
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar-light-active mesoporous Cr–TiO2 for photodegradation of spent wash: an in-depth study using QTOF LC-MS†
Shrikant P. Taklea,
Onkar A. Apineb,
Jalindar D. Ambekara,
Sukeshani L. Landgec,
Namdeo N. Bhujbalc,
Bharat B. Kale a and
Ravindra S. Sonawane*a
a and
Ravindra S. Sonawane*a
aCentre for Materials for Electronic Technology, Government of India, Panchavati, Off Pashan Road, Pune 411008, India. E-mail: sonawaner@yahoo.com; sonawane@cmet.gov.in
bDepartment of Biotechnology, Shivaji University, Kolhapur, 416004, India
cAnnasaheb Magar Mahavidyalaya, Hadapsar, Pune 411028, India
First published on 1st February 2019
Abstract
A dark-coloured effluent called “spent wash” is generated as an unwanted product in sugarcane-based alcohol distilleries. Most distilleries discharge this effluent into soil or water without any treatment, causing water and soil pollution. Herein, we report chromium-doped TiO2 (Cr–TiO2) as a photocatalyst for the degradation of spent wash colour under natural sunlight. Cr-doped TiO2 nanoparticles were prepared using an aqueous titanium peroxide-based sol–gel method with titanium isopropoxide as the Ti precursor and chromium nitrate as the Cr precursor. To observe the effect of dopant on sol–gel behaviour and physicochemical properties, the Cr concentration was varied in the range 0.5–5 wt%. The crystallization temperature and time were optimized to obtain the required phase of Cr–TiO2. The physicochemical characteristics of the Cr-doped TiO2 catalyst were determined using X-ray diffraction, FE-SEM, FETEM, TG, XPS, the Brunauer–Emmett–Teller (BET) method, FT-IR, Raman, PL, ICP-MS, and UV visible spectroscopy. A shift in the absorption edge of TiO2 by doping with chromium suggested an increase in visible light absorption due to a decrease in the effective band gap. The application potential of the Cr–TiO2 catalyst was studied in the degradation of sugar-based alcohol distillery waste under natural sunlight, and the results were compared with those of undoped TiO2 and Degussa P25 TiO2. Degradation of the spent wash solution was monitored using UV-visible, gel permeation chromatography (GPC), and QTOF LC-MS. GPC and LC-MS showed significant changes in the molecular weight of spent wash colour-forming compounds due to the degradation reaction. QTOF LC-MS analysis suggested that acids, alcohols, glucosides, ketones, lipids, peptides, and metabolites were oxidized to low-molecular-weight counterparts. From the results, 5% Cr–TiO2 showed the highest degradation rate among all Cr–TiO2 samples, undoped TiO2, and Degussa P25 TiO2 under identical reaction conditions, with nearly 68–70% degradation achieved in 5 h.
1. Introduction
An adequate supply of fresh and clean water is a basic need of all humans. Without clean water, neither humans nor the environment can survive. The demand for water has increased over the years, which has led to water scarcity in many regions. Furthermore, agricultural land loses its fertility due to the disposal of spent wash directly into rivers. Distilleries use different raw materials, such as sugarcane juice, sugarcane molasses, sugar beet molasses, cereals, and other agricultural products to produce alcohol and other beverages. The production of fermented and distilled beverages worldwide is based on materials that can be grown locally and are best suited to prevailing climatic conditions. During the distillation process, an average of 12–15 L of effluent is generated per litre of ethanol produced. Molasses is fermented by yeast after suitable dilution. The unwanted bottom part of the distillation is known as stillage, spent wash, or alcohol distillery waste. In addition to being coloured, this discharged distillery effluent contains high amounts of total dissolved solids, contributed to by an acidic pH and other pollutants. These solids are complex polymers containing heterocyclic nitrogenous aldehyde-amine compounds, various heavy metals, phenolic compounds, and plant-derived resins and fatty acids. The polluting strength of the effluent is very high owing to the presence of biodegradable organic materials, such as sugars, lignin, hemicellulose, dextrin, resin, and organic acid. In addition to its overall composition, distillery waste possesses a strong foul smell at high temperatures. The dark brown colour of spent wash is due to the presence of melanoidin, a natural condensation product of sugars and amino acids produced by the Maillard reaction, a non-enzymatic browning reaction.1 Melanoidins are widely distributed in food2 and drinks, and are discharged in large amounts by various agricultural and allied industries.3 These melanoidins are highly resistant to microbial attack and conventional biological processes, such as activated sludge treatment, are not sufficient to treat colorants containing wastewater released from distilleries and the fermentation industries. Treatment methods, such as ozonation, flocculation, chemical treatment, electrocoagulation, charcoal-based filtration, and activated carbon adsorption have been extensively studied for treating spent wash solution.4–14 However, less attention has been paid to environmentally benign photocatalytic degradation routes, which can potentially provide a better solution. Therefore we have investigated the photocatalytic route using a cheap, stable, and readily available photocatalyst, TiO2, and modified TiO2-based photocatalysts under UV and visible light.Titanium dioxide (TiO2) is among the most widely used photocatalysts in environmental treatment technologies. Several applications of TiO2 have been reported previously in the degradation of toxic organic compounds, removal of pollutants from contaminated water, air, and destruction of cells in harmful bacteria.15–18 The potential of TiO2 in these applications is mainly limited by its wide band gap (3.2 eV), meaning that only light below 387 nm in the solar spectrum can be utilized. Consequently, TiO2 photocatalysts that are active under visible light are promising for enhancing the photoactivity and widening the application potential of TiO2. Much effort has been made to increase the efficiency of TiO2 photocatalysts under UV and visible light. Surface modification19–23 and the introduction of dopants into the TiO2 lattice24–27 are recognized methods for improving TiO2 performance. Anionic or cationic dopants can be introduced into the crystal lattice to obtain visible-light activity. In the last decade, several researchers have studied visible-light anion-doped TiO2 (N, S, F, P) materials synthesized using physical and chemical methods, and assessed their photocatalytic performance under UV and visible light.
In addition to anion doping, metal-ion doping, particularly with transition metals such as Fe, Ni, Cu, Mn, Co, V, W, Nb, Mo, and Cr, has been reported by various groups.27–33 The doping of transition metal ions into TiO2 extends the absorption edge from the UV region to the visible region, resulting in an improved photoresponse due to additional energy levels introduced within the TiO2 band gap.27,33–36 Although transition-metal-doped TiO2 shows an improved photoresponse to visible light and a consequent increase in pollutant degradation activity, Cr3+ ion doping has proven to be most effective method for improving the photocatalytic activity of TiO2. Cr3+ ion doping helps to extend the absorption edge from UV to the visible-light region, making the photocatalyst active under visible light, which is the most abundant natural solar radiation that reaches the earth's surface.27,37–42 In most studies, the intension of doping was to insert chromium into the TiO2 lattice. Different synthesis methods, such as sol–gel,43 hydrothermal,44 flame pyrolysis,45 solid-state,46 physical vapour deposition (PVD),47 and ion implantation,48 have been reported for the preparation of Cr-doped TiO2 thin films and powders. Each method has its own merits and disadvantages. Previous studies have shown that CVD and sputtering are more effective for incorporating Cr into the TiO2 matrix and, as a result, shifting the absorption threshold towards the visible region.37 A hydrothermal method reported for the preparation of Cr-doped TiO2 also improved the photocatalytic performance.27 Mesoporous Cr-doped TiO2 synthesized using an evaporation-induced self-assembly technique also showed improved absorption in the visible region.42 In some previous studies, Cr–TiO2 synthesized by the sol–gel technique showed only a slight red shift in the absorption edge toward the visible region.38 However our group has successfully demonstrated that an aqueous titanium peroxide-based modified sol–gel method is suitable for synthesizing N- and transition-metal-doped TiO2 thin film and powder photocatalysts.49–56 A series of doped TiO2 photocatalysts have been synthesized using this technique and undoped and doped TiO2 have been applied to organic pollutant degradation and H2O splitting into H2 under UV and visible light, especially under naturally available energy sources such as sunlight. In most reports, Cr-doped TiO2 has been used for organic pollutant degradation, but the application of Cr–TiO2 to the degradation of spent wash, an industrial waste from sugar-based alcohol distilleries, has yet to be reported.
Therefore, in this study, we report the synthesis of Cr-doped mesoporous TiO2 using the aqueous titanium-peroxide-based sol–gel method and its application to the degradation of spent wash. Parameters including dopant concentration, aging time, and annealing temperature were optimised. The samples were characterized by XRD, the BET method, porosity, FE-SEM, TEM, FT-IR, Raman, TG, XPS, PL, ICP-MS, and UV-Vis absorption spectroscopy. The photocatalytic activity of sol–gel-derived chromium-doped mesoporous TiO2 has been tested in a unique application of industrial importance, namely, spent wash degradation under natural sunlight. The degradation of spent wash and its degradation products were monitored using UV-Vis, GPC, and LC-MS techniques. The change in colour was monitored using UV-Vis spectrophotometry, the molecular weight of degradation products was determined using GPC, and the untargeted group of compounds was monitored using QTOF LC-MS.
2. Experimental
2.1. Materials and reagents
Chemicals and raw materials were used as received without further treatment. Titanium(IV) tetraisopropoxide (Ti(OCH(CH3)2)4) was obtained from Acros Organics. Hydrogen peroxide (H2O2, 30% w/w) and chromium nitrate nonahydrate were obtained from Merck India. P25 TiO2 (Degussa) was purchased from a local supplier (M.M. Supplier, Pune, India). Milli-Q Water was obtained from a Q Pad filter with a conductivity of 0.05 μs cm−1 at 25 °C.2.2. Catalyst preparation
A series of Cr-doped TiO2 catalysts with Cr contents of 0.5, 1, 2, 3, 4, and 5 wt% were prepared by an aqueous sol–gel technique using chromium nitrate nonahydrate and titanium isopropoxide as the chromium(III) and titanium precursors, respectively. In a typical synthesis, titanium isopropoxide (4.02 g) was hydrolysed using Milli-Q water. Aqueous H2O2 (30%) was slowly added to get a transparent orange sol of titanium peroxide. Chromium nitrate nonahydrate (for example, 76.92 mg for 1.0% Cr–TiO2) was suspended in Milli-Q water (5 mL) and 30% aqueous H2O2 (5 mL) was added to obtain a clear green peroxochromic acid solution. This solution was added to the titanium peroxide solution with stirring for 30 min. The mixture of both sols slowly converted into a transparent greenish-yellow viscous gel. The chromium–titania peroxide gel was dried at ambient temperature to obtain a dry gel. The dry gel was then heated in a hot air oven at 105 °C, followed by annealing at 400 °C under static air flow in a muffle furnace. The heating/cooling rate was 5 °C min−1, with a 4 h dwell time at the selected temperature. Similarly, undoped TiO2 was prepared using the aqueous sol–gel method.2.3. Characterization techniques
X-ray diffraction (XRD) was performed on a Bruker Model-D8-Advance AXS diffractometer (Cu Kα radiation, λ = 1.5406 A) with an operating voltage of 40 kV and a current of 44 mA. Data were collected in the 2θ range of 20–80° with a step size of 0.028 and counting time of 15 s per step. Diffuse reflectance UV-Vis DRS spectra were recorded in the range 200–800 nm with a 0.5 nm spectral bandwidth in air at ambient temperature using a Shimadzu instrument (UV 3600) spectrophotometer. The powder samples were mixed with barium sulphate powder (NIST traceable) and pressed into the sample holder for UV-Vis measurements. Raman spectra were recorded using a Raman microscope (Renishaw 2000) at a wavelength of 532 nm and exposure time of 16 scan per s, with four accumulations. FT-IR spectra were recorded with a Perkin-Elmer (Spectrum Two, U-ATR) instrument using 32 scans and a 4 cm−1 spectral resolution in the range of 4000–450 cm−1. The thermal stability and behaviour of the samples were analysed using a TG/DTA instrument (Mettler-Toledo) with a heating rate of 5 °C min−1 under an air atmosphere. The total organic carbon (TOC) in spent wash samples was measured in grab mode using a GE TOC-500 RL analyzer. Elemental analysis was conducted by ICP-MS (Agilent Technologies 7700 spectrometer with ASX 500 series auto sampler) using Mass Hunter workstation software (version A.01.02). Calcined samples were digested in a digester (SP-Discover, CEM) by supra-grade concentrated hydrochloric acid at 180 °C for 10 min. After digestion, the sample is diluted to respective volume with Milli-Q water. The surface area of samples was obtained with the Brunauer–Emmett–Teller (BET) method using a Micromeritics Gemini VII (2390t) analyser. The pore size distribution was calculated from the desorption isotherm using the Barrett–Joyner–Halenda (BJH) method. Prior to measurements, the powder samples were degassed using UHP grade nitrogen gas at 90 °C for 1 h and 150 °C for 4 h. Sample porosity was measured using the mercury intrusion porosimetry (MIP) technique of a Micromeritics Autopore IV (9510 model). The surface morphology and microstructure were analysed using a FESEM and FETEM instruments. For these measurements, the powder samples were first sonicated in ethanol using an ultrasonic bath and then dropcast onto carbon tape. HRTEM measurements were performed with a JEOL instrument operated at an accelerating voltage of 300 kV. Samples for HRTEM analysis were prepared by vacuum drying the TiO2 nanoparticles solution dropcast onto carbon-coated copper grids. Image processing and interplanar distance (d) evaluation were performed using micrograph Gatan software. Spent wash samples after degradation were analysed using UV-Vis, GPC, and LC-MS techniques. Details of the LC-MS method used for analysis are provided below.3. Photocatalytic study
3.1. Photocatalytic decomposition of spent wash (SW)
The photocatalytic activity of the prepared photocatalysts was tested in the degradation of spent wash using a 100 mL quartz reactor with a water flow arrangement to maintain constant temperature. Spent wash solution (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm in water) was used as a model pollutant. A suspension was prepared by mixing Cr–TiO2 photocatalyst (0.025 g) in spent wash solution (100 mL, 10000 ppm). Before exposure to light, the suspension was kept in the dark for 1 h to achieve an adsorption–desorption equilibrium. For photocatalytic degradation of the spent wash, the Cr–TiO2 powder samples were used as photocatalysts. The suspension was irradiated at ambient conditions under natural solar light on sunny days (September–December) in Pune, Maharashtra, India, for specified time periods. At regular intervals, samples were collected, centrifuged, and allowed to settle to separate the photocatalyst particles. The supernatant was analysed for colour using UV-Vis spectrophotometry, MW using GPC, and organic compounds using QTOF LC-MS.
000 ppm in water) was used as a model pollutant. A suspension was prepared by mixing Cr–TiO2 photocatalyst (0.025 g) in spent wash solution (100 mL, 10000 ppm). Before exposure to light, the suspension was kept in the dark for 1 h to achieve an adsorption–desorption equilibrium. For photocatalytic degradation of the spent wash, the Cr–TiO2 powder samples were used as photocatalysts. The suspension was irradiated at ambient conditions under natural solar light on sunny days (September–December) in Pune, Maharashtra, India, for specified time periods. At regular intervals, samples were collected, centrifuged, and allowed to settle to separate the photocatalyst particles. The supernatant was analysed for colour using UV-Vis spectrophotometry, MW using GPC, and organic compounds using QTOF LC-MS.
The UHPLC-QTOF-MS instrument used in this study was an Agilent Series 1290 infinity rapid resolution LC system interfaced with electrospray ionization (ESI) to an Agilent 6540 UHD Accurate Mass QTOF LC-MS. A ZORBAX RRHD Eclipse Plus reversed phase C-18 column (100 mm × 2.1 mm × 1.8 μm particle size) was used at a temperature of 40 °C. The sample volume was 3 μL in both ionization modes and 5 μL for MS–MS analysis. The mobile phases used were 0.1% formic acid in water (eluent A) and 0.1% formic acid in methanol (eluent B). The following gradient elution was used: 3–20% B, 0–4 min; 20–43% B, 4–7 min; 43–50% B, 7–10 min; 50–65%, 10–15 min; 65–85% B, 14–18 min; 85–97% B, 18–21 min; 3% B, 21–27 min. The equilibration step was followed by a stop time 1 min at a flow rate of 0.4 mL min−1.
4. Results
4.1. Thermal and crystallization behaviour
The thermogravimetric (TG) analysis curve of air-dried 5% Cr–TiO2 gel is shown in Fig. S1 (ESI).† The sample subjected to TG showed a total weight loss of about 20–25% in two stages in the temperature range of 30–300 °C. The initial steep weight loss at around 90–100 °C might be due to the loss of absorbed water in the Cr–TiO2 gel. The weight loss in the temperature range of 100–300 °C might be due to decomposition of the titanium peroxide complex into titanium dioxide and titanium hydrous oxide. In this temperature range, most of the titanium peroxo complex is converted to titanium oxide and a much lower amount of hydrous oxide is left unconverted. Further weight loss between 300 to 900 °C, which was not steep but steady, was observed and might be attributed to the conversion of some remaining hydrous oxide into crystalline TiO2.4.2. Structural analysis using X-ray diffraction (XRD)
The gel was allowed to dry in an open atmosphere at ambient temperature and used for XRD analysis. The dried gel was further heated at 105 °C in an oven overnight and finally calcined at various temperatures in the range of 200–600 °C. The XRD pattern of Cr-doped TiO2 is shown in Fig. 1.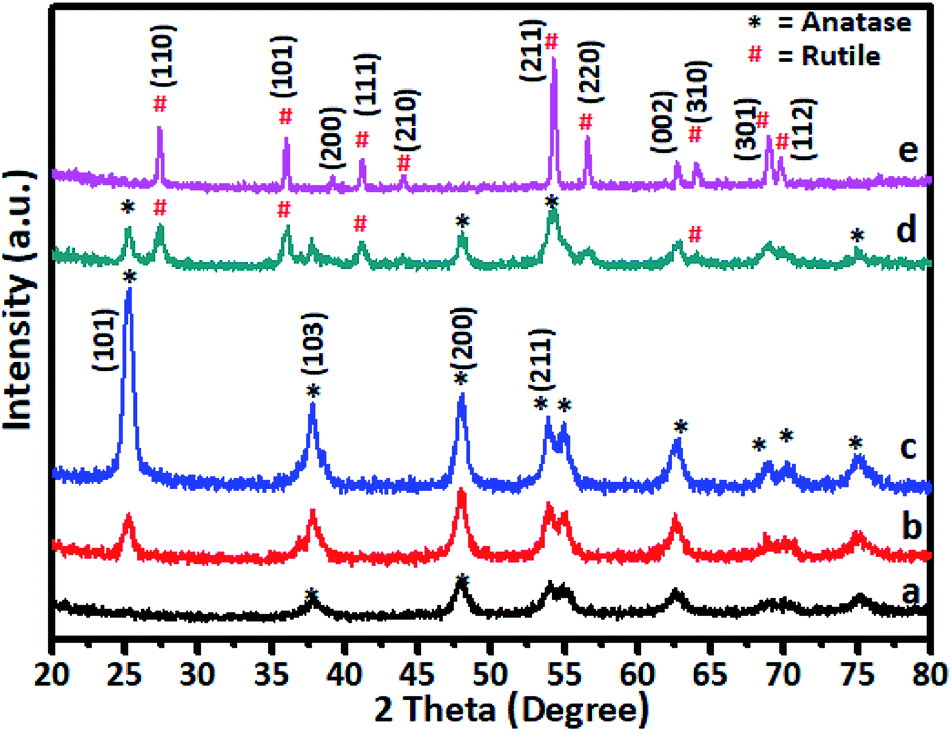 | ||
| Fig. 1 XRD pattern of 5% Cr–TiO2 calcined at (a) 200, (b) 300, (c) 400, (d) 500, and (e) 600 °C. | ||
Generally, as the sol–gel-derived TiO2 was amorphous, calcination was required to induce crystallization. As shown in Fig. 1a, the sample calcined at 200 °C showed very weak peaks symptomatic of the beginning of crystallization at this temperature. All diffraction peaks were indexed to anatase-phase TiO2 (JCPDS card no. 21-1272), suggesting the rearrangement of atoms in the amorphous gel to the anatase phase. As the annealing temperature was increased to 300 °C (curve b), further crystal phase growth occurred, as demonstrated by the increase in peak intensity observed at this temperature and peaks appearing more distinct. The peaks positioned at 2θ values of 25.36, 37.84, 48.11, 54.38, 55.07, and 62.88 are indexed as (101), (103), (200), (105), and (213) reflections of crystalline anatase phase. At 400 °C, the peak intensities corresponding to the anatase phase were increased to a level that indicated formation of fully grown anatase phase. This was confirmed by the sample annealed at 500 °C (curve d) showing some additional peaks in addition to the anatase phase, which were indexed to rutile-phase TiO2. For undoped TiO2 in the titanium peroxy complex-based gel system, the critical temperature at which the anatase-to-rutile phase transformation began was around 600 °C.52 However, in Cr-doped TiO2, rutile phase crystallization started at a much lower temperature of 500 °C. This lower crystallization temperature might be attributed to Cr doping in TiO2, which was consistent with previous reports.20 A further increase in annealing temperature to 600 °C showed growth of rutile TiO2 as all peaks in curve (e) were indexed to the rutile phase. The diffraction peaks at 2θ values of 27.53, 36.14, 39.24, 41.32, and 54.38 were indexed to the (110), (101), (200), (111), and (211) reflections of the crystalline rutile phase. No crystalline phase of chromium clusters, chromium oxides, or Cr–Ti oxide phases were observed, even at the highest Cr concentration of 5 wt%. This showed that, as a dopant in TiO2, Cr exhibited no tendency to segregate and/or precipitate in different phases during the synthetic process.27,33,57,58 The Cr ions can occupy interstitial positions or systematically substitute Ti ions in Cr–TiO2 without modifying the host TiO2 matrix. The XRD peak positions of 5% Cr-doped samples annealed at 600 °C were in good agreement with the standard JCPDS data for TiO2 rutile phase.37–41 As the radius of Cr3+ ions (0.76 Å) is very close to that of Ti4+ (0.75 Å), the substitution of Ti4+ by Cr3+ would not much alter the rutile lattice. This observation indicated that the probability of Cr occupying interstitial positions in the TiO2 matrix was low in our samples. The effect of Cr content on the crystallization behaviour of the doped samples was studied by varying the Cr concentration of in TiO2 while keeping the calcination temperature constant. The calcination temperature was kept constant at 400 °C because the samples annealed at this temperature showed the highest activity for spent wash degradation among Cr–TiO2 samples annealed at other temperatures. The XRD patterns of pure TiO2 and Cr–TiO2 samples containing different chromium concentrations are shown in Fig. S2 (ESI).† The diffraction patterns do not show much change in the TiO2 crystal phase, as most peaks were indexed to the pure anatase phase structure. However, as discussed earlier, the crystallization temperature of the rutile phase was lowered by doping Cr into TiO2.
4.3. Surface morphology using FESEM and HRTEM
The surface morphology/microstructure of undoped and doped samples analysed by FE-SEM showed that the powders contain TiO2 granules with densely packed particle agglomerates. The granules on the surface are spheroid-shaped particles (Fig. 2a and b). The interparticle distance was lower, suggesting that the sample has less porosity, which could be the reason for the lower surface area of undoped TiO2. In FESEM images of TiO2, although the particles had a granular texture, the particle morphology was not uniform in size and shape. However, Cr–TiO2 (Fig. 2c and d) showed granules with small empty spaces between particles, resulting in the sample texture appearing porous. The particle porosity was high in Cr–TiO2 samples, which might explain the higher surface area of these samples compared with undoped TiO2. The Cr and nitrate groups from the Cr precursor were present in the gel, which might be the reason for the porous nature of Cr–TiO2 samples. This ultimately resulted in an increase in the surface area of Cr–TiO2. The FESEM analysis results were in good agreement with the surface area analysis results. The sample porosity was confirmed using the mercury intrusion porosimetry (MIP) method.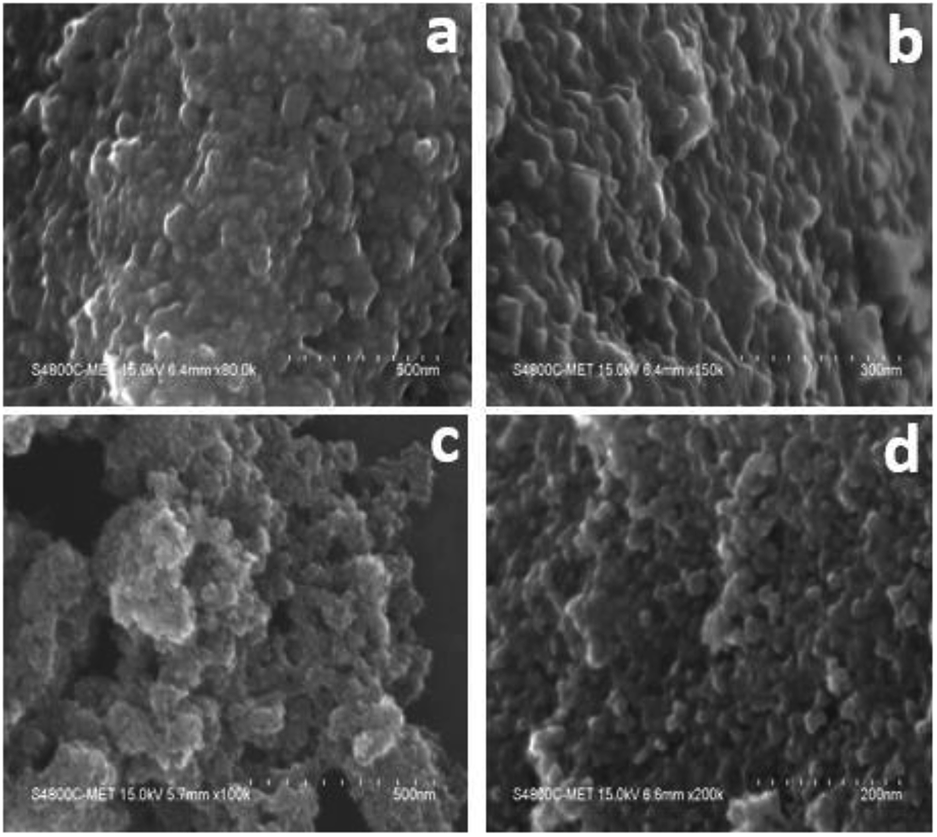 | ||
| Fig. 2 FESEM images: (a and b) high and low magnification images of undoped TiO2, and (c and d) high and low magnification images of 5% Cr–TiO2. | ||
The surface morphology was further confirmed using FETEM, with the results for Cr-doped TiO2 shown in Fig. 3. All results were collected from the 5% Cr–TiO2 sample as a representative example, and scanning was performed in different regions of the sample. The TEM images clearly indicated that the particles were small and identical shapes. No individual chromium oxide species were observed during analysis, which further validated the incorporation of chromium into TiO2. As shown in Fig. 3b, the aggregated nanoparticles with average sizes of ∼10–15 nm were observed with good uniformity in size and shape. The lattice image in Fig. 3c suggested that the measured d-spacing (0.34 nm) corresponded to the (101) plane of anatase TiO2. This domain exhibited smaller particles with pores evenly dispersed between particles. The SAED pattern in Fig. 3d clearly shows a dot-type diffraction pattern indexed to the (101) plane of anatase TiO2, suggesting that the nanoparticles were single crystals.
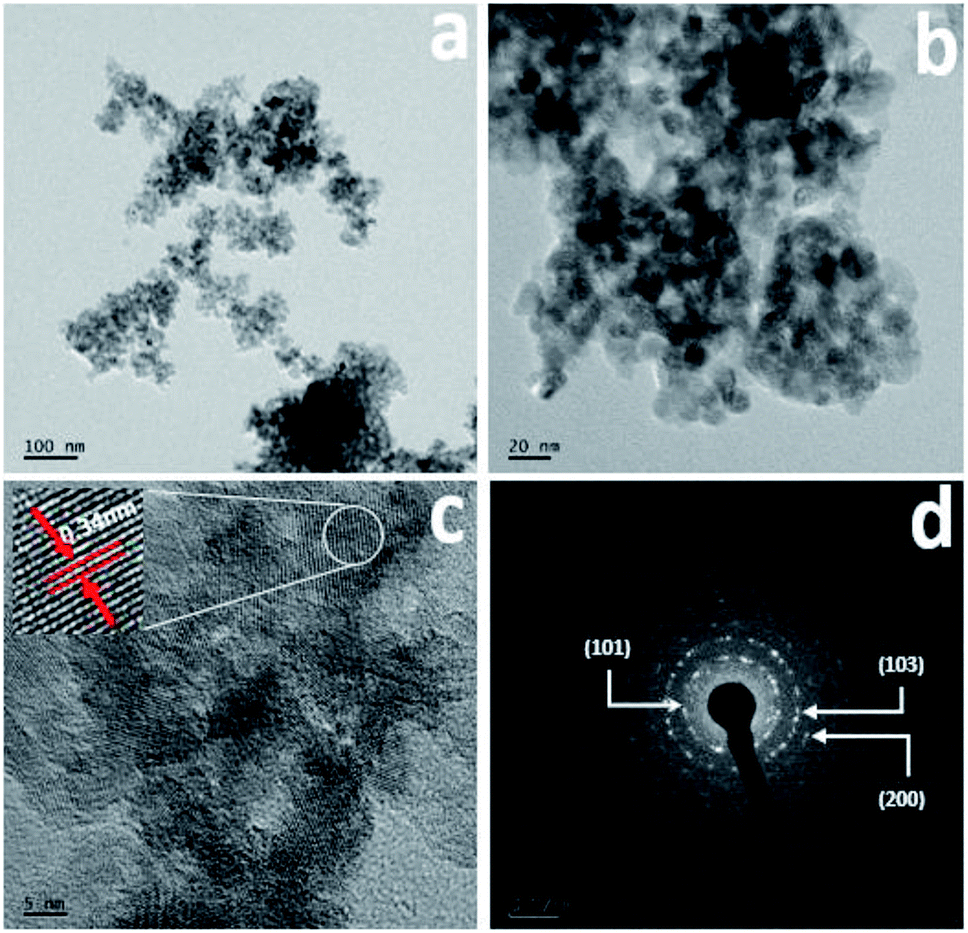 | ||
| Fig. 3 TEM images: (a and b) low and high magnification images, (c) lattice image, and (d) SAED pattern of 5% Cr–TiO2 sample. | ||
4.4. Nitrogen isotherms of catalysts
The N2 sorption overlay isotherms of undoped TiO2 and Cr–TiO2 catalysts annealed at 400 °C are shown in Fig. 4. The curves exhibited type IV isotherms with H2-type hysteresis loops according to the IUPAC classification.59 The isotherms were logged at 77 K with a Micromeritics Gemini VII 2390t instrument. The adsorption–desorption curves for undoped and doped catalysts at 400 °C showed hysteresis curves at relative pressures of 0.55 to 0.85 with anatase phase formation.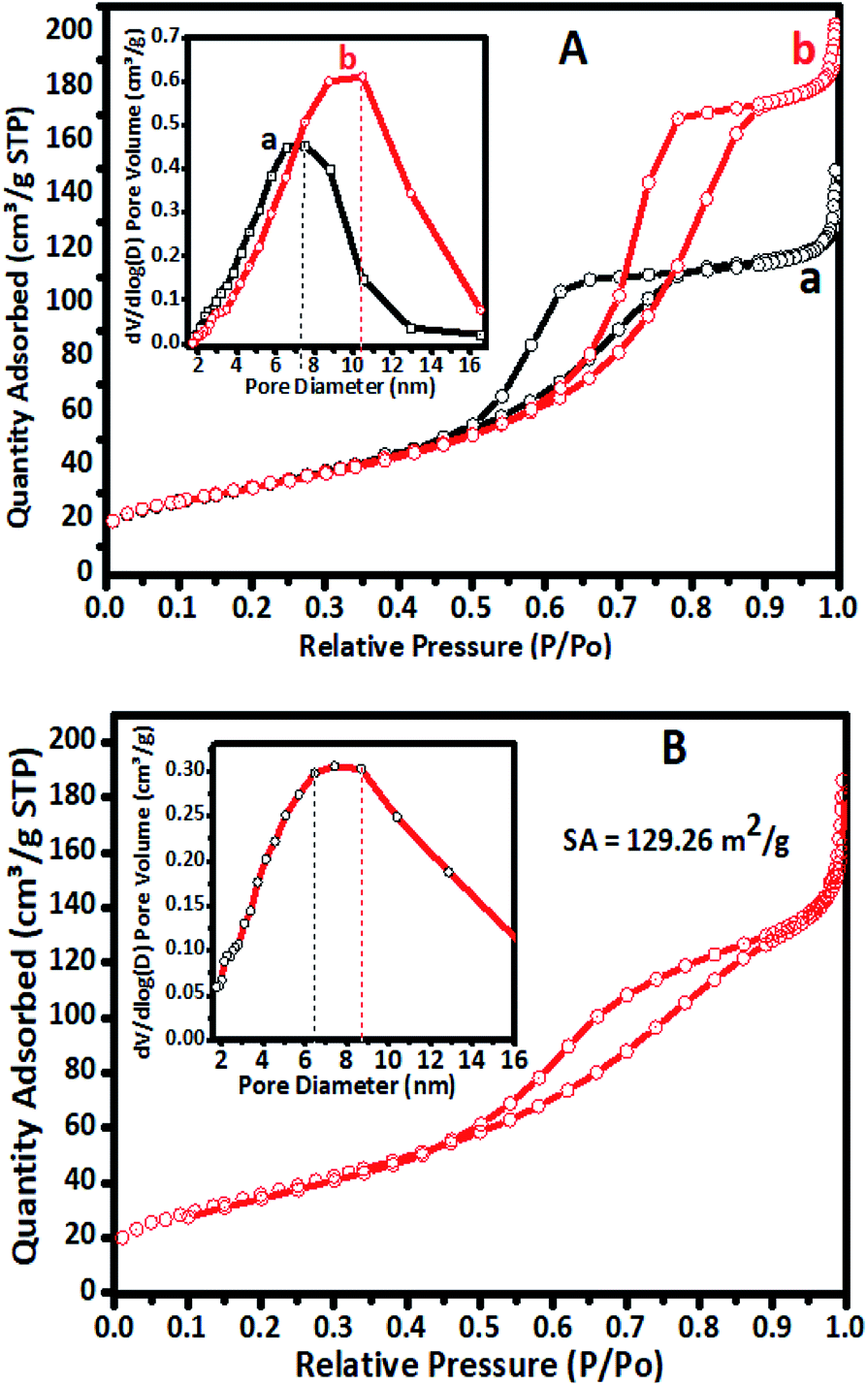 | ||
| Fig. 4 (A) Nitrogen physisorption isotherms of (a) undoped TiO2 and (b) 5% Cr–TiO2 calcined at 400 °C. Inset: BJH pore size distribution curves of (a) undoped TiO2 and (b) 5% Cr–TiO2 at 400 °C. (B) N2 sorption isotherm of 5% Cr–TiO2 recovered after the third cycle. | ||
Nitrogen adsorption–desorption isotherms of undoped TiO2, 5% Cr–TiO2, and 5% Cr–TiO2 recovered after the third cycle of the photodegradation reaction are shown in Fig. 4. A slight decrease in surface area from 148 to 129 m2 g−1 was observed from the first to third catalyst cycles, which might be due to particle agglomeration after recovery and drying. The adsorption and desorption curves of 5% Cr–TiO2 calcined at a higher temperature are shown in Fig. S3 (ESI).† The adsorption and desorption (using ASTM D4222-03(2015) e1) curves suggested that there was not much variation in the curve patterns at different calcination temperatures.
Micropore formation at 400 °C in the undoped and doped catalysts was not observed, as confirmed by isotherm t-plots (Fig. S4, ESI†).60 The t-plot is considered to be the graph of Vads vs. t. The plot of the gas quantity adsorbed versus thickness (nm) was linear. The isotherm data showed that the catalysts did not contain any micropores, indicating that the prepared Cr–TiO2 was mesoporous.
All samples were annealed at 400 °C prior to surface area and porosity measurements. The porosity of catalyst samples was measured using an ASTM method (ASTM-D4284-12(2017)e1) using the MIP technique.61 The surface area and porosity analysis results are summarized in Table 1.
| Sr. no. | Catalyst | Surface area (m2 g−1) | Porosity by MIP (%) |
|---|---|---|---|
| 1 | TiO2 | 72.59 | 46.70 |
| 2 | 0.5% Cr–TiO2 | 87.56 | 58.16 |
| 3 | 1% Cr–TiO2 | 95.71 | 60.45 |
| 4 | 2% Cr–TiO2 | 103.11 | 59.68 |
| 5 | 3% Cr–TiO2 | 104.65 | 60.47 |
| 6 | 4% Cr–TiO2 | 111.87 | 84.35 |
| 7 | 5% Cr–TiO2 | 148.06 | 84.44 |
The surface area and porosity results showed that undoped TiO2 had a surface area of 72.59 m2 g−1, while Cr-doped TiO2 showed a steadily increase in surface area with increasing chromium content, up to a doping concentration of 5%. Interestingly, the surface area of 5% Cr–TiO2 was highest among all doped Cr–TiO2 catalysts. The increase in surface area might be attributed to the decomposition of nitrate and peroxide groups. Nitrate groups from the Cr precursor in Cr–TiO2 gel acted as fuel for pore growth within the TiO2 microstructure, which ultimately helped to increase the surface area. There might also be another reason for the gel having this nature. Generally, the surface area of powder materials depends on the particle size, the pore size, number, and type, and the distribution of pores with voids. In sol–gel synthesis, the gel nature is important because pores in the gel are filled with solvent molecules. Upon careful removal of these solvent molecules, the porous structure remains undisturbed, which ultimately helps to increase the surface area. During synthesis, formation of the homogeneous gel was observed until a doping concentration of 5%, beyond which the gel was converted to a precipitate. For chromium doping up to 5%, the gel network was perfect, such that pores within the gel network were filled with solvent, which increased the surface area after calcination at higher temperatures. The porosity was affected by the size, packing, shape, and size distribution of intra-particle, inter-particle, inter-aggregation, inter-cluster, agglomerates, and clusters of agglomerate pores. The MIP results suggested that the pore size distribution for undoped TiO2 and Cr–TiO2 was in the range of 90–100 Å.
4.5. FT-IR and Raman spectra of Cr–TiO2 catalysts
The FT-IR spectra of undoped TiO2 and Cr–TiO2 catalysts annealed at 400 °C are shown in Fig. S5 (ESI).† In IR spectra, a sharp band centred at 500–750 cm−1 was attributed to metal oxygen (Ti–O) bond and bridging Ti–O–Ti or Ti–O–Cr stretching modes.54–62 The IR signal in the range of 400–1250 cm−1 was characteristic of O–Ti–O lattice formation. The broad peak at 3300–3750 cm−1 was assigned to fundamental stretching vibrations of monodentate O–H (free or bonded) and surface adsorbed water molecules or hydroxyl (–OH) groups. The weak band located at 1620–1630 cm−1 corresponded to O–H group bending vibrations. Peaks corresponding to hydrocarbons were absent from the IR spectra. Furthermore, the intensity of peaks at 764, 940, and 1625 cm−1 in Cr-doped TiO2 were stronger than those in TiO2. Raman spectra confirmed the doping of chromium into titanium dioxide. Raman spectra of undoped TiO2 and Cr–TiO2 catalysts calcined at 400 °C are shown in Fig. 5.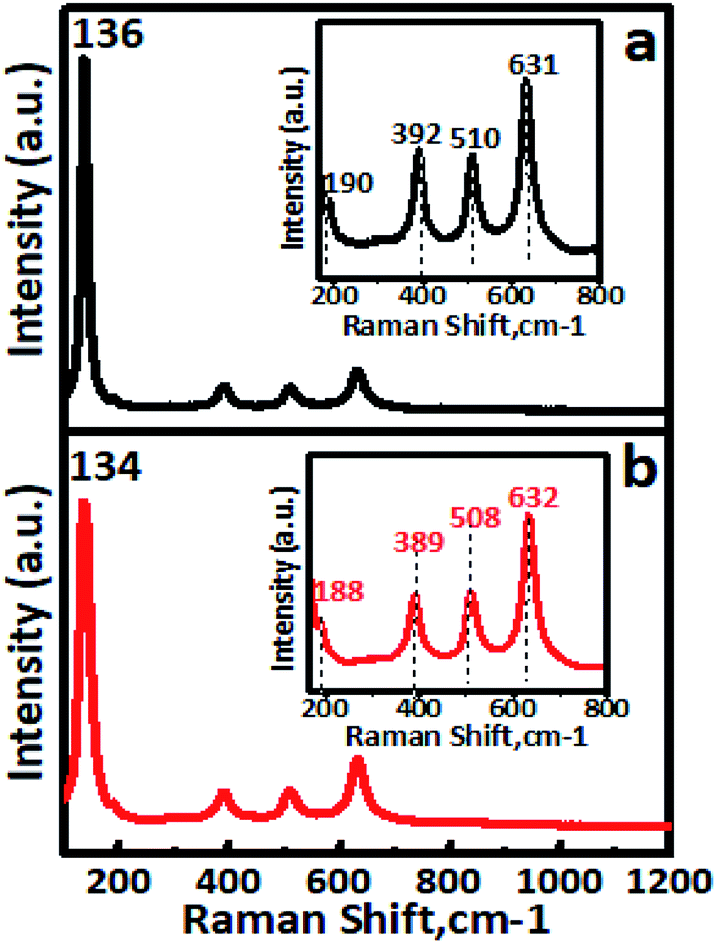 | ||
| Fig. 5 Room-temperature Raman spectra of (a) undoped TiO2 and (b) 5% Cr–TiO2 samples. Inset shows wavenumber shifting. | ||
Raman spectroscopy is extremely sensitive and extensively used for detecting phase changes and nano-oxide clusters in samples. Fig. 5a shows the Raman spectrum of undoped TiO2, with typical anatase vibrational modes at 136, 190, 392, 510, and 631 cm−1. The Raman spectrum of the 5% Cr–TiO2 sample (Fig. 5b) showed all peaks indexed to the anatase phase, with no appearance of individual chromium oxide peaks.63 The absence of individual chromium oxide peaks suggested that the doped metal occupied substitutional or interstitial positions in Cr–TiO2 rather than existing as an impurity. This observation was in agreement with the XRD results and further supported lattice substitution. Cr–TiO2 annealed at 400 °C showed fundamental vibrational modes of the anatase phase. However, a slight shift in the position of the main peak at 134 cm−1 and other peaks was observed with marginal peak broadening (Fig. 5b) compared with the undoped TiO2 sample (Fig. 5a). This might be attributed to Cr insertion into the TiO2 matrix in the case of Cr–TiO2 samples.
4.6. XPS spectra
XPS analysis was performed to investigate the valence states of titanium and chromium, and the presence of oxygen vacancies. The 5% Cr–TiO2 sample was used as an example for XPS measurements because it exhibited a stronger signal than samples with lower Cr contents. A typical high resolution survey spectrum and individual element spectra of Ti2p, Cr2p, and O1s are shown in Fig. 6.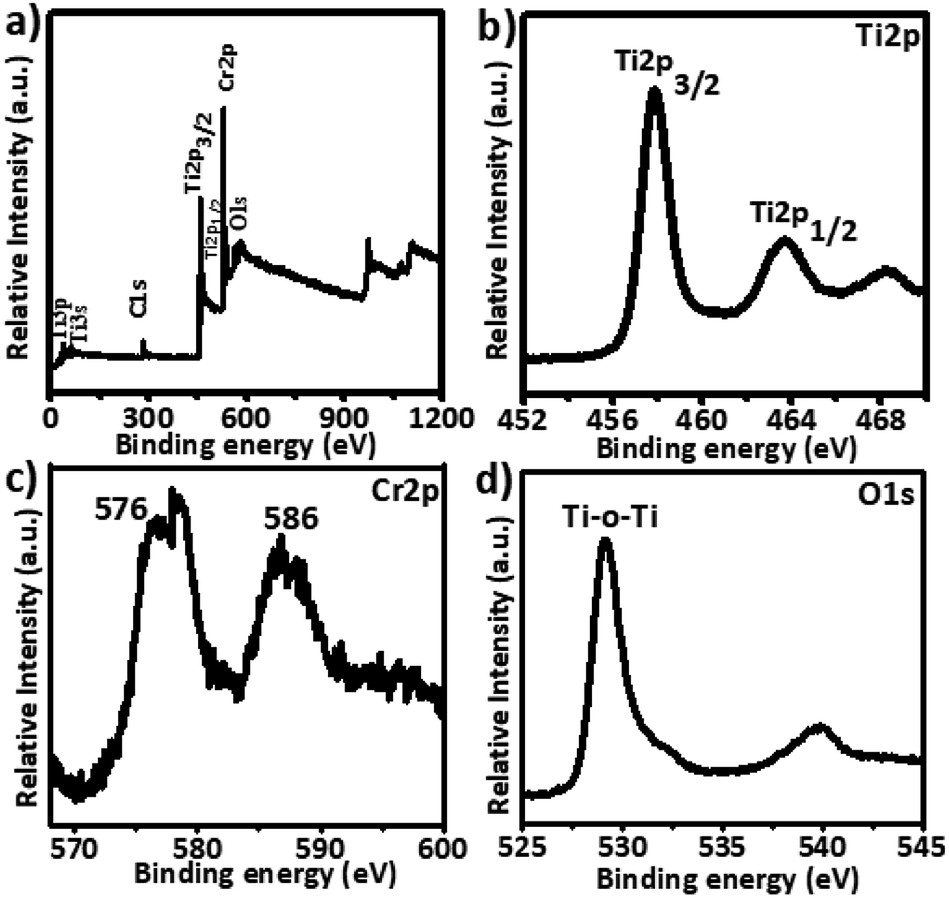 | ||
| Fig. 6 X-ray photoelectron spectra of Cr–TiO2: (a) survey spectra, (b) Ti2p, (c) Cr2p, and (d) O1s. | ||
The full XPS spectrum (Fig. 6a) of 5% Cr-doped TiO2 showed that Cr-doped TiO2 contained only Ti, O, and Cr elements. As shown in Fig. 6b, peaks at 458.25 and 463.95 eV were attributed to Ti2p (3/2) and Ti2p (1/2) spin–orbital splitting, and these peaks appeared at slightly lower binding energies in Cr-doped TiO2 than the standard positions. A difference of nearly 5.7 eV was observed in both peaks, supporting the +4 valence state of Ti. The Cr2p (3/2) spin–orbital splitting photoelectrons for 5% Cr–TiO2, located at binding energies of 586.0 and 576 eV in Fig. 6c, were assigned to Cr3+.64 The O1s spectrum of Cr-doped TiO2 (Fig. 6d) clearly showed two peaks, one at 530 eV and a weak shoulder peak at around 532 eV. The O1s peak at about 530 eV, indicating the presence of oxygen according to the binding energy, was assigned to crystal lattice oxygen (Ti–O–Ti) in TiO2. As the peak shoulders at around 532 eV and 539 eV are generally absent in the XPS spectra of pure TiO2 samples, they were assigned to oxygen vacancies and surface hydroxyls. The O1s peaks at about 532 and 539 eV suggested an increase in oxygen vacancies by doping Cr into TiO2.
4.7. Photoluminescence (PL) spectra
PL spectra are used to investigate doping behaviour. The PL performance was closely related to defects in TiO2, which can be influenced greatly by metal/ion doping. Photoluminescence spectra of undoped TiO2 and Cr-doped TiO2 samples recorded using a pulsed N2 laser (λex = 300 and 350 nm) were used to determine the radiative recombination efficiency of electron hole charge carriers, as shown in Fig. 7. The PL emission spectrum of the TiO2 sample consisted of a broad PL band with a well-resolved fine structure. This depended on the carrier density of upper energy states, density of lower energy states, and transition probability of upper energy states to lower vacant states.63,65 The emission peak at around 342 nm in the UV range originated from the near band edge (NBE) free excitation emission, which is the direct transition from the VB to CB. The latter additional emission signals at 436, 449, 468, 481, and 491 nm originated from the charge-transfer transition from electrons trapped in oxygen vacancies.66 Emission signals of Cr-doped TiO2 samples were observed at 348, 443, 455, 473, 487, and 497 nm. Quenching of the PL spectrum was observed for Cr-doped TiO2 samples. This effective quenching in the PL spectra was attributed to two pathways: (i) created electrons being trapped by oxygen vacancies and holes being trapped by doped chromium in the TiO2 lattice; and (ii) excited electron transferring from the valence band to new existing energy levels introduced by chromium incorporation.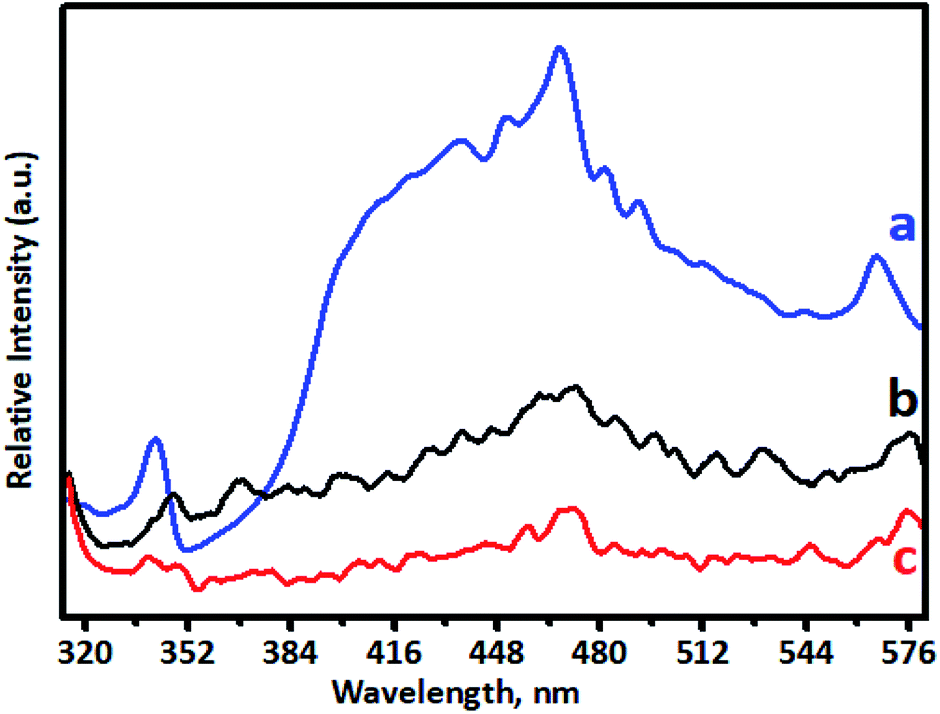 | ||
| Fig. 7 Photoluminescence spectra of (a) undoped TiO2, (b) 1% Cr–TiO2, and (c) 5% Cr–TiO2. | ||
4.8. Optical properties of Cr–TiO2 powder
To observe the application potential and discuss the effects of photocatalysis, it was necessary to study the optical properties of undoped and Cr–TiO2 nanoparticles to clearly express the effect of dopant-induced electronic properties. The activity of a semiconductor photocatalyst is strongly associated with its optical absorption ability in the visible region. The effect of Cr-doping on the UV-vis absorption spectra of TiO2 is shown in Fig. 8. The spectra clearly showed a shift in the absorption band edge toward longer wavelengths with increasing Cr concentration. The red-shift in the absorption band edge reached a maximum of about 800 nm in the 5% Cr–TiO2 sample. The absorption edge of undoped TiO2 is known to appear below 400 nm in the bulk sample and nanosize TiO2, which is lower than that of the bulk material (blue shift) owing to size effects. Furthermore, undoped TiO2 showed an absorption edge at around 370 nm because the particle size was in the sub-nanometre region due to the intrinsic band gap absorption of TiO2. However, in Cr–TiO2 samples, a shift in the absorption edge toward the visible region (red shift) and the origin of visible light sensitivity in Cr-doped TiO2 might be attributed to the appearance of additional energy levels, such as the Cr2p level, and oxygen vacancies created by Cr doping. The absorption band appearing at 450–500 nm for Cr–TiO2 samples might be attributed to charge transfer between Cr3+ → Ti4+ or 4A2g → 4T1g levels in Cr3+ ions. Furthermore, an additional band appeared between 600 and 800 nm, which might be due to 4A2g→ 4T2g d–d transitions of Cr3+ ions. Furthermore, the oxygen vacancies at the Cr2p level might play significant roles in improving visible-light absorption. As discussed above, Cr doping helped to create oxygen vacancies in the TiO2 lattice, reported previously as being located at 0.75–1.18 eV below the conduction band minimum of TiO2.67 This band narrowing due to the formation of oxygen vacancies induced visible-light absorption ranging from 500 to 600 nm. With increasing Cr concentration in TiO2, (curves ‘b’, ‘c’, ‘d’, ‘e’, ‘f’, and ‘g’), absorption in the visible region improved and 5% Cr–TiO2 showed maximum visible light absorption. However, as stated in previous sections, a further increase in Cr concentration beyond 5% led to disturbance of the gel network and the formation of gelatinous precipitates with separation of precipitates at the bottom and solvent at the top. Therefore, Cr doping of above 5% in TiO2 has not been studied and reported. These results indicated that the appropriate amount of Cr doping was possible using this method and helped to significantly improve TiO2 absorption in the visible light region, thereby allowing possible applications for TiO2 activated by UV and visible light. The photocatalytic activity of Cr-doped TiO2 powder was tested in the degradation of spent wash, a sugarcane-based distillery waste. The results were quite promising and indicated the good application potential of Cr–TiO2 photocatalysts in degradation reactions under natural sunlight. | ||
| Fig. 8 UV-visible DRS spectra of powder samples: (a) undoped TiO2, (b) 0.5% Cr–TiO2, (c) 1% Cr–TiO2, (d) 2% Cr–TiO2, (e) 3% Cr–TiO2, (f) 4% Cr–TiO2, and (g) 5% Cr–TiO2. | ||
4.9. Element analysis by ICP-MS
Elemental analysis was conducted using ICP-MS. An Agilent 7700 model instrument with Mass Hunter Software was used for data processing. The NIST traceable standard & internal standard (ISTD) solutions were used for calibration curves and the recovery study. The instrument was calibrated with NIST traceable tuning solution with a concentration of 1 μg L−1 (USA) and containing components including Ce, Co, Li, Mg, Tl, and Y. The calibration graph was plotted at 1 ppb, 5 ppb, 10 ppb, 50 ppb 100 ppb, and 200 ppb using an internal standard solution (Sc, 100 μg mL−1). The wash water blank (ASTM-Type 1 water) was used as blank. The chromium recoveries for standard and doped TiO2 catalyst were in the range of 90–100% with a linear correlation coefficient. The Cr concentrations in prepared Cr–TiO2 catalysts obtained by ICP-MS analysis are shown in Table 2.| Sr. no. | Catalyst | Cr content, % |
|---|---|---|
| 1 | TiO2 | Not detected |
| 2 | 0.5% Cr–TiO2 | 0.48 |
| 3 | 1% Cr–TiO2 | 1.02 |
| 4 | 2% Cr–TiO2 | 1.99 |
| 5 | 3% Cr–TiO2 | 2.98 |
| 6 | 4% Cr–TiO2 | 3.97 |
| 7 | 5% Cr–TiO2 | 4.99 |
5. Photocatalytic study
5.1. Degradation of spent wash
The photocatalytic activity was tested in the degradation of sugarcane distillery spent wash solution (10000 ppm solution) under solar radiation using Cr–TiO2 powder samples as catalysts. The change in colour intensity and concentrations of colour-forming compounds in the samples irradiated for different time intervals under solar radiation were monitored using UV-Vis spectrophotometry (absorbance at 280 nm) and UHPLC-QTOF-LC-MS by comparing with blank solution. Typical UV-Vis spectra of the as-prepared spent wash solution and samples obtained after solar light irradiation at different time intervals are shown in Fig. S6 (ESI).† From UV-Vis analysis, the percentage degradation of spent wash colour at different time intervals was calculated and plotted. The percentage degradation of spent wash using undoped and Cr–TiO2 with irradiation time was also plotted. The graph of percentage degradation vs. time is shown in Fig. 9. The photodegradation results suggested that undoped TiO2 showed the lowest degradation rate, with nearly 27% degradation after 5 h, while, under identical conditions, P-25 TiO2 showed nearly 35% degradation in the spent wash colour. In contrast, Cr–TiO2 photocatalysts showed better photocatalytic activity, with 40–68% degradation of the spent wash solution colour. Among the Cr-doped TiO2 photocatalysts, 5% Cr–TiO2 showed the highest activity and nearly 68% degradation was achieved within a 5 h duration. As shown in Fig. 9, an increase in the photodegradation reaction rate with increasing Cr concentration up to a 5% doping level was clearly observed, while a decreasing trend was observed for higher doping concentrations, such as 6% Cr–TiO2 and 7% Cr–TiO2, which showed lower degradation rates than 5% Cr–TiO2. Therefore, the effect of calcination temperature on photocatalytic activity was studied using 5% Cr–TiO2 photocatalyst only. For this study, 5% Cr–TiO2 samples annealed at different temperatures (200–600 °C) were used. The graph of percentage degradation of spent wash vs. calcination temperature using 5% Cr–TiO2 photocatalysts is shown in Fig. S7 (ESI).† The photocatalytic results suggested that, among all samples, 5% Cr–TiO2 annealed at 400 °C showed the highest activity, with nearly 68% degradation of spent wash within 5 h. The highest activity of Cr–TiO2 might be attributed to the extension of the absorption edge towards visible region, increased porosity, and higher surface area of the doped catalysts compared with undoped TiO2 and P25 TiO2. The degradation study results were in good agreement with the physicochemical properties. Detailed analysis of the degradation products was conducted using GPC and LC-MS techniques. The molecular weight of the degradation products was obtained by GPC analysis using PL gel Aqua OH-30 (300 × 7.5 mm, 10A) and PL gel Aqua OH-40 (300 × 7.5 mm, 100A) columns. The results are shown in Table 3.
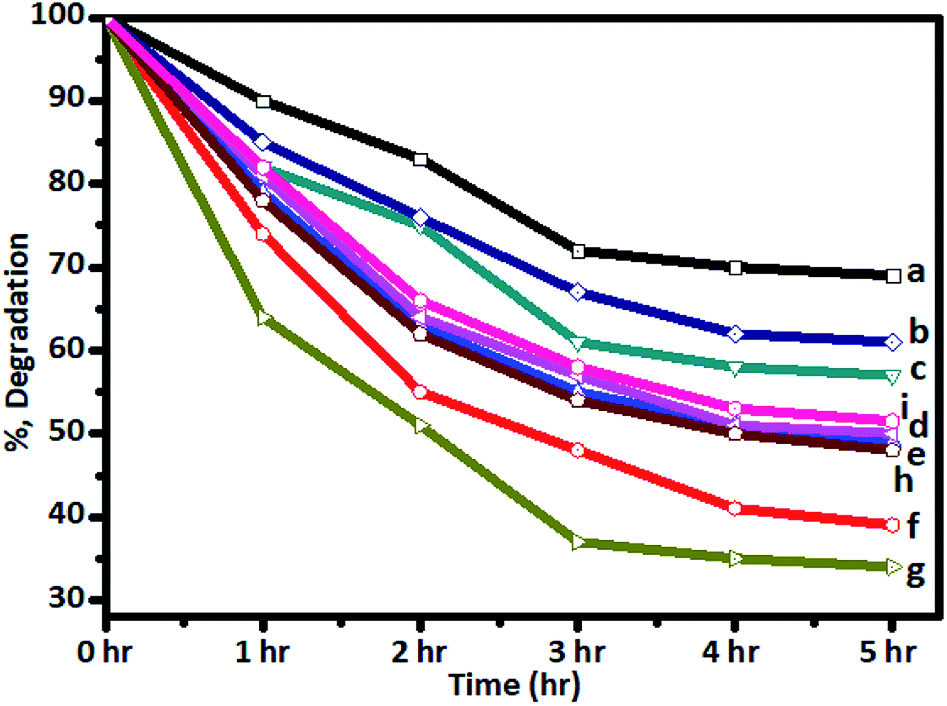 | ||
| Fig. 9 Degradation of spent wash solution after irradiation with sunlight using (a) undoped TiO2, (b) P-25 Degussa, and (c) 1%, (d) 2%, (e) 3%, (f) 4%, (g) 5%, (h) 6%, and (i) 7% Cr–TiO2 catalysts. | ||
| Retention time (min) | MW (Da) of spent wash solution before sunlight irradiation (0 h) | MW (Da) after sunlight irradiation (5 h) with undoped TiO2 | MW (Da) after sunlight irradiation (5 h) with Degussa P-25 | MW (Da) after sunlight irradiation (5 h) with Cr–TiO2 |
|---|---|---|---|---|
| 11.258 | 142665 |
139875 |
136649 |
93351 |
| 13.125 | 32200 |
31054 |
30309 |
16714 |
| 15.633 | 4363 | 4217 | 3963 | 3008 |
| 19.767 | 162.9 | 161 | 161 | 142 |
| 21.167 | 53 | 52 | 52 | 35 |
The high and low molecular weight (MW) polymeric compounds in spent wash samples degraded under sunlight in the presence of undoped TiO2, Degussa P-25 TiO2, and the Cr-doped TiO2 photocatalysts. The high molecular weight compounds (142665 Da) were degraded to compounds of 136,649, 136, 649, and 93,351 Da using undoped TiO2, Degussa P-25 TiO2, and Cr–TiO2, respectively, while compounds with molecular weights of 32200 Da were converted to compounds of 31054, 30309, and 3008 Da using undoped TiO2, Degussa P-25 TiO2, and Cr–TiO2 photocatalysts, respectively. Therefore, degradation was faster using Cr–TiO2 than Degussa P-25 TiO2 and undoped TiO2. For low MW compounds, no appreciable change in MW was observed using Degussa P-25 and undoped TiO2 photocatalysts, indicating good photocatalytic activity for high MW compounds only. Generally, high MW compounds tend to disintegrate easily in the presence of light and a photocatalyst, so the rate of degradation of such compounds is higher compared with those of low MW compounds.55
Photocatalytic oxidation is an important tool for the degradation and release of compounds during degradation of distillery spent wash. To identify the organic contents in spent wash, it was subjected to LC-MS analysis. The spent wash, as received from industry, and aliquots of spent wash after photocatalytic treatment under natural sunlight conditions, were analysed using Q-TOF LC-MS. The spectral data obtained from LC-MS were analysed under negative and positive ionization modes in the METLIN database (Fig. S8 and S9, ESI†).
Before injecting the samples into the mass spectrometer, certain generic settings were applied for data acquisition. Untargeted analysis aimed to acquire all possible compounds, irrespective of their concentrations. Compounds generally represent all compositions in different chemical forms in a particular sample at the time of analysis. To acquire all possible compounds, the mass spectrometer was operated in positive and negative ionization modes. An acidic mobile phase was maintained during positive ionization to encourage positive ion formation. In negative ionization mode, the aim was to deprotonate the molecules or metabolites by applying operating conditions well above their isoelectric points. This type of analysis is very helpful for understanding the complete chemical composition of a sample. Based on the data obtained, the types of chemical shifts present resulting from certain treatments of a particular sample could be determined. Within this framework, the received spent wash and aliquots of spent wash obtained after photocatalytic treatment under natural sunlight, were analysed using Q-TOF LC-MS to identify the compound complexity. The resulting chromatograms obtained in negative and positive modes are shown in Fig. S8 and S9 (ESI).† As mentioned earlier, the acquired data were processed qualitatively to understand the change in compounds after respective treatments. A change in the number of compounds was observed after every treatment, providing a tentative database matching that identified the nature of these compounds. The number of compounds was classified from different chemical groups, and their changes in number are shown in Table 4. Certain masses in the samples did not match with the database and were, therefore, denoted as metabolites.
| Compounds | Spent wash solution | Undoped TiO2 | Degussa P-25 | Doped 5% Cr–TiO2 |
|---|---|---|---|---|
| Organic compounds (acids) | 3338 | 2907 | 2461 | 1272 |
| Peptides | 373 | 303 | 243 | 135 |
| Lipids | 181 | 133 | 93 | 62 |
| Ketones | 570 | 319 | 232 | 70 |
| Glucosides | 74 | 68 | 64 | 22 |
| Alcohol compounds | 1385 | 1079 | 1020 | 727 |
| Metabolites | 73 | 70 | 64 | 34 |
Spent wash sample analysis revealed a total of 5994 compounds, with around 3088 and 2906 compounds obtained from negative and positive ionization modes, respectively, identified using the METLIN database. Based on database matching, the identified compounds were classified according to their functional groups, including acids, alcohols, peptides, lipids, ketones, and glucosides. All of these compounds showed major changes upon solar light irradiation in the presence of photocatalysts owing to degradation reactions. The major compound fraction was acids and phenols, comprising approximately 55–56% of the compounds in spent wash. Almost 50% of acids in the spent wash sample belonged to different subgroups, including mineral, sulfonic, carboxylic, halogenated, vinylogous, nucleic, and phenolic acids, with different concentrations. Similarly, organic alcohols comprised nearly 23–24% of the compounds in spent wash, while the other compounds comprised nearly 10–12% by weight. The results of spent wash degradation using different photocatalysts are shown in Table 4. Degradation of spent wash samples containing organic compounds, such as acids, peptides, lipids, ketones, glucosides, alcohols, and metabolites was achieved using undoped TiO2, Degussa P-25 TiO2, and Cr–TiO2 photocatalysts. Faster photocatalytic degradation was observed with Cr–TiO2 than with undoped TiO2 and Degussa P-25 photocatalysts. Nearly 60–70% of acids and alcohols were reduced after degradation using the Cr–TiO2 photocatalyst. This was attributed to the extended absorption edge of Cr–TiO2 in the visible region, improved porosity, and higher surface area. Both undoped TiO2 and P-25 TiO2 were UV-light active, while Cr–TiO2 as UV and visible-light active. As natural sunlight comprises less than 5% UV light and 40–45% visible light, Cr–TiO2 is a good candidate for improved activity in natural sunlight. Therefore, the results were as expected. In addition, the photocatalyst efficiency was based on the recombination rate of electron/hole pairs. In this study, the Cr–TiO2 photocatalyst showed higher activity under visible light irradiation (see Fig. 10 for a schematic representation of the proposed mechanism) because Cr3+ ions acted as electron trapping sites. The presence of Cr3+ reduced the rate of electron–hole recombination to prolong the lifetime of the photogenerated carriers (e−/h+) and increase the photocatalytic reactivity.68 The reduction potential of the Cr3+/Cr4+ couple (2.1 V vs. NHE) was just above the valence band (VB) edge of TiO2 (3.2 eV), which allowed Cr3+ to interact with holes present in VB, resulting in oxidation to Cr4+. These Cr4+ ions (or trapped holes in the VB) reacted with hydroxide ions adsorbed on the surface of the photocatalyst (−OHads) to regenerate Cr3+ ions and produce hydroxyl radicals (·OH), which are strong oxidizing agents.
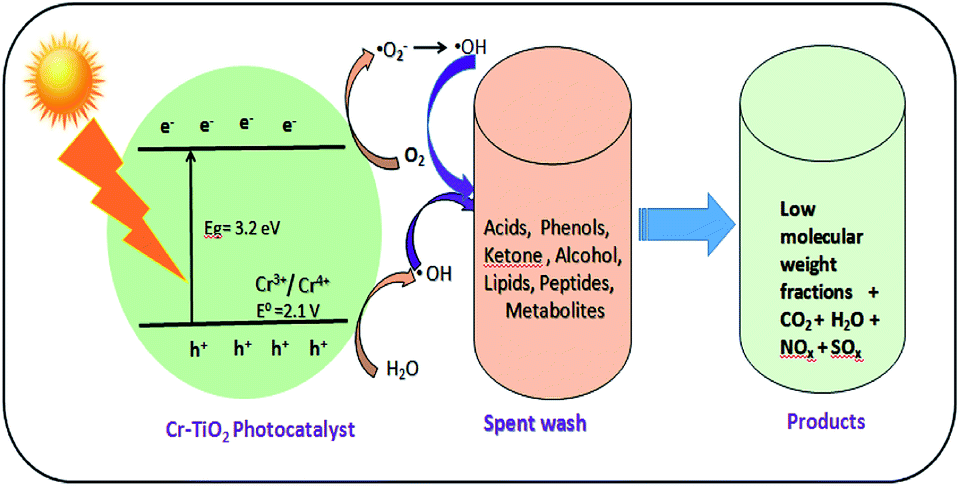 | ||
| Fig. 10 Scheme of photocatalytic degradation of spent wash solution using Cr–TiO2 under natural sunlight. | ||
The photocatalytic oxidation of spent wash samples resulted in low molecular weight counterparts, CO2, H2O, and other gas molecules. The slight increase in the pH of spent wash solution from 3.26 to 5.12 after degradation was indicative of the mineralization of organic compounds, especially organic acids, into CO2 and H2O. This increase in pH will help reduce the toxicity of the spent wash sample, making its disposal into soil or water less toxic compared with as-received spent wash from alcohol distilleries. The degradation of organic compounds in spent wash was also confirmed by measuring the total organic carbon (TOC). The TOC content of the as-received spent wash sample was 328 ppm, which was significantly reduced to 298 ppm, 246 ppm, and 112 ppm after degradation using undoped TiO2, Degussa P-25, and 5% Cr–TiO2 (Fig. S10, ESI†). Based on the photocatalytic results, a reaction scheme was proposed, as shown in Fig. 10.
Catalyst stability is a very important parameter in catalytic reactions because it directly affects the process cost and final product. The stability was confirmed by performing XRD analysis and Cr leaching tests in degraded spent wash using ICP-MS. The XRD spectrum of 5% Cr–TiO2 before photocatalytic use (curve a) and recovered after three cycles of photodegradation (curve b) are shown in Fig. S11 (ESI).† The XRD spectra showed no change in structural behaviour, with all peaks and their positions identical to those of the 5% Cr–TiO2 photocatalyst before use. The Cr leaching test was performed by analysing the Cr content in the spent wash recovered after photodegradation by ICP-MS. The spent wash samples recovered after photodegradation showed Cr contents below the detection limit, suggesting that no Cr leaching occurred during the reaction. The XRD and Cr leaching test results suggested that this photocatalyst had good stability under the given experimental conditions.
The repeatability and reproducibility of this degradation reaction were accessed by conducting the degradation reaction for three repeated cycles under identical conditions. The results of this recyclability test for the 5% Cr–TiO2 catalyst are shown in Fig. S12 (ESI).† Only a small decrease in the performance of the photocatalyst was observed after the 2nd and 3rd cycles, suggesting good recyclability and stability of the photocatalyst in this reaction. The recovery test was performed by recovering the photocatalyst after each cycle of the experiment. The plot of photocatalyst quantity recovered vs. number of cycles is shown in Fig. S13 (ESI).† Only a 6% loss in catalyst weight after three degradation reaction cycles confirmed that the majority of the catalyst was easily recovered.
6. Conclusions
In this work, Cr-doped TiO2 was prepared using an aqueous sol–gel synthesis method. Undoped TiO2 and Cr–TiO2 were characterized and evaluated for their application potential under natural sunlight. The performance of the prepared catalyst (Cr–TiO2) was tested in the degradation of spent wash, a sugarcane-based ethanol industry waste product, as a model pollutant. In-depth analysis of the spent wash and degradation products obtained after the photocatalytic study was performed using UV-Vis, GPC, and LC-MS techniques. LC QTOF provided the highest performance to meet the needs of challenging qualitative and quantitative applications. The results of the photocatalytic study were compared with undoped TiO2 synthesized using same method and Degussa P-25 photocatalyst. The Cr–TiO2 powder photocatalysts were more effective than undoped and Degussa P-25 in the photocatalytic degradation of spent wash under natural sunlight. Among the photocatalysts investigated, 5% Cr–TiO2 was the most effective catalyst for the degradation of spent wash. Its activity under visible light was attributed to Cr species incorporated into the TiO2 matrix and the subsequent shift in absorption of the doped catalyst in the visible region. The degradation was also monitored using gel permeation chromatography, in which the breakdown of large MW compounds into low MW compounds was observed. The additional dimensions offered by MS analysis allowed the identification of individual components. The Cr–TiO2 photocatalyst shows application potential and could be a good candidate for the elimination of pollutants from industrial wastewater.Conflicts of interest
No conflict of interest.Acknowledgements
Mr Shrikant P. Takle would like to thank the Director General of C-MET, Pune, Department of Biotechnology, Shivaji University, Kolhapur and the Department of Chemistry, Annasaheb Magar Mahavidyalaya, Pune, for their encouragement, the Centre for Material for Electronics Technology (C-MET) for providing research facilities, and the nanocrystalline materials group C-MET, Pune, for their support.References
- M. Plavšić, B. Ćosović and C. Lee, Sci. Total Environ., 2006, 366, 310–319 CrossRef PubMed.
- T. J. Painter, Carbohydr. Polym., 1998, 36, 335–347 CrossRef CAS.
- P. Kumar and R. Chandra, Bioresour. Technol., 2006, 97, 2096–2102 CrossRef CAS PubMed.
- M. D. A. Belina, D. M. I. de Roxas, M. G. E. Guardian and E. J. del Rosario, Journal of Environmental Science and Management, 2006, 9(1), 44–59 Search PubMed.
- R. Chandra and H. Singh, Indian J. Environ. Prot., 1999, 19, 833–837 CAS.
- S. B. Kim, F. Hayase and H. Kato, Agric. Biol. Chem., 1985, 49, 785–792 CAS.
- R. M. Bhise, A. A. Patil, A. R. Raskar, P. J. Patil and D. P. Deshpande, Res. J. Recent Sci., 2012, 1, 66–69 CAS.
- Y. Satyawali and M. Balakrishnan, Bioresour. Technol., 2007, 98, 2629–2635 CrossRef CAS PubMed.
- M. Bernal, M. O. Ruiz, R. M. Geanta, J. M. Benito and I. Escudero, Chem. Eng. J., 2016, 283, 313–322 CrossRef CAS.
- M. Wagh, P. D. Nemade, M. S. R. Dhasal, S. B. Principal and P. Indapur, Int. J. Eng. Res. Gen. Sci., 2015, 3, 1159–1173 Search PubMed.
- P. R. Chittaragi and A. S. Byakodi, International Research Journal of Engineering and Technology, 2018, 5(6), 2907–2911 Search PubMed.
- M. N. Vineetha, M. Matheswaran and K. N. Sheeba, Sol. Energy, 2013, 91, 368–373 CrossRef CAS.
- M. Chandrakant and R. Kedar, Res. J. Chem. Sci., 2013, 3, 53–56 Search PubMed , (ISSN 2231-606X).
- C. G. Alfafara, V. P. Migo, J. A. Amarante, R. F. Dallo and M. Matsumura, Water Sci. Technol., 2000, 42, 193–198 CrossRef CAS.
- D. Beydoun, R. Amal, G. Low and S. McEvoy, J. Nanopart. Res., 1999, 1, 439–458 CrossRef CAS.
- W. F. Zhou, Q. J. Liu, Z. Q. Zhu and J. Zhang, J. Phys. D: Appl. Phys., 2010, 43, 035301 CrossRef.
- E. Sthathatos, D. Tsiourvas and P. Lianos, Colloids Surf., A, 1999, 149, 49 CrossRef.
- R. Comparelli, E. Fanizza, M. L. Curri, P. D. Cozzoli, G. Mascolo and A. Agostiano, Appl. Catal., B, 2005, 60, 1–11 CrossRef CAS.
- S. Mu, Y. Z. Long, S. Z. Kang and J. Mu, Catal. Commun., 2010, 11, 741–744 CrossRef CAS.
- L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef.
- R. Beranek, J. M. Macak, M. Gartner, K. Meyer and P. Schmuki, Electrochim. Acta, 2009, 54, 2640–2646 CrossRef CAS.
- A. Ghicov, J. M. Macak, H. Tsuchiya, J. Kunze, V. Haeublein, L. Frey and P. Schmuki, Nano Lett., 2006, 6, 1080–1082 CrossRef CAS.
- R. S. Sonawane, B. B. Kale and M. K. Dongare, Mater. Chem. Phys., 2004, 85, 52–57 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- V. Kiisk, V. Reedo, M. Karbowiak, M. G. Brik and I. Sildos, J. Phys. D: Appl. Phys., 2009, 42, 125107 CrossRef.
- Y. V. Lim, H. M. Fan and Z. X. Shen etal, Appl. Phys. A: Mater. Sci. Process., 2009, 95, 555–562 CrossRef CAS.
- J. F. Zhu, Z. G. Deng, F. Chen, J. Zhang, H. Chen, M. Anpo and L. Zhang, Appl. Catal., B, 2006, 62, 329–335 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 108, 79–84 CrossRef CAS.
- A. Di Paola, G. Marci, L. Palmisano, M. Schiavello, K. Uosaki, S. Ikeda and B. Ohtani, J. Phys. Chem. B, 2002, 106, 637–645 CrossRef CAS.
- X. B. Li, L. L. Wang and X. H. Lu, J. Hazard. Mater., 2010, 177, 639 CrossRef CAS PubMed.
- S. Li and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 201204 CrossRef.
- L. Sangaletti, M. C. Mozzati, P. Galinetto, C. B. Azzoni, A. Speghini, M. Bettinelli and G. Calestani, J. Phys.: Condens. Matter, 2006, 18, 7643 CrossRef CAS PubMed.
- C.-C. Pan and J. C. S. Wu, Mater. Chem. Phys., 2006, 100, 102–107 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- W. Y. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 13669–13679 CrossRef.
- M. I. Litter and J. A. Navıo, J. Photochem. Photobiol., A, 1996, 98, 171–181 CrossRef CAS.
- F. Cracia, J. P. Holgado, A. Caballero and A. R. Gonzalez-Elipe, J. Phys. Chem. B, 2004, 108, 17466 CrossRef.
- R. Dholam, N. Patel, M. Adami and A. Miotello, Int. J. Hydrogen Energy, 2009, 34, 5337–5346 CrossRef CAS.
- H. T. Jun and L. S. Kee, Mater. Lett., 2010, 64, 2287–2289 CrossRef.
- L. Pan, J. J. Zou, X. Zhang and L. Wang, Ind. Eng. Chem. Res., 2010, 49, 8526–8531 CrossRef CAS.
- R. Dholam, N. Patel, A. Santini and A. Miotello, Int. J. Hydrogen Energy, 2010, 35(18), 9581–9590 CrossRef CAS.
- X. Fan, X. Chen and S. Zhu etal, J. Mol. Catal. A: Chem., 2008, 284(1–2), 155–160 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- E. Borgarello, J. Kiwi, M. Graetzel, E. Pelizzetti and M. Visca, J. Am. Chem. Soc., 1982, 104, 2996–3002 CrossRef CAS.
- J.-M. Herrmann, J. Disdier and P. Pichat, Chem. Phys. Lett., 1984, 108, 618–622 CrossRef CAS.
- T. Ikeda, T. Nomoto, K. Eda, Y. Mizutani, H. Kato, A. Kudo and H. Onishi, J. Phys. Chem. C, 2008, 112(4), 1167–1173 CrossRef CAS.
- G. K. Larsen, R. Fitzmorris, J. Z. Zhang and Y. Zhao, J. Phys. Chem. C, 2011, 115(34), 16892–16903 CrossRef CAS.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216(1–2), 505–516 CrossRef CAS.
- R. S. Sonawane and S. Ramakrishna, Mater. Sci. Eng., B, 2012, 177(9), 652–660 CrossRef CAS.
- S. K. Khore, N. V. Tellabati, S. K. Apte, S. D. Naik, P. Ojha, B. B. Kale and S. Ravindra, RSC Adv., 2017, 7(52), 33029–33042 RSC.
- R. S. Sonawane and M. K. Dongare, J. Mol. Catal. A: Chem., 2006, 243, 68–76 CrossRef CAS.
- R. S. Sonawane, S. G. Hegde and M. K. Dongare, Mater. Chem. Phys., 2003, 77, 744–750 CrossRef CAS.
- R. Akbarzadeh, S. B. Umbarkar, R. S. Sonawane, S. Takle and M. K. Dongare, Appl. Catal., A, 2010, 374, 103–109 CrossRef CAS.
- T. C. Jagadale, S. P. Takale, R. S. Sonawane, H. M. Joshi, S. I. Patil, B. B. Kale and S. B. Ogale, J. Phys. Chem. C, 2008, 112, 14595–14602 CrossRef CAS.
- S. P. Takle, S. D. Naik, S. K. Khore, S. A. Ohwal, N. M. Bhujbal, S. L. Landge, B. B. Kale and R. S. Sonawane, RSC Adv., 2018, 8, 20394–20405 RSC.
- S. K. Khore, S. R. Kadam, S. D. Naik, B. B. Kale and R. S. Sonawane, New J. Chem., 2018, 42, 10958 RSC.
- N. H. Hong, A. Ruyter, W. Prellier and J. Sakai, Appl. Phys. Lett., 2004, 85(25), 6212–6214 CrossRef CAS.
- T. Droubay, S. Heald, V. Shutthanandan, S. Thevuthasan and S. Chambers, J. Appl. Phys., 2005, 97, 046103 CrossRef.
- H. Sutrisno, Ariswan and D. Purwaningsih, Chiang Mai J. Sci., 2017, 44(3), 1056–1064 CAS.
- S. Storck, H. Bretinger and W. F. Maier, Appl. Catal., A, 1998, 174, 137–146 CrossRef CAS.
- M. Boualleg and B. Guichard, US Pat. 15/318151, 2017.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. ElAasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- B. Choudhury and A. Choudhury, Mater. Chem. Phys., 2012, 132, 1112–1118 CrossRef CAS.
- G. Cappelletti, C. L. Bianchi and S. Ardizzone, Appl. Catal., B, 2008, 78, 193–201 CrossRef CAS.
- M. I. Zaki, A. Katrib, A. I. Muftah, T. C. Jagadale, M. Ikram and S. B. Ogale, Appl. Catal., A, 2013, 452, 214–221 CrossRef CAS.
- H. Jensen, A. Soloviev, Z. Li and E. G. Søgaard, Appl. Surf. Sci., 2005, 246, 239–249 CrossRef CAS.
- D. C. Cronemeyer, Phys. Rev., 1959, 113, 1222 CrossRef CAS.
- K. B. Jaimy, S. Ghosh, S. Sankar and K. G. K. Warrier, Mater. Res. Bull., 2011, 46, 914–921 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10026h |
| This journal is © The Royal Society of Chemistry 2019 |