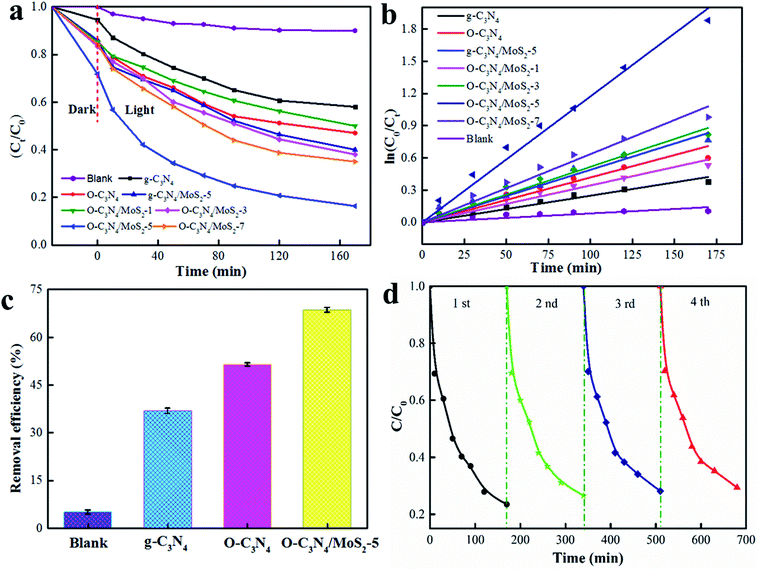 |
| | Fig. 8 (a and b) Photodegradation of glyphosate and kinetic curves for the photodegradation of glyphosate under simulated sunlight irradiation, (c) photodegradation of glyphosate under solar light, and (d) cycling runs in the photodegradation of glyphosate over O-C3N4/MoS2-5 under simulated sunlight irradiation. | |
The photodegradation of glyphosate over different materials under solar light for 3 h is illustrated in Fig. 8c. It is clearly seen that O-C3N4/MoS2-5 exhibited the highest removal efficiency of about 68%, which was twice that of pristine g-C3N4, which is in good agreement with the photodegradation of glyphosate under solar light. Because the photocatalysts received light of a lower intensity under solar light in comparison with that of the simulated sunlight, the photodegradation of glyphosate over O-C3N4/MoS2-5 was less effective under solar light than that under the simulated sunlight. The stability of the photodegradation of glyphosate over O-C3N4/MoS2-5 was studied for 4 cycles under simulated sunlight. As seen from Fig. 8d, after 4 cycles the removal rate still indicated no obvious loss of activity and reached an acceptable value of about 75%, which demonstrated the high photostability of the photodegradation of glyphosate.
3.4 Photodegradation mechanism
In order to confirm the presence of reactive species (ROS) that evolved during the photocatalytic reactions over pure g-C3N4 and O-C3N4/MoS2-5, ESR spin-trapping spectra with 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the trapping agent were recorded. As seen in Fig. 9a and b, no signals due to DMPO-·O2− or DMPO-·OH were observed in the dark over pure g-C3N4 and O-C3N4/MoS2-5. However, O-C3N4/MoS2-5 exhibited obvious signals due to DMPO-·O2− and DMPO-·OH under visible-light irradiation for the same period, in comparison with pure g-C3N4, which indicated that more ·O2− and ·OH radicals were produced, which may enhance the photodegradation of glyphosate. As depicted in Fig. 9c, it is obvious that the photodegradation rate decreased after the addition of BQ; however, the photodegradation rate did not decrease markedly after the addition of EDTA-2Na and IPA, which suggested that ·O2− was the major reactive species in the photodegradation of glyphosate over O-C3N4/MoS2-5.38
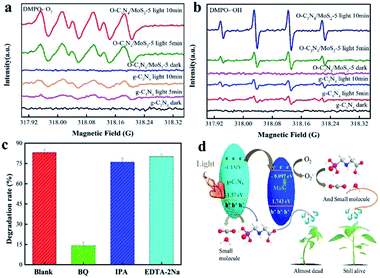 |
| | Fig. 9 ESR spectra of radical adducts trapped by DMPO in dispersions of pure g-C3N4 and O-C3N4/MoS2-5 both in the dark and under light irradiation: (a) in methanol under irradiation for DMPO-·O2− and (b) in an aqueous dispersion for DMPO-·OH; (c) photocatalytic photodegradation of glyphosate over O-C3N4/MoS2-5 with the addition of hole, ·O2− and ·OH radical scavengers under simulated sunlight; (d) proposed mechanism of the photocatalytic photodegradation of glyphosate over O-C3N4/MoS2-5. | |
A proposed mechanism of the photocatalytic photodegradation of glyphosate is illustrated in Fig. 9d. The MoS2 and g-C3N4 nanosheets could lead to significant synergetic effects on the photodegradation of glyphosate under simulated sunlight, owing to the effect of the electrons generated by charge redistribution on the band alignment between MoS2 and g-C3N4. On the basis of the above experimental results and the CB/VB edge potentials, the CB of g-C3N4 was more negative than that of MoS2, and the photogenerated electrons in the g-C3N4 nanosheets could be quickly transferred to the interface of g-C3N4 and MoS2. In addition, the CBs of g-C3N4 and MoS2 were more negative than E(O2/·O2−) = −0.33 eV, and hence the accumulated electrons could be trapped by O2 to form superoxide radicals (·O2−), which played a vital role in photodegradation. As a result, the interlayer structure can shorten the transfer distance of the electrons, which finally accumulate in the CB of MoS2, which can prolong the fluorescence lifetime of electrons and promote more efficient charge separation of e−/h+ pairs.26,39 Moreover, the holes in the VBs of g-C3N4 and MoS2 were more negative than E(OH−/·OH) = 2.40 eV, and hence the photogenerated holes (h+) could not react with H2O to produce ·OH over g-C3N4 and MoS2. The ·OH radicals that were generated may be attributed to the conversion of ·O2−. According to the above analysis, the reaction process can be expressed as the following equations:31,40
| | |
g-C3N4 + hv → g-C3N4 (h+ + e−)
| (5) |
| | |
·O2− + e− + 2H+ → H2O2
| (7) |
| | |
H2O2 + e− → OH− + ·OH
| (8) |
3.5 Toxicity test
As shown in Fig. 10, 20 mL glyphosate and its photodegradation products were sprayed on the leaves of pepper seedlings, and the photodegradation products were obtained by separating liquids from a mixture of glyphosate and O-C3N4/MoS2-5 exposed to simulated sunlight for 170 min. In this figure, the pots marked with “D” indicate that the plant was sprayed with the photodegradation products and the pots marked with “G” indicate that the plant was sprayed with glyphosate (9 g L−1). Interestingly, the plants became withered 5 h after being sprayed with glyphosate and almost died after 48 h. Nevertheless, the plants that were sprayed with the photodegradation products were still alive after 48 h, which provides convincing evidence that glyphosate had been almost completely degraded to small molecules by O-C3N4/MoS2-5 under simulated sunlight for 170 min. This may have occurred because ·O2− radicals played a vital role in the process of cleaving the C–P bond and C–N bonds in glyphosate to form small molecules such as PO3−, NO3−, NH4− and NO2−, which could not kill the plants.
 |
| | Fig. 10 (a–c) Pepper seedlings 0 min, 4 h and 48 h, respectively, after being sprayed with glyphosate and its photodegradation products. | |
4. Conclusions
In summary, an O-C3N4/MoS2 interlayer composite photocatalyst has been successfully constructed. In this architecture, the interlayer photocatalyst possesses abundant surface active sites and high stability, effectively utilizes visible light, and reduces the recombination efficiency of electrons and holes. When the mass ratio of MoS2 was 5%, O-C3N4/MoS2-5 exhibited remarkably enhanced activity in the photodegradation of glyphosate under simulated sunlight irradiation and solar light, which was twice that achieved using pure g-C3N4. The average fluorescence lifetime of the composite photocatalysts was extended from 6.67 ns to 7.30 ns in comparison with that of g-C3N4. This work provides a feasible strategy for synthesizing an interlayer photocatalyst for the highly enhanced photodegradation of glyphosate to a less toxic or non-toxic small molecule.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported financially by funding from the National Natural Science Foundation of China (21868034) and the International Science and Technology Cooperation Program of Shihezi University (GJHZ201601).
Notes and references
- J. Dechartres, J. L. Pawluski, N. Nmezu, C. Renoult and T. D. Charlier, Toxicol. Lett., 2017, 280, S151 CrossRef.
- T. Poiger, I. J. Buerge, A. Bachli, M. D. Muller and M. E. Balmer, Environ. Sci. Pollut. Res., 2017, R.24(2), 1588–1596 CrossRef PubMed.
- X. Chen, Z. Wu, D. Liu and Z. Gao, Nanoscale Res. Lett., 2017, 12, 143 CrossRef PubMed.
- D. Liu, Z. Wu, F. Tian, B. Ye and Y. Tong, J. Alloys Compd., 2016, 676, 489–498 CrossRef CAS.
- Y. Zhou, L. Zhang, J. Liu, X. Fan, B. Wang, M. Wang, W. Ren, J. Wang, M. Li and J. Shi, J. Mater. Chem. A, 2015, 3(7), 3862–3867 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27(13), 2150–2176 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116(12), 7159–7329 CrossRef CAS PubMed.
- P. Qiu, J. Yao, H. Chen, F. Jiang and X. Xie, J. Hazard. Mater., 2016, 317, 158–168 CrossRef CAS PubMed.
- D. Zheng, G. Zhang, Y. Hou and X. Wang, Appl. Catal., A, 2016, 521, 2–8 CrossRef CAS.
- W. Xing, C. Li, G. Chen, Z. Han, Y. Zhou, Y. Hu and Q. Meng, Appl. Catal., B, 2017, 203, 65–71 CrossRef CAS.
- J. W. Zhang, S. Gong, N. Mahmood, L. Pan, X. Zhang and J. J. Zou, Appl. Catal., B, 2018, 221, 9–16 CrossRef CAS.
- X. He, Z. Wu, Z. Sun, X. Wei, Z. Wu, X. Ge and G. Cravotto, J. Mol. Liq., 2018, 255, 160–167 CrossRef CAS.
- A. Lannoy, R. Bleta, C. Machut-Binkowski, A. Addad, E. Monflier and A. Ponchel, ACS Sustainable Chem. Eng., 2017, 5(5), 3623–3630 CrossRef CAS.
- S. Susarla, P. Manimunda, Y. Morais Jaques, J. A. Hachtel, J. C. Idrobo, S. A. S. Amnulla, D. S. Galvao, C. S. Tiwary and P. M. Ajayan, ACS Nano, 2018, 12(4), 4036–4044 CrossRef CAS PubMed.
- M. A. R. Anjum, H. Y. Jeong, M. H. Lee, H. S. Shin and J. S. Lee, Adv. Mater., 2018, 30(20), 1707105 CrossRef PubMed.
- B. Ren, W. Shen, L. Li, S. Wu and W. Wang, Appl. Surf. Sci., 2018, 447, 711–723 CrossRef CAS.
- Y. Liu, H. Zhang, J. Ke, J. Zhang, W. Tian, X. Xu, X. Duan, H. O Sun, M. Tade and S. Wang, Appl. Catal., B, 2018, 228, 64–74 CrossRef CAS.
- H. Tian, M. Liu and W. Zheng, Appl. Catal., B, 2018, 225, 468–476 CrossRef CAS.
- M. A. Mohamed, J. M. Jaafar, M. F. Zain, L. J. Minggu, M. B. Kassim, M. N. I. Salehmin, M. S. W. Rosmi, W. N. Salleh and M. H. D. Othman, Scr. Mater., 2018, 142, 143–147 CrossRef CAS.
- X. Xu, R. Zhao, W. Ai, B. Chen, H. Du, L. Wu, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2018, 30(27), 1800658 CrossRef PubMed.
- C. Zhou, C. Lai, D. Huang, G. Zeng, C. Zhang, M. Cheng, L. Hu, J. Wan, W. Xiong, M. Wen, X. Wen and L. Qin, Appl. Catal., B, 2018, 220, 202–210 CrossRef CAS.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S. Z. Qiao, Adv. Mater., 2018, 30(25), 1800128 CrossRef PubMed.
- Y. Zou, X. W. Shi, D. Ma, Z. Fan, L. Cheng, D. Sun, W. Zeyan and N. Chunming, ChemSusChem, 2018, 11, 1187–1197 CrossRef CAS PubMed.
- Q. Liu, X. Wang, Q. Yang, Z. Zhang and X. Fang, Appl. Catal., B, 2018, 225, 22–29 CrossRef CAS.
- X. An, S. Wu, Q. Tang, H. Lan, Y. Tang, H. Liu and J. Qu, Catal. Commun., 2018, 112, 63–67 CrossRef CAS.
- L. Shi, W. Ding, S. Yang, Z. He and S. Liu, J. Hazard. Mater., 2018, 347, 431–441 CrossRef CAS PubMed.
- Q. Xu, B. Zhu, C. Jiang, B. Cheng and J. Yu, Sol. RRL, 2018, 2(3), 1800006 CrossRef.
- Y. Cao, Q. Gao, Q. Li, X. Jing, S. Wang and W. Wang, RSC Adv., 2017, 7(65), 40727–40733 RSC.
- Q. Guo, Y. Zhang, J. Qiu and G. Dong, J. Mater. Chem., 2016, C4(28), 6839–6847 Search PubMed.
- S. Li, S. Hu, W. Jiang, Y. Liu, Y. Liu, Y. Zhou, L. Mo and J. Liu, Front. Chem., 2018, 6, 255 CrossRef PubMed.
- S. Li, S. Hu, W. Jian, Y. Liu, Y. Zhou, J. Liu and Z. Wang, J. Colloid Interface Sci., 2018, 530, 171–178 CrossRef CAS PubMed.
- G. Swain, S. Sultana, J. Moma and K. Parida, Inorg. Chem., 2018, 57(16), 10059–10071 CrossRef CAS PubMed.
- B. Ren, W. Shen, L. Li, S. Wu and W. Wang, Appl. Surf. Sci., 2018, 447, 711–723 CrossRef CAS.
- Z. Xie, Y. Feng, F. Wang, D. Chen, Q. Zhang, Y. Zeng, W. Lv and G. Liu, Appl. Catal., B, 2018, 229, 96–104 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- Z. Liu, Y. Liu, P. Xu, Z. Ma, J. Wang and H. Yuan, ACS Appl. Mater. Interfaces, 2017, 9(24), 20620 CrossRef CAS PubMed.
- A. Wu, C. Tian, Y. Jiao, Q. Yan, G. Yang and H. Fu, Appl. Catal., B, 2017, 203, 955–963 CrossRef CAS.
- S. Liu, D. Li, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, J. Colloid Interface Sci., 2016, 468, 176–182 CrossRef CAS PubMed.
- S. Li, S. Hu, W. Jiang, Y. Liu, J. Liu and Z. Wang, J. Alloys Compd., 2017, 501, 156–163 CAS.
- F. Tian, Z. Wu, Q. Chen, Y. Yan, G. Cravotto and Z. Wu, Appl. Surf. Sci., 2015, 351, 104–112 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Yongtao Xuea,
Zhenzhen Gaoa and
Xia Yangb
*ab,
Yongtao Xuea,
Zhenzhen Gaoa and
Xia Yangb