
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel approach coupling ferrous iron bio-oxidation and ferric iron chemo-reduction to promote biomineralization in simulated acidic mine drainage†
Ning Wang‡
,
Di Fang‡,
Guanyu Zheng ,
Jianru Liang and
Lixiang Zhou*
,
Jianru Liang and
Lixiang Zhou*
College of Resources and Environmental Sciences, Nanjing Agricultural University, Nanjing 210095, P. R. China. E-mail: lxzhou@njau.edu.cn
First published on 11th February 2019
Abstract
A novel Acidithiobacillus ferrooxidans-mediated approach coupling biological oxidation and chemical reduction for treating acid mine drainage (AMD) was investigated. The results showed that controlled addition of zero valent iron (ZVI) into the coupling system did not exhibit a significant adverse influence on the bacterial activity of Acidithiobacillus ferrooxidans but markedly increased the formation of secondary Fe-minerals. Nutrition did not affect the efficiency of coupling process, except for the bacteria density of A. ferrooxidans. 2 days cyclic treatment performed better than that of 4 and 8 days. After 14 cycles of the coupling process, 89.4% of total iron (2.23 g L−1) was transferred into Fe-minerals finally. In addition, the combined system was highly effective in removing sulfate (63%) from a simulated AMD that contained soluble Cu, Zn, Al, and Mn. Valuable iron-sulfate material e.g. schwertmannite was formed with little co-precipitation of other metals. Therefore, the integration of A. ferrooxidans into the reduction by ZVI may have considerable potential in the enhancement of biomineralization efficiency, which may further decrease soluble TFe and sulfate loads in AMD before lime neutralization.
1. Introduction
Acidic mine drainage (AMD) are ubiquitously generated in many mines all over the world, including North America, Spain, Australia, South Africa, and southern China.1–7 AMD is always acidic (pH < 4) and contains high concentrations of toxic metals/metalloids (i.e., aluminum, manganese, copper, zinc, arsenic, etc.), soluble iron (Fe2+, Fe3+), and sulfate (SO42−).8,9 The cost-effective treatment of AMD has become an environmental challenge that requires increased attention.Neutralization with lime or other reagents (e.g., limestone, sodium hydroxide, and sodium carbonate) is a frequently used method for treating large volume of AMD, particularly those from small mines, because of its simplicity.10,11 However, there are several limitations for this method. On one hand, the conversion of ferrous ions (Fe2+) into ferrous hydroxides occurs at pH > 9, indicating that a large amount of alkalinity is needed to increase the pH of AMD from 2–4 to >9.12 On the other hand, applied chemicals (e.g. limestone) can be rapidly coated with newly formed ferric hydroxides, inhibiting the further interactions of these chemicals with other metals in AMD.8 Thus, the amount of lime required in neutralization is far more than the theoretical dose because the direct treatment of AMD by neutralization process may be inefficient.12 Besides, a massive amount of toxic solid wastes, a mixture of waste gypsum and metallic hydroxide, are usually generated during neutralization process.13 These secondary wastes require highly regulated and costly disposal.14 Therefore, it is important to remove soluble iron and sulfate as much as possible before lime neutralization in order to reduce the amount of toxic sludge generated and to improve the neutralization efficiency in AMD (Fig. S1†).
The hydrolysis of ferric iron (Fe3+) and sulfate leads to formation of secondary minerals, such as schwertmannite (Fe8O8(OH)8−2x(SO4)x, eqn (1)) and jarosite (MFe3(OH)6(SO4)2, M: K, Na, NH4, H3O, eqn (2)), which is an effective approach to remove iron and sulfate from AMD.15,16 In AMD treatment, the advantages of mineralization before neutralization are as follows: (1) decrease in lime dosage for neutralization; (2) simultaneous decrease in iron and sulfate load of AMD by precipitation; (3) hydrolysis-induced generation of minerals that are prospective resources for water treatment (as adsorbent, catalyst, etc.).17,18
| 8Fe3+ + xSO42− + (16 − 2x)H2O = Fe8O8(OH)8−2x(SO4)x + (24 − 2x)H+, (1 ≤ x ≤ 1.75) | (1) |
| M+ + 3Fe3+ + 2SO42− + 6H2O = (M)Fe3(SO4)2(OH)6 + 6H+, (M: K/Na/NH4/H3O) | (2) |
| 4Fe2+ + O2 + 4H+ = 4Fe3+ + 2H2O | (3) |
It has been documented that biological Fe2+ oxidation at approximately pH 3 promotes the removal of iron and sulfate in AMD via the precipitation of iron hydroxysulfate minerals.19–22 The vital role of Acidithiobacillus ferrooxidans in biomineralization has been investigated extensively. In A. ferrooxidans system, Fe2+ oxidation rate is substantially improved by millions of times compared with the abiotic oxidation (eqn (3)), and mineral is produced spontaneously under acidic conditions.15,16,23–27 Crystal seed is able to improve mineral production, and the initial concentrations of Fe2+, K+ and NH4+ play important roles in controlling the formation of different type of mineral precipitates (schwertmannite or/and jarosite).28,29 However, a growing number of evidences suggest that only limited amount of iron can be integrated into minerals in A. ferrooxidans biomineralization system, even under the optimal conditions, because of the finally achieved equilibrium between Fe2+ bio-oxidation and Fe3+ mineralization.30–33 Our previous studies demonstrated that about 70% and 40% of Fe3+ (about 9 g L−1 iron initially) failed to enter into schwertmannite and jarosite phases, respectively.22,28,34 Therefore, a high removal efficiency of TFe may be achieved by reducing the residual Fe3+ to Fe2+ to restart the bio-oxidation of Fe2+ and coupling biomineralization for several cycles.
Song et al.35 used a combination process of electrolytic reduction and biological oxidation to enhance the removal of TFe from a simulated AMD, but the system was electric power-dependent and time-consuming, and could suffer from the aging of the electrodes. Zero-valent iron (ZVI), a partial electron donor, is an active metal capable of reducing ions as well as organic and inorganic compounds with oxidation activity.36 ZVI-based technologies have been widely used in remediating various contaminants for the treatment of groundwater and industrial wastewater due to their features of rapid, simple, controllable, and inexpensive.36–40 Fe3+ can be reduced by ZVI rapidly (eqn (4)), and the newly formed Fe2+ may be bio-oxidized by A. ferrooxidans again followed by the hydrolysis of Fe3+ to mineral precipitate (Fig. S2†). Therefore, we speculate that the integration of A. ferrooxidans into the reduction by ZVI may have considerable potential in enhancing the biomineralization efficiency, which may further decrease soluble TFe and sulfate loads in AMD before lime neutralization.
Some recent studies reported that biogenic schwertmannite would be of great promise for wastewater treatments due to its capability in adsorbing heavy metals/metalloids and catalysing advanced oxidation.18,41–47 For instance, As(III) in simulated groundwater can be effectively removed by schwertmannite biosynthesized using A ferrooxidans, with a maximum adsorption capacity of 113.9 mg As(III) g−1 in the optimal pH range of 7–10;32 Fenton-like reaction catalysed by biosynthetic schwertmannite could completely degrade phenanthrene (1 mg L−1) within only 3 h;48 and biogenic schwertmannite exhibited a much higher catalytic capacity than biogenic jarosite for the degradation of phenol in heterogeneous Fenton-like reactions, because of its shorter induction period and higher kinetic rate (k) of the second-stage.49 Therefore, the production of schwertmannite during the precipitation of soluble iron and sulfate in A. ferrooxidans biomineralization system is advantageous over the production of other types of minerals.
In this work, a combined system was initially set up for the coupling of bio-oxidation and chemical reduction by optimizing periodic ZVI treatment. Subsequently, the system was tested to investigate the effects of ZVI supplement and nutrition on the removal and mineralization performance of soluble TFe and sulfate. The contents of toxic metals in mineral precipitates in simulated AMD were also analysed. The outcomes of this study may provide a promising approach to drastically enhance the biomineralization of soluble iron and sulfate in AMD via the formation of secondary Fe-minerals in order to improve lime neutralization treatment efficiency of AMD.
2. Materials and methods
2.1 Bacteria and culture conditions
A. ferrooxidans LX5 (CGMCC no. 0727) was obtained from China General Microbiological Culture Collection Centre (CGMCC). The bacterium was cultured in 500 mL Erlenmeyer flasks containing 250 mL of modified 9 K medium with the following composition: 3.50 g L−1 (NH4)2SO4, 0.119 g L−1 KCl, 0.058 g L−1 K2HPO4, 0.0168 g L−1 Ca(NO3)·4H2O, 0.583 g L−1 MgSO4·7H2O, and 44.2 g L−1 FeSO4·7H2O.50 Sulfuric acid (50%, v/v) was used to adjust the pH to approximately 2.5. 10% (v/v) of A. ferrooxidans culture was inoculated into the medium and then incubated at 28 °C and 180 rpm for around 60 h. The bacterial cultures were filtered with filter paper (Whatman no. 4) to remove the precipitates, while filtrates containing bacterial cells were then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g (Beckman Avanti J-E, USA) for 10 min at room temperature. After being washed twice with dilute sulfuric acid solution (pH 2.2), the bacterial cells were resuspended in dilute sulfuric acid solution (pH 2.5) to approximately 5 × 109 cells per mL.
000 × g (Beckman Avanti J-E, USA) for 10 min at room temperature. After being washed twice with dilute sulfuric acid solution (pH 2.2), the bacterial cells were resuspended in dilute sulfuric acid solution (pH 2.5) to approximately 5 × 109 cells per mL.
2.2 Mineralization efficiency by coupling chemo-reduction and bio-oxidation
A. ferrooxidans cell suspensions were incubated in 250 mL coupling culture solution (CCS) with the following composition: 3.50 g L−1 (NH4)2SO4, 0.119 g L−1 KCl, 0.058 g L−1 K2HPO4, 0.0168 g L−1 Ca(NO3)·4H2O, 0.583 g L−1 MgSO4·7H2O, and 11.2 g L−1 FeSO4·7H2O. Cultures were incubated in 500 mL Erlenmeyer flasks at 28 °C on a rotary shaker at 180 rpm. ZVI powder (about 50% amount of Fe3+ in system) was added to system once every 2 days, starting from the third day (Table S1,† Exp#1). Reductive reaction was allowed to proceed for 15 min, and then ZVI was removed from the culture solution by a magnetic rod. A. ferrooxidans-mediated oxidation continued on a rotary shaker for another 2 days. Soluble TFe and pH was monitored periodically before ZVI addition. After 31 days, the precipitates were collected and analysed with a powder X-ray diffraction (XRD) system (Thermo X'TRA, Germany), using Cu-Kα radiation (40 kV, 40 mA).Copper powder (ZVC), a reductant of Fe3+ (eqn (5)), is added to A. ferrooxidans biomining system as a reference to compare with iron powder, so as to reflect the dynamic changes of initial iron ions. ZVI and ZVC (about 50% amount of Fe3+ in system) were added to 250 mL coupling culture solution (CCS) containing 4 × 107 cells per mL every 2 days, respectively, starting from the third day (Table S1,† Exp#2), at 28 °C on a rotary shaker at 180 rpm. Reductive reaction was allowed to proceed for 1 hour, then ZVI and ZVC was removed by filtration (Waterman no. 4). A. ferrooxidans-mediated oxidation continued on a rotary shaker for another 2 days. Soluble TFe, sulfate and pH was monitored before the addition of ZVI or ZVC.
To investigate the effect of nutrient on coupling system, ZVI (about 50% amount of Fe3+ in system) was added to nutrition (N) and nutrition free (NF) system, respectively, every 2 days, starting from the third day (Table S1,† Exp#3). Nutrition culture (N) contains 3.50 g L−1 (NH4)2SO4, 0.119 g L−1 KCl, 0.058 g L−1 K2HPO4, 0.0168 g L−1 Ca(NO3)2·4H2O, 0.583 g L−1 MgSO4·7H2O, and 11.2 g L−1 FeSO4·7H2O. Nutrition free system (NF) contains only 11.2 g L−1 FeSO4·7H2O. Considering the toxicity of ZVI, 2 mL A. ferrooxidans cell suspension (1010 cells) was inoculated into the nutrient free medium every 2 days, starting from the ninth day.
| 2Fe3+ + Fe0 = 2Fe2+ + Fe2+ | (4) |
| 2Fe3+ + Cu0 = 2Fe2+ + Cu2+ | (5) |
To investigate the effect of treatment frequency on the coupling system, ZVI (about 50% amount of Fe3+ in system) was added to CCS system every 2 days, 4 days, or 8 days, respectively (Table S1,† Exp#4). Reductive reaction was allowed to proceed for 15 minutes and followed by A. ferrooxidans-mediated oxidation continued on a rotary shaker for another 2 days, 4 days, and 8 days, respectively.
Different amount of bacterial cells were inoculated into reaction system only containing 2.23 g L−1 FeSO4 to investigate relationship between oxidation rate and mineralization. Toxicity of ZVI powder (1.12–4.47 g L−1) added to A. ferrooxidans density in bio-mineralization system was conducted.
2.3 Performance of the combined system on simulated AMD
Total of 300 mL simulated AMD was incubated in 500 mL Erlenmeyer flasks containing 10% (v/v) A. ferrooxidans culture with nutrition (CCS) and metals (Fe2+ 2234 mg L−1, Cu2+ 3.1 mg L−1, Zn2+ 261 mg L−1, Al3+ 54 mg L−1, and Mn2+ 55 mg L−1). When Fe2+ was completely oxidized by A. ferrooxidans after 3 days, the reduction by ZVI was allowed to proceed for 15 min followed by 2 days of A. ferrooxidans-mediated biomineralization (Table S1,† Exp#5). Precipitates were collected at the end of the experiment. For the measurement of the metal content in the precipitates, the collected precipitates were digested with nitric acid, hydrofluoric acid, and perchloric acid. Then, the digested solution was analyzed for metal concentration by using an inductively coupled plasma-atomic emission spectrometry system (Agilent Technologies 700 ICP-OEC, Agilent Technologies, Inc., USA).2.4 Analytical procedures
The pH values were determined using a pHS-3C pH meter (Shanghai, China). The concentrations of Fe2+, TFe ions, and sulfate were measured by spectrophotometry in accordance with previously reported standard methods.51,52Results were expressed as the mean ± standard deviation (SD) of at least three replicate measurements. Significant differences between the treatments were statistically evaluated by SD and one-way analysis of variance (ANOVA) using SPSS 2.0. Data between two different treatments were compared statistically by ANOVA, followed by F-test if the ANOVA result was significant at P < 0.05. For multiple comparison analysis, least significant difference (LSD) tests were performed on all the data following the ANOVA tests for significant (P < 0.05) differences between two different treatment.
3. Results and discussion
3.1 Effects of ZVI on bio-oxidation and biomineralization
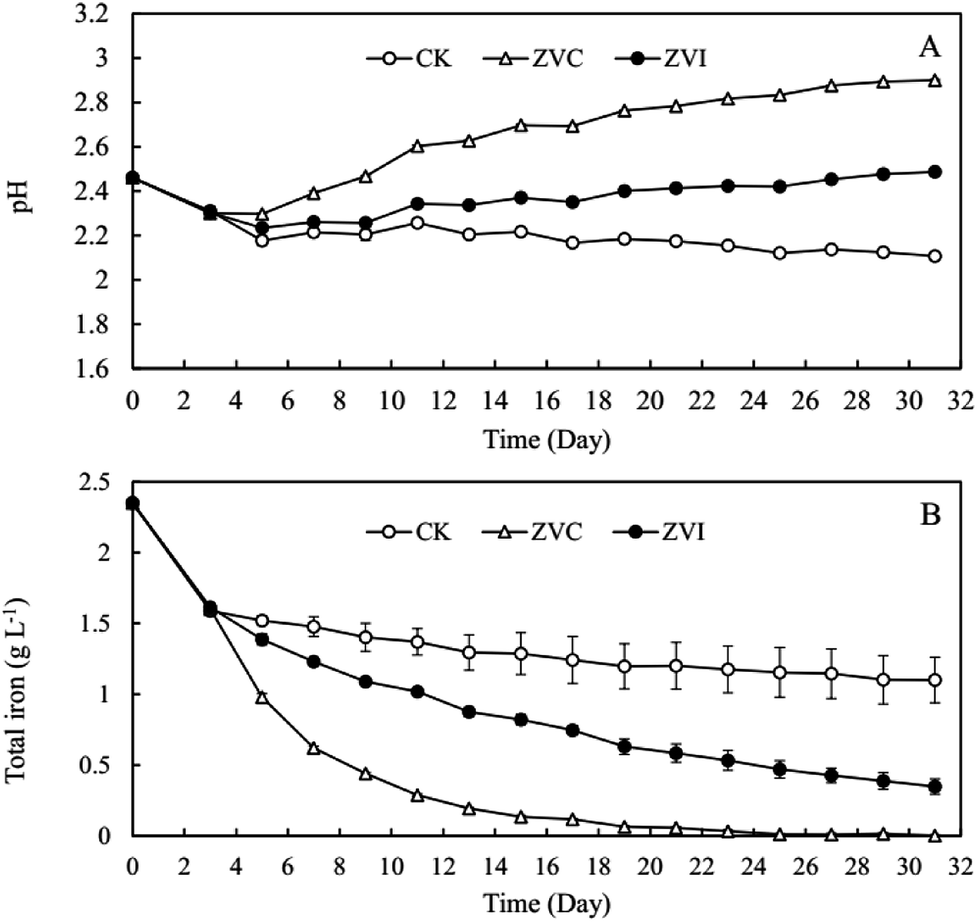
Our preliminary study demonstrated that excessive ZVI remained in the reaction system was toxic to A. ferrooxidans (Fig. S3†), restricting the oxidization of ferric ion in the system. Lee et al.53 reported that ZVI could disrupt bacterial cell activity by reductively decomposing the functional groups in the proteins and lipopolysaccharides on outer membranes. Consequently, ZVI was added cyclically to avoid the toxic effect of excessive ZVI on A. ferrooxidans.During coupling process, the pH in ZVI system declined continuously to 2.28 at the end of reaction for 31 days, higher than the pH of 1.95 in the control system (Fig. 1A). After 14 cyclic reductions, TFe concentration decreased significantly from 2.24 g L−1 to 0.24 g L−1 (31 days) in ZVI system and 1.05 g L−1 in the control system without ZVI (Fig. 1B). The removal efficiency of iron under ZVI treatment was 89.4%, which was much higher than that in the control system (53.2%). ZVI treatment significantly resulted in much more mineral precipitation as compared to the control (Fig. 2). During the whole experimental process, a total amount of 1.2 g ZVI powder was discontinuously added into the tested AMD system (250 mL) and finally 3.2 g mineral precipitates was accordingly collected from the system after 31 days. And compared with the control system without addition of ZVI (using Fe-oxidizing bacteria only), the final amount of iron minerals increased by 2.17 times. These results demonstrated that the coupling of bio-oxidation and reduction improved biomineralization effectively. In addition, the much higher pH and much lower TFe concentration in ZVI system is beneficial for reducing the dosage of reagents during the subsequent lime neutralization step.
 | ||
| Fig. 1 Change of (A) pH and (B) total iron concentration in A. ferrooxidans system (ZVI) with or (CK) without zero-valent iron reduction. | ||
 | ||
| Fig. 2 Final weight of mineral precipitates after 31 days in A. ferrooxidans system. The mean values of three replicates followed by asterisk (*) indicated significance of difference between the treatment and control (P < 0.05). | ||
XRD pattern suggested that ZVI treatment altered the composition of the final mineral precipitates, as exhibiting that the ZVI treatment resulted in a significantly higher content of schwertmannite with comparison to that of the control system (Fig. 3). Schwertmannite has been demonstrated to be a possible precursor of jarosite in acidic sulfate-rich environments, providing a suitable cation that is available to occupy the “A” site in the jarosite structure.29 High ratio of K+/Fe or NH4+/Fe impacted mineral transformation from schwertmannite to jarosite.28 The periodic addition of ZVI enhanced Fe/K in system, which limited the final formation of jarosite. In addition, NH4+ and K+ in 9 K medium may be not sufficient to account for jarosite type precipitation with the increase in mineral formation. Therefore, it is possible that only schwertmannite precipitated when NH4+ and K+ were inadequacy at later stage. Furthermore, because of higher Fe/S ratio in schwertmannite and jarosite, much more minerals produced from coupling process (bio-oxidation and reduction) lead to a drop of sulfate load in solution along with iron.29
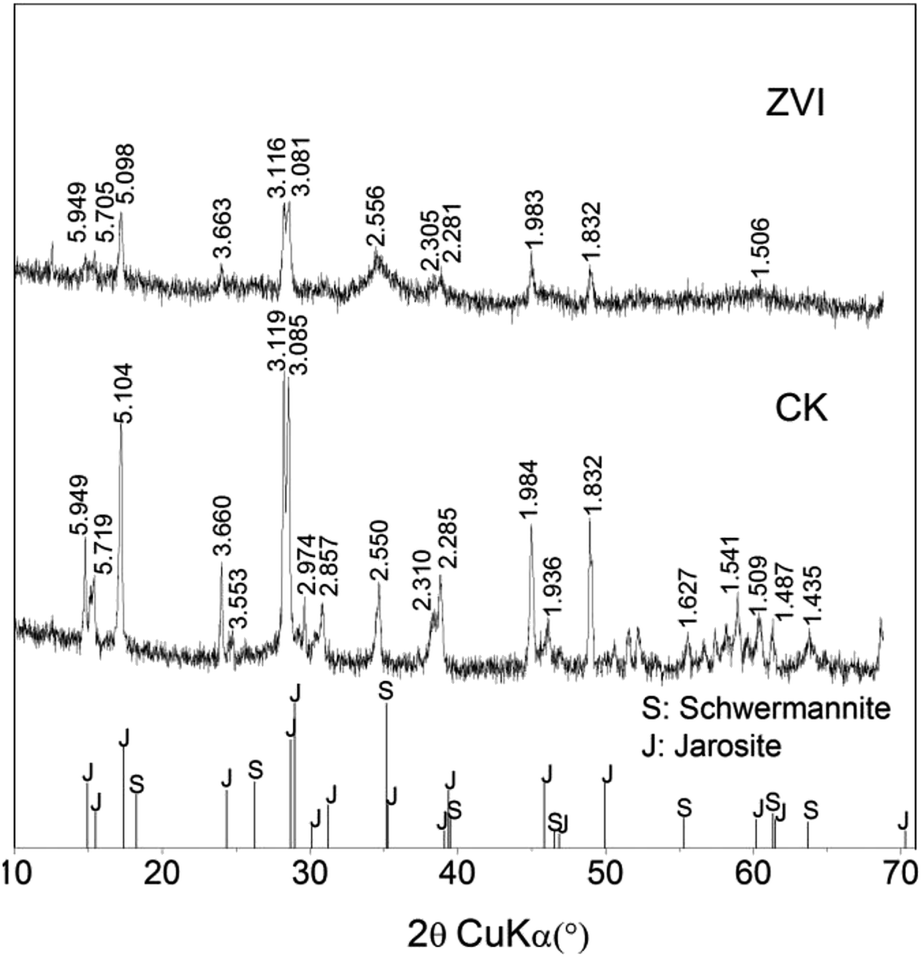 | ||
| Fig. 3 XRD patterns of mineral precipitates formed in A. ferrooxidans system (ZVI) with or (CK) without periodic reduction by zero-valent iron after 31 days. Standard XRD patterns of schwertmannite (PDF-47-1775) and jarosite (PDF-22-0827) were from the International Centre for Diffraction Data (ICDD) Powder Diffraction Files (PDF) database, respectively. | ||
Because ZVI addition introduced extra iron into the system, it is necessary to inspect the behaviour of iron ions during coupling process. A typical reducer ZVC was used as an object of reference. TFe removal efficiency were 80.9%, 91.6%, and 97.2% after 3 (9 days), 5 (13 days), and 8 (19 days) cyclic additions of ZVC, respectively (Fig. 4B). Meanwhile, reductive rate ([Fe3+ before reductive − Fe3+ after reductive]/Fe3+ before reductive) of first five reactions (3 days, 5 days, 7 days, 9 days and 11 days) in cultural solution was similar in both ZVI (49–71%) and ZVC (38–64%) systems (Table S2†). These results suggest that the initial soluble iron ions, but not new iron ions from ZVI, were precipitated at approximately 90% after 5 cycles in ZVI treatment, as same as in ZVC treatment. Furthermore, 3.20 g L−1 soluble SO42− were removed from solution after coupling process of ZVI (Fig. 5). Thus, the introduced iron ions by ZVI reduction could contribute to further improve sulfate precipitation into mineral.
 | ||
| Fig. 4 Change of (A) pH and (B) total iron concentration in A. ferrooxidans system (ZVI) with or (CK) without zero-valent iron reduction, and (ZVC) with zero-valent cooper reduction, respectively. | ||
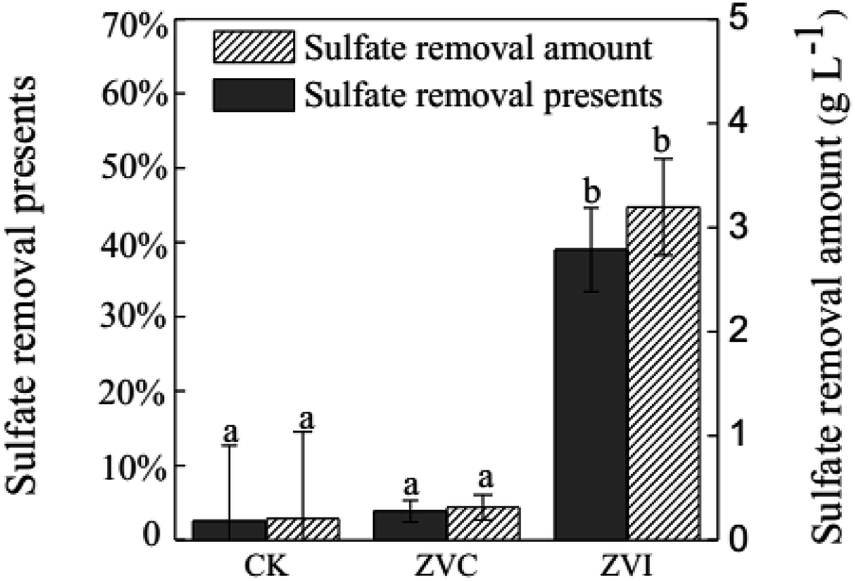 | ||
| Fig. 5 Amount and percentage of sulfate removal in A. ferrooxidans system (ZVI) with or (CK) without zero-valent iron reduction, and (ZVC) with zero-valent cooper reduction, respectively. | ||
ZVC treatment showed higher removal rate of iron than that of ZVI system, because extra copper ions entered into system by reduction reaction instead of introducing extra iron ions by ZVI system. Besides, the final pH of ZVC system was much higher than that of ZVI system (Fig. 4A). However, ZVC may not be suitable to be used as a reducing agent during practical application, because it introduces copper ion during the process, the release of which into the environment poses serious threat to both human health and plant growth.54,55
3.2 Influence of frequency and nutrition on ZVI system
The highest frequency of ZVI addition (2 d per cycle) resulted in the largest removal of TFe (Fig. 6A). Obviously, more frequent addition of ZVI provided continuous reducing power to produce more Fe2+, further entering into the mineral phase during A. ferrooxidans-mediated biomineralization. However, before each addition of ZVI, it is to be ensured that Fe2+ has been oxidized completely and reaction hydrolysis precipitation achieve equilibrium. Under acidic conditions, the biological oxidation rate of Fe2+ by A. ferrooxidans is known to be significantly higher (105–106 times) as compared to natural abiotic oxidation reactions.56 In addition, a experiment, inoculated with different bacterial density, indicated that a more rapid biological oxidation process not only facilitated the transformation of Fe2+ to Fe3+ but also significantly accelerated the production of Fe-mineral by A. ferrooxidans during 3 days (Fig. S4†). | ||
| Fig. 6 Changes of total iron in A. ferrooxidans system (A) with different frequency of zero-valent iron addition and, and (B) with nutrient or not. ZVI-2d, ZVI-4d, and ZVI-8d indicate reduction by zero-valent iron followed by 2 days, 4 days, and 8 days of biological mineralization by A. ferrooxidans, respectively. N-CK and N-ZVI indicate A. ferrooxidans-nutrient system (ZVI) with or (CK) without periodic zero-valent iron reduction. NF-CK and NF-ZVI indicate A. ferrooxidans-nutrient free system (ZVI) with or (CK) without periodic zero-valent iron reduction. 2 mL bacteria suspensions (1 × 1010) were inoculated into the nutrition free system (NF) from the third cycle after removing zero-valent iron powder by filtration per cycle. | ||
Accelerated reaction can shorten the bio-mineralization time. Except for bacterial density, oxidation rate may be affected by several other factors, such as pH, temperature, Fe2+ or Fe3+ concentration, dissolved O2 (DO), and CO2 level in the solution.34,57–60 Future studies are needed to examine the effect of these factors on ZVI-A. ferrooxidans system. These results illustrated a relationship between oxidation and mineralization, which provided evidence for the determination of appropriate frequency of ZVI addition.
Nutrition did not affect the removal of soluble TFe after 14 cycles (31 days) treatment of ZVI (Fig. 6B). Because the density of A. ferrooxidans cell declined (1.32 × 107 cell per mL) in nutrition-free solution at the end of 3 cycles (9 days), Fe2+ was not oxidized (1.05 g L−1) completely. Total of 2 mL suspend cells (1 × 1010 cells) were inoculated into the nutrition free system from ninth day after removing ZVI powder by filtration per cycle to compensate the loss of bacteria. Nutrition did not affect the efficiency of biological mineralization, but nutrition did affect bacteria density, which is crucial in determining the oxidation and mineralization rates. In practical treatment projects, biological mineralization reaction process can be separated from chemical reduction reaction process to lower ZVI toxicity for the protection of A. ferrooxidans cells.
3.3 Sulfate and toxic metal removal in simulated AMD by periodic ZVI treatment coupled with biomineralization
Compared with the control (AMD-CK) that was free of ZVI, cyclic addition of ZVI significantly decreased the concentrations of free iron ion and sulfate in simulated AMD by promoting the formation of iron-sulfate minerals (Fig. 7). At the end of the reaction (after 9 cycles), the removal efficiency of iron and sulfate reached 83% and 63% in simulated AMD, respectively (Fig. 7). The concentration of metal ions including Cu, Zn, Al, and Mn in this trial did not affect the bio-oxidation and hydrolytic mineralization mediated by A. ferrooxidans. Since sulfate and iron ions precipitated in the form of mineral precipitates, the high removal efficiency of both iron and sulfate could be achieved when more Fe hydroxylsulfate precipitates were generated. Extra introduction of iron by ZVI resulted in higher production of precipitates, which in turn enhanced the efficiency of sulfate removal.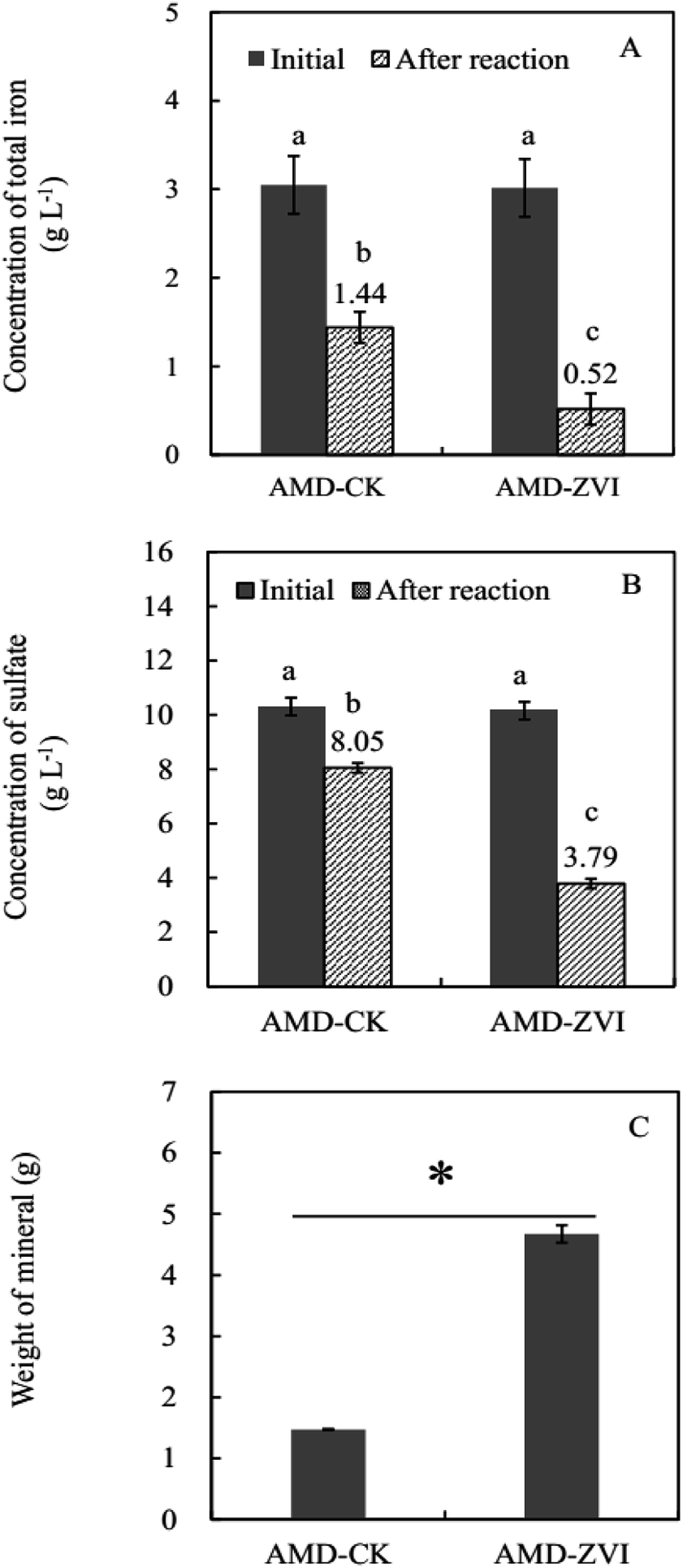 | ||
| Fig. 7 Changes of total iron (A) and sulfate (B) before (initial) and after (after reaction) in A. ferrooxidans system at the end of 21 days. (C) Weight of mineral precipitate from A. ferrooxidans system. AMD-CK and AMD-ZVI indicate synthetic acid mine drainage system (AMD-ZVI) with or (AMD-CK) without periodic reduction by zero-valent iron inoculated by A. ferrooxidans. The mean values of three replicates followed by letters (a and b) in (A and B) indicated significance of difference among the treatments (P < 0.05). The mean values of three replicates followed by asterisk (*) in (C) indicated significance of difference between two designated treatments (P < 0.05). | ||
Metal ions (e.g., Zn, Al, and Mn) in simulated AMD did not decrease significantly after periodic ZVI treatments, except for Cu with the lowest concentration (Fig. 8A). Zn content in precipitates obtained from simulated AMD in control and ZVI system were 0.425 mg g−1 and 0.456 mg g−1, respectively (Fig. 8B). The concentration of Zn was the highest in stimulated AMD as compared to other metals. The removal of total Zn reached 2.13 mg, which only accounted for 3% of its initial quantity. This finding is consistent with previous reports by Hedrich and Johnson,21 who described a modular bioremediation reactor system for the selective iron removal and precipitation of schwertmannite from mine waters. They found that some metals, such as Cu, Zn, Al, and Mn, remained in the solution throughout the process and exhibited minimal changes. It may be due to low pH (<2.5), ionic group (Fe3+, OH− and SO42−) and structure of mineral crystal lattice.61,62 In addition, some investigations demonstrated that at relatively higher pH (2.5–4.0), lesser amounts of other Fe-minerals (e.g., goethite) was simultaneously formed along with the predominant generation of schwertmannite, and trace levels of Zn, Ni and Co (around at μ mol g−1 concentrations) were accumulated in these secondary iron-minerals.63,64 In fact, for the treatment of the real AMD, once soluble total Fe is efficiently removed by ZVI-assisted biomineralization process, other metal pollutants such as Cu2+, Zn2+, Al3+, and Mn2+ can be readily removed by conventional approaches, such as sulfide precipitation (Cu, Zn), oxidation (Mn), and neutralization.65 All of these findings suggest that the coupling of biomineralization and chemo-reduction could selectively recover considerable amounts of soluble total iron from AMD as the form of Fe hydroxylsulfate precipitates separating from other metal ions in acidic solution.
 | ||
| Fig. 8 (A) Content of metal (Cu, Zn, Al and Mn) in simulated AMD and (B) mineral by ICP determination. The mean values of three replicates followed by asterisk (*) in (B) indicated significance of difference between two designated treatments (P < 0.05). | ||
4. Conclusion
In this study, a novel combined system of Fe2+ bio-oxidation by A. ferrooxidans and Fe3+ chemo-reduction by ZVI was developed for enhanced biomineralization efficiency in AMD. The successive addition of ZVI into A. ferrooxidans-mediated biomineralization could regenerate Fe2+ and restart the low-pH Fe2+ bio-oxidation. Through this combined system, the residual amount of soluble total iron in AMD was significantly decreased and, thus, the formation efficiency of secondary Fe-minerals (e.g., schwertmannite) was improved accordingly. Therefore, it is a promising approach to improve, to a great extent, the subsequent lime neutralization efficiency through this combined system for AMD treatment. For the scale-up application of this ZVI-assisted biomineralization strategy for treatment of real AMD, industrial scrap iron, may be chosen as a potential reductive agent, instead of relatively expensive ZVI. The practical treatment performance of the system described here should be further evaluated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 21637003 and No. 21477054) and the National Key R&D Program of China (2018YFD0800700).References
- R. A. Davis Jr, A. T. Welty, J. Borrego, J. A. Morales, J. G. Pendon and J. G. Ryan, Environ. Geol., 2000, 39, 1107–1116 CrossRef.
- J. S. España, E. L. Pamo, E. S. Pastor, J. R. Andrés and J. A. M. Rubí, Environ. Geol., 2005, 49, 253–266 CrossRef.
- A. Chen, C. Lin, W. Lu, Y. Wu, Y. Ma, J. Li and L. Zhu, Chemosphere, 2007, 70, 248–255 CrossRef CAS PubMed.
- J. Oyarzún, D. Castillo, H. Maturana, N. Kretschmer, G. Soto, J. M. Amezaga, T. S. Rötting, P. L. Younger and R. Oyarzún, J. Geochem. Explor., 2012, 115, 47–58 CrossRef.
- J. L. Kuang, L. N. Huang, L. X. Chen, Z. S. Hua, S. J. Li, M. Hu, J. T. Li and W. S. Shu, ISME J., 2013, 7, 1038–1050 CrossRef CAS PubMed.
- A. K. Parbhakar-Fox, M. Edraki, K. Hardie, O. Kadletz and T. Hall, J. Geochem. Explor., 2014, 137, 11–28 CrossRef CAS.
- S. Naidoo, Environment, Development and Sustainability, 2015, 17, 1045–1063 CrossRef.
- A. Akcil and S. Koldas, J. Cleaner Prod., 2006, 14, 1139–1145 CrossRef.
- A. H. Kaksonen, M. Riekkola-Vanhanen and J. A. Puhakka, Water Res., 2003, 37, 255–266 CrossRef CAS PubMed.
- V. R. K. Vadapalli, J. N. Zvimba, M. Mathye, H. Fischer and L. Bologo, Environ. Technol., 2015, 36, 2515–2523 CrossRef CAS PubMed.
- A. Qureshi, Y. Jia, C. Maurice and B. Öhlander, Environ. Sci. Pollut. Res. Int., 2016, 23, 17083–17094 CrossRef CAS PubMed.
- E. T. Tolonen, A. Sarpola, T. Hu, J. Rämö and U. Lassi, Chemosphere, 2014, 117, 419–424 CrossRef CAS PubMed.
- F. Liu, J. Zhou, L. Zhou, S. Zhang, L. Liu and M. Wang, J. Hazard. Mater., 2015, 299, 404–411 CrossRef CAS PubMed.
- W. Wang, Z. H. Xu and J. Finch, Environ. Sci. Technol., 1996, 30, 2604–2608 CrossRef CAS.
- C. Drouet and A. Navrotsky, Geochim. Cosmochim. Acta, 2003, 67, 2063–2076 CrossRef CAS.
- J. Majzlan, A. Navrotsky and U. Schwertmann, Geochim. Cosmochim. Acta, 2004, 68, 1049–1059 CrossRef CAS.
- X. M. Dou, D. Mohan and C. U. Pittman Jr, Water Res., 2013, 47, 2938–2948 CrossRef CAS PubMed.
- H. Duan, Y. Liu, X. Yin, J. Bai and J. Qi, Chem. Eng. J., 2016, 283, 873–879 CrossRef CAS.
- H. Olem and R. F. Unz, Biotechnol. Bioeng., 1977, 19, 1475–1491 CrossRef CAS.
- E. Heinzel, E. Janneck, F. Glombitza, M. Schlomann and J. Seifert, Environ. Sci. Technol., 2009, 43, 6138–6144 CrossRef CAS PubMed.
- S. Hedrich and D. B. Johnson, Bioresour. Technol., 2012, 106, 44–49 CrossRef CAS PubMed.
- M. Wang and L. Zhou, Hydrometallurgy, 2012, 125–126, 152–156 CrossRef CAS.
- D. T. Lacey and F. Lawson, Biotechnol. Bioeng., 1970, 7, 29–50 CrossRef.
- K. J. Edwards, P. L. Bond, T. M. Gihring and J. F. Banfield, Science, 2000, 287, 1796–1799 CrossRef CAS PubMed.
- D. B. Johnson and K. B. Hallberg, Sci. Total Environ., 2005, 338, 3–14 CrossRef CAS PubMed.
- K. B. Hallberg, Hydrometallurgy, 2010, 104, 448–453 CrossRef CAS.
- S. Eisen, A. Poehlein, D. B. Johnson, R. Daniel, M. Schlömann and M. Mühling, Genome Announc., 2015, 3, e00383–e003815 Search PubMed.
- S. Bai, Z. Xu, M. Wang, Y. Liao, J. Liang, C. Zheng and L. Zhou, Mater. Sci. Eng., C, 2012, 32, 2323–2329 CrossRef CAS.
- F. Sandy Jones, J. M. Bigham, J. P. Gramp and O. H. Tuovinen, Mater. Sci. Eng., C, 2014, 44, 391–399 CrossRef CAS PubMed.
- J. Daoud and D. Karamanev, Miner. Eng., 2006, 19, 960–967 CrossRef CAS.
- S. Huang and L. Zhou, Mater. Sci. Eng., C, 2012, 32, 916–921 CrossRef CAS.
- Y. H. Liao, J. R. Liang and L. X. Zhou, Chemosphere, 2011, 83, 295–301 CrossRef CAS PubMed.
- J. Liu, X. Xiu and P. Cai, Procedia Earth Planet. Sci., 2009, 1, 706–712 CrossRef.
- Q. Hou, D. Fang, J. Liang and L. Zhou, PLoS One, 2015, 10, e0120966 CrossRef PubMed.
- Y. Song, M. Wang, J. Liang and L. Zhou, Hydrometallurgy, 2014, 143, 23–27 CrossRef CAS.
- X. H. Guan, Y. K. Sun, H. J. Qin, J. Li, I. M. C. Lo, D. He and H. Dong, Water Res., 2015, 75, 224–248 CrossRef CAS PubMed.
- B. Lai, Z. Chen, Y. Zhou, P. Yang, J. Wang and Z. Chen, J. Hazard. Mater., 2013, 250–251, 220–228 CrossRef CAS PubMed.
- P. Pourrezaei, A. Alpatova, K. Khosravi, P. Drzewicz, Y. Chen, P. Chelme-Ayala and M. Gamal El-Din, J. Environ. Manage., 2014, 139, 50–58 CrossRef CAS PubMed.
- C. Ou, S. Zhang, J. Liu, J. Shen, Y. Liu, X. Sun, J. Li and L. Wang, Phys. Chem. Chem. Phys., 2015, 17, 22072–22078 RSC.
- H. Xu, D. Zhao, Y. Li, P. Liu and C. Dong, Environ. Sci. Pollut. Res. Int., 2014, 21, 5132–5140 CrossRef CAS PubMed.
- D. Baron and C. D. Palmer, Geochim. Cosmochim. Acta, 2002, 66, 2841–2853 CrossRef CAS.
- C. Drouet, D. Baron and A. Navrotsky, Am. Mineral., 2003, 88, 1949–1954 CrossRef CAS.
- P. Acero, C. Ayora, C. Torrentó and J. M. Nieto, Geochim. Cosmochim. Acta, 2006, 70, 4130–4139 CrossRef CAS.
- J. Antelo, S. Fiol, D. Gondar, C. Pérez, R. López and F. Arce, Geochim. Cosmochim. Acta, 2013, 119, 149–163 CrossRef CAS.
- S. Paikaray, J. Essilfie-Dughan, J. Göttlicher, K. Pollok and S. Peiffer, J. Hazard. Mater., 2014, 265, 208–216 CrossRef CAS PubMed.
- J. Guo, C. Dong, J. Zhang and Y. Lan, Sep. Purif. Technol., 2015, 143, 27–31 CrossRef CAS.
- J. Xu, Z. Xu, M. Zhang, J. Xu, D. Fang and W. Ran, Mater. Chem. Phys., 2015, 152, 4–8 CrossRef CAS.
- X. Q. Meng, S. Yan, W. Z. Wu, G. Y. Zheng and L. X. Zhou, RSC Adv., 2017, 7, 21638–21648 RSC.
- S. Yan, G. Y. Zheng, X. Q. Meng and L. X. Zhou, Sep. Purif. Technol., 2017, 185, 83–93 CrossRef CAS.
- L. X. Zhou, D. Fang, S. M. Wang, J. W. C. Wong and D. Z. Wang, Environ. Technol., 2005, 26, 277–284 CrossRef CAS PubMed.
- A. D. Eaton, A. E. Greenberg, L. S. Clesceri and M. Franson, Standard Methods for The Examination of Water & Wastewater, American Public Health Association, Washington, 1985 Search PubMed.
- A. Kolmert, P. Wikström and K. B. Hallberg, J. Microbiol. Methods, 2000, 41, 179–184 CrossRef CAS PubMed.
- C. Lee, J. Y. Kim, W. I. Lee, K. L. Nelson, J. Yoon and D. L. Sedlak, Environ. Sci. Technol., 2008, 42, 4927–4933 CrossRef CAS PubMed.
- M. Adrees, S. Ali, M. Rizwan, M. Ibrahim, F. Abbas, M. Farid, M. Zia-ur-Rehman, M. K. Irshad and S. A. Bharwana, Environ. Sci. Pollut. Res., 2015, 22, 8148–8162 CrossRef CAS PubMed.
- L. M. Gaetke, H. S. Chow-Johnson and C. K. Chow, Arch. Toxicol., 2014, 88, 1929–1938 CrossRef CAS PubMed.
- K. Bosecker, FEMS Microbiol. Rev., 1997, 20, 591–604 CrossRef CAS.
- M. Nemati, S. T. L. Harrison, G. S. Hansford and C. Webb, Biochem. Eng. J., 1998, 1, 171–190 CrossRef CAS.
- Y. Kawabe, C. Inoue, K. Suto and T. Chida, J. Biosci. Bioeng., 2003, 96, 375–379 CrossRef CAS.
- G. Meruane and T. Vargas, Hydrometallurgy, 2003, 71, 149–158 CrossRef CAS.
- C. G. Bryan, C. S. Davis-Belmar, N. van Wyk, M. K. Fraser, D. Dew, G. F. Rautenbach and S. T. L. Harrison, Biotechnol. Bioeng., 2012, 109, 1693–1703 CrossRef CAS.
- J. M. Blgham, U. Schwertmann, K. Carlson and E. Murad, Geochim. Cosmochim. Acta, 1990, 54, 2743–2758 CrossRef.
- S. Regenspurg and S. Peiffer, Appl. Geochem., 2005, 20, 1226–1239 CrossRef CAS.
- W. D. Burgos, T. Borch, L. D. Troyer, F. Luan, L. N. Larson, J. F. Brown, J. Lambson and M. Shimizu, Geochim. Cosmochim. Acta, 2012, 76, 29–44 CrossRef CAS.
- Y. Z. Sheng, B. kaley, K. Bibby, C. Grettenberger, J. L. Macalady, G. C. Wang and W. D. Burgos, RSC Adv., 2017, 7, 35962–35972 RSC.
- K. B. Hallberg and D. B. Johnson, Sci. Total Environ., 2005, 338, 115–124 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09887e |
| ‡ These authors contribute equally to the paper. |
| This journal is © The Royal Society of Chemistry 2019 |