
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen and sulfur-codoped porous carbon derived from a BSA/ionic liquid polymer complex: multifunctional electrode materials for water splitting and supercapacitors†
Xiaojun Liub,
Junrui Yub,
Honghong Songb,
Pengfei Songb,
Rongming Wang b and
Yubing Xiong*ab
b and
Yubing Xiong*ab
aDepartment of Chemistry, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China. E-mail: yubing_xiong@163.com
bKey Laboratory of Eco-Environment-Related Polymer Materials, Ministry of Education, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, P. R. China
First published on 11th February 2019
Abstract
Bovine serum albumin (BSA) was complexed with a hydrophobic ionic liquid polymer (PIL) via electrostatic interaction to fabricate a carbon precursor. Then, a novel nitrogen (N) and sulfur (S) codoped micro-/mesoporous carbon (NSPC) was obtained via direct carbonization of the interpolyelectrolyte BSA@PIL complex. The newly developed NSPC materials exhibited excellent HER/OER electrocatalytic activity and stability, as well as outstanding capacitance performance. Remarkably, NSPC pyrolyzed at 1000 degrees (NSPC-1000) presented an overpotential as low as 172 mV vs. RHE (without iR correction) to achieve a current density of 10 mA cm−2 and a Tafel slope of 44.3 mV dec−1 in 0.5 M H2SO4 for HER, as well as a low overpotential of 460 mV vs. RHE in 0.1 M KOH for OER. Furthermore, NSPC-1000 offers a specific capacitance as high as 495 F g−1 at a current density of 0.1 A g−1. Such excellent performance of NSPC in electrocatalytic water splitting and supercapacitors originates from the synergistic effects of its N/S-codoping and micro-/mesoporous hierarchical architecture. Our facile protocol through combining biomacromolecules and synthetic polymers offers a new strategy in the development of effective, readily scalable and metal-free heteroatom-doped carbon materials for energy-related applications.
Introduction
Spurred by the urgent demand for green and renewable energy technologies, great efforts have been applied worldwide for the exploration of alternatives to traditional fossil fuels.1,2 Electrocatalytic water splitting to H2 and O2 via the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) affords an appealing solution for addressing the issues associated with global warming and the energy crisis.3,4 Hydrogen, as an abundant and renewable clean fuel, is an ideal candidate to replace fossil fuels. Simultaneously, producing O2 from water splitting is also related to a variety of energy storage systems.5–7 As benchmark electrocatalysts, platinum is sufficiently active in the HER and ruthenium/iridium oxides are highly efficient for OER. However, both of them suffer from the weaknesses of high cost and non-renewability.8–12 Therefore, highly efficient and robust earth-abundant electrocatalyst materials are urgently needed for achieving scalable and sustainable hydrogen fuel production by water splitting.13 In this context, a variety of non-noble metal catalysts, such as transition metal compounds including Mo, W, Co, Ni, and other metal ions, have been designed as promising candidates for water splitting.14–25 However, in light of sustainable hydrogen production and environmental benignity, it is highly desired to develop efficient and scalable metal-free electrocatalysts.26Heteroatom-doped carbon materials have been widely investigated for energy-conversion/storage due to their large specific surface area, tunable electronic structure, abundance and strong tolerance in acidic/alkaline environments.27–30 Nevertheless, solely heteroatom-doped carbon materials showed very poor activity in water splitting.13,31 Recently, some advances demonstrated that multi-heteroatom (such as N, S, P, and F) codoped carbon materials offer acceptable electrocatalytic activity for water splitting by tuning the Gibbs free energy of H* absorption.13,32–36 Up to date, most of the developed metal-free carbon electrocatalysts are based on the low dimension carbon materials, such as graphene, carbon nanotubes (CNT), and C3N4 composites, etc.13,31,37,38 In this regard, synthetic polymers are more suitable for fabrication of multi-heteroatom-codoped carbon materials due to their versatile structures and large-scale preparation. As a result, ionic liquid polymers (PIL) have been explored as the precursors for the preparation of heteroatom-doped porous carbon, stemming from their high mass yield, designable structure and variable multi-heteroatoms codoping.39–42
Herein, we reported a facile strategy to fabricate novel N/S co-doped micro-/mesoporous carbon (NSPC) material that was achieved by directly carbonization of bovine serum albumin (BSA) and PIL complex (BSA@PIL). The results demonstrated that the resulting NSPC materials have efficient overall water splitting electrocatalytic activity and excellent supercapacitor performance. Specifically, NSPC pyrolyzed at 1000 degrees (NSPC-1000) presented an overpotential as low as 172 mV vs. RHE (without iR correction) to achieve a current density of 10 mA cm−2 with a small Tafel slope of 44.3 mV dec−1 in 0.5 M H2SO4 for HER and a low overpotential of 460 mV vs. RHE in 0.1 M KOH for OER. Moreover, beyond applications in water splitting, NSPC-1000 is also a promising candidate for supercapacitor, offering a specific capacitance as high as 495 F g−1 at a current density of 0.1 A g−1. Such excellent performances of NSPC-1000 in water splitting and supercapacitor originate from the synergistic effects of N/S-codoping and micro-/mesoporous hierarchical architecture.
Experimental
Synthesis of 1-vinyl-3-cyanomethylimidazolium bromine (CMVImBr)
1-Vinyl-3-cyanomethylimidazolium bromine (CMVImBr) was synthesized via quaternization of N-vinylimidazole with bromoacetonitrile.40 In a typical procedure, a 50 mL flask, equipped with a magnetic stirring bar, was fed with methanol (2 mL), N-vinylimidazole (9.411 g, 0.1 mol) and bromoacetonitrile (11.995 g, 0.1 mol). The mixture was stirred at ambient temperature to give a white solid. Then, the white solid was dissolved in methanol and the reaction mixture was purified by precipitating three times in ether. Finally, white crystals was obtained and dried at room temperature under vacuum to a constant weight. Yield: 70%. 1H NMR (hexadeuterodimethyl sulfoxide (DMSO-d6), 400 MHz, δ ppm, Fig. S1†): 9.58(s, 1H), 8.26(t, 1H), 8.01(t, 1H), 7.36(dd, 1H), 5.99(dd, 1H), 5.63(s, 2H), 5.46(dd, 1H). 13C-NMR (DMSO-d6, 100 MHz, Fig. S2†), δ (ppm): 137.64, 129.40, 123.92, 120.16, 114.99, 110.21, 37.81.Synthesis of poly(1-vinyl-3-cyanomethylimidazolium bromine) (PIL-Br)
Radical polymerization of CMVImBr was performed in dimethyl sulfoxide using 2,2-azobisisobutyronitrile (AIBN) as an initiator.40 In a typical procedure, a 250 mL round-bottomed flask equipped with a reflux condenser, magnetic stirring bar, and thermometer was charged with 10.0 g of CMVImBr, 200 mg of AIBN, and 200 mL of DMSO. The flask was flushed with nitrogen after three freeze–thaw cycles. The mixture was heated at 75 °C for 24 h. After cooling down, the reaction mixture was purified by precipitating twice from THF. A light yellow solid was obtained and dried at 65 °C under vacuum to a constant weight. Yield: 65%. 1H-NMR (DMSO-d6, δ ppm, Fig. S3†): 8.9–9.6 (1H), 7.0–8.4 (2H), 5.2–5.7 (2H), 3.5–4.6 (1H), 1.9–2.7 (2H).Preparation of poly(3-cyanomethyl-1-vinyl imidazolium bis(trifluoromethanesulfonyl)imide) (PIL-Tf2N)
PCMVImTf2N was prepared through the anion exchange of PIL-Br in water.41 Typically, PIL-Br (3.0 g) was added to 390 mL of water under gentle heating. After complete dissolution and cooling down, 120 mL of an aqueous solution containing 1.05 molar equivalents of lithium bis(trifluoromethanesulfonyl)imide salt was added under vigorous stirring. The precipitate was filtered and washed with water several times. The product was dried at 50 °C to constant weight under high vacuum. Yield: 50%.Fabrication of the BSA@PIL-Tf2N complex and NSPC materials
BSA@PIL-Tf2N complex was fabricated according to the reported methods.42 PIL-Tf2N and BSA (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, w/w) were fully dissolved in N,N-dimethylformamide (DMF) and water mixed solvent (5:2, v/v). The as-formed homogeneous solution was then cast onto a pre-cleaned glass slide and dried at 75 °C for 3 h. Then, the complex was immersed into an ammonia aqueous solution (0.5 wt%). The free COOH groups on BSA were immediately deprotonated by excessive NH3 into carboxylate anions (COO−NH4+), triggering the spontaneous ionic cross-linking between BSA and PIL-Tf2N. This material is termed as BSA@PIL.
:1, w/w) were fully dissolved in N,N-dimethylformamide (DMF) and water mixed solvent (5:2, v/v). The as-formed homogeneous solution was then cast onto a pre-cleaned glass slide and dried at 75 °C for 3 h. Then, the complex was immersed into an ammonia aqueous solution (0.5 wt%). The free COOH groups on BSA were immediately deprotonated by excessive NH3 into carboxylate anions (COO−NH4+), triggering the spontaneous ionic cross-linking between BSA and PIL-Tf2N. This material is termed as BSA@PIL.
For carbonization, the as-prepared BSA@PIL-Tf2N complex was loaded into an alumina crucible in an oven and carbonized under argon flow. The sample was heated up to 800–1000 °C (10 °C min−1) and kept for 1 h at this temperature. Then, the furnace was cooled down to room temperature. The samples are defined as NSPC-800, NSPC-900 and NSPC-1000 in accordance with the carbonization temperatures.
Characterizations
Proton and carbon nuclear magnetic resonance spectroscopy (1H and 13C NMR) were recorded using a Brucker AM 400 MHz spectrometer at 25 °C. Gel permeation chromatography (GPC) was conducted at 35 °C in a Waters-column with DMF as the eluent (Flow rate: 1.00 mL min−1, PEO standards using an RI detector). A field emission scanning electron microscope (FESEM, FEI Quanta 600 FEG) was used to acquire SEM images. Transmission electron microscope (TEM), high-resolution TEM (HRTEM) images, selected-area electron diffraction (SAED) patterns, and the HAADF-STEM-EDS data were taken using a Tecnai G20 transmission electron microscope operated at 200 kV. X-ray diffraction (XRD) patterns were collected on a Philips X'Pert Pro diffractometer using Cu Kα (λ = 1.541 Å) radiation source. X-ray photoelectron spectroscopy (XPS) data were collected using a PerkinElmer PHI-5702 multifunctional X-ray photoelectron spectroscope (Physical Electronics, Waltham, Massachusetts) with Al Kα radiation (1486.6 eV) as the excitation source. The adventitious carbon 1s peak was calibrated at 285 eV and used as an internal standard to compensate for any charging effects. Raman measurements were performed using a Horiba JY-HR800 with the excitation wavelength of 532 nm and laser power of 100 mW at room temperature. Elemental analysis (EA) was conducted on Vario EL analyzer (Elementar, Germany). Nitrogen adsorption–desorption isotherm measurements were conducted using a Micromeritics ASAP 2020 volumetric adsorption analyzer at 77 K. The Brunauer–Emmett–Teller method was utilized to calculate the surface area of each sample. Pore size distributions were determined from the adsorption branches of the isotherms based on density functional theory.Electrochemical measurements
Electrochemical measurements were conducted using a conventional three-electrode electrochemical cell at room temperature on a CHI 660e station (Chenhua, Shanghai, China). A glassy carbon electrode (GCE) with a diameter of 3 mm was used as the working electrode. The counter electrode and reference electrode were a platinum wire electrode and a saturated calomel electrode (SCE, 0.24 V vs. a reversible hydrogen electrode (RHE)), respectively. The working electrodes were fabricated similar to the reported method.43 A catalyst ink was prepared using one part (by mass) of catalysts and 11 parts of 0.5 wt% Nafion in ethanol. After sonicating thoroughly, 10 μL of the ink was dispensed onto GCE to completely cover the current collector with a thin film of catalyst. The catalyst loading was 0.47 mg cm−2 on the working electrode. Linear sweep voltammetry (LSV) measurements were performed from 0.1 V to −0.7 V with a scan rate of 5 mV s−1 in 0.5 M H2SO4 solution for HER, −0.95 V to −1.45 V with a scan rate of 2 mV s−1 in 1.0 M KOH solution for HER, and 0 V to 0.8 V with a scan rate of 2 mV s−1 in 0.1 M KOH solution for OER. To fabricate CC-supported catalyst, carbon cloth (0.8 × 0.5 cm) was utilized as the current collector. Typically, NSPC-1000 (10 mg) and Nafion ethanol dispersion solution (1 mL, 0.1 wt%) were mixed thoroughly by ultrasonicating for 20 min. Then, the uniform suspension was dropped on CC and left to dry at room temperature (this yielded a loading of 0.25 mg cm−2 on CC). All applied potentials, measured against an SCE electrode, are reported versus the reversible hydrogen electrode (RHE) according to E(vs. RHE) = E(vs. SCE) + 0.24 + 0.059 × pH without iR correction. To compare the influence of the component of catalysts to HER and OER activities, the electrocatalytic activities of Pt/C and the control were measured under similar conditions, and the corresponding electrocatalytic results are included in the main text. Electrochemical impedance spectroscopy (EIS) was performed over the frequency range from 100 kHz to 0.1 Hz under the potentiostatic mode at the cell voltage close to the overpotentials of 175 mV, 309 mV, and 417 mV (vs. RHE) corresponding to NSPC-1000, NSPC-900 and NSPC-800, respectively. The amperometric i–t curve and open circuit potential (time curve) were measured with the voltage corresponding to their overpotential. Cyclic voltammetry (CV) was measured between 0–1.0 V at scan rates of 20, 30, 50, 100, 200, 300, and 500 mV s−1 in 0.5 M H2SO4 solution. The galvanostatic charge/discharge (GCD) property was characterized at the current densities of 0.1, 0.2, 0.3, 0.5, 1, 2, 3, and 5 A g−1 with a cutoff voltage of 0–0.8 V.Results and discussion
The structures of the monomers and polymer were characterized using 1H NMR (Fig. S1–S3 in the ESI†). The results indicated that PIL-Br was prepared successfully. GPC measurement (Fig. S4†) demonstrated that the apparent number-average molecular weight and polydispersity index of PIL-Br was 1.06 × 106 g mol−1 and 1.36, respectively. Therefore, the apparent number-average molecular weight of PIL-Tf2N is calculated to be 1.3 × 106 g mol−1. The following three steps are involved in the synthesis of NSPC: (i) BSA@PIL-Tf2N complex was prepared by blending BSA and PIL-Tf2N with a ratio of 1:7 (w/w) in N,N-dimethyl dimethylformamide (DMF)/water (5:2, v/v) mixed solvent. (ii) The as-prepared BSA@PIL-Tf2N composite was further treated by immersing into ammonia aqueous (0.5 wt%) for 2 h to form interpolyelectrolyte complex. (iii) Direct pyrolysis of BSA@PIL-Tf2N composite at different temperatures under an argon flow yielded NSPCs. These samples are denoted as NSPC-800, NSPC-900 and NSPC-1000, labeled with different carbonization temperatures (Scheme 1).
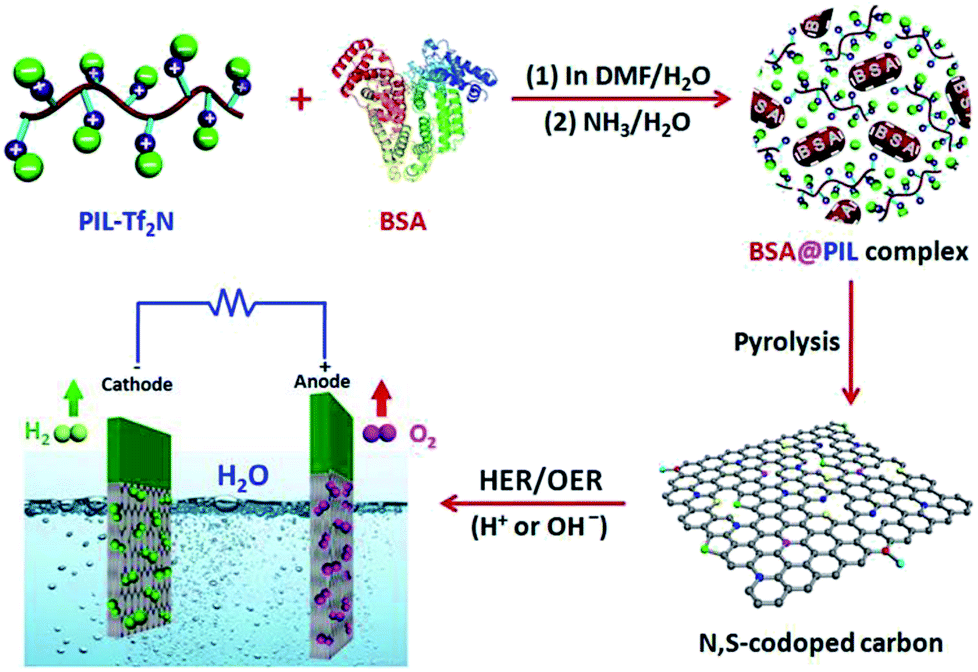 | ||
| Scheme 1 Schematic synthesis of NSPC and its application in electrocatalytic water splitting via HER and OER. | ||
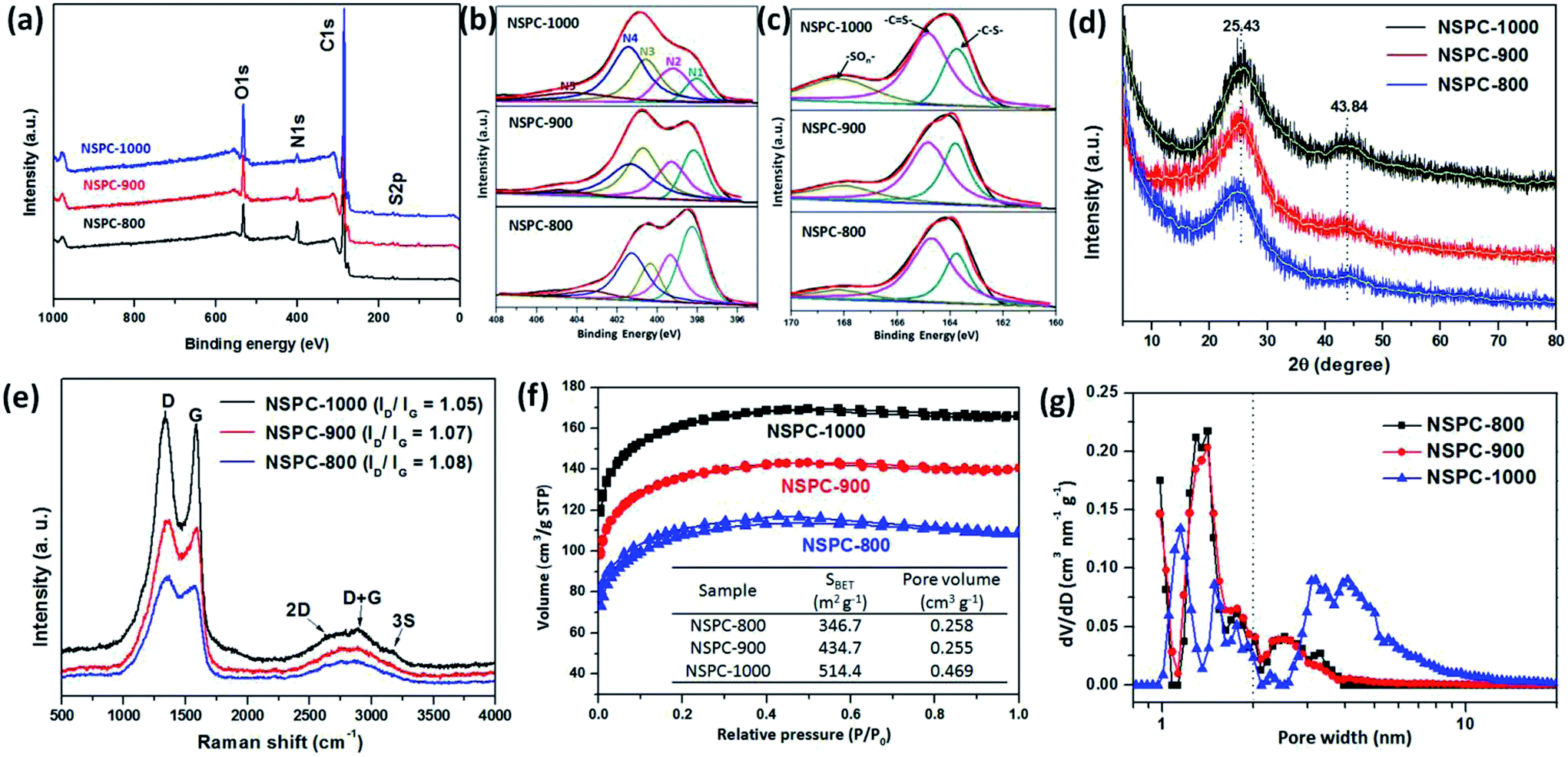
Fig. 1a–c shows the scanning electron microscopy (SEM) images of as-prepared samples. As demonstrated in Fig. 1a and b, spherical or irregular particles were produced when BSA or PIL-Tf2N were pyrolyzed separately at 1000 °C (the samples are denoted as BSA-1000 and PIL-Tf2N-1000, respectively). However, the particles with rough surfaces can be observed in NSPC-1000 (Fig. 1c). The elemental compositions of NSPC materials were examined using energy dispersive X-ray (EDX) and elemental analysis (EA). As revealed in Fig. 1d and S5,† NSPCs mainly consist of C, O, N and S elements. The populations of both N and S decreased with enhancing the carbonization temperature from 800 to 1000 °C (Table S1† and the inset in Fig. 1d). It should be stated that 3.58% of N and 2.01% of S still existed in NSPC-1000. These values are comparable to some well-defined N, S-codoped porous carbon.34,44
 | ||
| Fig. 1 Morphologies and the elemental compositions of the carbon materials. (a) BSA-1000; (b) PIL-Tf2N-1000; (c) NSPC-1000; (d) energy dispersive X-ray (EDX) spectrum of NSPC-1000. Inset table summarizes the corresponding N and S contents in NSPC-800/900/1000 by EA; (e–j) SEM elemental mapping images of C, N, O and S within NSPC-1000; TEM images of (k): NSPC-800; (l): NSPC-900; (m): NSPC-1000. Inset SAED pattern indicates the poorly crystallized structure of NSPC-1000. | ||
Energy-filtered scanning electron microscopy mappings for C, N and S (Fig. 1e–j) show a uniform distribution of N and S in the carbon matrix, which is expected due to in situ molecular doping of NSPC with N and S.42
Transmission electron microscopy (TEM) images of NSPC materials demonstrate that all of NSPCs are porous structure (Fig. 1k–m), which will be favorable for the internal mass transport, a mandatory requirement for efficient electrocatalysis.45 A selected area electron diffraction (SAED) image (inset in Fig. 1m) suggests that NSPC-1000 is poorly crystallized, which can be partially attributed to the high concentration of heteroatoms. Despite the poorly crystallized characteristic of NSPCs, some ordered concentric nanostructures can still be observed (Fig. S6–S8†). It can be seen that more nanostructures can be observed in NSPC-1000, indicating progressive graphitization of NSPC at elevated temperatures from 800 to 1000 °C. The synergy between the N and S lone pairs and the π-system of the C lattice can dramatically modulate the physicochemical properties of porous carbon materials (e.g., oxidative stability and catalytic activity).46
In accordance with the EDX and EA results, the X-ray photoelectron spectroscopy (XPS) spectra also confirm that there exist primarily C, O, N and S elements in the NSPCs (Fig. 2a). As illustrated in Fig. 2b, the fitted XPS peaks for the N1s orbit of NSPCs can be deconvoluted into five different bands at around 398.2, 399.8, 400.5, 401.5, and 404.6 eV, corresponding to pyridinic (N1), pyrrolic (N2), graphitic (N3), oxidized pyridinic (N4) and chemisorbed oxidized nitrogen (N5), respectively. These various N species lead to different chemical/electronic environments of the neighboring carbon atoms and hence different electrocatalytic activities.42 The curve fitting and the corresponding normalized results indicate that pyridinic N gradually turned into graphitic N with increasing carbonization temperature. Additionally, the XPS S2p spectrum can be deconvoluted into two predominate peaks at 163.7 eV (–C–S–) and 164.8 eV (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S–) (Fig. 2c), suggesting the successful doping of S atoms into the carbon skeleton in the carbonization process.34
S–) (Fig. 2c), suggesting the successful doping of S atoms into the carbon skeleton in the carbonization process.34
 | ||
| Fig. 2 (a) XPS spectra of NSPC-800/900/1000; fitted XPS peaks for N1s (b) and S2p (c) (N1: pyridinic, N2: pyrrolic, N3: graphitic, N4: oxidized pyridinic, N5: chemisorbed oxidized nitrogen); XRD patterns (d) and Raman spectra (e) of NSPC-800/900/1000; N2 absorption–desorption isotherms (f) and corresponding pore size distribution (PSD) curves (g) of NSPC-800/900/1000; the inset table presents the data for the specific surface area (SBET) and pore volume of NSPC-800/900/1000 based on N2 absorption–desorption isotherm curves. | ||
X-ray diffraction (XRD) spectra of NSPC materials (Fig. 2d) shows that all of as-prepared samples have two broad peaks located at 2θ = 25° and 44°. They are attributed to the graphite (002) and (101) facets of carbon, respectively.47 The intensity of the peaks enhanced with increasing carbonization temperature, indicating the improved graphitization degree of NSPC at the elevated temperature. The Raman spectra shows the D band (1350 cm−1), G band (1575 cm−1), 2D band (2685 cm−1), D + G band (2902 cm−1), and 3S band (3153 cm−1) of NSPC samples (Fig. 2e). The G-band is associated with the E-vibration mode of the sp2 electron structure, corresponding to the aromatic ring structure carbon of the graphite layer, while the D-band is the A-vibrational mode of the diamond-like carbon sp3 electronic structure, corresponding to the defective/disordered sp3 hybridized carbon.48 As shown in Fig. 2e, the ID/IG relative ratio decreases a little as the temperature increases from 800 to 1000 °C, confirming the better graphitic nature of the N, S-codoped carbon skeleton at higher temperature.49
Furthermore, the behaviors of nitrogen adsorption–desorption isotherms and the pore size distribution of NSPC materials are shown in Fig. 2f and g, respectively. It can be seen that all NSPC materials exhibit a type I adsorption curve and H2-type hysteresis loop, which clearly confirms that NSPCs are hierarchical in nature, traversing the micro- to mesopore range NSPC-1000 presents a Brunauer–Emmett–Teller (BET) surface area of 514.4 m2 g−1 and a total pore volume of 0.469 cm3 g−1, contributed by a substantial quantity of tiny mesopores around 2.5–8.0 nm and micropores around 1.5 nm. The meso-/micropores are probably induced by the leaving small molecules, including H2O, CO2, NH3, H2S, and CS2, during the carbonization and decomposition process of BSA@PIL-Tf2N complexes.50 This also accounts for the observed pyrolysis temperature-dependent BET surface areas of different samples. Based on the results discussed above, our synthetic protocol enables a facile fabrication of N- and S-enriched and hierarchical micro-/mesoporous carbon material. It should be noted that our protocol avoids the postsynthetic treatment and simultaneously retains a large surface area in conjunction with abundant dopant species.
To evaluate the electrocatalytic performance of NSPCs for HER, linear sweep voltammetry (LSV) curves were firstly recorded in a conventional three-electrode electrochemical cell in N2-saturated 0.5 M H2SO4 solution. The electrocatalysts were cast on a glass carbon electrode (GCE) as the working electrode. As shown in Fig. 3a, NSPC-1000 demonstrates an unprecedentedly low overpotential of 172 mV (vs. RHE, without iR correction) to achieve a current density of 10 mA cm−2 and a small Tafel slope of 44.3 mV dec−1 (Fig. 3c), which are much lower than that of NSPC-800 (416 mV and 108.9 mV dec−1) and NSPC-900 (309 mV and 65.7 mV dec−1). At the same time, a large number of bubbles produced on the NSPC-1000-loaded working electrode (Fig. S9†). As a controlled experiment, PIL-Tf2N-1000 exhibits the lowest activity among all of these electrocatalysts, while BSA-1000 offers the activity closed to NSPC-900. Furthermore, when BSA-1000 was mixed with PIL-Tf2N-1000 (1:7, w/w), the mixture offers a much higher activity than that of PIL-Tf2N-1000, and very close to that of BSA-1000. The results demonstrate that the presence of BSA-1000 play a significant role in achieving higher HER activity.
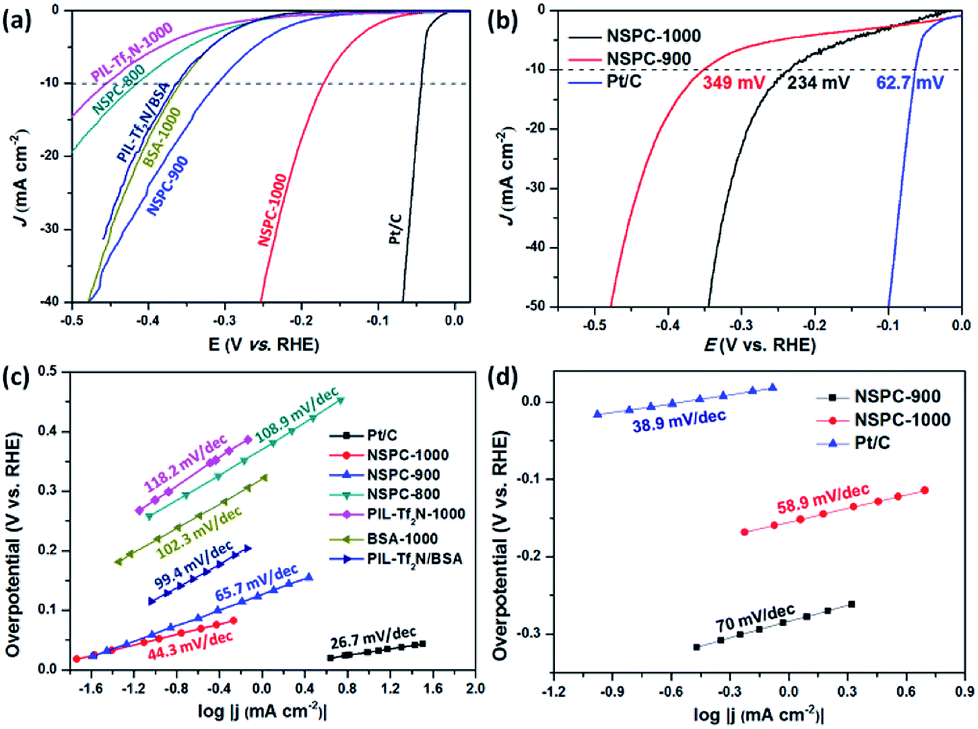 | ||
| Fig. 3 Electrocatalytic performances of different catalysts in acidic and alkaline conditions. (a) LSV curves of NSPCs in 0.5 M H2SO4 with a sweep rate of 5 mV s−1, where PIL-Tf2N/BSA was prepared by mixing PIL-Tf2N-1000 and BSA-1000 (7:1, w/w). (b) LSV curves of NSPC-900, NSPC-1000, and Pt/C in 1.0 M KOH with a sweep rate of 2 mV s−1 (c) and (d) Tafel plots for the data presented in (a) and (b), respectively. All catalysts were loaded on the GCE for HER tests. | ||
Generally, HER in acidic electrolyte on the electrode surfaces proceeds via a three-step reaction, which includes a primary discharge step (Volmer reaction, with a Tafel slope of 120 mV dec−1) followed by an electrochemical desorption step (Heyrovsky reaction, with a Tafel slope of 40 mV dec−1) or a recombination step of adsorbed hydrogen on catalysts (Tafel reaction, with a Tafel slope of 30 mV dec−1).51–53 Typically, a lower Tafel slope predicates a greatly enhanced HER rate at a moderate increase of overpotential. Therefore, the Tafel slope of 44.3 mV dec−1 for NSPC-1000 indicates that the corresponding rate-determining step most probably involved a combination of both electroreduction of protons into H* and the subsequent formation of 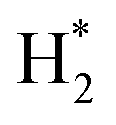 , that is, the Volmer–Tafel reactions.54 In addition, S and N codoping into sp2 hybridized carbon can produce a unique electronic structures, which will enhance the chemisorptions of intermediate H* for HER. As a result, a much lower overpotential and smaller Tafel slope could be produced by NSPC-1000.
, that is, the Volmer–Tafel reactions.54 In addition, S and N codoping into sp2 hybridized carbon can produce a unique electronic structures, which will enhance the chemisorptions of intermediate H* for HER. As a result, a much lower overpotential and smaller Tafel slope could be produced by NSPC-1000.
Furthermore, NSPC materials also exhibit efficient electrocatalytic activity in alkaline conditions. NSPC-1000 and NSPC-900 show the overpotential of 234 mV and 349 mV at the current density of 10 mA cm−2, respectively (Fig. 3b). Tafel plots shown in Fig. 3c and d show a small Tafel slope of NSPC-1000, demonstrating more favorable kinetics toward HER on the NSPC-1000 electrode. All of these performances presented by NSPC-1000 are comparable to those of many state-of-the-art metal-free and non-noble metal electrocatalysts (Table S2†), as well as some biomass-derived metal-free carbon materials (Table S3†).
LSV curves were also conducted to evaluate the electrocatalytic activity of NSPC for OER in 0.1 M KOH solution. As indicated in Fig. 4a, only NSPC-1000 shows efficient electrocatalytic performance under the above conditions. Specifically, NSPC-1000 shows an overpotential of 460 mV at the current density of 10 mA cm−2 and the Tafel slope of 88 mV dec−1. This value is comparable to those of some well-defined metal-free electrocatalysts (Table S4†). Moreover, continuous HER and OER at a given potential were performed to investigate the durability of the NSPC-1000 electrocatalyst. As shown in Fig. 4b, the current density is stable without an obvious reduction after long-term cycling for over 3.5 h. The robust stability of NSPC-1000 provides great potential for sustainable hydrogen and oxygen production in practice.
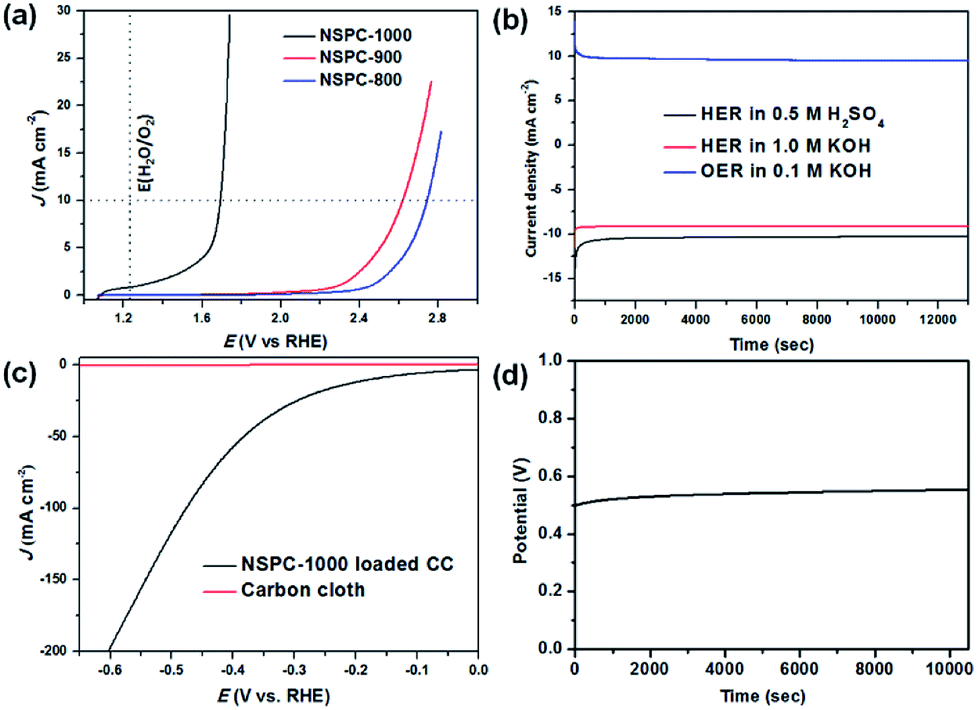 | ||
| Fig. 4 Electrocatalytic performances of NSPC materials. (a) LSV curves of NSPC-800/900/1000, carbon cloth (CC) and CC-loaded NSPC-1000 in 0.1 M KOH (for OER tests); (b) long-term stability test of NSPC-1000 catalyst for HER (−0.45 V in 0.5 M H2SO4, −1.38 V in 1.0 M KOH) and OER (0.8 V in 0.1 M KOH); (c) LSV curve of water electrolysis using NSPC-1000 as HER and OER catalysts (both loaded into CC at a loading of 0.25 mg cm−2 based on the active mass) in 0.5 M H2SO4; (d) the stability test of the electrolyzer at 10 mA cm−2. | ||
To gain insight into the electrochemical behavior under HER operating conditions, electrochemical impedance spectroscopy (EIS) measurements were conducted for frequencies from 100 MHz to 0.01 Hz at a constant overpotential. From the Nyquist plot, the charge-transfer resistance (Rct) and solution resistance (Rs) can be deduced from the semicircles in the low and high frequency zones, respectively. Rs is used to correct the polarization curves, and Rct is related to the electrocatalytic kinetics.32,55 The result depicted in Fig. S10† reveals a small internal transmission resistance of the NSPCs. The small Rct enables simple and effective electrical integration that minimized parasitic ohmic losses and enhanced the reaction rate.32 Using the Rs obtained from EIS, the iR-corrected polarization curves of the catalysts are shown in Fig. S11.† In the iR-corrected data, NSPC-1000 exhibits a current density of 10 mA cm−2 at the lower overpotential of 165 mV in 0.5 M H2SO4 solution. Therefore, the high catalytic activity of NSPC-1000 resulted from the synergy between its N and S codoping, micro/mesoporous and graphitic architecture.
Since NSPC-1000 has been shown to be an efficient and stable bifunctional electrocatalyst for both HER and OER, we loaded NSPC-1000 into a commercially available carbon cloth (CC) and used it as both the anode and the cathode for electrochemical water splitting in 0.5 M H2SO4 solution. As demonstrated in Fig. 4c, the current density increases gradually with increase of potential, and the current density of 10 mA cm−2 can be achieved at 175 mV, which is very close to that by NSPC-1000 loaded on GCE. As demonstrated in Fig. S12 and Video S1†, a large number of bubbles were produced from both of the electrodes under the experimental potential, indicating that NSPC-1000 is an efficient electrocatalyst for water splitting. The long-term stability of NSPC-1000-loaded CC electrodes was also evaluated, and the duration of the electrolyzer was conducted at 10 mA cm−2 in 0.5 M H2SO4 solution at room temperature. We can see that the cell shows excellent stability at nearly 3 h (Fig. 4d). Consequently, NSPC materials derived from biomacromolecules and PIL complex, as a novel and versatile metal-free highly efficient electrocatalyst, pave a new pathway for designing water splitting catalysts and show substantial potential for use in renewable energy applications.
In addition to the promising electrocatalytic activity for HER and OER, heteroatom-doped carbonaceous materials have also been demonstrated to be of great potential in supercapacitor. As a proof-of-concept demonstration of the potential application of NSPC in supercapacitor, a symmetric supercapacitor was fabricated using NSPCs as the electrode material. First, cyclic voltammetric (CV) scanning of different NSPCs were conducted in 0.5 M H2SO4 solution between 0 and 1.0 V, with the obtained curves shown in Fig. S13.† The results show that all of these CV curves exhibit rectangular-like shapes (Fig. S13a and b†), indicating the ideal capacitive behavior and good charge propagation within the electrodes.56 Under identical conditions, the NSPC-1000 curve provides the largest encircled area among these electrode materials, suggesting that NSPC-1000 shows better capacitance behavior. The CV curves measured with scan rates ranging from 20 to 500 mV s−1 (Fig. S13c†) also exhibit a rectangular-like shape, and the encircled area shrinks simultaneously. Additionally, when the potential window is negatively shifted and still kept within 1.0 V, the curves become increasingly sickle-like, similar to the behavior of the metal catalysts (Fig. S13d†).57 Meanwhile, the working electrode loaded with NSPC-1000 began to bubble during the test, and the bubbling is accelerated with the negative voltage shifting. This phenomenon also demonstrates that NSPC-1000 is an excellent electrocatalyst for water splitting.
Then, galvanostatic charge/discharge (GCD) measurements were conducted to evaluate the capacitance performance of the above materials at 0.1 A g−1 in a stable voltage window between 0 and 0.8 V. As illustrated in Fig. 5a and b, the linear voltage–time profiles and the triangular shape of the charge/discharge plots without an obvious iR drop illustrate the small mass transfer resistance and good charge propagation behavior in the electrode materials. The specific capacitance (Cs) can be calculated from the discharge curves according to the following equation:58
| Cs = C/m = (IΔt)/(ΔVm) |
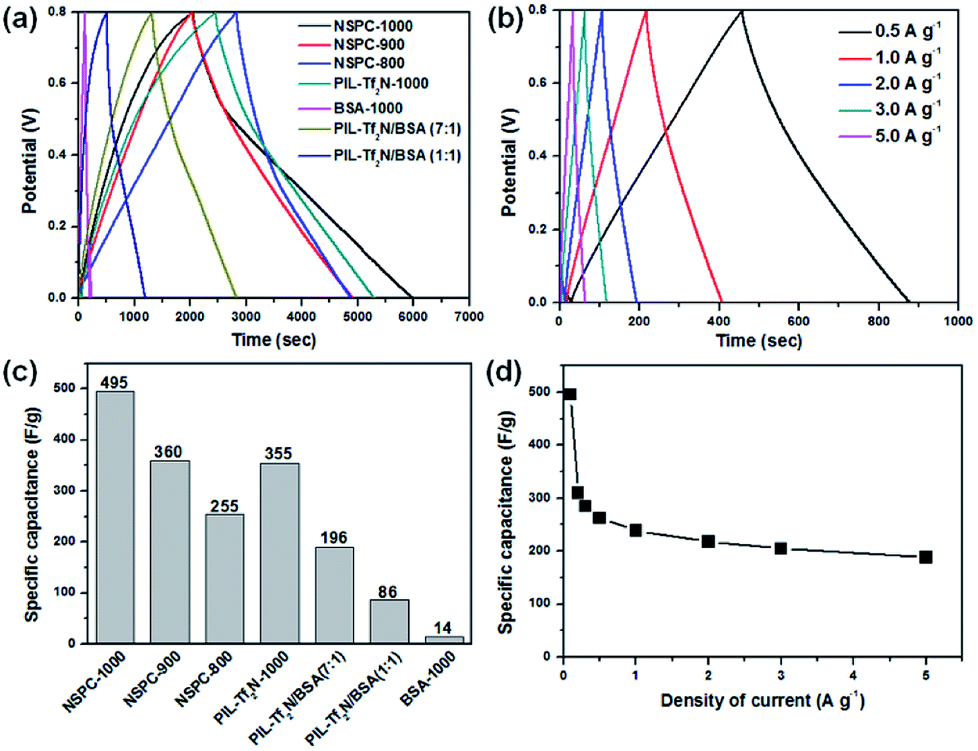 | ||
| Fig. 5 Electrochemical performances of carbon materials for supercapacitor. (a) Galvanostatic charge/discharge (GCD) curves of different carbon materials at a current density of 0.1 A g−1. (b) GCD curves of NSPC-1000 electrode material at different current densities. (c) Discharge capacitances of different carbon materials corresponding to the data in (a). (d) Discharge capacitances at various current densities corresponding to the data in (b). | ||
Conclusions
In summary, a facile protocol for fabrication of N, S-codoped porous carbon was developed via carbonization of the BSA@PIL-Tf2N complex. Our studies reveal that the novel N, S-codoped carbons featured micro-/mesoporous architectures. Due to their large surface area and good electron/electrolyte transport properties, NSPC materials are demonstrated to be effective bifunctional HER/OER electrocatalysts with excellent activity and stability. In particular, NSPC-1000 affords the lower overpotential and smaller Tafel slope, outperforming many well-defined non-precious metals and metal-free electrocatalysts. Simultaneously, the newly developed NSPC materials also exhibit outstanding capacitance performance. In view of the low cost of biomacromolecules and the facile synthesis of PIL, NSPC material is a promising alternative to (non)noble metal-based catalysts for water splitting, as well as for supercapacitor and other electrochemistry applications. Our facile protocol via the combination of biomacromolecules and synthetic polymers opens up a new avenue for devising effective, readily scalable and metal-free N, S-codoped carbon electrode materials for multi-functional applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support given by the National Natural Science Foundation of China (Project No. 21774101, 21474080).Notes and references
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. Volk and T. M. L. Wigley, Science, 2002, 298, 981–987 CrossRef CAS PubMed.
- Y. Zhou, M. Hejazi, S. Smith, J. Edmonds, H. Li, L. Clarke, K. Calvina and A. Thomsona, Energy Environ. Sci., 2015, 8, 2622–2633 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- S. Dunn, Int. J. Hydrogen Energy, 2002, 27, 235–264 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- N. C. Cheng, N. B. Banis, J. Liu, A. Riese, X. Li, R. Y. Li, S. Y. Ye, S. N. Knights and X. L. Sun, Adv. Mater., 2015, 27, 277–281 CrossRef CAS PubMed.
- H. F. Lv, Z. Xi, Z. Z. Chen, S. J. Guo, Y. S. Yu, W. L. Zhu, Q. Li, X. Zhang, M. Pan, G. Lu, S. C. Mu and S. H. Sun, J. Am. Chem. Soc., 2015, 137, 5859–5862 CrossRef CAS PubMed.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1500985 CrossRef.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P. C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 4345 CrossRef CAS PubMed.
- W. F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896–8909 RSC.
- L. A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- M. R. Gao, J. X. Liang, Y. R. Zheng, Y. F. Xu, J. Jiang, Q. Gao, J. Li and S. H. Yu, Nat. Commun., 2015, 6, 5982 CrossRef CAS PubMed.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- H. Vrubel and X. L. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- Y. Yang, H. L. Fei, G. D. Ruan, C. S. Xiang and J. M. Tour, Adv. Mater., 2014, 26, 8163–8168 CrossRef CAS PubMed.
- Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, Angew. Chem., Int. Ed., 2013, 52, 13638–13641 CrossRef CAS PubMed.
- D. Y. Wang, M. Gong, H. L. Chou, C. J. Pan, H. A. Chen, Y. P. Wu, M. C. Lin, M. Y. Guan, J. Yang, C. W. Chen, Y. L. Wang, B. J. Hwang, C. C. Chen and H. J. Dai, J. Am. Chem. Soc., 2015, 137, 1587–1592 CrossRef CAS PubMed.
- Z. L. Wang, X. F. Hao, Z. Jiang, X. P. Sun, D. Xu, J. Wang, H. X. Zhong, F. L. Meng and X. B. Zhang, J. Am. Chem. Soc., 2015, 137, 15070–15073 CrossRef CAS PubMed.
- Z. W. Liu, F. Wang, M. Li and Z. H. Ni, RSC Adv., 2016, 6, 37500–37505 RSC.
- Z. Xiao, X. Y. Gao, M. H. Shi, G. Y. Ren, G. Z. Xiao, Y. Zhu and L. Jiang, RSC Adv., 2016, 6, 86401–86409 RSC.
- J. Deng, M. M. Li and Y. Wang, Green Chem., 2016, 18, 4824–4854 RSC.
- S. J. Yuan and X. H. Dai, RSC Adv., 2015, 5, 45827–45835 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 128, 2270–2274 CrossRef.
- C. Hu and L. Dai, Adv. Mater., 2017, 29, 1604942 CrossRef PubMed.
- M. Zhang and L. Dai, Nano Energy, 2012, 1, 514–517 CrossRef CAS.
- K. Qu, Y. Zheng, Y. Jiao, X. Zhang, S. Dai and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1602068 CrossRef.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- J. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003–5012 CrossRef CAS.
- J. Yuan, A. G. Márquez, J. Reinacher, C. Giordano, J. Janek and M. Antonietti, Polym. Chem., 2011, 2, 1654–1657 RSC.
- H. Wang, S. Min, C. Ma, Z. Liu, W. Zhang, Q. Wang, D. Li, Y. Li, S. Turner, Y. Han, H. Zhu, E. Abou-hamad, M. N. Hedhili, J. Pan, W. Yu, K. W. Huang, L. J. Li, J. Yuan, M. Antonietti and T. Wu, Nat. Commun., 2017, 8, 13592 CrossRef CAS PubMed.
- M. Lei, P. G. Li, L. H. Li and W. H. Tang, J. Power Sources, 2011, 196, 3548–3552 CrossRef CAS.
- Z. Pei, H. Li, Y. Huang, Q. Xue, Y. Huang, M. Zhu, Z. Wang and C. Zhi, Energy Environ. Sci., 2017, 10, 742–749 RSC.
- J. C. Lytle, J. M. Wallace, M. B. Sassin, A. J. Barrow, J. W. Long, J. L. Dysart, C. H. Renninger, M. P. Saunders, N. L. Brandell and D. R. Rolison, Energy Environ. Sci., 2011, 4, 1913–1925 RSC.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 RSC.
- J. Liang, Y. Jiao, M. Jaroniec and S.-Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- J. Duan, Y. Zheng, S. Chen, Y. Tang, M. Jaroniec and S.-Z. Qiao, Chem. Commun., 2013, 49, 7705–7707 RSC.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 3892–3896 CrossRef CAS PubMed.
- J. G. N. Thomas, Trans. Faraday Soc., 1961, 57, 1603–1611 RSC.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- Z. Chen, D. Cummins, B. N. Reinecke, E. Clark, M. K. Sunkara and T. F. Jaramillo, Nano Lett., 2011, 11, 4168–4175 CrossRef CAS PubMed.
- X. Liu, W. Zhou, L. Yang, L. Li, Z. Zhang, Y. Ke and S. Chen, J. Mater. Chem. A, 2015, 3, 8840–8846 RSC.
- S. Mao, Z. Wen, S. Ci, X. Guo, K. Ostrikov and J. Chen, Small, 2015, 11, 414–419 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- C. C. Hu and T. W. Tsou, Electrochem. Commun., 2002, 4, 105–109 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- X. A. Chen, X. H. Chen, X. Xu, Z. Yang, Z. Liu, L. J. Zhang, X. Q. Xu, Y. Chen and S. M. Huang, Nanoscale, 2014, 6, 13740–13747 RSC.
- L. Sun, J. Liu, Z. Liu, T. Wang, H. Wang and Y. Li, RSC Adv., 2018, 8, 19964–19970 RSC.
- S. Liu, Y. Cai, X. Zhao, Y. Liang, M. Zheng, H. Hu, H. Dong, S. Jiang, Y. Liu and Y. Xiao, J. Power Sources, 2017, 31, 373–382 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, GPC curve, EDX, element analysis, SEM and HRTEM images, photographs, electrochemical impedance spectroscopy and CV profiles. See DOI: 10.1039/c8ra09700c |
| This journal is © The Royal Society of Chemistry 2019 |