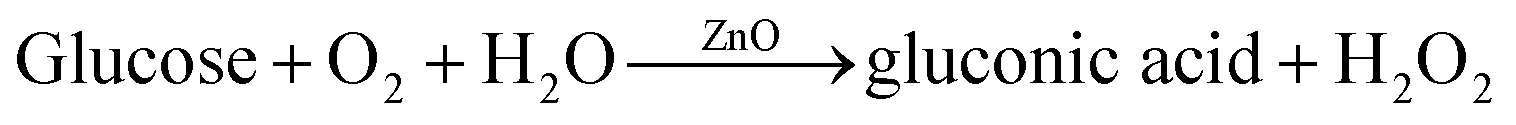DOI:
10.1039/C8RA09599J
(Paper)
RSC Adv., 2019,
9, 12134-12145
Hybrid ZnO–graphene electrode with palladium nanoparticles on Ni foam and application to self-powered nonenzymatic glucose sensing
Received
22nd November 2018
, Accepted 3rd April 2019
First published on 17th April 2019
Abstract
A self-powered nonenzymatic glucose sensor electrode boasts the advantages of both a glucose sensor and fuel cell. Herein, an electrode composed of ZnO–graphene hybrid materials on nickel foam (NF) is prepared by electrodeposition of Pd NPs. The electrode is characterized systematically and the dependence of electrocatalytic oxidation of glucose on the concentrations of KOH and glucose, temperature, and potential limit in the anodic direction is investigated. The Pd/NF-ZnO–G electrode shows high catalytic activity, sensitivity, stability, and selectivity in glucose detection, as exemplified by an electrocatalytic glucose oxidation current of 222.2 mA cm−2 under alkaline conditions, high linearity in the glucose concentration range from 5 μM to 6 mM (R2 = 0.98), and high sensitivity of 129.44 μA mM−1−1 cm−2. The Pd/NF-ZnO–G electrode which exhibits superior electrocatalytic activity under alkaline conditions has large potential in nonenzymatic glucose sensing and direct glucose fuel cells and is suitable for miniaturized self-powered nonenzymatic glucose sensing.
1. Introduction
Glucose monitoring is one of the most important sensing technologies in medical science, clinical diagnostics, and food industry. In order to facilitate monitoring of the blood glucose concentration, the sensor should be miniaturized and portable. However, most glucose sensors need an external power supply which inevitably increases the size and complexity. A self-powered sensor which is able to generate its own energy either by itself or from the surroundings does not require batteries and can be reduced in size. Self-powered systems such as piezoelectric-powered,1 triboelectric-powered,2 thermoelectric-powered,3 and photovoltaic-powered systems4 and fuel cells have been proposed as potential glucose sensors. So far, enzymatic glucose fuel cells are the dominant self-powered sensors because of the simple configuration and capabilities of providing sustainable energy from renewable environmental sources under mild conditions.5 However, the limited lifetime and stringent requirement for enzyme immobilization impose restrictions and impact the stability.6 Hence, nonenzymatic glucose sensors and fuel cells using metal materials as catalysts have been designed to overcome the drawbacks of enzymatic electrodes.
Pt, Pd, Au and their based binary or ternary catalysts such as Pd–Rh,7 Pt–Bi,8 Au–Ni,9 Pd–Ni,10 and Pt–Pd–Au11 have been found to have good electro-catalytic activity in the glucose oxidation reaction (GOR). Although platinum is the most common catalyst in glucose sensors and direct glucose fuel cells (DGFC), it is quite expensive. As Pd is 50 times more abundant on earth and commands only half the price of Pt, it is a more active, stable, and poison-tolerant electrocatalyst compared to Pt in glucose oxidation in alkaline media. Consequently, Pd is a promising alternative electrocatalyst to Pt or enzymatic sensor. Nickel foam (NF), a commercial material with unique electronic conductivity, light weight, and 3D cross-linked grid structure providing high porosity and surface area has been proposed as an excellent support for electrocatalysts.12 Moreover, Pd–Ni binary catalysts have higher electrocatalytic activity and poison-tolerance than the individual metal in GOR.10 Owing to the excellent electron mobility and large specific surface area, graphene (G) can boost the electrode activity as an electron mediator to promote the charge separation efficiency. In addition, ZnO nanoparticles (NPs) have been extensively investigated because of their high electron mobility and well-controlled morphology. The ZnO–graphene hybrid may constitute an advanced nonenzymatic electrode with high electrocatalytic activity.
Herein, a self-powered nonenzymatic glucose sensor capable of generating electrical power from the biochemical energy stored in glucose is designed and fabricated. The novel electrode composed of the NF substrate and ZnO–G hybrid is prepared by step electro-deposition of Pd NPs. The structure and morphology of the electrode are characterized by scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), and Brunauer–Emmett–Teller (BET) method. The electrocatalytic characteristics are evaluated by cyclic voltammetry (CV), amperometry analysis, and electrochemical impedance spectroscopy (EIS).
2. Experimental details
2.1 Raw materials
The NF (thickness of 1.5 mm, pore density of 100 PPI, and areal density of 380 g m−2) was purchased from Changsha Lyrun New Materials Corporation (Changsha, China) and oily slurry of graphene (5 wt%) was acquired from Suzhou Graphene Nanotechnology Co. Ltd (Suzhou, China). Nano-zinc oxide was obtained from Shanghai Hansi Chemical Industry Co. Ltd (Shanghai, China). KOH, palladium chloride (PdCl2), ammonia chloride (NH4Cl), ammonia (NH3·H2O), ethanol and glucose were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Ascorbic acid (AA), uric acid are (UA) and dopamine (DA) was purchased from Aladdin industrial Co. Ltd (Shanghai, China). Fructose, D(+)-sucrose and lactose was obtained from Tianjin Kermel Chemical Reagent Co. Ltd (Tianjin, China). All the other reagents were analytical grade and used without further purification. All the solutions were prepared with deionized (DI) water.
2.2 Preparation of NF-ZnO–G
The NF was cut into squares (1 cm2) and washed in acetone for 10 minutes for degreasing. The NF was taken out and a large amount of ethanol was applied to remove the organic solvents from the NF surface followed by thorough rinsing with DI water. The NF was soaked in 6 M HCl for 15 minutes to remove the oxide layer on the surface and then rinsed with a large amount of DI water repeatedly. Silver was applied to the cleaned NF and stick to copper wires to form the electrode followed by fixing and protection with epoxy resin.
Nano-ZnO powders (0.1 g) were dispersed into 50 mL of ethanol ultrasonically for 10 min to obtain solution A (2 g L−1). 1 g of the oily slurry of graphene was dispersed into 50 mL of absolute ethanol by mild bath-sonication to produce solution B. 1 mL of A was gradually added to 5 mL of B under stirring for 1 min and then sonicated for 20 min at room temperature. Excess product was introduced to the cleaned NF electrode by dropping, which makes electrode surface is completely saturated. Finally, the NF was dried at 60 °C for 12 h to form the NF-ZnO–G electrode.
2.3 Electro deposition of palladium nanoparticles on NF-ZnO–G
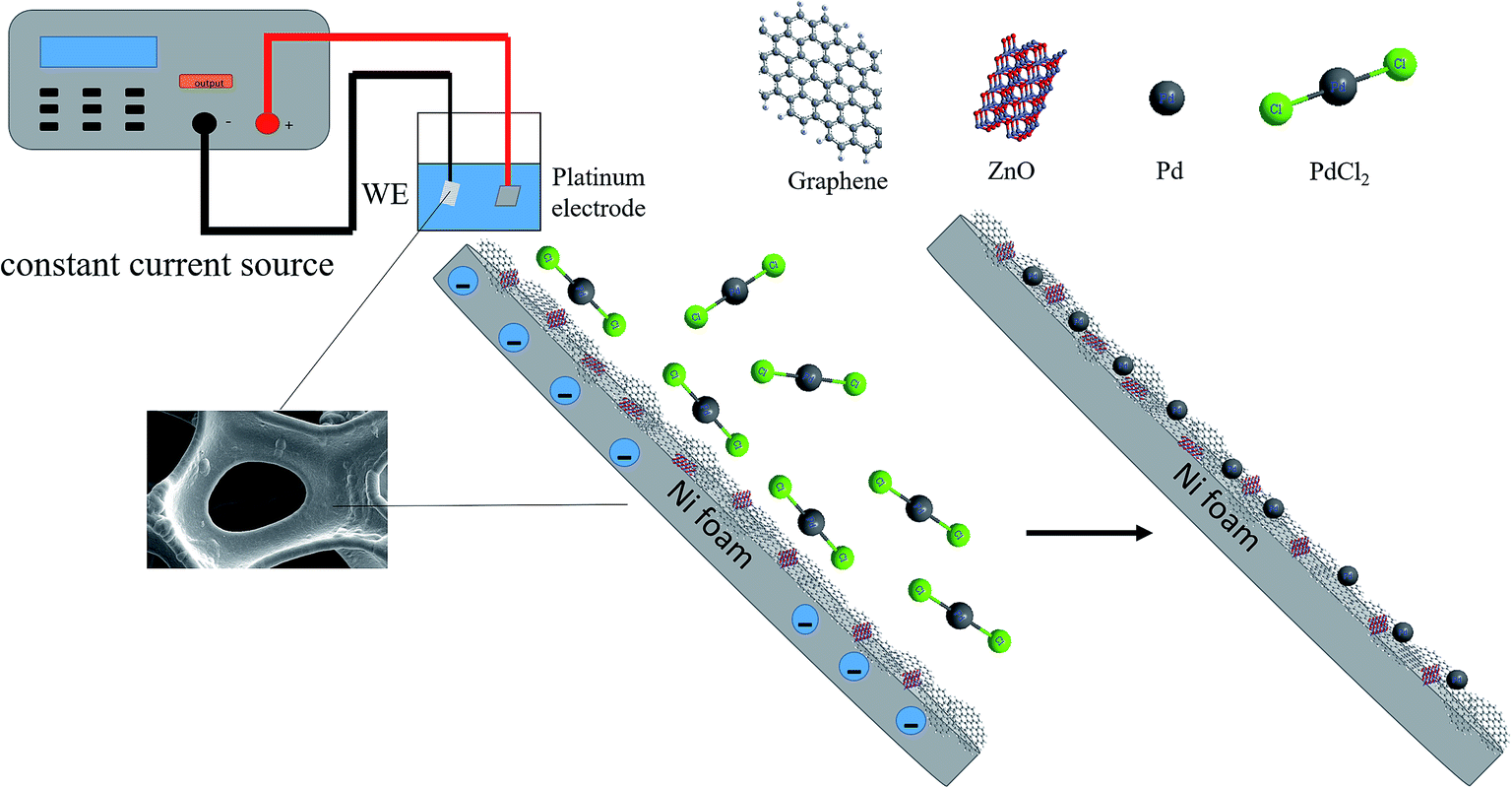
50 mL of the electroplating solution containing 2.7 mmol palladium chloride and 0.35 mol NH4Cl were adjusted to a pH of 8 with ammonia. The electroplating apparatus is described in Fig. 1. The power supply was set to the constant current mode. The current density was 10 mA cm−2 and the selected electroplating time was 30 min.
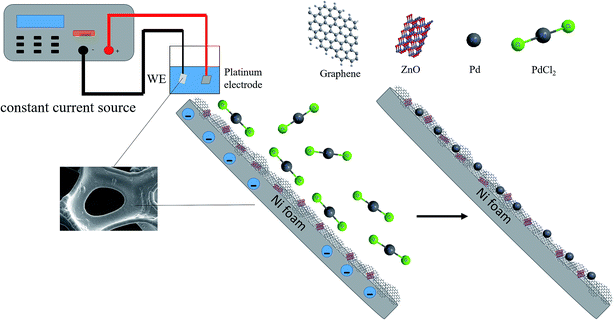 |
| | Fig. 1 Schematic depiction of electrodeposition of PdNPs on NF-ZnO–G electrode. | |
2.4 Characterization and measurements
The structure and morphology of the electrode were characterized by SEM at 20 kV (Hitachi S-3400). XRD was conducted on the Gigaku SmartLab using Cu Kα radiation (λ = 1.54056 Å, 45 kV, 200 mA) and XPS was performed on the Thermo Scientific Escalab 250Xi at 15 kV. The BET specific surface area was determined by nitrogen adsorption at 77.35 K using the Nova2000E. The electrochemical measurements were performed on a CHI604E electrochemical workstation (Chenhua, China) based on the conventional three-electrode system composed of the platinum auxiliary, a Hg/HgO (saturated KCl) reference, and a G–ZnO/NF working electrodes. The electrolyte was KOH and deoxygenated in every experiment.
3. Results and discussion
3.1 Characterization of Pd/NF-ZnO–G
Fig. 2(a) shows the 300× image of the Pd/NF-ZnO–G electrode revealing a three-dimensional network on the NF substrate. The loose and spongy 3D macroporous structure is arranged in a crisscross pattern with more pores and larger specific surface area. The inset in Fig. 2(a) is the image of pure NF showing flake-like and bulges on the surface of the Pd/NF-ZnO–G compared with pure NF. The image of NF-ZnO–G is shown in Fig. 2(b) and stacking of the ZnO–G nanocomposite on the NF is clearly observed. The inset in Fig. 2(b) is the image of pure ZnO NPs. The size of ZnO nanoparticles is 50–500 nm and the ZnO shows a hexagonal prism structure. The magnified image of Fig. 2(a) is shown in Fig. 2(c). Stacked of graphene sheets is observed and the size of the graphene nano-platelets is 1–5 μm. The spots are dispersed on the sheet surface homogeneously and may be ZnO NPs and aggregation of Pd PNs. The surface of NF becomes rougher perhaps due to Pd NPs electrodeposition on the NF directly. The edge of the electrode shows dendritic bulges as shown in Fig. 2(d). The Pd nanocolumns are formed under the partial discharge because of the electrodeposition current is not completely uniform. The 5,000× and 12,000× images of the Pd/NF-ZnO–G electrode are depicted in Fig. 2(e) and (f), respectively. The Pd/NF-ZnO–G surface is decorated with dense Pd NPs and slight Pd NPs aggregation is observed. The results confirm successful anchoring of PdNPs on the Pd/NF-ZnO–G surface. Fig. 2(f) shows the magnified dendritic bulges which may contribute to GOR.
 |
| | Fig. 2 (a) SEM image of Pd/NF-ZnO–G with inset showing the SEM image of clean NF; (b) SEM image of NF-ZnO–G with inset showing the SEM image of pure ZnO NPs; (c–f) SEM images of Pd/NF-ZnO–G. | |
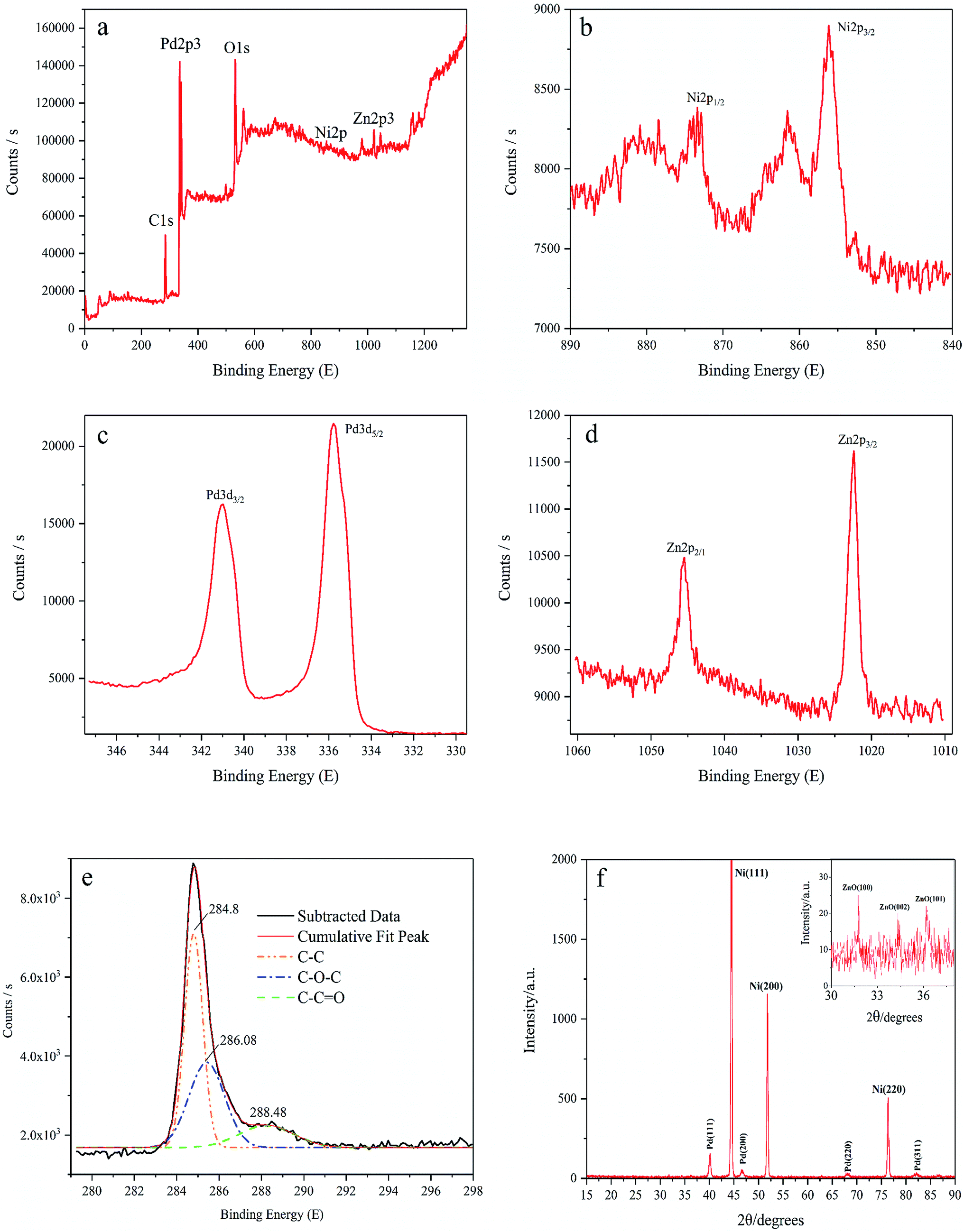
XPS is carried out to determine the surface chemical states of Pd/NF-ZnO–G. We were only able to make a rough estimate of the amount of element of Pd/NF-ZnO–G by XPS experiment, atomic% data given directly through instruments: Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd:Ni:C = 3.73:27.85:2.07:62.3. Silver and chlorine were introduced in the experiment as impurity elements, accounting for 4.05%. As shown in the survey spectrum (Fig. 3(a)), Pd, Zn, O, and C are presented but the Ni signal is indistinct because NF is covered by the Pd/graphene–ZnO nanocomposite and the sampling depth of XPS is about 5–10 nm. O should be assigned to the ZnO from air exposure. The Ni 2p3/2 and Ni 2p1/2 peaks are at 856.08 eV and 872.58 eV (Fig. 3(b)), respectively. There are some satellite peaks around the Ni 2p3/2 and Ni 2p1/2 signals in the Ni 2p region. The high-resolution peak of Pd 3d shows two broad peaks related to the Pd 3d5/2 and Pd 3d3/2 spin orbital states (Fig. 3(c)). For Pd/NF-ZnO–G, strong signals at 335.78 eV and 340.98 eV corresponding to Pd0 are observed indicating significant Pd3d signal corresponding to the binding energy of Pd further supporting that PdNPs are assembled on the NF-ZnO–G electrode. As shown in Fig. 3(d), the Zn 2p3 peak of ZnO nanoparticles shows two componential peaks at 1021.58 and 1004.68 eV and the Zn 2p3 data reveal that ZnO is loaded on the NF substrate successfully. The graphene spectrum (Fig. 3(e)) exhibits three peaks at 284.7, 285.8, and 287.8 eV, corresponding to C–C, C–O, and C
:Pd:Ni:C = 3.73:27.85:2.07:62.3. Silver and chlorine were introduced in the experiment as impurity elements, accounting for 4.05%. As shown in the survey spectrum (Fig. 3(a)), Pd, Zn, O, and C are presented but the Ni signal is indistinct because NF is covered by the Pd/graphene–ZnO nanocomposite and the sampling depth of XPS is about 5–10 nm. O should be assigned to the ZnO from air exposure. The Ni 2p3/2 and Ni 2p1/2 peaks are at 856.08 eV and 872.58 eV (Fig. 3(b)), respectively. There are some satellite peaks around the Ni 2p3/2 and Ni 2p1/2 signals in the Ni 2p region. The high-resolution peak of Pd 3d shows two broad peaks related to the Pd 3d5/2 and Pd 3d3/2 spin orbital states (Fig. 3(c)). For Pd/NF-ZnO–G, strong signals at 335.78 eV and 340.98 eV corresponding to Pd0 are observed indicating significant Pd3d signal corresponding to the binding energy of Pd further supporting that PdNPs are assembled on the NF-ZnO–G electrode. As shown in Fig. 3(d), the Zn 2p3 peak of ZnO nanoparticles shows two componential peaks at 1021.58 and 1004.68 eV and the Zn 2p3 data reveal that ZnO is loaded on the NF substrate successfully. The graphene spectrum (Fig. 3(e)) exhibits three peaks at 284.7, 285.8, and 287.8 eV, corresponding to C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively. The C–O and CO components are absent confirming a small concentration of oxygen-containing groups in graphene and that graphene has not been converted into graphene oxide.
O, respectively. The C–O and CO components are absent confirming a small concentration of oxygen-containing groups in graphene and that graphene has not been converted into graphene oxide.
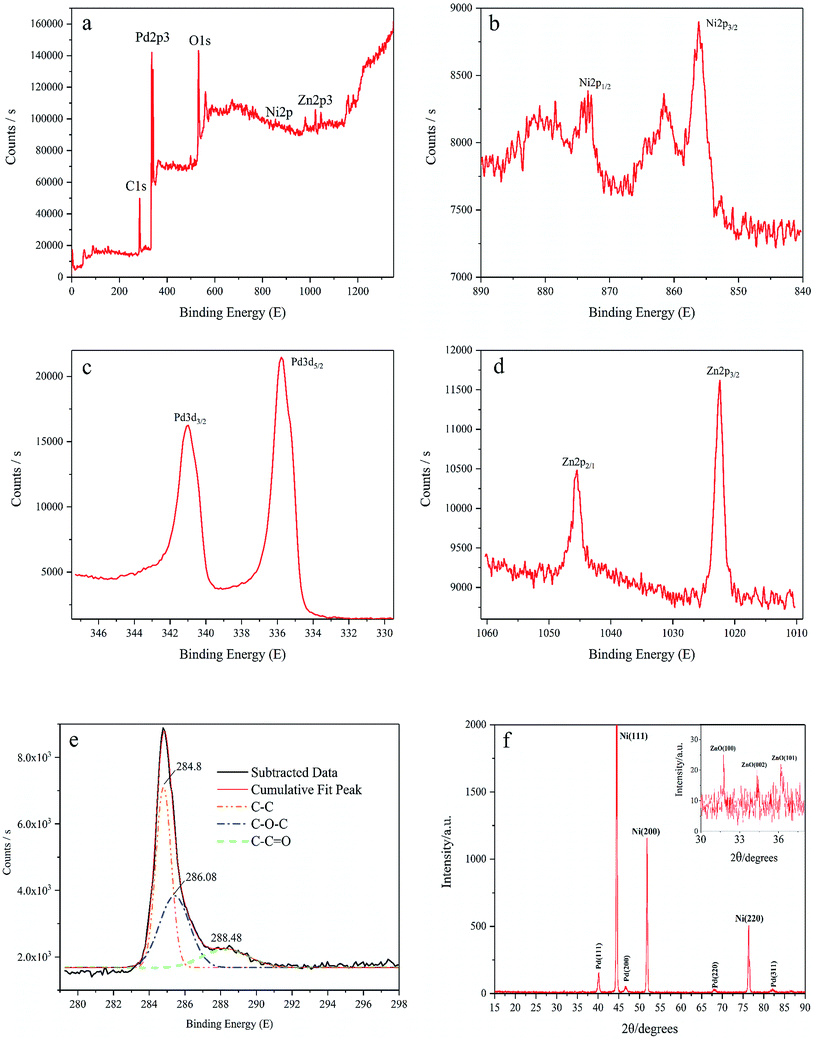 |
| | Fig. 3 XPS spectra of (a) Pd/NF-ZnO–G, (b) XPS spectrum of NF, (c) Pd NPs, and (d) ZnO NPs, (e) graphene, and (f) XRD spectrum of Pd/NF-ZnO–G. Inset: magnified XRD spectrum of the Pd/NF-ZnO–G (2θ from 30° to 38°). | |
Fig. 3(f) shows the XRD results of the Pd/NF-ZnO–G electrode. The peaks at 40.2°, 46.5°, 68.1°, and 82.2° can be assigned to the (111), (200), (220) and (311) planes of the face-centered cubic Pd metal, respectively. The nickel substrate shows the main characteristic peaks of the face-centered cubic structure at 2θ = 44.5°, 51.8°, and 76.4°, associated with the (111), (200), and (220) planes confirming that Pd NPs are electrodeposited on the NF. ZnO–G is loaded on the NF substrate covered by high-density Pd NPs, even though the diffraction peaks are not obvious. In order to identify the diffraction peak of ZnO, the magnified XRD pattern (2θ from 30° to 38°) is shown in the inset of Fig. 3(f). Partial diffraction peaks of ZnO are indeed observed at 2θ of 31.7°, 34.4°, and 36.2° corresponding to the (100), (002) and (101) planes of the hexagonal wurtzite structure.
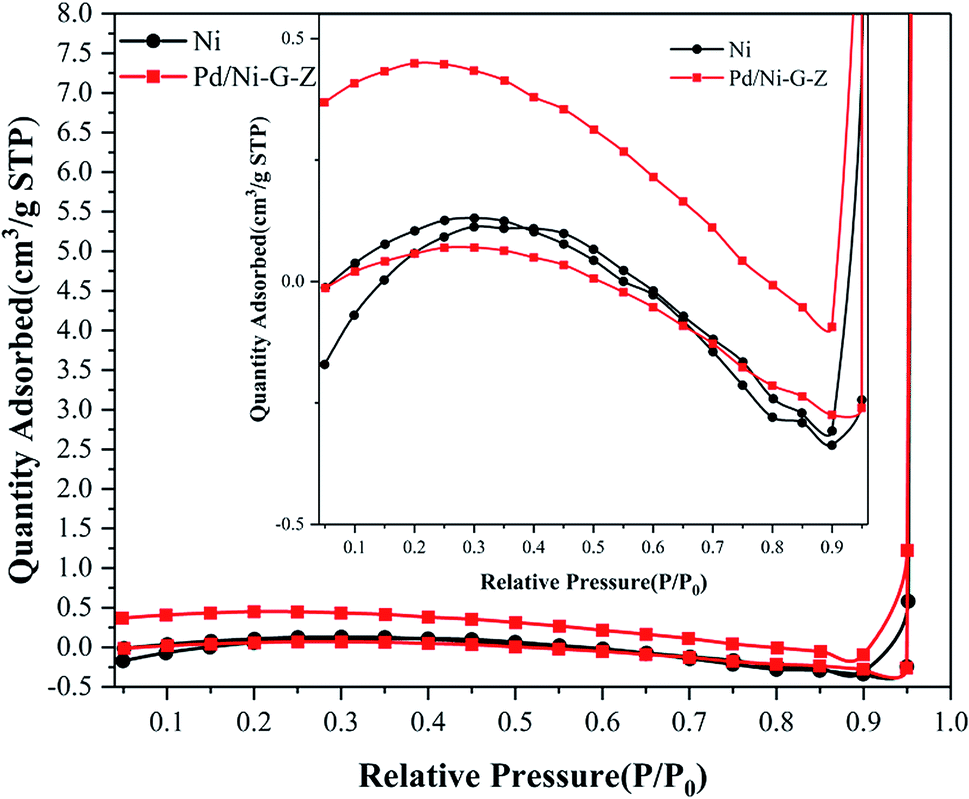
N2 adsorption–desorption was conducted using the Brunauer–Emmett–Teller (BET) method to determine the surface area of the electrode. As shown in Fig. 4, the non-limiting adsorption at high P/P0 for Pd/NF-ZnO–G is characteristic of a Type IV loop. The inset in Fig. 4 corresponds to those of Pd/NF-ZnO–G (triangular spots) and pure NF (square spots), respectively, revealing that Pd/NF-ZnO–G has a surface area of 0.974 m2 g−1 that is 4.6 times larger than 0.212 m2 g−1 of the pure NF.
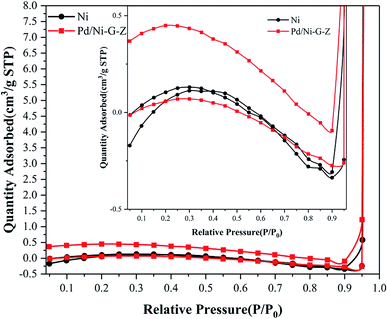 |
| | Fig. 4 N2 adsorption–desorption isotherm of Pd/NF-ZnO–G. Inset: magnified isotherm compared with Pd/NF-ZnO–G and pure NF. | |
3.2 Electrochemical response of Pd/NF-ZnO–G in GOR
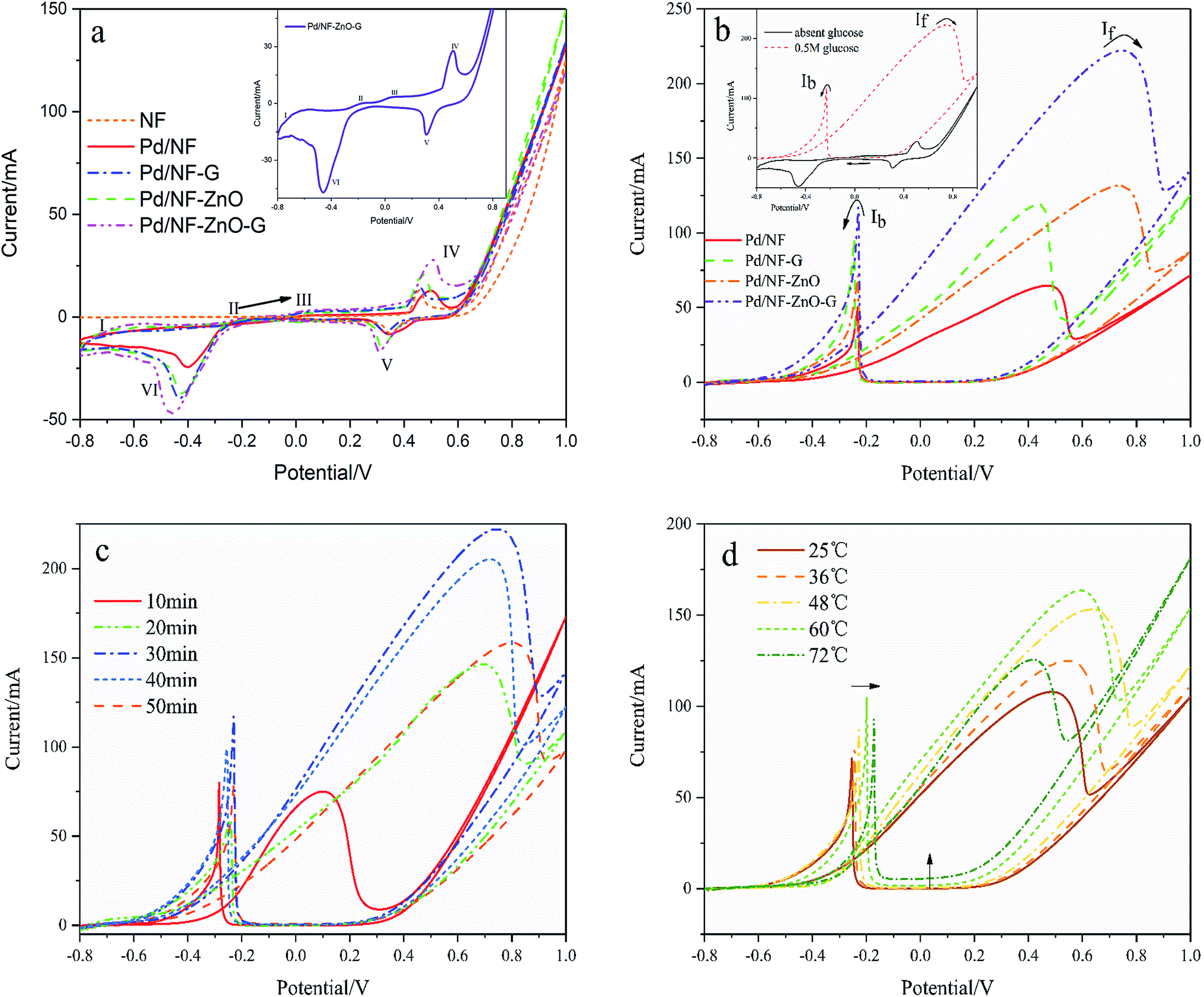
Fig. 5(a) shows the electrochemical behavior of the pure NF and Pd/Ni based electrocatalyst in 1 M KOH at a scanning rate of 50 mV s−1 and the inset is the magnified image of the Pd/NF-ZnO–G electrode under the same conditions. As shown in Fig. 5(a), during the positive-going sweep, there are four oxidation peaks corresponding to different electrochemical processes on the surface of the electrode. Peak I at the potential range between −0.8 and −0.7 V can be assigned to oxidation of the adsorbed and absorbed hydrogen (eqn (1)). Peak II which appears from −0.25 to −0.1 V is attributed to formation of PdOH− (eqn (2)) and the third peak from −0.05 to 0.3 V stems from the Pd(II) oxide on the surface of the catalyst (eqn (3) and (4)). When the potential is changed to more positive values, the Pd(II) oxide layer is converted into higher-valence oxides, Pd(III) and Pd(IV) or Pd(OH)x, from the partial discharge of OH− at the electrocatalyst surface in the alkaline solution (eqn (5)). It is probable that the peaks of formation of Pd (OH)x species partially overlaps Peak IV. The possible mechanism is shown in the following:13| | |
Pd − Habs/ads + OH− → Pd + H2O + e−
| (1) |
| | |
Pd + OH− ⇄ Pd − OHads + e−
| (2) |
| | |
Pd − OHads + OH− ⇄ Pd − O + H2O + e−
| (3) |
| | |
Pd − OHads + Pd − OHads ⇄ Pd − O + H2O
| (4) |
| | |
Pd + xOH− → PdOHx + xe−
| (5) |
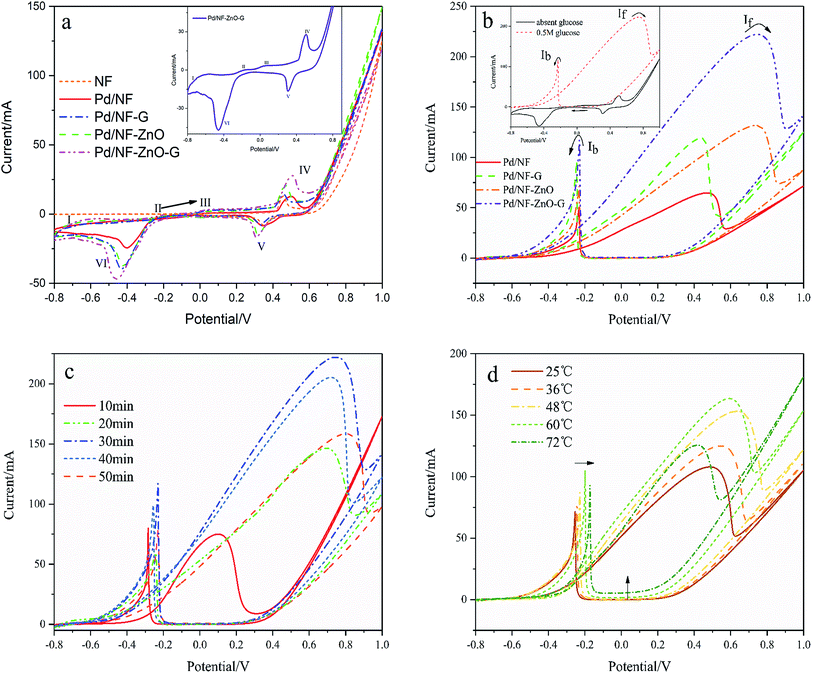 |
| | Fig. 5 (a) CVs of Pd/NF, Pd/NF-G, Pd/NF-ZnO, and Pd/NF-ZnO–G in 1 M KOH at a scanning rate of 50 mV s−1. Inset: magnified CV of Pd/NF-ZnO–G in 1 M KOH at a scanning rate of 50 mV s−1. (b) CVs of Pd/NF, Pd/NF-G, Pd/NF-ZnO and Pd/NF-ZnO–G in 1 M KOH containing 0.5 M glucose at a scanning rate of 50 mV s−1. Inset: magnification CV of Pd/NF-ZnO–G in 1 M KOH in the absence (solid line) and presence (dash line) of 0.5 M glucose at a scanning rate of 50 mV s−1. (c) CVs of 5 Pd/NF-ZnO–G for different plating time of 10, 20, 30, 40 and 50 minutes in 1 M KOH containing 0.5 M glucose at a scanning rate of 50 mV s−1. (d) CVs of Pd/NF-ZnO–G in 1 M KOH containing 0.5 M glucose at a scanning rate of 50 mV s−1 at different temperature of T = 25, 36, 48, 60, and 72 °C. | |
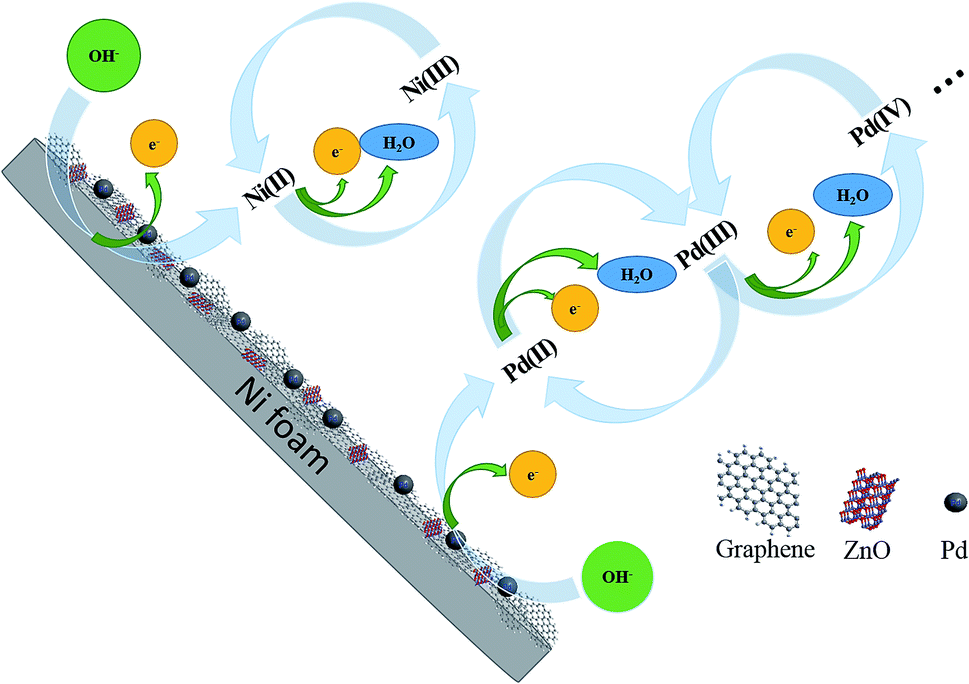
Formation of Pd NPs oxides is shown in Fig. 6. Peak IV from 0.4 V to 0.7 V is related to the formation of Ni(II) and Ni(III) according to eqn (6) and (7):
| | |
Ni + 2OH− → Ni(OH)2 + 2e−
| (6) |
| | |
Ni(OH)2 + OH− → NiOOH + H2O + e−.
| (7) |
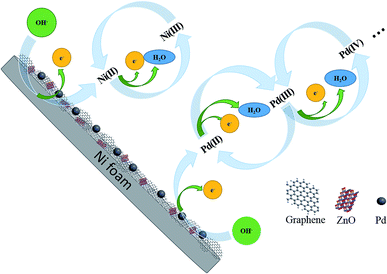 |
| | Fig. 6 Mechanism of activation of Pd/NF-ZnO–G in KOH. | |
For the negative peak V, a pair of symmetrical peaks with Peak VI can be assigned to reduction of Ni (III and II) and Pd (III and IV). Peak VI at −0.4 V arises from reduction of the Pd(II) species according to eqn (8):14
| | |
Pd − O + H2O + 2e− → Pd + 2OH−.
| (8) |
The characteristics of NF, Pd/NF, Pd/NF-G, Pd/NF-ZnO, and Pd/NF-ZnO–G are shown in Fig. 5(a). For Peak I, the adsorption/absorbed hydrogen peak current of the Pd/NF is less than those of other electrodes. The pair of symmetrical peaks (Peak IV and Peak V) are mainly attributed to oxidation-reduction of Ni and therefore, pure NF has only the oxidation peak associated with Ni. Pd/NF has the minimum peak current because ZnO NPs and graphene provides a large surface area that improve the ability of the electrode to adsorption of ions. In the reduction region of Pd(II) (Peak VI), the Pd/NF-ZnO–G electrode delivers the best performance due to advantages rendered by ZnO and graphene.
To investigate the electrocatalytic activity of the different electrodes for glucose, CVs are recorded in 1 M KOH containing 0.5 M glucose as shown in Fig. 5(b) with the inset showing the CVs of Pd/NF-ZnO–G under the same conditions. By adding glucose (0.5 M), the CVs of the Pd/NF-ZnO–G electrode change significantly. The maximum peak (anodic peak) at 0.742 V in the forward direction corresponds to direct glucose oxidation to form intermediates. One main reason is oxidation of glucose by palladium. With regard to the anodic peak, the electrochemical behavior of Pd should be attributed to the electrosorption of glucose to form intermediates by releasing one proton.14 When the intermediates accumulate on the Pd/NF-ZnO–G surface and occupy active sites of the electrocatalyst, the current decreases. The potential moving to more positive values and Pd (OH)x species are generated from partial discharge of OH− on the electrocatalyst surface in the alkaline solution.
For the cathode peak, reduction of surface palladium oxide occur at a potential more negative than −0.2 V. The active sites are renewed and available for direct oxidation of glucose. Therefore, there is a sharp increase in the current for the peak at −0.23 V. Based on the previously reported oxidation mechanism for glucose,15 a possible catalytic mechanism for Pd on the Pd/NF-ZnO–G electrode for oxidation of glucose is described as follows:
| | |
Pd + glucose → Pd − H + intermediates
| (9) |
| | |
Pd + xOH− → PdOHx + xe−
| (10) |
| | |
PdOHx + intermediates → Pd + glucolactone or gluconic acid
| (11) |
| | |
PdOHx + glucose → Pd + glucolactone or gluconic acid.
| (12) |
NF also plays a significant role in oxidation of glucose to glucolactone in the upper potential region (potential at 0.8 V) as follows:
| | |
Ni + 2OH− → Ni(OH)2 + 2e−
| (6) |
| | |
Ni(OH)2 + OH− → NiOOH + H2O + e−
| (7) |
| | |
NiOOH + glucose → Ni(OH)2 + glucolactone
| (13) |
In addition, during the electrocatalytic reaction in the alkaline solution, ZnO reacts with glucose to form glutonic acid:16
| |
 | (14) |
| | |
H2O2 → O2 + 2H+ + 2e−.
| (15) |
Therefore, ZnO nanoparticles on the graphene sheets are directly involved in the electrocatalytic reaction. The graphene sheets increase the surface area of the electrode to enhance adsorption of ions and the electrocatalytic reaction.
Concerning the positive potential scan, the Pd/NF-ZnO–G electrode shows an obvious maximum peak current. It means that Pd/NF-ZnO–G delivers the best electrocatalytic performance in GOR and ZnO–G with well-distributed Pd NPs on the surface helps electron transport to the electrode. With the addition of nano-ZnO, the potential of the oxidation peaks moves to a more positive value (about 0.3 V) and the process may be associated with ZnO reacting with glucose. Moreover, the ratio of the forward anodic peak current density (If) to the reverse anodic peak current density (Ib), If/Ib, can be used to describe the tolerance to carbonaceous species accumulation. A higher ratio indicates more effective removal of the poisoning species on the catalyst surface during the anodic scan and little accumulation of carbonaceous residues on the electrode surface, a low If/Ib ratio shows the reverse case.17,18 The If/Ib ratio of the Pd/NF-ZnO–G electrocatalyst (1.96) is 1.46, 1.54 and 1.07 times larger than those of Pd/NF (1.34), Pd/NF-ZnO (1.27), and Pd/NF–G (1.84), suggesting that Pd/NF-ZnO–G has better poison tolerance.
The plating time affects the thickness and morphology of the Pd NPs layer and catalytic performance of the electrode. Therefore, 5 electrodes are prepared using plating time of 10, 20, 30, 40 and 50 minutes. As shown in Fig. 5(c), with increasing of electroplating time, the amount of Pd NPs deposited on the electrode surface increases and the electrocatalytic effect in GOR is enhanced as well. However, the current of the oxidation peak for plating time over 30 minutes decreases and the potential of the oxidation peak moves forward because the Pd NPs on the NF-ZnO–G electrode begin to condense into larger spheres leading to the reduction of the actual active area. The optimal plating time is thus 30 minutes.
The effects of temperature (T = 25–72 °C) on the electro-oxidation of glucose are investigated and Fig. 5(d) shows the CVs at the Pd/NF-ZnO–G electrode in GOR at different temperature. Increasing the temperature causes the peak current to increase until 60 °C. As the temperature exceeds 48 °C, a negative shift in the oxidation peak potential is observed. Furthermore, by increasing the temperature, the baseline of the CVs curves shifts upward and there is a positive shift in the re-oxidation peak potential. Since glucose degrades rapidly above 40 °C as evidenced by a rapid change of color to yellow-brown and caramel like smell,13 the same degradation phenomenon is observed here. A higher temperature facilitates electro-oxidation of glucose but the current density decreases for T > 60 °C.
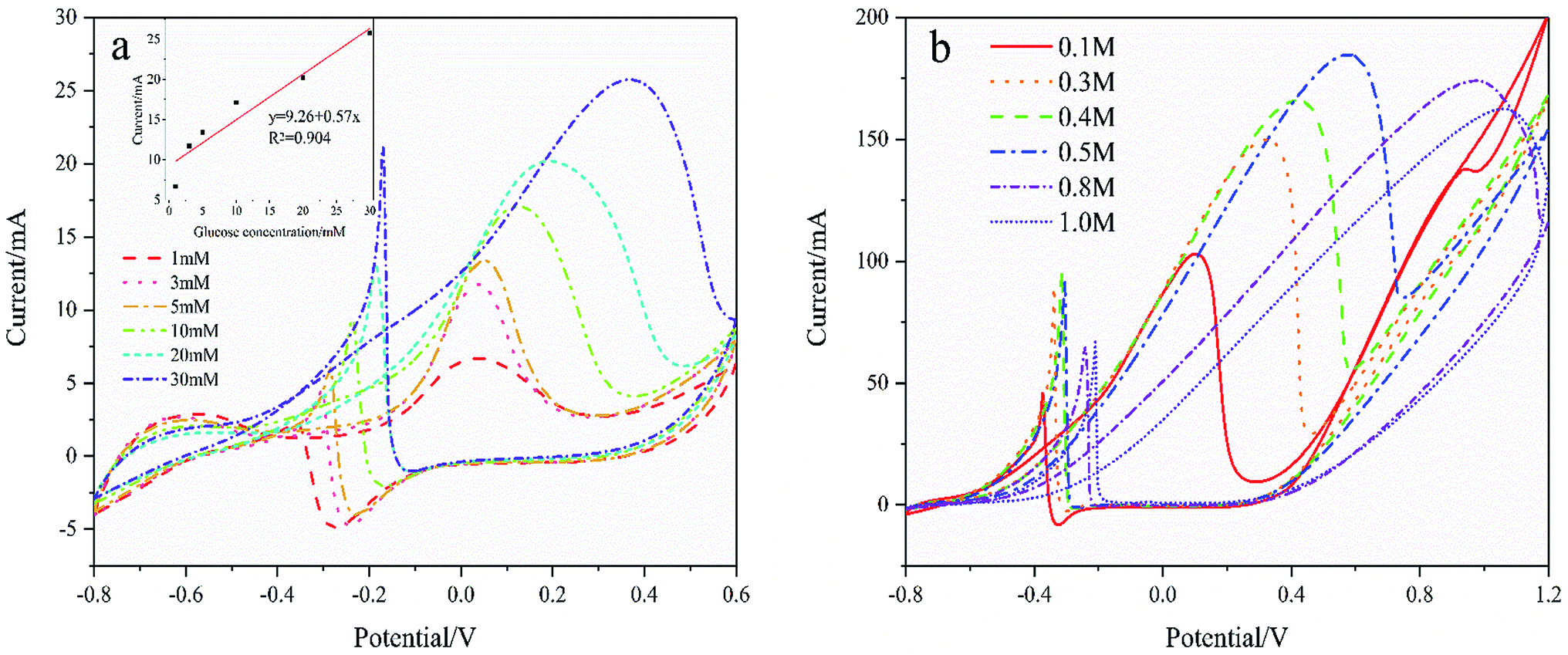
Fig. 7(a) shows the CVs curves of Pd/NF-ZnO–G in the presence of different concentrations of glucose. The CVs are recorded in 0.1 M KOH and the inset shows the calibration plots of glucose concentration versus oxidation peak current. As the glucose concentration increases, the current increases linearly reaching 25.75 mA cm−2 for 30 mmol L−1 glucose. As the glucose concentrations increases further, the peak potential shows a positive shift. The performance enhancement can be attributed to that more reactants are involved in the reaction as the glucose concentration is increased. On the other hand, by further increasing the glucose concentration in 1 M KOH at 50 mV s−1 as shown in Fig. 7(b), the peak potential shows a positive shift and the peak current increases until the concentration of glucose reaches 0.5 M. When the concentration exceeds 0.5 M, the oxidation peak current decreases. The deteriorated performance may be due to the following reasons.22 Firstly, the electrode surface cannot provide adequate active sites for GOR and secondly, the intermediates may cover and poison the electrode surface to decrease its catalytic activity. Thirdly, a higher concentration of glucose gives rise to higher solution viscosity and less effective mass transfer.
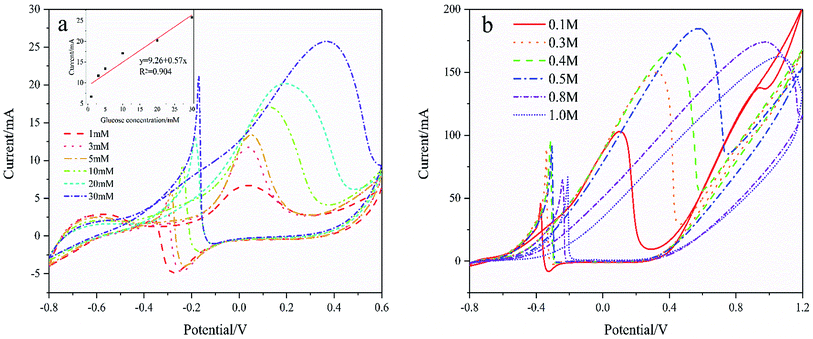 |
| | Fig. 7 (a) CVs of Pd/NF-ZnO–G in 0.1 M KOH for different concentrations of glucose of 1, 3, 5, 10, 20, and 30 μM. Inset: corresponding calibration plots of anodic peak current vs. concentration of glucose. (b) CVs of Pd/NF-ZnO–G in 1 M KOH for different concentrations of glucose of 0.1, 0.3, 0.4, 0.5, 0.8, and 1.0 M. | |
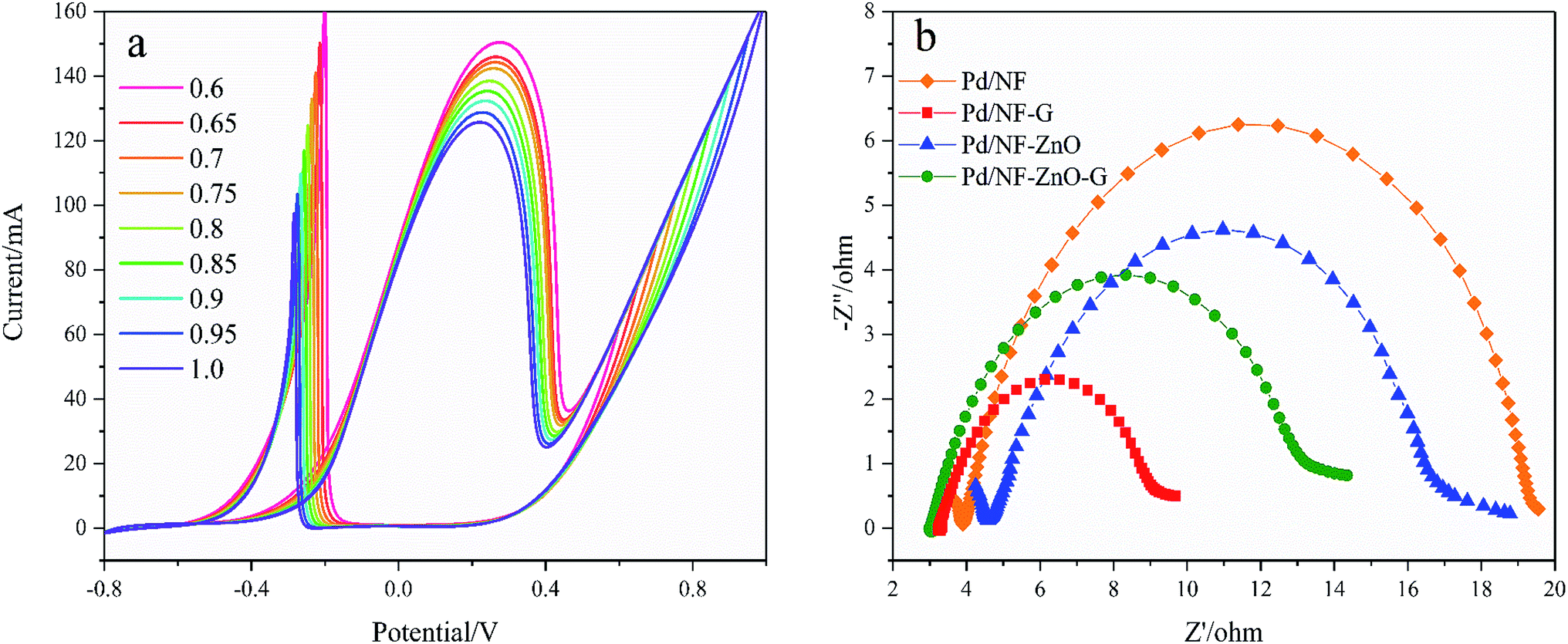
Fig. 8(a) shows the CVs of the Pd/NF-ZnO–G electrode in 1 M KOH containing 0.5 M glucose for different upper scanning limits at a scanning rate of 50 mV s−1. The lower limit is fixed at −0.8 V. The currents of the oxidation peak and re oxidation peak decrease and move negatively as the upper scanning limit is increased indicating that with a positive shift in the upper scan limit, GOR becomes easier but the upper scan limit weakens the GOR intensity. It is noteworthy that If/Ib increases meaning that the intermediate species can be effectively oxidized and the electrode poisoning tolerance is improved.
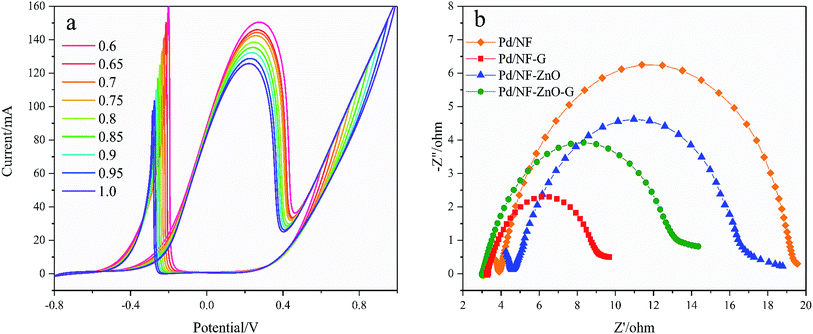 |
| | Fig. 8 (a) CVs of Pd/NF-ZnO–G in 1 M KOH containing 0.5 M glucose for different upper scanning limits at a scanning rate of 50 mV s−1; (b) Nyquist plots of Pd/NF, Pd/NF-ZnO, Pd/NF-G and Pd/NF-ZnO–G in 0.1 M KOH + 0.1 M glucose at 0.3 V in the frequency range between 0.05 Hz and 105 Hz. | |
Electrochemical impedance spectroscopy (EIS) is used to obtain information on electron transfer between the electrolyte and electrode. Fig. 8(b) shows the Nyquist plots of Pd/NF, Pd/NF-ZnO, Pd/NF-G, and Pd/NF-ZnO–G at the applied potential of 0.3 V in 1 M KOH containing 0.5 M glucose in the frequency range between 0.05 Hz and 105 Hz. In the high frequency region, data fitting shows that Rct follows the order of Pd/NF-G < Pd/NF-ZnO–G < Pd/NF-ZnO < Pd/NF. The diameter of the semicircle for Pd/NF-G is the smallest demonstrating that the Pd/NF-G has higher electrical conductivity and enhanced electron transfer rate. The performance of Pd/NF-ZnO–G is second to Pd/NF-G. Addition of ZnO not only improves Rct, but also increases the inherent impedance (Rs).
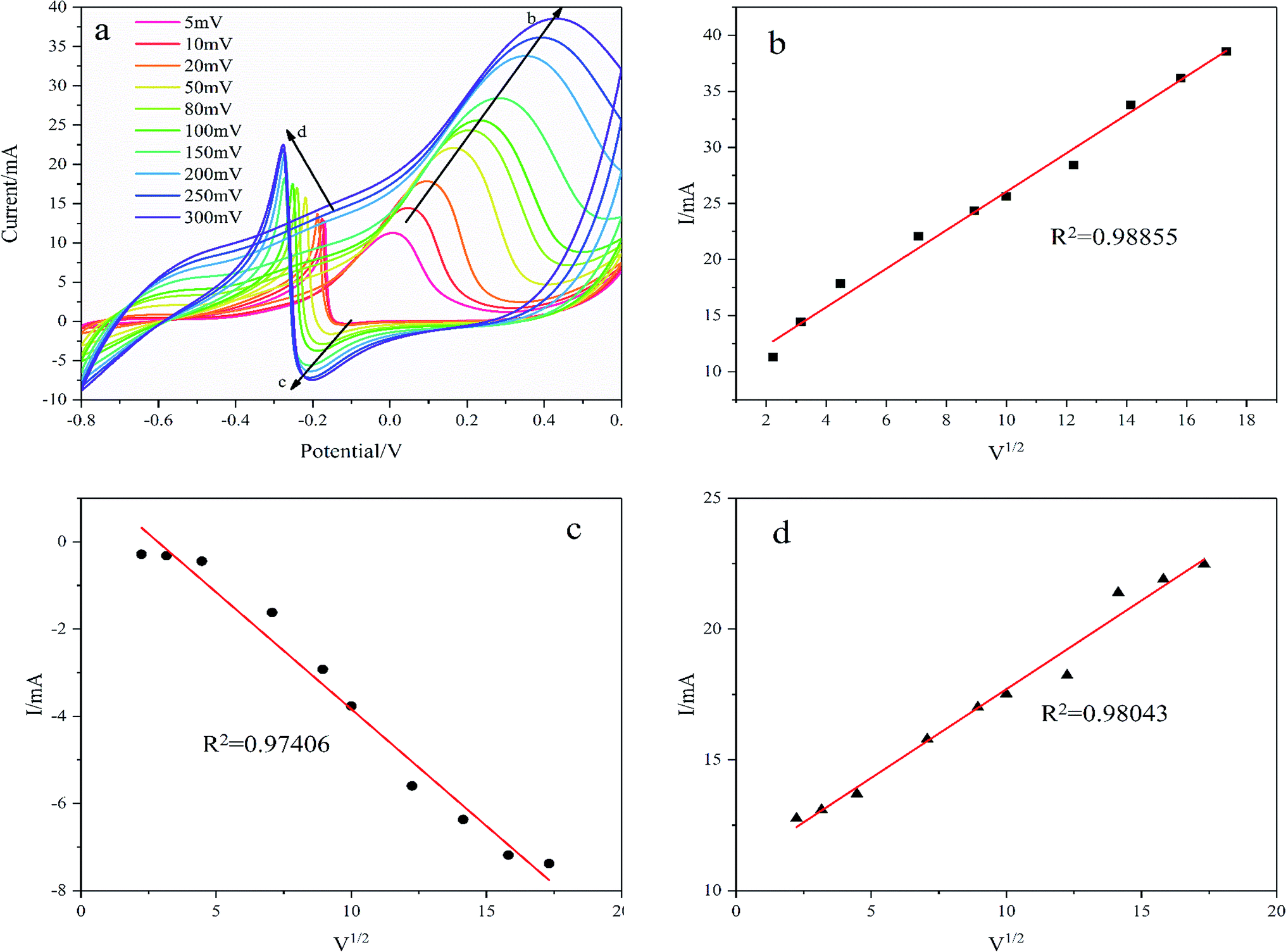
As shown in Fig. 9, the oxidation reduction peak current increases with scanning rates from 5 mV s−1 to 300 mV s−1 demonstrating a diffusion-controlled redox reaction on the Pd/NF-ZnO–G electrode. The linear relationship between the square root of the potential scan rate (v1/2) and peak current (I) verifies that the overall glucose oxidation reaction is controlled by diffusion as shown in Fig. 9(b–d). The R2 values of the three oxidation reduction peaks are 0.98855, 0.97406, and 0.98043 respectively showing that glucose oxidation is controlled by mass transport of glucose from the bulk solution to electrode surface.
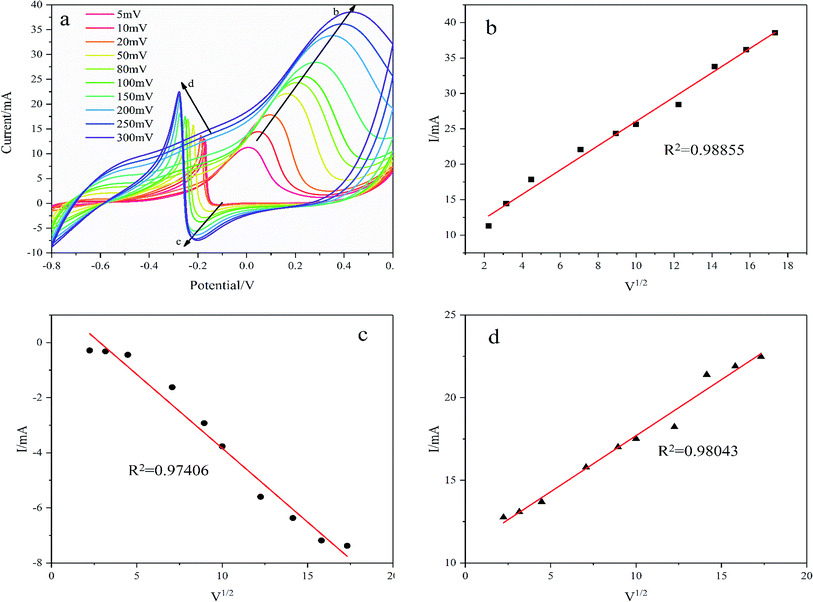 |
| | Fig. 9 (a) CVs of Pd/NF-ZnO–G at scanning rates of 5, 10, 20, 50, 80, 100, 150, 200, 250, and 300 mV in 0.1 M KOH containing 0.5 M glucose; (b), (c) and (d) calibration plots showing the linear relationship between the square root of the potential scan rate (v1/2) vs. peak current (I). | |
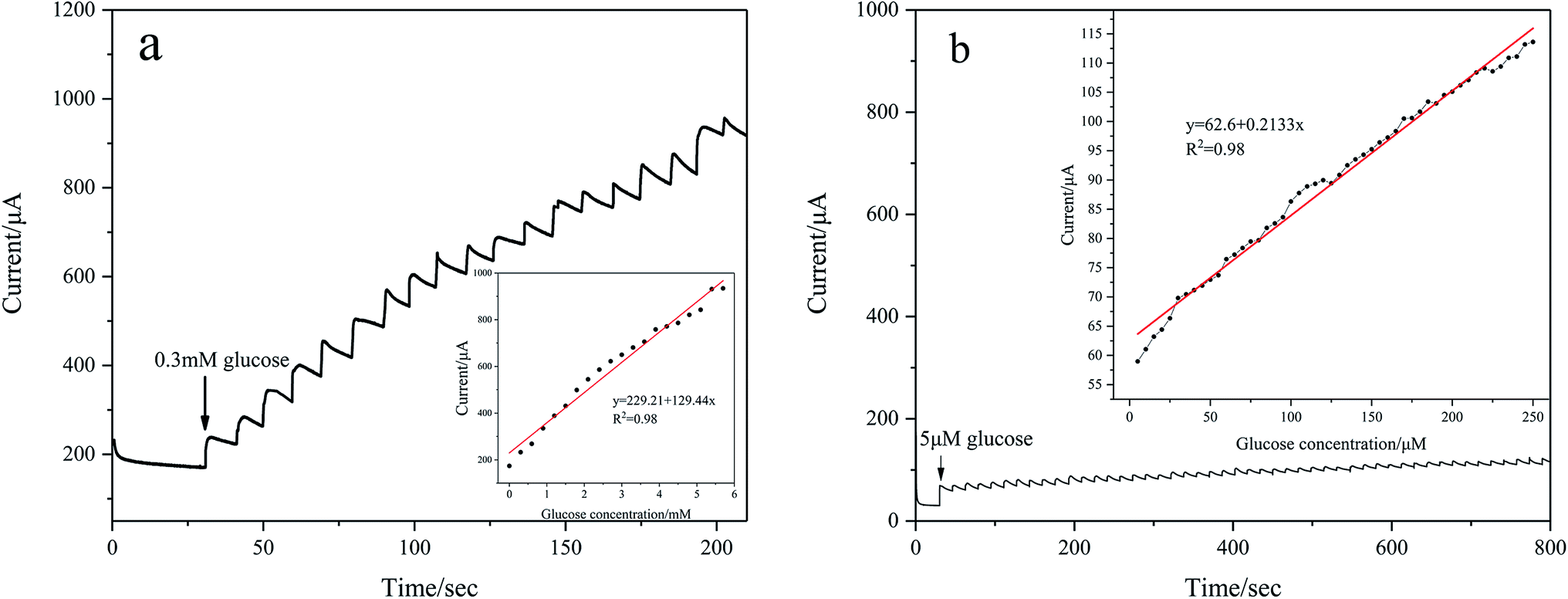
To evaluate amperometric glucose monitoring and sensing, the current response is measured at different concentrations of glucose in 0.1 M KOH at a fixed potential. As shown in Fig. 7(a), the CV of Pd/NF-ZnO–G shows that the oxidation peaks appear from 0.05 V to 0.2 V and so the operating potential is chosen to be 0.2 V. Fig. 10(a) shows the typical amperometric current–time response of Pd/NF-ZnO–G upon successive stepwise addition of 0.3 mM glucose from 0 to 6 mM into 0.1 M KOH at the optimal potential of 0.2 V. The corresponding calibration plot is shown in the inset in Fig. 10(a). The sensitivity is 129.44 μA mM−1 cm−2 with a lowest detection limit of 0.184 M (S/N ratio = 3) and there is excellent linearity in the wide range from 0 to 6 mM. The amperometric response to for lower concentrations with stepwise addition of 5 μM glucose in the range from 0 to 250 μM is shown in Fig. 10(b). The sensitivity is 213.3 μA mM−1 cm−2 with a lowest detection limit of 0.056 M (S/N ratio = 3). Comparing the two experiments, the amperometric response for a lower concentration shows higher sensitivity. The lower sensitivity of glucose at high concentrations arises from adsorption of intermediates from glucose oxidation.21 The performance of Pd/NF-ZnO–G is compared to that of other Pd-based electrodes in Table 1.
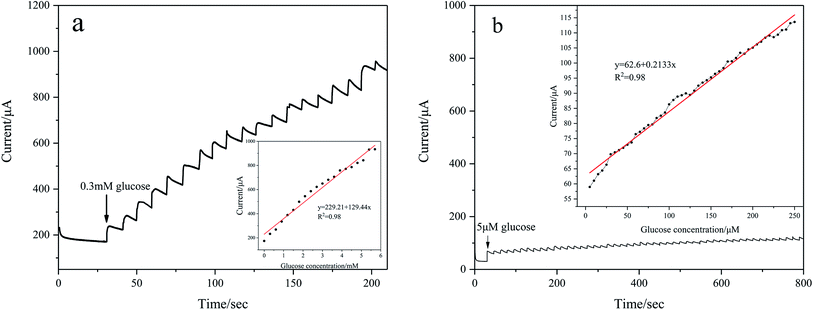 |
| | Fig. 10 Amperometric current–time response of Pd/NF-ZnO–G upon successive addition of different concentrations of glucose to 0.1 M KOH at 0.2 V: (a) step of 0.3 mM glucose added in the range from 0 to 6 mM and (b) step of 5 μM glucose added in the range from 0 to 250 μM. Inset: corresponding calibration plots of the linear relationship between current vs. concentration of glucose. | |
Table 1 Comparison of the analytical performance of the Pd/NF-ZnO–G electrode with other Pd-based non-enzymatic glucose sensors
| Working electrode |
Linear rang (μM) |
Sensitivity (μA mM−1 cm−2) |
Detection limit (μM) |
Detection potential |
Ref. |
| Pd/NF-ZnO–G |
5 μM to 12 mM |
213.3 |
0.056 |
0.2 V |
This work |
| PdNPs/Ni5P4-NF |
2 μM to 4.65 mM |
242.5 |
0.91 |
0.3 V |
10 |
| Ni-Pd@AC/GCE |
10 μM to 1.65 mM |
90 |
0.014 |
0.5 V |
21 |
| Pd–Ni/SiNW |
2–20 mM |
190.72 |
2.88 |
−0.06 V |
23 |
| PdNPs–CNTs |
1–20 mM |
11 |
— |
0.025 V |
20 |
| Pd30Au70/C |
1 mM-X |
2.372 |
— |
−0.2 V |
19 |
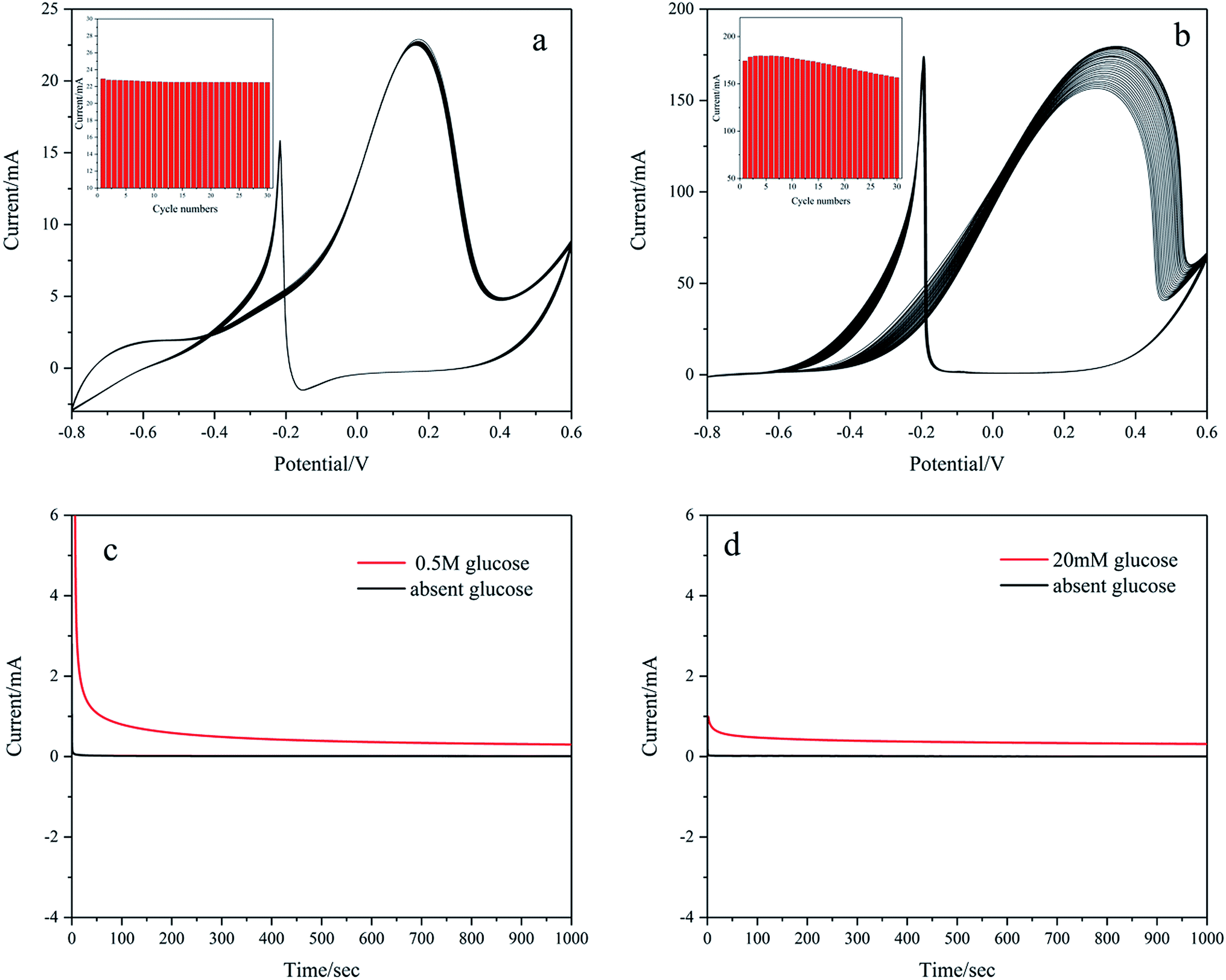
The stability of Pd/NF-ZnO–G is assessed by obtaining CVs for 30 cycles in 0.1 M KOH containing 20 mM glucose and 1 M KOH containing 0.5 M glucose as shown in Fig. 11(a) and (b), respectively. The peak current of Pd/NF-ZnO–G with cycle number is shown in the inset. Fig. 11(a) shows no noticeable change and the peak current remains at 98.2% after 30 cycles. Fig. 11(b) shows that the peak current increases with cycle number for the first 3 cycles because the electrolyte needs to penetrate the surface of the electrode. At the 4th cycle, the peak current shows a large value of 179.6 mA and then decreases gradually finally reaching 87.15% after 30 cycles. The results demonstrate that the Pd/NF-ZnO–G electrode has good stability. The loss of catalytic activity can be explained in terms of poisoning during glucose oxidation. The CA response to Pd/NF-ZnO–G in 1 M KOH + 0.5 M glucose and 0.1 M KOH + 20 mM glucose at 0.2 V for 1000 s is presented in Fig. 11(c) and (d), respectively. The results suggest that Pd/NF-ZnO–G has good stability in glucose sensing application and DGFCs.
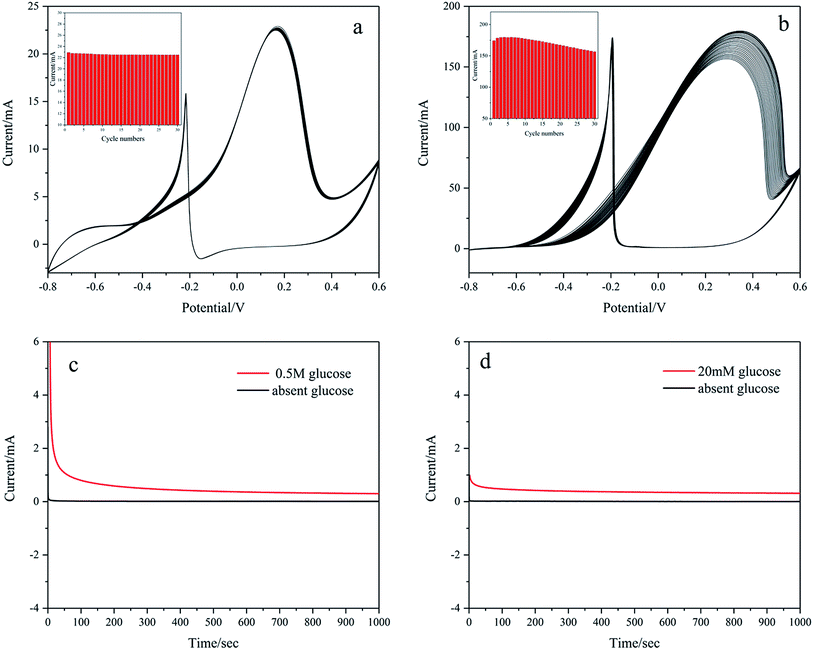 |
| | Fig. 11 Stability of Pd/NF-ZnO–G electrode: 30 cycles CVs of Pd/NF-ZnO–G in (a) 0.1 M KOH containing 20 mM glucose and (b) 1 M KOH containing 0.5 M glucose. Inset: corresponding relationship between peak current vs. number of cycles. CA response of Pd/NF-ZnO–G in (c) 1 M KOH + 0.5 M glucose and (d) 0.1 M KOH + 20 mM glucose at 0.2 V for 1000 s. | |
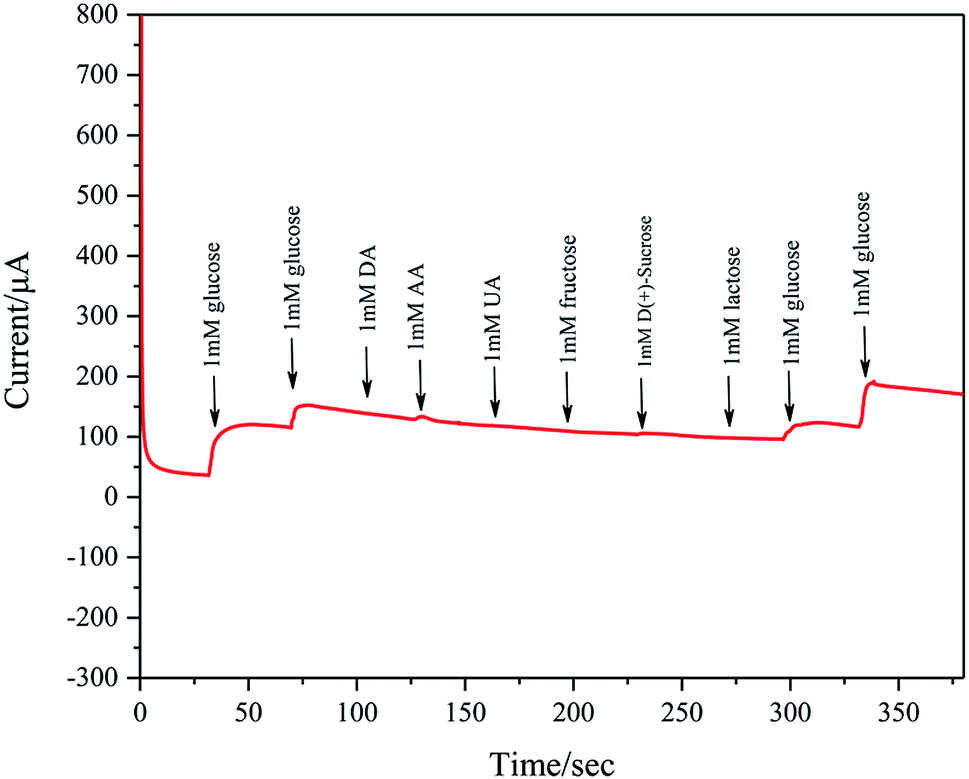
Ascorbic acid (AA), uric acid are (UA), dopamine (DA), and various carbohydrates are recognized as major interferences for detection of glucose. Considering that the glucose concentration is about 30–50 times of interfering species in human serum, the amperometric response to interfering species is also studied by successive addition of 1 mM glucose, 1 mM glucose, 1 mM DA, 1 mM AA, 1 mM UA, 1 mM fructose, 1 mM D(+)-sucrose, 1 mM lactose, 1 mM glucose and 1 mM glucose to 0.1 M KOH at a fixed potential of 0.2 V. These concentrations exceed those in normal human blood. As shown in Fig. 12, Pd/NF-ZnO–G does not show clear response to AA, DA, UA, and various carbohydrates, further verifying the excellent sensitivity and selectivity in nonenzymatic glucose sensing.
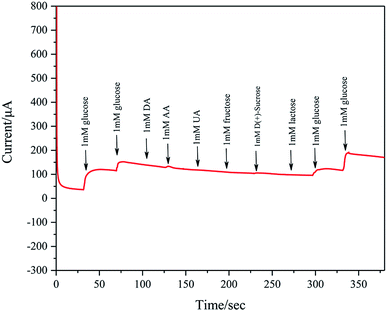 |
| | Fig. 12 (a) Interference test performed on the Pd/NF-ZnO–G electrodes in 0.1 M KOH at 0.2 V with successive addition of 1 mM glucose, 1 mM glucose, 1 mM DA, 1 mM AA, 1 mM UA, 1 mM fructose, 1 mM D(+)-sucrose, 1 mM lactose, 1 mM glucose and 1 mM glucose. | |
4. Conclusion
A DGFC-powered glucose sensor encompassing the advantages of both glucose sensor and fuel cell is designed and fabricated. The electrochemical response of the Pd/NF-ZnO–G electrode reveals high catalytic activity, sensitivity, stability, and selectivity in GOR. This electrode exhibits an electrocatalytic glucose oxidation current as high as 222.2 mA cm−2. The sensitivity of the Pd/NF-ZnO–G electrode in 0.1 M KOH is 129.44 μA mM−1 cm−2 and 213.3 μA mM−1 cm−2 (at a smaller concentration of glucose). It delivers superior electro-oxidation performance. As the operating temperature increases, a strong current response to glucose is observed even for long-term oxidation of glucose. The Pd/NF-ZnO–G electrode has potential in self-powered glucose sensors and the self-powered nonenzymatic glucose sensing scheme provides insights into the design of portable medical diagnosis devices such as self-powered miniaturized glucose sensors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank the technology Innovation Center of Agricultural Multi-Dimensional Sensor Information Perception, Heilongjiang Province. This work was jointly supported by the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (Grant No. UNPYSCT-2016087), Scientific Research Foundation for the Returned Overseas Chinese Scholars in Heilongjiang Province, Project of Plant food processing technology-Heilongjiang Province superiority and characteristic discipline (Grant No. YSTSXK201873), Graduate Innovation Project of Qiqihar University (Grant No. YJSCX2017-ZD14), Fundamental Research Funds in Heilongjiang Provincial Universities (No. 135109244, 135309115, 135309211), Hong Kong Research Grants Council (RGC) General Research Funds (GRF) No. CityU 11301215 and 11205617, as well as City University of Hong Kong Strategic Research Grant (SRG) No. 7005105.
References
- R. Yu, C. Pan, J. Chen, G. Zhu and Z. L. Wang, Adv. Funct. Mater., 2013, 23, 5868–5874 CrossRef CAS.
- H. Wang, Z. Xiang, P. Giorgia, X. Mu, Y. Yang, Z. L. Wang and C. Lee, Nano Energy, 2016, 23, 80–88 CrossRef CAS.
- S. Kim, K. Choi and A. Tazebay, et al., ACS Nano, 2014, 8, 2377–2386 CrossRef CAS PubMed.
- B. Cai, W. Mao, Z. Ye and J. Huang, Appl. Phys. A: Mater. Sci. Process., 2016, 122 Search PubMed.
- Z. Liu, B. Cho, T. Ouyang and B. Feldman, Anal. Chem., 2012, 84, 3403–3409 CrossRef CAS PubMed.
- S. K. Chaudhuri and D. R. Lovley, Nat. Biotechnol., 2003, 21, 1229–1232 CrossRef CAS PubMed.
- A. Brouzgou, L. L. Yan, S. Q. Song and P. Tsiakaras, Appl. Catal., B, 2014, 147, 481–489 CrossRef CAS.
- D. Basu and S. Basu, Electrochim. Acta, 2011, 56, 6106–6113 CrossRef CAS.
- J. Chen, H. Zheng, J. Kang, F. Yang, Y. Cao and M. Xiang, RSC Adv., 2017, 7, 3035–3042 RSC.
- M. Wang, Z. Ma, J. Li, Z. Zhang, B. Tang and X. Wang, J. Colloid Interface Sci., 2018, 511, 355–364 CrossRef CAS PubMed.
- D. Basu and S. Basu, Int. J. Hydrogen Energy, 2012, 37, 4678–4684 CrossRef CAS.
- C.-W. Kung, Y.-H. Cheng and K.-C. Ho, Sens. Actuators, B, 2014, 204, 159–166 CrossRef CAS.
- A. Brouzgou, S. Song and P. Tsiakaras, Appl. Catal., B, 2014, 158–159, 209–216 CrossRef CAS.
- M. Grdeń and A. Czerwiński, J. Solid State Electrochem., 2008, 12, 375–385 CrossRef.
- Z.-x. Cai, C.-c. Liu, G.-h. Wu, X.-m. Chen and X. Chen, Electrochim. Acta, 2013, 112, 756–762 CrossRef CAS.
- M. Sreejesh, S. Dhanush, F. Rossignol and H. S. Nagaraja, Ceram. Int., 2017, 43, 4895–4903 CrossRef CAS.
- Y. Zhao, L. Zhan, J. Tian, S. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967–1972 CrossRef CAS.
- X. Yang, Q. Yang, J. Xu and C.-S. Lee, J. Mater. Chem., 2012, 22, 8057–8062 RSC.
- L. Yan, A. Brouzgou, Y. Meng, M. Xiao, P. Tsiakaras and S. Song, Appl. Catal., B, 2014, 150–151, 268–274 CrossRef CAS.
- Z.-x. Cai, C.-c. Liu, G.-h. Wu, X.-m. Chen and X. Chen, Electrochim. Acta, 2013, 112, 756–762 CrossRef CAS.
- G. H. Wu, X. H. Song, Y. F. Wu, X. M. Chen, F. Luo and X. Chen, Talanta, 2013, 105, 379–385 CrossRef CAS PubMed.
- Y. Koskun, A. Şavk, B. Şen and F. Şen, Anal. Chim. Acta, 2018, 1010, 37–43 CrossRef CAS PubMed.
- S. Hui, J. Zhang, X. Chen, H. Xu, D. Ma, Y. Liu and B. Tao, Sens. Actuators, B, 2011, 155, 592–597 CrossRef CAS.
Footnote |
| † Wenyi Wu and Fengjuan Miao are joint first authors. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yu Zangb,
Lei Zhua,
Cuiping Shia and
Paul K. Chuc
*a,
Yu Zangb,
Lei Zhua,
Cuiping Shia and
Paul K. Chuc