
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMigration of mesenchymal stem cells tethered with carbon nanotubes under a chemotactic gradient
Jun Zhang and
Ching-An Peng *
*
Department of Biological Engineering, University of Idaho, Engineering Physics Building 421, 875 Perimeter Drive, Moscow, ID 83844-0904, USA. E-mail: capeng@uidaho.edu; Tel: +1-208-885-7461
First published on 1st March 2019
Abstract
Carbon nanotubes (CNTs) have been extensively studied for photothermal ablation of malignant cells due to their ability to absorb near-infrared (NIR) laser light and convert it to thermal energy for the lysis of tumor cells. Functionalizing CNTs with tumor-targeting moieties can facilitate the delivery to tumor sites. Instead of using targeting moieties, mesenchymal stem cells (MSCs) have been considered as vehicles to deliver therapeutic agents to cancer cells. In this study, the effects of attaching CNTs to MSCs on cell migration in response to a chemotactic gradient were investigated. Multiwalled carbon nanotubes (MWCNTs) were functionalized with streptavidin–fluorescein isothiocyanate (SA–FITC). The surface of human MSCs was biotinylated by culturing MSCs with biotin–lipid containing medium. CNTs were then attached on the outer cell membrane of biotinylated MSCs through SA–biotin binding. Fluorescence microscopy confirmed CNTs were located on the surface of MSCs. Various amounts of CNTs anchored on the membrane of MSCs were used to examine the effects of CNTs on MSC proliferation and migration. Our transwell migration assay showed that 4.26 ng CNT per cell is the threshold value that would not affect the migration speed of CNT-tagged MSCs toward the established gradient of chemoattractant SDF-1α.
1. Introduction
Carbon nanotubes (CNTs) have been well studied in cancer therapy and drug delivery due to their unique structures and properties such as high aspect ratio, large surface area, mechanical strength and thermal conductivity.1,2 In the field of cancer nanotechnology, CNTs have been demonstrated to be one of the potential photothermal absorbers for the ablation of malignant cells under near infrared (NIR) laser exposure.3–6 In photothermal therapy, an engineered substance such as CNT absorbs NIR light and then converts it to thermal energy near tumor sites. The strong absorbance of CNTs in the NIR region enables the optical stimulation of CNTs and the transmission of external heat inside body.6 Compared to traditional radio-chemotherapy, photothermal therapy has the advantage of low toxicity to body and less injuries to non-malignant cells and tissues.7,8Due to their tendency to aggregate in aqueous solvents, CNTs have been chemically modified to enhance their aqueous solubility. For example, CNTs have been treated with strong acids to shorten the length and endow with carboxyl groups to increase their suspension in water.9,10 Amino acids and peptides have been used for enhanced CNT's aqueous solubility and biocompatibility.11 CNTs have also been functionalized with poly(ethylene glycol)12 and hyaluronic acid13 for increased solubility and prolonged circulation time. In addition, functionalization of CNTs could enhance targeted delivery capabilities. For example, CNTs have been tethered with folic acid to target HeLa cells for the release of doxorubicin.14 Magnetic nanoparticles incorporated CNTs were fabricated for cancer diagnosis and drug delivery.15,16 CNTs have also been conjugated with monoclonal antibody CD133 for targeted delivery of CNTs to CD133+ stem-like glioblastoma cells.3 However, efficient delivery of nanoparticles to tumor sites remains challenging such as ingesting by the reticuloendothelial system and eliciting inflammatory response. To circumvent the antecedent issues, mesenchymal stem cells (MSCs), due to their low immunogenicity and intrinsic tumor-tropism, have been considered as vehicles to deliver therapeutic agents to cancer cells.17,18
Mesenchymal stem cells (MSCs) are stromal cells which can be isolated from bone marrow and adipose tissue.19 Their multipotency to differentiate into several mesenchymal lineages has great promise for regenerative medicine20 and their tumor homing capacity has initiated interest in using MSCs as cell carriers for cancer therapy.21,22 Using MSCs as tumor-targeted delivery vehicles is associated with less risks for clinical complications.23 The migration of MSCs is mediated by homing factor and receptor pairs such as SDF-1/CXCR4, HGF/c-Met and VEGF/VEGFR,24,25 among which SDF1/CXCR4 plays the most critical role in migration and homing of MSCs.26,27 Recently, avidin attached nanodrug has been incorporated in MSCs by endocytosis as well as cell membrane anchoring.28 In that study, MSCs were first chemically modified with avidin, then treated with biotinylated DOX conjugates. The stemness and migration ability of nanodrug-loaded MSCs were both retained. Kang et al. used pH-sensitive gold nanoparticles (AuNPs) internalized MSCs in photothermal therapy for enhanced tumor-targeting efficiency.29 The use of MSC significantly increased tumor-targeting efficiency as well as local hyperthermia.
In the current study, CNTs were anchored on the outer membrane of human MSCs as a potential cancer photothermal therapy system. The surface of MSCs was first biotinylated by biotin–lipid treatment, and then MSCs were tethered with streptavidin (SA)-conjugated CNTs. Due to the rapid binding of biotin and SA, the anchorage of CNTs on the surface of MSCs occurred within 1 h in order to minimize the concern of particle internalization. A variety of weights of CNTs hitchhiking on MSC surface were harnessed to evaluate the effects of CNTs on MSC motility and proliferation. According to the transwell migration assay, 4.26 ng CNTs per cell is the threshold amount that would not affect the migration speed of CNT-tethered MSCs toward the established concentration gradient of chemoattractant SDF-1α. The growth kinetics of re-seeded CNT-tethered MSCs showed no cytotoxicity of CNTs treated MSCs.
2. Materials and methods
2.1 Materials
Multiwalled carbon nanotube (MWCNT) was obtained from CNT Co., LTD (Incheon, Korea). Hydrogen peroxide (30%) and acetic acid were purchased from J. T. Baker (Philipsburg, NJ, USA). Succinic anhydride was purchased from Acros Organics (New Jersey, USA). Streptavidin–fluorescein isothiocyanate (SA–FITC) was purchased from Invitrogen (Frederick, MD, USA). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and collagen (bovine achilles tendon, Type I) were purchased from Sigma-Aldrich (St. Louis, MO, USA). N-Hydroxysuccinimide (NHS) and paraformaldehyde were purchased from Alfa Aesar (Haverhill, MA, USA). Human bone marrow-derived mesenchymal stem cells were purchased from RoosterBio (Frederick, MD, USA). Minimum essential medium, alpha (αMEM), L-glutamine, 0.25% trypsin/EDTA, and methanol were purchased from Fisher Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was purchased from Gibco (Grand Island, NY, USA). 1,2-Dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-cap biotinyl (biotin–lipid) was purchased from Avanti (Alabaster, AL, USA). Trypan blue was purchased from BTC Beantown chemical (Hudson, NH, USA). 8 μm transwell inserts for 24-well plates were purchased from Corning Inc. (Kennebunk, ME, USA). SDF-1α was purchased from Tonbo Biosciences (San Diego, CA, USA). Hematoxylin was purchased from VWR (Radnor, PA, USA).2.2 Cutting and functionalization of CNTs
As schematically illustrated in Fig. 1A, a series of chemical reactions were performed to conjugate SA–FITC with CNT. First, based on previously reported protocols30 to produce CNT-OH from pristine CNTs, 10 mg of CNTs were dissolved in 10 mL hydrogen peroxide (30%). The solution was autoclaved at 15 psi and 121 °C for 3 h. The solution underwent sonication at 4 watts for 4 h in an ice bath using a tip sonicator (Misonix XL-2000, Farmingdale, NY). Hydroxyl functionalized CNTs were collected by centrifugation. Secondly, the collected CNT-OH was re-dissolved in 50 mL of tetrahydrofuran (THF) in a 3-neck round bottom flask. 50 mg of succinic anhydride was added to the flask, and the solution was refluxed at 85 °C under agitation for 48 h to form carboxylated CNTs (CNT-COOH). The CNT-COOH was collected by centrifugation, and then washed three times with deionized (DI) water. The CNT-COOH product was re-suspended in 10 mL of DI water for further treatment. Finally, 10 mg of EDC and 10 mg of NHS were added to the CNT-COOH solution. The solution was stirred with a magnetic stir bar for 2 h at room temperature. 50 μL of SA–FITC solution was added to the reaction mixture, and mixture was stirred at 4 °C for overnight to obtain CNT-SA–FITC. The CNT-SA–FITC was collected by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 min, and then washed three times with DI water to remove excess SA–FITC. The washed CNT-SA–FITC was re-suspended in cell culture medium (see section below). The chemical analyses of pristine CNTs, CNT-COOH and CNT-SA–FITC were characterized by Fourier transform infrared spectroscopy (FT-IR). The FT-IR spectra were recorded by a FT-IR spectrometer (Nicolet iS10, ThermoFisher Scientific, Waltham, MA, USA).
000 × g for 10 min, and then washed three times with DI water to remove excess SA–FITC. The washed CNT-SA–FITC was re-suspended in cell culture medium (see section below). The chemical analyses of pristine CNTs, CNT-COOH and CNT-SA–FITC were characterized by Fourier transform infrared spectroscopy (FT-IR). The FT-IR spectra were recorded by a FT-IR spectrometer (Nicolet iS10, ThermoFisher Scientific, Waltham, MA, USA).
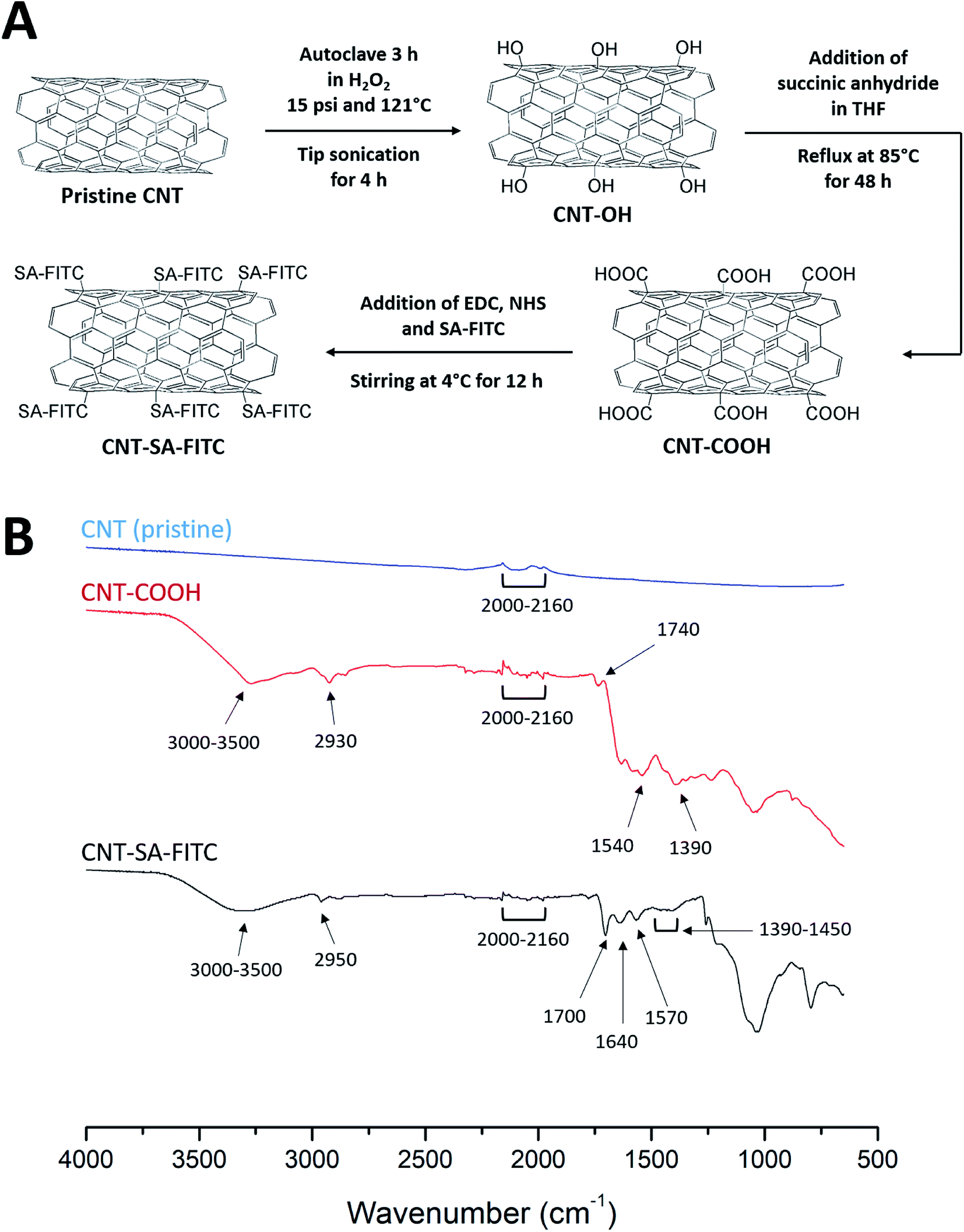 | ||
| Fig. 1 (A) A schematic diagram of a series of chemical reactions to produce the final product CNT-SA–FITC from pristine CNT. (B) FT-IR spectra of pristine CNTs, COOH-functionalized CNTs and SA–FITC-functionalized CNTs. | ||
2.3 Surface biotinylation of MSCs
Human MSCs were cultivated at 37 °C in 5% CO2 balanced with humidified air. The MSCs were cultured with αMEM supplemented with 16.5% FBS, 1% penicillin–streptomycin and 2 mM L-glutamine. MSCs were incubated in culture medium containing 0.01 and 0.02 mg mL−1 biotin–lipid for different timings of up to 60 h in 24-well plates. Following biotinylating, MSCs were washed with 1× phosphate buffered saline (PBS). Biotinylated MSCs were treated with SA–FITC for 1 h for SA–biotin binding. After 1 h, unbound SA–FITC was discarded and each well was washed with 1× PBS for 3 times. The fluorescence intensity of MSCs at 525 nm emission with 485 nm excitation was measured by a microplate reader (SpectraMax M2e, Molecular Devices, Sunnyvale, CA) to obtain a biotinylation standard curve. The MSC biotinylation standard curve was constructed by plotting fluorescence intensity versus time.2.4 Preparation and characterization of CNT-tagged MSCs
To estimate the weight of CNTs attached on the surface of MSCs, MSCs cultured in T-25 flasks were treated with 0.01 mg mL−1 biotin–lipid for different timings up to 48 h. Non-treated MSCs were used as control. Biotinylated MSCs were then treated with culture medium containing 20 μg mL−1 CNT-SA–FITC for 1 h. After 1 h, the culture medium was discarded, and the CNT-MSCs were washed three times with 1× PBS. Phase contrast and fluorescence images of CNT-MSCs were taken with Leica DMi8 microscope equipped with Leica EC3 camera (Leica Microsytems, Wetzlar, Germany). To assess the degree of anchored CNTs internalized by MSCs, FITC-CNT-MSCs were imaged at right after tethering CNT-SA–FITC on biotinylated MSC surface. The cultures were maintained for additional 12 and 24 h and then quenched by 0.01 mg mL−1 trypan blue, respectively. Phase contrast and fluorescence images were taken accordingly.A variety of weights of CNTs anchored on MSCs were determined by the following approach. Control MSCs and CNT-MSCs were suspended by treating the cells with 0.25% trypsin/EDTA, and the cell numbers were counted using a hemocytometer. The cells were collected by centrifugation at 400 × g for 8 min. The pellet was washed with 1× PBS, and the cells were centrifuged again to collect cell pellets. The cell pellets were taken up by a pipettor and transferred to a microbalance (XS3DU, Mettler Toledo, Columbus, OH). The weight of control MSCs and CNT-MSCs was divided by the cell numbers to obtain the weight of MSCs and CNT-MSCs. The weight of CNTs on MSCs was estimated by subtracting the weight of control MSCs from CNT-MSCs. All of the experiments were performed in triplicate.
After CNT-SA–FITC attached to the MSC surface, the excess CNTs were washed away, and cells were suspended by treating the cells with 0.25% trypsin/EDTA. The MSCs were seeded on 24-well plates at the seeding density of 3000 cells per cm2. The cells were allowed to grow for 8 days, and the cell numbers were counted every 2 days using a hemocytometer. Phase contrast images were taken with a Leica DMi8 microscope equipped with Leica EC3 camera (Leica Microsytems, Wetzlar, Germany).
2.5 Migration assay of CNT-MSCs
The migration capacity of CNT-MSCs was assessed using transwell plates with 8 μm pore size inserts (Corning Incorporated, Corning, NY). The transwell inserts were immersed in 10 μg mL−1 collagen solution at 4 °C for 6 to 8 h. The inserts were then rinsed with 1× PBS. 2 × 105 CNT-MSCs were suspended in 200 μL serum-free αMEM and added to the top chamber of each transwell inserts. The lower chamber was loaded with 600 μL αMEM containing 10% FBS and 200 ng mL−1 chemoattractant SDF-1α. CNT-MSCs were allowed to migrate for 24 h at 37 °C in a 5% CO2-balanced and humid culture chamber. The CNT-MSCs that migrated to the bottom of the insert were fixed with 4% paraformaldehyde at room temperature for 15 min. The cells were then stained with hematoxylin at room temperature for 20 min. Cells in the top chamber were removed with a cotton swab. Cells in lower chamber were counted manually by phase contrast microscopy.3. Results and discussion
3.1 FT-IR of functionalized CNTs
Fig. 1B shows the FT-IR spectra of pristine CNTs, CNT-COOH, and CNT-SA–FITC (note: the spectrum of CNT-OH was given in ref. 30). The FT-IR spectra of both pristine CNT and functionalized CNTs exhibited bending vibrations from 2000 to 2160 cm−1. This bending vibration was due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the cyclic carbons in the CNTs. In the spectrum of COOH-functionalized CNTs, the broad peak from 3000 to 3500 cm−1 was due to O–H stretching from the carboxyl group. The peak at 2930 cm−1 was from C–H stretching of the carboxyl group. The peak at 1740 cm−1 was due to the carboxylic CO stretching. The peak at 1540 cm−1 was probably due to CC stretching of the CNTs. The C–O stretching was observed at 1390 cm−1. These peaks confirmed the functionalization of COOH on CNTs. In the spectrum of CNTs functionalized with SA–FITC, the peaks at 1700 and 1570 cm−1 were related to the stretching vibration of CO and bending vibration of N–H caused by the formation of amide linkage via the EDC crosslinking of amines of SA with COOH of CNT. The peak at 1640 cm−1 was due to the unreacted amines on SA showing N–H bending vibration. It should be noted that the characteristic peaks of FITC were not clearly observed because they were merged with peaks of CNT and SA. For example, the NCS peak at 2100–2150 cm−1, O–H peak at 3000–3500 cm−1, and aromatic keto group around 1715 cm−1. However, the weak peaks at 1390–1450 cm−1 associated with the stretching vibration of the FITC aromatic ring were observed. Through the FT-IR analysis, the functionalization of SA–FITC on CNTs is confirmed.
C of the cyclic carbons in the CNTs. In the spectrum of COOH-functionalized CNTs, the broad peak from 3000 to 3500 cm−1 was due to O–H stretching from the carboxyl group. The peak at 2930 cm−1 was from C–H stretching of the carboxyl group. The peak at 1740 cm−1 was due to the carboxylic CO stretching. The peak at 1540 cm−1 was probably due to CC stretching of the CNTs. The C–O stretching was observed at 1390 cm−1. These peaks confirmed the functionalization of COOH on CNTs. In the spectrum of CNTs functionalized with SA–FITC, the peaks at 1700 and 1570 cm−1 were related to the stretching vibration of CO and bending vibration of N–H caused by the formation of amide linkage via the EDC crosslinking of amines of SA with COOH of CNT. The peak at 1640 cm−1 was due to the unreacted amines on SA showing N–H bending vibration. It should be noted that the characteristic peaks of FITC were not clearly observed because they were merged with peaks of CNT and SA. For example, the NCS peak at 2100–2150 cm−1, O–H peak at 3000–3500 cm−1, and aromatic keto group around 1715 cm−1. However, the weak peaks at 1390–1450 cm−1 associated with the stretching vibration of the FITC aromatic ring were observed. Through the FT-IR analysis, the functionalization of SA–FITC on CNTs is confirmed.
3.2 Surface biotinylation of MSCs
Instead of using conjugating chemicals (e.g., sulfo–NHS–LC–biotin) to biotinylated MSCs, an alternative mild approach was employed by having MSCs pre-fed with biotinylated lipid. MSCs were treated with culture medium containing 0.01 and 0.02 mg mL−1 biotin–lipid followed by SA–FITC treatment. It has been reported using 0.02 mg mL−1 biotin–lipid containing culture medium to biotinylate the surface of Vero cells.31 In that study, biotinylated Vero cells were able to proliferate without obvious cell toxicity. However, in the present study, treating human MSCs with 0.02 mg mL−1 biotin–lipid followed by SA–FITC caused an uneven distribution of SA–FITC on MSC cell surfaces as well as some cell morphology change (Fig. 2A). This is probably because MSCs are more sensitive to biotin–lipid than Vero cells and high concentration of biotin–lipid lead to uneven distribution of biotin on MSC surface. Decreasing the concentration of biotin–lipid to 0.01 mg mL−1 resulted in a more uniform distribution of biotin on the cell surfaces (Fig. 2B). In any case, 0.01 mg mL−1 biotin–lipid was chosen to biotinylate the surface of MSCs. Fig. 2C shows the MSC biotinylation standard curve where fluorescence intensity was plotted against time. From 0 to 24 h, the fluorescent intensity showed an initial burst increase, and the intensity then slowly increased from 24 to 60 h.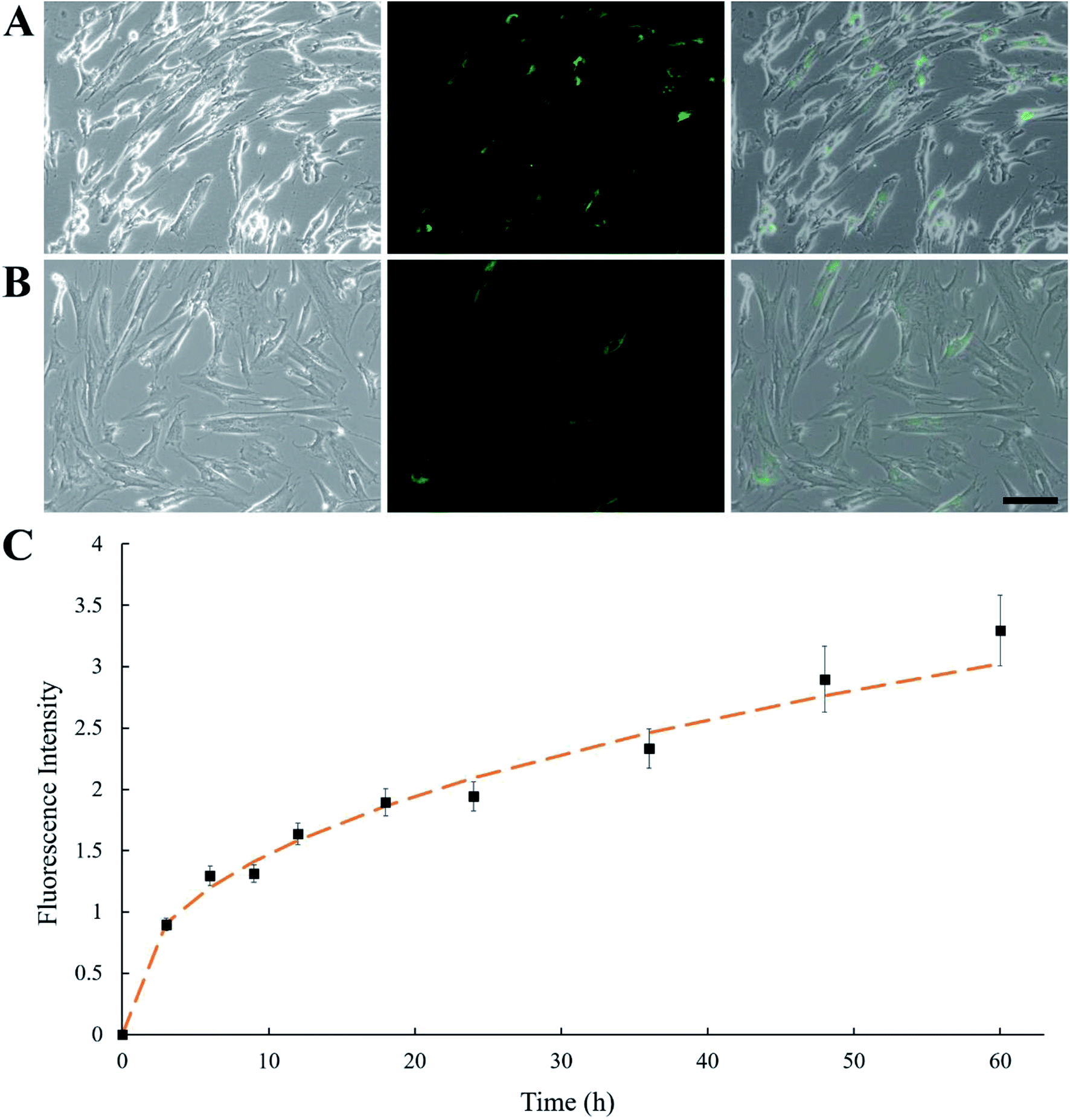 | ||
| Fig. 2 Phase contrast, fluorescence and overlay images of human MSCs treated with (A) 0.02 mg mL−1 biotin–lipid and (B) 0.01 mg mL−1 biotin–lipid for 48 h, followed by SA–FITC treatment. Scale bar denotes 100 μm. (C) MSCs were treated with 0.01 mg mL−1 biotin–lipid for different time, up to 60 h. MSCs were treated with SA–FITC, and fluorescence intensity was plotted against time. Error bars indicate standard deviation (n = 3). | ||
3.3 Verification of CNT-loaded MSCs
In Fig. 3A, fluorescence microscopy confirmed that CNTs were attached to MSCs. In order to minimize cytotoxicity and cellular interference (e.g., migration and proliferation), keeping CNTs external to MSCs could be a better option than being internalized. However, nanoparticles are prone to be endocytosed when located in the vicinity of cell membrane, hence CNTs might not anchor long enough on the surface of MSCs. To test whether CNTs were internalized by MSCs, fluorescence on CNT-MSCs were quenched by treating the cells with trypan blue solution. In Fig. 3B, fully quenching of FITC fluorescence indicated the CNTs were indeed located on the surfaces of the MSCs via the strong affinity of biotin–SA. The cells were imaged 12 and 24 h respectively after CNT anchored on MSC surface. As shown in Fig. 3C and D, the fluorescence intensity was extremely low to be detected indicating just very small amount of CNTs (if there was any) was internalized by MSCs and majority of CNTs remained tethered on the outer membrane of MSCs, and therefore their labeled FITC fluorescence was quenched by trypan blue. According to published accounts, internalization of CNTs depends on various factors such as size, aspect ratio and concentration of CNTs.32 Since a substantial amount of CNTs were still anchored on the outer cell membrane over 24 h (the time of used for the transwell migration study), it is conjectured that the reduced endocytosis of CNTs into MSCs was due to their large aspect ratios compared to spherical nanoparticles. Moreover, Chen et al. prepared glycopolymer-coated CNTs and anchored CNTs to Chinese hamster ovary cell surface via crosslinking agent Helix pomatia agglutinin (HPA).33 They found that at low CNT concentration in the bulk solution, CNTs efficiently bound cell surface; however, when the bulk concentration was high, cellular internalization of CNTs were observed. In the current study, extremely low amount of CNTs on cell surface was internalized by MSCs probably because the concentration of CNTs near the cell surface was low. Yao et al. have used sulfo–NHS–LC–biotin to chemically conjugate the surface of MSCs, and then treated MSCs with an avidin solution.28 The avidinylated MSCs were then treated with biotinylated DOX conjugates, and nanodrugs were observed on both cell surface and cytoplasm. In this study, the approach is through the incorporation of biotin–lipid into lipid membrane of MSCs which is much mild than the chemical conjugation method via N-hydroxysuccinimide. Moreover, the steps involved in anchoring nanomaterials are much less than the ones reported by Yao et al.28 | ||
| Fig. 3 MSCs were biotinylated by treating cells with 0.01 mg mL−1 biotin–lipid for 48 h. Biotinylated MSCs were then treated with CNT-SA–FITC for 1 h. Cells were washed with 1× PBS for 3 times. (A) Phase contrast (left) and fluorescent (right) images were taken after washing with PBS. (B–D) After anchoring of CNTs on MSCs, fluorescence was quenched by treating cells with 0.01 mg mL−1 trypan blue at different timing (0, 12 and 24 h). Scale bar denotes 100 μm. | ||
Cytotoxicity is one of the major concerns when nanoparticles such as AuNPs or CNTs were internalized by MSCs for 24 h or longer.34,35 A previous study showed that culture medium containing over 32 μg mL−1 CNTs causes toxicity to MSCs after 6 days of culture, while reducing concentration to 6.4 μg mL−1 showed lower toxicity effect.36 Another study showed that cellular uptake of AuNPs can cause a dose-dependent cytotoxicity to MSCs.37 MSCs treated with 500 μg mL−1 AuNPs could lead to decreased cell viability.29 In the present study, due to the rapid attachment of CNT-SA to biotinylated MSCs (less than 1 h), fewer CNTs had the chance of getting internalized by MSCs. The control MSCs and CNT-MSCs were seeded on 24-well plates at the seeding density of 3000 cells per cm2. After re-seeding, both control MSCs and CNT-MSCs were able to proliferate to confluency after an 8 day culture period (Fig. 4). The growth of CNT-MSCs were not affected compared to the control group. The amount of CNTs observed on cell surfaces decreased over the course of cultivation.
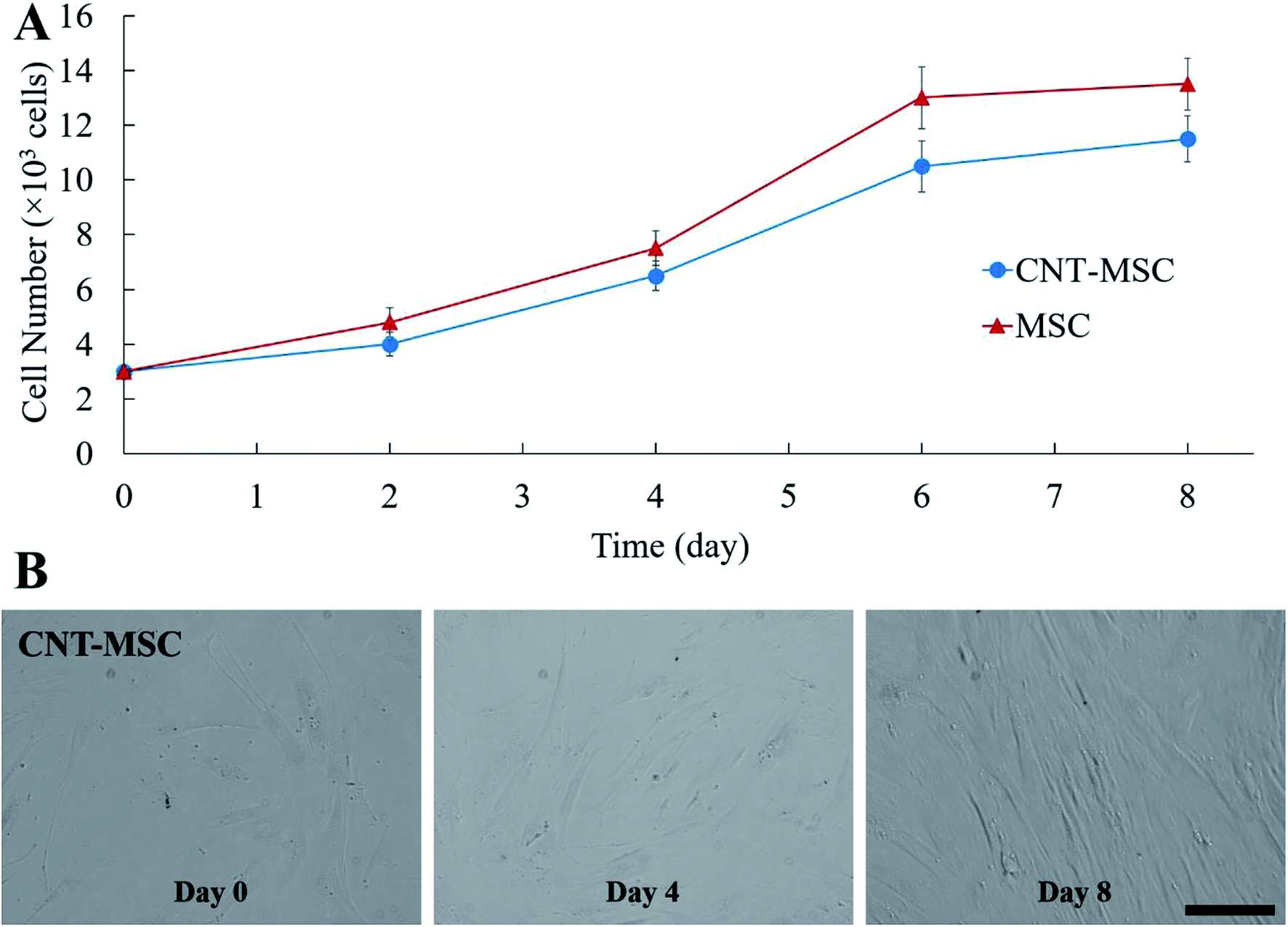 | ||
| Fig. 4 (A) Growth kinetics of MSCs from the control group and CNT-MSC seeded on 24-well plates at the seeding density of 3000 cells per cm2. (B) Phase contrast microscope images for CNT-MSCs at day 0, 4 and 8. Scale bar denotes 100 μm. Error bars indicate standard deviation (n = 3). | ||
3.4 Effect of CNTs on MSC migration toward chemoattractant SDF-1α
The weights of CNTs on MSCs was estimated by a microbalance. The amount of CNT attached on the MSC increased with duration of cell surface biotinylation up to 48 h. The average weight of individual MSC used in the control group was 17.65 ± 0.75 ng. When MSC were treated with biotin–lipid for 12, 24, 36, and 48 h, the estimated weight of CNT on MSCs was 2.47 ± 0.53, 4.26 ± 0.62, 7.71 ± 1.97 and 7.96 ± 2.29 ng per cell, respectively. The effect of CNT amount on the migration capacity of MSCs was evaluated by in vitro transwell migration assay. With respect to the transwell migration assay, our results demonstrated that SDF-1α induced migration of CNT-MSCs depends on the amount of CNTs on the MSC surfaces. The cell number of migrated control MSCs toward 200 ng mL−1 SDF-1α was 48.9 ± 9.5 cells per microscopic field. The higher rate of migration was observed with MSCs treated with 0.01 mg mL−1 biotin–lipid for 12 h and 24 h (Fig. 5A). The average observed cell number was 52.4 ± 10.6 and 46.2 ± 7.4 cells per field, respectively (Fig. 5B). When time of biotinylation was increased to 36 h or longer, the rate of migration toward the chemoattractant decreased significantly. The average observed cell number was 18.7 ± 5.5 and 16.3 ± 4.3 cells per field for 36 h and 48 h of biotin treatment time, respectively. It is speculated that the weight of CNTs on MSCs over the threshold of 4.26 ng per cell hindered the migration of the cells. Nold et al. reported that the migration capacity of MSCs was retained when MSCs were exposed to 10 nM AuNPs.38 However, when the concentration was increased to 25 nM or above, the migration capacity of MSCs significantly decreased. Therefore, finding an optimized nanoparticle concentration is crucial for MSCs to retain their proliferation and migration capacity. In the present study, prolonged biotinylation time resulted in more CNT anchoring on MSC surfaces. The migration ability of MSCs was significantly decreased with more than 4.26 ng CNTs per cell anchored on the surface of MSCs. The reason that MSCs could not retain their motility toward the established chemotactic gradient is speculated to be (1) CNT amount over the threshold that a cell could move with normal speed, and/or (2) CNTs loaded on cell surface increasing the friction force for cells to pass through the 8 μm membrane pores of transwell inserts.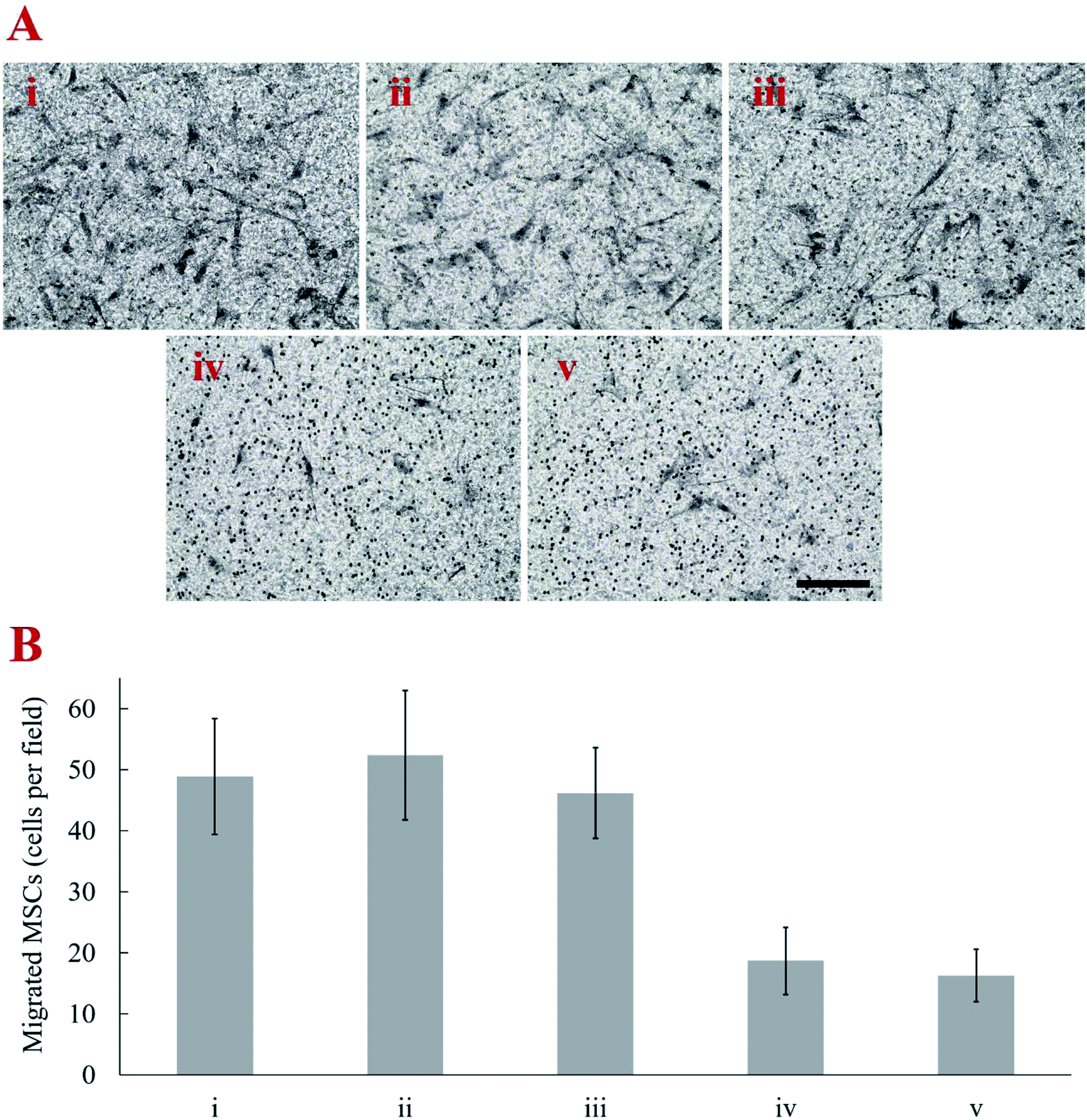 | ||
| Fig. 5 (A) Representative images of transwell migration assay of (i) control MSCs (ii–v) CNT-MSCs with 12, 24, 36, 48 h biotinylation time toward 200 ng mL−1 SDF-1α over 24 h exposure. Cell were fixed and stained with hematoxylin. Scale bar denotes 100 μm. (B) Transwell migration results showed a decreased number per microscopic field of MSCs migrating toward SDF-1α when MSCs were treated with biotin–lipid for 48 h. Error bars indicate standard deviation (n = 3). | ||
4. Conclusions
In this study, we demonstrated that CNT-hitchhiking MSC can be manufactured while retaining cell viability and their tumor-tropic property. When triggered by the chemotactic concentration gradient established by SDF-1α, MSCs could loaded up to ∼4.26 ng of CNT per cell without decreasing their migration capability. The MSCs tethered with CNTs reported here can be considered for future applications in tumor-targeted photothermal therapy.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We are grateful for financial support from the Idaho INBRE (NIH Grant No. P20 GM103408) and the help of Dustin Pierce and Xutu Wang for the cutting of CNTs and transwell migration assay.References
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- A. M. A. Elhissi, W. Ahmed, I. U. Hassan, V. R. Dhanak and A. D'Emanuele, J. Drug Delivery, 2012, 2012, 837327 Search PubMed.
- C. H. Wang, S. H. Chiou, C. P. Chou, Y. C. Chen, Y. J. Huang and C. A. Peng, Nanomedicine, 2011, 7, 69–79 CrossRef CAS PubMed.
- C. J. Gannon, P. Cherukuri, B. I. Yakobson, L. Cognet, J. S. Kanzius, C. Kittrell, R. B. Weisman, M. Pasquali, H. K. Schmidt, R. E. Smalley and S. A. Curley, Cancer, 2007, 110, 2654–2665 CrossRef CAS PubMed.
- F. Zhou, D. Xing, Z. Ou, B. Wu, D. E. Resasco and W. R. Chen, J. Biomed. Opt., 2009, 14, 021009 CrossRef PubMed.
- N. W. Kam, M. O'Connell, J. A. Wisdom and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11600–11605 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotech., 2007, 2, 751 CrossRef CAS PubMed.
- K. H. Son, J. H. Hong and J. W. Lee, Int. J. Nanomed., 2016, 11, 5163–5185 CrossRef CAS PubMed.
- J. Liu, A. G. Rinzler, H. Dai, J. H. Hafner, R. K. Bradley, P. J. Boul, A. Lu, T. Iverson, K. Shelimov, C. B. Huffman, F. Rodriguez-Macias, Y. S. Shon, T. R. Lee, D. T. Colbert and R. E. Smalley, Science, 1998, 280, 1253–1256 CrossRef CAS PubMed.
- T. Saito, K. Matsushige and K. Tanaka, Phys. B, 2002, 323, 280–283 CrossRef CAS.
- A. Bianco, K. Kostarelos, C. D. Partidos and M. Prato, Chem. Commun., 2005, 571–577 RSC.
- Z. Liu, C. Davis, W. Cai, L. He, X. Chen and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1410–1415 CrossRef CAS PubMed.
- J. Shi, R. Ma, L. Wang, J. Zhang, R. Liu, L. Li, Y. Liu, L. Hou, X. Yu, J. Gao and Z. Zhang, Int. J. Nanomed., 2013, 8, 2361–2373 CrossRef PubMed.
- X. Zhang, L. Meng, Q. Lu, Z. Fei and P. J. Dyson, Biomaterials, 2009, 30, 6041–6047 CrossRef CAS PubMed.
- H. Wu, G. Liu, X. Wang, J. Zhang, Y. Chen, J. Shi, H. Yang, H. Hu and S. Yang, Acta Biomater., 2011, 7, 3496–3504 CrossRef CAS PubMed.
- T. Peci, T. J. S. Dennis and M. Baxendale, Carbon, 2015, 87, 226–232 CrossRef CAS.
- M. Lee, S. Y. Jeong, J. Ha, M. Kim, H. J. Jin, S.-J. Kwon, J. W. Chang, S. J. Choi, W. Oh, Y. S. Yang, J.-S. Kim and H. B. Jeon, Biochem. Biophys. Res. Commun., 2014, 446, 983–989 CrossRef CAS PubMed.
- S. Kidd, E. Spaeth, J. L. Dembinski, M. Dietrich, K. Watson, A. Klopp, V. L. Battula, M. Weil, M. Andreeff and F. C. Marini, Stem Cells, 2009, 27, 2614–2623 CrossRef CAS PubMed.
- A. J. Friedenstein, R. K. Chailakhyan, N. V. Latsinik, A. F. Panasyuk and I. V. Keiliss-Borok, Transplantation, 1974, 17, 331–340 CrossRef CAS PubMed.
- J. S. Park, S. Suryaprakash, Y. H. Lao and K. W. Leong, Methods, 2015, 84, 3–16 CrossRef CAS PubMed.
- L. Kucerova, S. Skolekova, M. Matuskova, M. Bohac and Z. Kozovska, BMC Cancer, 2013, 13, 535 CrossRef PubMed.
- M. Roger, A. Clavreul, M. C. Venier-Julienne, C. Passirani, L. Sindji, P. Schiller, C. Montero-Menei and P. Menei, Biomaterials, 2010, 31, 8393–8401 CrossRef CAS PubMed.
- J. L. Santos, D. Pandita, J. Rodrigues, A. P. Pego, P. L. Granja and H. Tomas, Curr. Gene Ther., 2011, 11, 46–57 CrossRef CAS PubMed.
- S. Park, H. Jang, B. S. Kim, C. Hwang, G. S. Jeong and Y. Park, PLoS One, 2017, 12, e0184595 CrossRef PubMed.
- K. Shah, Adv. Drug Delivery Rev., 2012, 64, 739–748 CrossRef CAS PubMed.
- N. M. Moll and R. M. Ransohoff, Expert Rev. Hematol., 2010, 3, 315–322 CrossRef CAS PubMed.
- T. J. Myers, F. Granero-Molto, L. Longobardi, T. Li, Y. Yan and A. Spagnoli, Expert Opin. Biol. Ther., 2010, 10, 1663–1679 CrossRef CAS PubMed.
- S. Yao, X. Li, J. Liu, Y. Sun, Z. Wang and Y. Jiang, Drug Delivery, 2017, 24, 1372–1383 CrossRef CAS PubMed.
- S. Kang, S. H. Bhang, S. Hwang, J. K. Yoon, J. Song, H. K. Jang, S. Kim and B. S. Kim, ACS Nano, 2015, 9, 9678–9690 CrossRef CAS PubMed.
- E. J. Weydemeyer, A. J. Sawdon and C.-A. Peng, Chem. Commun., 2015, 51, 5939–5942 RSC.
- B.-H. Huang, Y. Lin, Z.-L. Zhang, F. Zhuan, A.-A. Liu, M. Xie, Z.-Q. Tian, Z. Zhang, H. Wang and D.-W. Pang, ACS Chem. Biol., 2012, 7, 683–688 CrossRef CAS PubMed.
- K. Maruyama, H. Haniu, N. Saito, Y. Matsuda, T. Tsukahara, S. Kobayashi, M. Tanaka, K. Aoki, S. Takanashi, M. Okamoto and H. Kato, BioMed Res. Int., 2015, 2015, 9 Search PubMed.
- X. Chen, U. C. Tam, J. L. Czlapinski, G. S. Lee, D. Rabuka, A. Zettl and C. R. Bertozzi, J. Am. Chem. Soc., 2006, 128, 6292–6293 CrossRef CAS PubMed.
- H. Ahmadi, M. Ramezani, R. Yazdian-Robati, B. Behnam, K. Razavi Azarkhiavi, A. Hashem Nia, A. Mokhtarzadeh, M. Matbou Riahi, B. M. Razavi and K. Abnous, Chem.-Biol. Interact., 2017, 275, 196–209 CrossRef CAS PubMed.
- L. A. Lara-Martínez, F. Massó, E. Palacios González, I. García-Peláez, A. Contreras–Ramos, M. Valverde, E. Rojas, F. Cervantes-Sodi and S. Hernández-Gutiérrez, Int. J. Nanomed., 2017, 12, 7695–7707 CrossRef PubMed.
- E. Mooney, P. Dockery, U. Greiser, M. Murphy and V. Barron, Nano Lett., 2008, 8, 2137–2143 CrossRef CAS PubMed.
- N. M. Schaeublin, L. K. Braydich-Stolle, A. M. Schrand, J. M. Miller, J. Hutchison, J. J. Schlager and S. M. Hussain, Nanoscale, 2011, 3, 410–420 RSC.
- P. Nold, R. Hartmann, N. Feliu, K. Kantner, M. Gamal, B. Pelaz, J. Hühn, X. Sun, P. Jungebluth, P. del Pino, H. Hackstein, P. Macchiarini, W. J. Parak and C. Brendel, J. Nanobiotechnol., 2017, 15, 24 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |