
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long noncoding RNA PTPRG-AS1 regulates growth of glioma cells by sponging miR-185-5p
Chenyang Xu,
Zhenjiang Li,
Tao He,
Bingjian Yuan and
Bingqian Ding *
*
Department of Neurosurgery, Huaihe Hospital of Henan University, No. 1, North Baogong Rd, Gulou, Kaifeng, Henan 475000, China. E-mail: 18539986279@163.com; Tel: +86-0371-2390-6516
First published on 8th April 2019
Abstract
Previous studies have found that long noncoding RNA (lncRNA) protein tyrosine phosphatase, receptor type, G, antisense (PTPRG-AS1) was upregulated in glioma cells. Our study aimed to explore the detailed molecular mechanisms of PTPRG-AS1 involved in glioma progression. qRT-PCR assay was performed to measure the expressions of PTPRG-AS1 and microRNA-185-5p (miR-185-5p). Cell viability, migration, invasion, and apoptosis were determined by CCK-8 assay, colony formation assay, transwell assay, and flow cytometry assay. Autophagy was evaluated using GFP-LC3 puncta analysis and western blot. Luciferase reporter and RIP assays were employed to explore the association between PTPRG-AS1 and miR-185-5p. Our data showed PTPRG-AS1 was upregulated in glioma cells and tissues. Besides, high expression of PTPRG-AS1 was positively associated with a low survival rate. Upregulation of PTPRG-AS1 promoted proliferation, migration, invasion, colony formations, and autophagy, and inhibited cell apoptosis in U373-MG cells. By contrast, PTPRG-AS1 downregulation had the inverse effect in SHG44 cells. PTPRG-AS1 negatively regulated the expression of miR-185-5p in U373-MG and SHG44 cells and the expression of miR-185-5p was decreased in glioma tissues and cells. In addition, miR-185-5p overexpression suppressed proliferation, metastasis, colony formations, and autophagy, while inducing cell apoptosis in SHG44 cells. As expected, miR-185-5p depletion exhibited the inverse effect in U373-MG cells. Enhanced expression of miR-185-5p attenuated the effect of PTPRG-AS1 upregulation on U373-MG cells, while silencing of miR-185-5p undermined the effect of downregulation of PTPRG-AS1 on SHG44 cells. Our data disclosed that LncRNA PTPRG-AS1 was upregulated in glioma cells and tissues. PTPRG-AS1 regulated glioma proliferation, invasion, migration, apoptosis and autophagy by sponging miR-185-5p in vitro. A new signaling pathway PTPRG-AS1/miR-185-5p was first observed in glioma.
Introduction
Glioma is the most common and aggressive primary malignant intracranial tumor in the central nervous system.1–3 Glioma is graded from I to IV based on morphology and malignant behavior as specified by World Health Organization (WHO) classification.1,4 Most patients with glioma have poor prognosis and high death rates.5–7 Yet the molecular mechanisms during malignant progression of glioma remain largely unknown.Long noncoding RNAs (LncRNAs) are a set of RNAs which are longer than 200 nucleotides (nts). Many lncRNAs have been demonstrated to exert crucial regulation activities in biological processes of human cancers,8–12 including glioma.13 Recently, a growing number of lncRNAs have been found to be linked with invasion and migration of glioma. LncRNA urothelial carcinoma associated 1 (UCA1) was upregulated in glioma tissues and UCA1 knockdown inhibited proliferation, migration, and invasion in glioma cell lines U87 and U251, which was correlated with poor prognosis.14 The transcripts of the gene Colorectal Neoplasia Differentially Expressed (CRNDE) was also reported to be elevated in both glioma cell lines and tissues. Besides, CRNDE promoted cell growth, migration and invasion in vivo.15 In addition, the abnormal autophagy was related to a variety of cancers.16 For example, lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) promoted tumor proliferation and metastasis through the stimulation of autophagy in vitro.17 LncRNA HOX transcript antisense RNA (HOTAIR) promoted activation of autophagy in hepatocellular carcinoma (HCC) cells by upregulating the expression of the autophagy-related genes ATG3 and ATG7.18 In glioma, lncRNA growth arrest-specific 5 (GAS5) regulated cell growth, invasion, and migration. Meanwhile, GAS5 decreased the resistance of glioma cells to cisplatin by suppressing excessive autophagy through the activation of mammalian target of rapamycin (mTOR) signaling.19 Although many studies about the function of most lncRNAs in glioma have been conducted, many lncRNAs have not been completely understood. Recently, lncRNA protein tyrosine phosphatase, receptor type, G, antisense (PTPRG-AS1), the antisense of PTPRG tumor suppressor gene, which was reported to be aberrantly expressed in breast cancer and associated with cancer progression.20,21 However, the expression of lncRNA PTPRG-AS1 and its molecular mechanisms in glioma are poorly investigated.
The lncRNA-miRNA axis was involved in many cellular processes, such as cell development, proliferation, and cell autophagy through controlling gene expression patterns.22 It has been reported that miR-188-3p participated in the regulation of ATG7 expression and lncRNA autophagy promoting factor (APF) participated in the autophagy regulation by directly binding to miR-188-3p in vivo and vitro.23 However, whether lncRNA PTPRG-AS1 is involved in cell growth and autophagy by sponging microRNA-185-5p (miR-185-5p) in glioma needs to be further explored.
In our study, we reported that lncRNA PTPRG-AS1 was significantly upregulated in glioma cells compared with that of human astroglia cell line. Moreover, we found that PTPRG-AS1 affected glioma cell proliferation, invasion, migration, apoptosis and autophagy by sponging miR-185-5p in vitro. Thus, our study provided evidence that PTPRG-AS1/miR-185-5p axis was implicated in glioma progression.
Materials and methods
Clinical samples
Glioma tissues samples and matched adjacent normal tissues were obtained from glioma patients (n = 50) at Huaihe Hospital of Henan University. All patients have written informed consent and the study was approved by the Ethics Committee of Ethics Committee of Huaihe Hospital of Henan University. The correlation between PTPRG-AS1 expression and clinicopathological features of glioma patients was displayed in Table 1. This study was performed in strict accordance with the guidelines for the care and use of laboratory animals and was approved by the Institutional Animal Care and Use Committee of Huaihe Hospital of Henan University.| Clinicopathological features | Cases | The relative expression of PTPRG-AS1 | P value | |
|---|---|---|---|---|
| Low expression (%) | High expression (%) | |||
| Age | P>0.05 | |||
| <50 | 31 | 16 (51.6) | 15 (48.4) | |
| ≥50 | 19 | 8 (42.1) | 11 (57.9) | |
| Sex | P>0.05 | |||
| Male | 27 | 14 (51.9) | 13 (48.1) | |
| Female | 23 | 13 (56.5) | 10 (43.5) | |
| WHO stage | P<0.05 | |||
| I–II | 16 | 11 (68.8) | 5 (31.3) | |
| III–IV | 34 | 7 (20.6) | 27 (79.4) | |
| Tumor volume (cm) | P>0.05 | |||
| T < 3 | 20 | 11 (55.0) | 9 (45.0) | |
| T ≥ 3 | 30 | 14 (46.7) | 16 (53.3) | |
Cell lines and culture
Human embryonic kidney (HEK) 293T cell line, glioma cells (U373-MG, U87-MG, U251, and SHG44) and human astroglia cell line HEB were purchased from American Tissue Culture Collection (ATCC). U373-MG and U251 cell lines were p53 mutant phenotype, whereas U87-MG and SHG-44 were p53 wild type phenotype. The cells were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin. All cells were incubated at 37 °C in a humidified incubator with 5% CO2.Reagent and cell transfection
Cells (1 × 105) were seeded into each well of 6-well plates and incubated for 24 h. Cells were then transfected with PTPRG-AS1 small interfering RNA (si-PTPRG-AS1), siRNA negative control (si-NC), miR-185-5p mimic (miR-185-5p), mimic negative control (miR-NC), miR-185-5p inhibitor (anti-miR-185-5p), inhibitor negative control (anti-NC), pcDNA3.0 vector (NC) or pcDNA PTPRG-AS1 (PTPRG-AS1) using lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The transfected cells were prepared for the following experiments.Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay
Total RNAs were extracted from cultured cells or tissues using Trizol (Takara Biomedical Technology, Dalian, China). MiRNAs were isolated using miRNeasy Mini Kits (Sangon Biotech, Shanghai, China) reversely transcribed into complementary DNA (cDNA) using TaqMan® MicroRNA Reverse Transcription kit (Biosystems, Forster City, CA, USA). qPCR was performed using the TaqMan® Universal PCR Master Mix II (Biosystems), the specific primer sequences were as follows: PTPRG-AS1-F, 5′-GGGTAAGCGATCTACGCCC-3′, PTPRG-AS1-R, 5′-GTGGTTGCCCTCCTTAGGTC-3′; miR-185-5p-F, 5′-GAAGGATCCGCATGAGAGGGTGTTGGAATGC-3′, miR-185-5p-R, 5′-GGAGAATTCGTGCAGGGGCAGCAGACC-3′; U6-F, 5′-CTCGCTTCGGCAGCACA-3′, U6-R, 5′-AACGCTTCACGAATTTGCGT-3′; fold changes were calculated using the relative quantification (2−ΔΔCt) method.CCK assay
Cell proliferation assay was performed using Cell Counting Kit-8 (CCK8, Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer's instructions. Cells transfected with above-mentioned plasmids or RNAs were seeded into 96-well plates for 24 h, 48 h, 72 h and 96 h, 20 μL per well of CCK8 solution was added and incubated for another 4 h. Absorbance was determined at 490 nm using an Elx800 reader (Bio-Tek, Winooski, VT, USA).Colony formation assay
After transfection for 12 h, cells were seeded into 6-well plates at a density of 500 per well and cultured to form colonies in complete medium for another 14 days. The colonies were immobilized with 4% paraformaldehyde (Sigma, St. Louis, MO, USA) for 10 min, stained with crystal violet (Sigma) for 20 min and washed with PBS for 3 times. The total number of colonies (>50 cells per colony) was counted manually and the stained colonies were photographed using a microscope (Leica, Wetzlar, Germany).Transwell assay
Cell migration and invasion were evaluated using 24-well transwell chambers with 8 μm pore size polycarbonate membranes (Corning, NY, USA) according to the manufacturer's instructions. Harvested cells were re-suspended in serum-free medium and then seeded in the upper chamber on the top side of membrane, or the upper chambers were pre-coated with Matrigel solution (BD Biosciences, Franklin Lakes, NJ, USA) and incubated at 37 °C for 4 h before the invasion assay was performed. Migrated and invaded cells on the lower membrane surface were fixed with 4% paraformaldehyde (Sigma) and stained with crystal violet (Sigma). The number of migrated cells were counted in 5 randomly picked view under microscope (Leica).Flow cytometry
Cell apoptosis was analyzed using FITC Annexin V Apoptosis Detection Kit (BD Biosciences). Cells transfected with above-mentioned plasmids or oligos were collected and digested with trypsin, washed with PBS. Then 5 μL annexin V-FITC and 5 μL propidium iodide (PI) were added in cells in dark for 15 min at room temperature. Cell apoptotic rate was detected by a FACS Calibur flow cytometer with Cell Quest software (BD Biosciences).GFP-LC3 puncta assay
SHG44 cells were co-transfected with GFP-LC3 plasmid (Genomeditech, Shanghai, China) and miR-185-5p mimic or mimic negative control. Non-treated cells were acted as control. After 48 h post-transfection, cells were fixed by 4% paraformaldehyde (Sigma) and stained with 4′, 6-diamidino-2-phenylindole (DAPI, 1 μg mL−1, Sigma). Number of GFP-LC3 positive cells (>20 puncta as positive cell) as well as total cells in 5 randomly selected fields were counted and the autophagosome formation observed by using a fluorescence microscope (Olympus, Tokyo, Japan) was indicated by the percentage of GFP-LC3 positive cells.Western blot assay
Cells treated with above-mentioned plasmids or oligos were lysed in RIPA buffer (Beyotime, Shanghai, China) supplemented with protease inhibitor (Beyotime) at 4 °C for 30 min. The protein concentration was determined using the BCA™ Protein Assay Kit (Beyotime). Total protein samples (25 μg per lane) were separated using SDS-PAGE (Beyotime) and then transferred into polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% skim milk for 1 h at room temperature, the membranes were then incubated with the indicated primary antibodies against β-actin, p62, and LC3 (Abcam, Cambridge, UK) at 4 °C overnight. The membranes were then washed with PBST buffer and incubated with the secondary antibodies (HRP-conjugated goat anti-rabbit IgG, Abcam) for 1 h at room temperature. Protein bands were visualized using Pierce™ ECL Western Blotting Substrate (Beyotime) with the intensity analysis via Quantity One software (Bio-Rad, Hercules, CA, USA).Luciferase reporter assay
A wild-type (WT) fragment of PTPRG-AS1 harboring the miR-185-5p binding site and their mutated (MUT) seed sequences were purchased from Genomeditech and cloned into the pGL3 luciferase promoter plasmid (Promega, Madison, WI, USA), namely PTPRG-AS1-WT and PTPRG-AS1-MUT, which were co-transfected with mimic negative control, miR-185-5p mimic, miR-185-5p inhibitor, and inhibitor negative control into 293T cells using lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA). Cells were harvested at 48 h after transfection. The luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega) according the protocols.RNA immunoprecipitation (RIP) assay
The RIP assay was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore) according to manufacturer's instructions. Total cell lysates were incubated with magnetic beads and anti-Ago2 antibody (Abcam), and anti-IgG antibody (Abcam) was used as a negative control. The beads were washed and incubated with Proteinase K to digest the protein at 24 h after incubation. The RNA was purified by the phenol-chloroform-isoamyl alcohol reagent (Sigma) and detected by reverse transcription and PCR assays.Statistical analysis
All data were expressed as the mean ± SD with at least three independent replicates. Differences between groups were analyzed using the Student's t test. A p-value of less than 0.05 (denoted by * or #) was considered to be statistically significant.Results
LncRNA PTPRG-AS1 was involved in proliferation and apoptosis in glioma cells
Firstly, the expression of PTPRG-AS1 in glioma patients and normal volunteers (normal) were examined. We found that PTPRG-AS1 was highly expressed in glioma group compared with that of normal group (Fig. 1A). Moreover, survival rate of glioma patients with PTPRG-AS1 high expression was reduced compared with those with low level of PTPRG-AS1 (Fig. 1B). Besides, we also showed that the high expression of PTPRG-AS1 may be correlated with a high-grade tumor stage (Table 1). To investigate the role of PTPRG-AS1 in glioma in vitro, we examined the expression of PTPRG-AS1 in glioma cells (U373-MG, U87-MG, U251, and SHG44) and human astroglia cell line HEB using qRT-PCR (Fig. 1C). We found that PTPRG-AS1 was upregulated in glioma cells. The U373-MG cells with lower expression and SHG44 cells with higher expression were selected as objectives. Then, the results of transfection efficiency revealed that the expression of PTPRG-AS1 was enhanced in U373-MG cells transfected with pcDNA PTPRG-AS1, while PTPRG-AS1 was decreased in SHG44 cells transfected with si-PTPRG-AS1 (Fig. 1D and E). Our results showed that upregulation of PTPRG-AS1 promoted proliferation and increased the number of cell colony formations in U373-MG cells, whereas downregulation of PTPRG-AS1 inhibited proliferation and decreased the number of cell colony formations in SHG44 cells (Fig. 1F–I). In addition, we also disclosed that upregulation of PTPRG-AS1 decreased the apoptosis in U373-MG cells, whereas PTPRG-AS1 depletion exhibited an opposite effect in SHG44 cells (Fig. 1J and K). These results suggested that PTPRG-AS1 may be an oncogene in the development of glioma.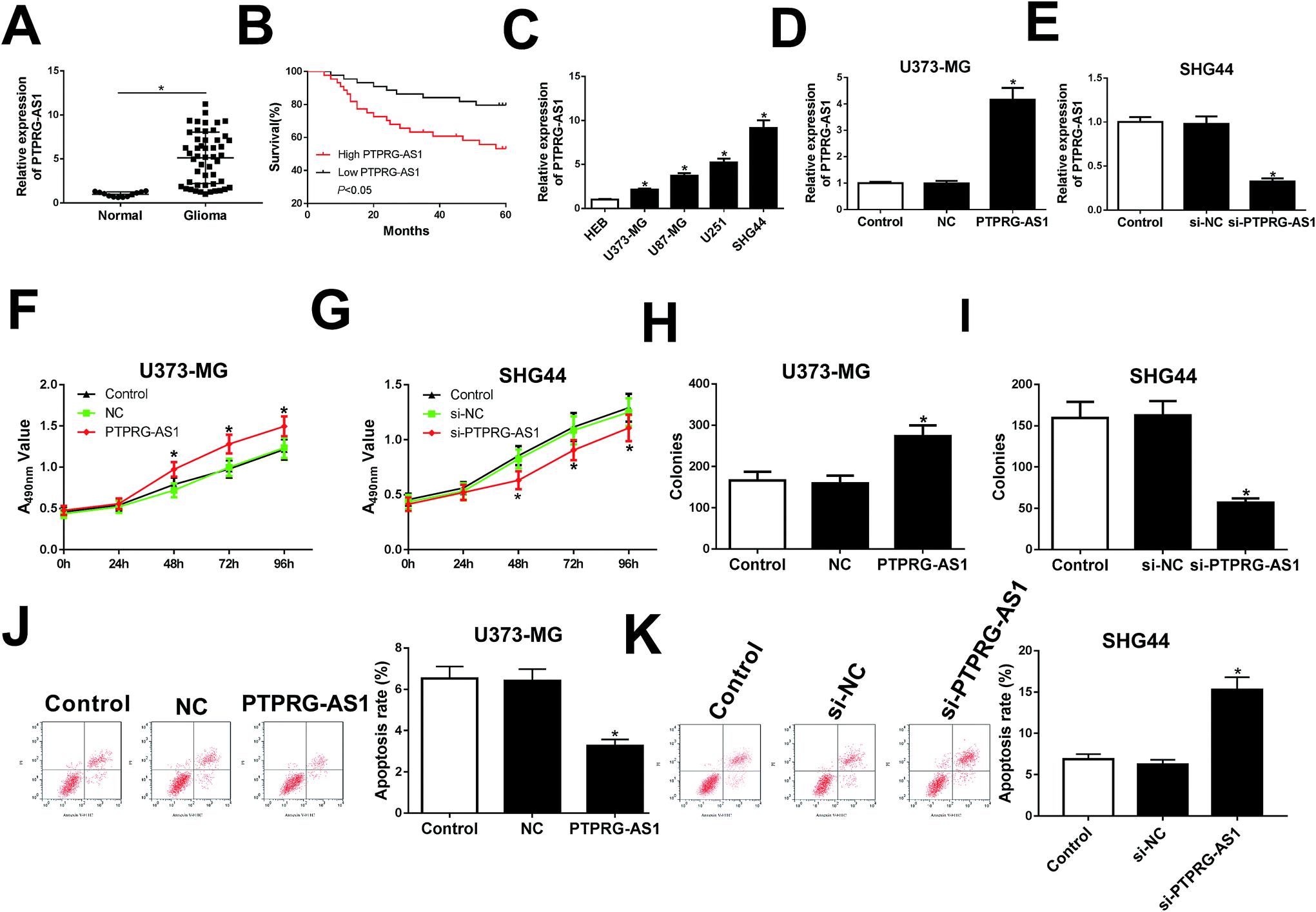 | ||
| Fig. 1 LncRNA PTPRG-AS1 was involved in proliferation and apoptosis in glioma cells. (A) The expression of PTPRG-AS1 in glioma tissues and matched adjacent normal tissues. (B) The correlation between the PTPRG-AS1 gene expression and overall survival in 50 patients with glioma. (C) qRT-PCR assay was performed to measure the expression of PTPRG-AS1 in glioma cells (U373-MG, U87-MG, U251, and SHG44) and human astroglia cell line HEB. (D and E) U373-MG cells were transfected with pcDNA3.0 vector or pcDNA PTPRG-AS1 and SHG44 cells were transfected with small interfering RNA negative control or si-PTPRG-AS1. (F–I) Cell proliferation ability was evaluated by CCK8 and colony formation assays. (J and K) Cell apoptotic rate was evaluated by flow cytometry. *P < 0.05. | ||
LncRNA PTPRG-AS1 regulated cell migration and invasion in U373-MG and SHG44 cells
To further explore the effect of PTPRG-AS1 on migration and invasion of glioma cells, U373-MG cells and SHG44 cells were introduced with pcDNA PTPRG-AS1 or si-PTPRG-AS1, respectively. We found that upregulation of PTPRG-AS1 promoted migration and invasion in U373-MG cells, whereas loss of PTPRG-AS1 suppressed migration and invasion in SHG44 cells (Fig. 2A and B). | ||
| Fig. 2 LncRNA PTPRG-AS1 regulated cell migration and invasion in U373-MG and SHG44 cells. U373-MG cells and SHG44 cells were treated as Fig. 1B and C. Cell migration (A) and invasion (B) abilities were evaluated by transwell assay. *P < 0.05. | ||
LncRNA PTPRG-AS1 was required for autophagy in U373-MG and SHG44 cells
Then, whether autophagy is a contributing factor to glioma cell growth was determined in the following experiments. Our results showed that forced expression of PTPRG-AS1 increased the green fluorescent protein microtubule-associated protein 1 light chain 3 (GFP-LC3) positive cells in U373-MG cells (Fig. 3A). Western blot further indicated that the expression of p62 was inhibited while the ratio of LC3II/LC3I was increased in U373-MG cells (Fig. 3B). By contrast, loss of PTPRG-AS1 decreased the GFP-LC3 positive cells in SHG44 cells (Fig. 3C). Moreover, downregulation of PTPRG-AS1 enhanced p62 expression and decreased the ratio of LC3II/LC3I in SHG44 cells (Fig. 3B and D). Those data further demonstrated that PTPRG-AS1 participated in cell autophagy in glioma cells.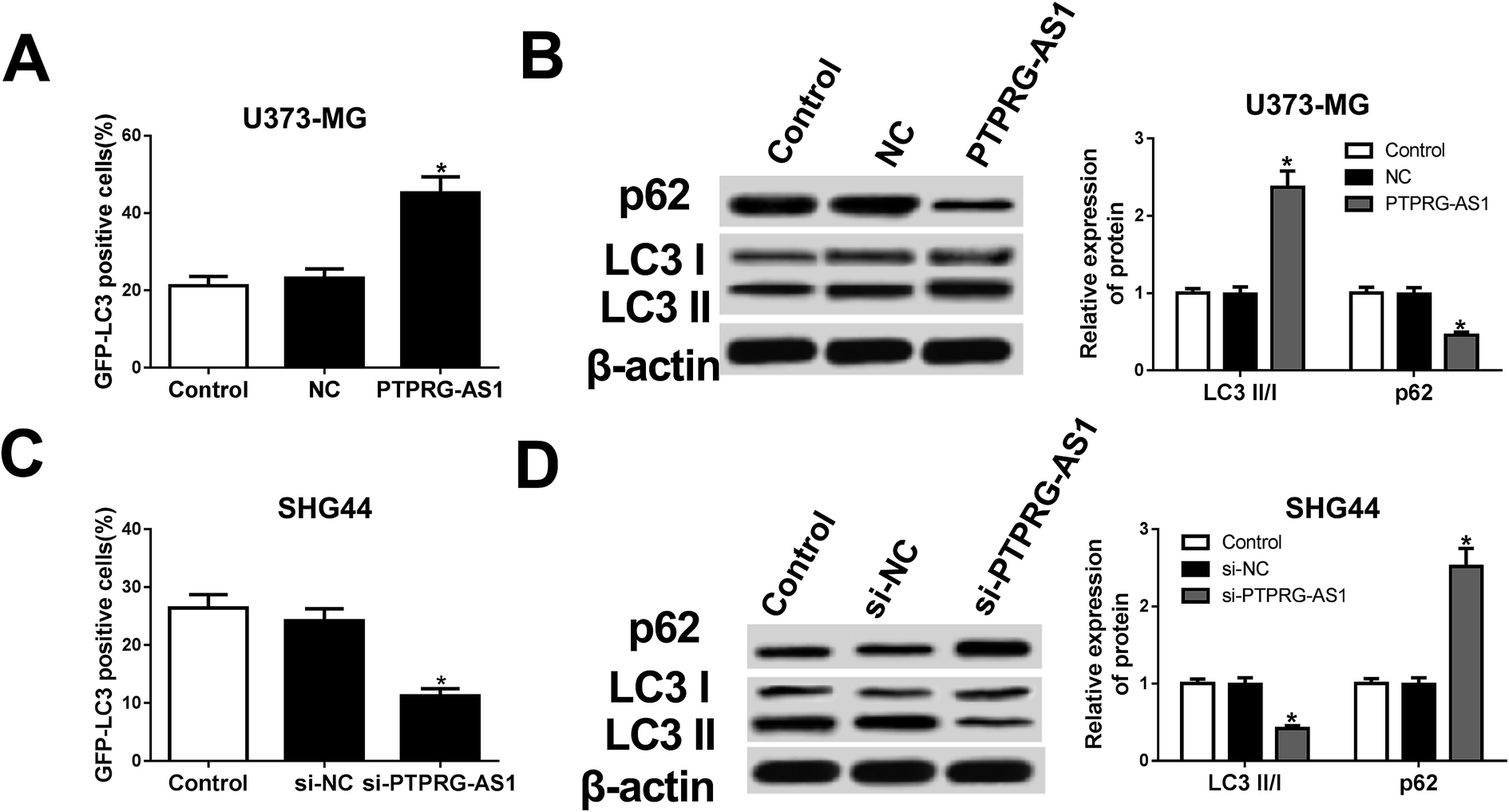 | ||
| Fig. 3 LncRNA PTPRG-AS1 was involved in autophagy in U373-MG and SHG44 cells. U373-MG cells and SHG44 cells were treated as Fig. 1B and C, respectively. (A) Autophagy formation was analyzed by transfection of GFP-LC3 into U373-MG cells. (B) Western blot was performed to detect the protein expressions of p62 and LC3 in U373-MG cells. (C) Autophagy formation was analyzed by transfection of GFP-LC3 into SHG44 cells. (D) Western blot was performed to detect the protein expressions of p62 and LC3 in SHG44 cells. *P < 0.05. | ||
LncRNA PTPRG-AS1 was interacted with miR-185-5p
To determine whether PTPRG-AS1 is interacted with miR-185-5p, bioinformatic software predicted that miR-185-5p has binding sites with PTPRG-AS1 (Fig. 4A). Luciferase reporter and RIP assays confirmed the relationship between them (Fig. 4B and C). In addition, upregulation of PTPRG-AS1 inhibited the expression of miR-185-5p in U373-MG cells, whereas PTPRG-AS1 knockdown promoted it in SHG44 cells (Fig. 4D and E). In addition, the expression of miR-185-5p in glioma patients was also detected. As demonstrated in Fig. 4F, decreased expression of miR-185-5p was observed in glioma group compared with that of normal group. Interestingly, the level of miR-185-5p was negatively correlated with that of PTPRG-AS1 in glioma tissues (Fig. 4G). Those findings suggested that miR-185 may be associated with the function of PTPRG-AS1 in glioma. | ||
| Fig. 4 LncRNA PTPRG-AS1 was interacted with miR-185-5p. (A) The binding sites between PTPRG-AS1 and miR-185-5p was predicted by bioinformatic software and the luciferase reporter plasmids containing the wild-type (WT) or mutated (MUT) PTPRG-AS1 binding sites of miR-185-5p were established. (B) The Luciferase activity was measured in 293T cells co-transfected with PTPRG-AS1-WT or PTPRG-AS1-MUT luciferase reporter and miR-185-5p mimic, mimic negative control, inhibitor negative control or miR-185-5p inhibitor. (C) The RIP assay was performed and expression of PTPRG-AS1 was detected in the samples bound to the Ago2 antibody or IgG in SHG44 cells. (D and E) U373-MG and SHG44 cells were treated as Fig. 1B and C, respectively. qRT-PCR assay was performed to measure the expression of miR-185-5p. (F) The level of miR-185-5p in glioma tissues and matched adjacent normal tissues. (G) The correlation between miR-185-5p and PTPRG-AS1. *P < 0.05. | ||
miR-185-5p regulated cell proliferation, migration, invasion, apoptosis, and autophagy in U373-MG and SHG44 cells
To understand the role of miR-185-5p in glioma cells, we transfected miR-185-5p mimic or mimic negative control into SHG44 cells. Meanwhile, miR-185-5p inhibitor and inhibitor negative control were introduced into U373-MG cells (Fig. 5A). Our results showed that increasing of miR-185-5p inhibited proliferation, migration, and invasion, while increased cell apoptotic rate in SHG44 cells. Interestingly, miR-185-5p depletion enhanced proliferation, migration, and invasion while blocked cell apoptosis in U373-MG cells (Fig. 5B–F). The GFP-LC3 positive cells and the ratio of LC3II/I were decreased, while expression of p62 was increased in SHG44 cells transfected with miR-185-5p mimic. However, the GFP-LC3 positive cells and the ratio of LC3II/I were increased, while expression of p62 was decreased in U373-MG cells transfected with miR-185-5p inhibitor (Fig. 5G and H).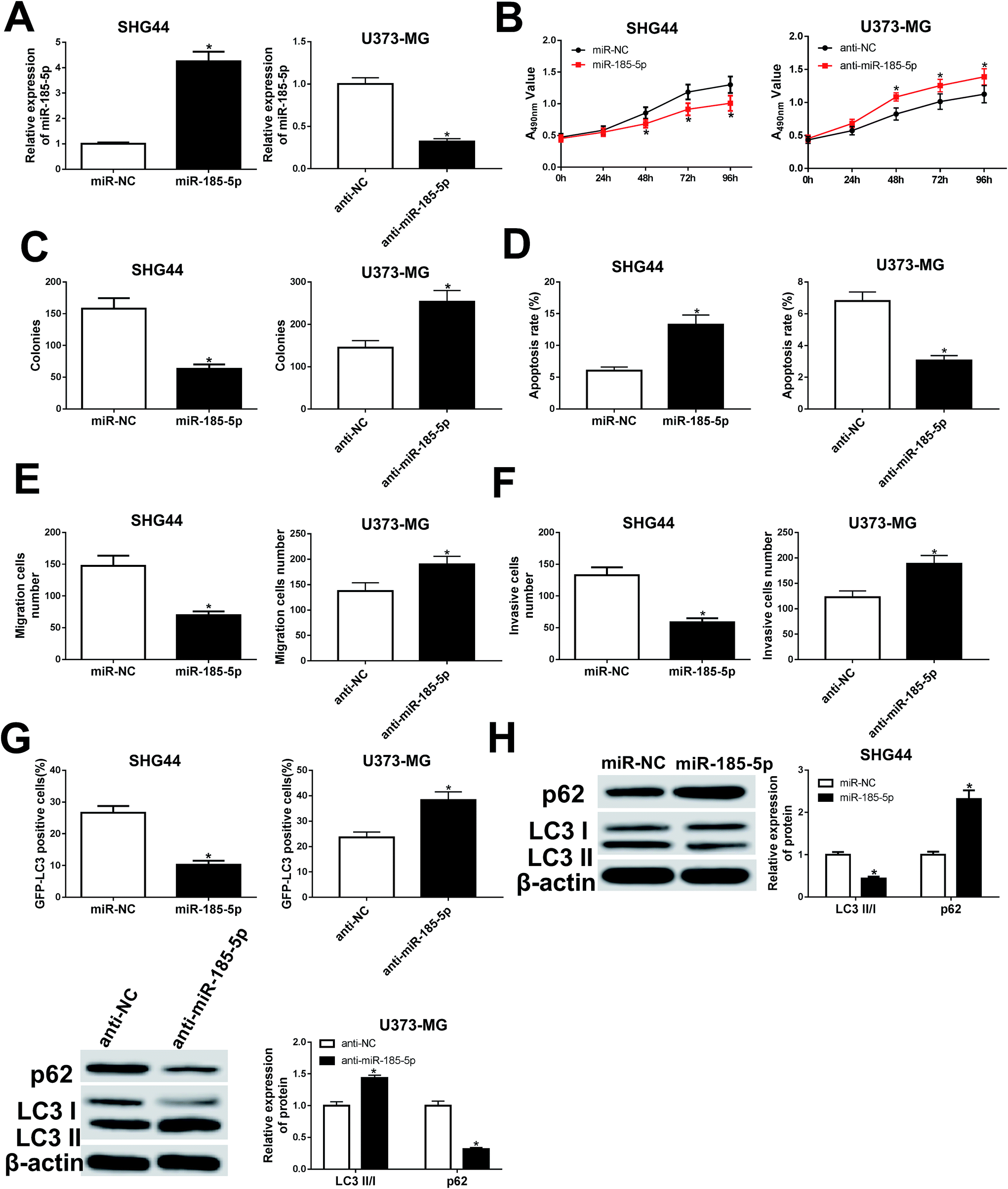 | ||
| Fig. 5 miR-185-5p regulated cell proliferation, migration, invasion, apoptosis, and autophagy in U373-MG and SHG44 cells. SHG44 cells were transfected with miR-185-5p mimic or mimic negative control. U373-MG cells were transfected with miR-185-5p inhibitor or inhibitor negative control. (A) qRT-PCR was performed to measure the expression of miR-185-5p. (B and C) Cell proliferation ability was evaluated by CCK8 and colony formation assays. (D) Cell apoptotic rate was evaluated by flow cytometry. (E and F) Cell migration and invasion abilities were evaluated by transwell assay. (G) Autophagy formation was analyzed by transfection of GFP-LC3 into SHG44 cells. (H) Western blot was performed to detect the protein expression of p62 and LC3. *P < 0.05. | ||
LncRNA PTPRG-AS1 participated in cell proliferation, migration, invasion, and apoptosis by sponging miR-185-5p in U373-MG and SHG44 cells
To further clarify whether PTPRG-AS1 plays oncogene role by sponging miR-185-5p, we co-transfected pcDNA PTPRG-AS1 and miR-185-5p mimic into U373-MG cells (Fig. 6A). Moreover, si-PTPRG-AS1 and miR-185-5p inhibitor were also co-transfected into SHG44 cells (Fig. 6A). Our results demonstrated that upregulation of PTPRG-AS1 promoted proliferation, invasion, and migration, while decreased apoptosis in U373-MG cells, which was attenuated by miR-185-5p mimic transfection (Fig. 6B–F). On the contrary, downregulation of PTPRG-AS1 inhibited proliferation, invasion, and migration, while increased apoptosis in SHG44 cells, which was also rescued by miR-185-5p inhibitor transfection (Fig. 6B–F).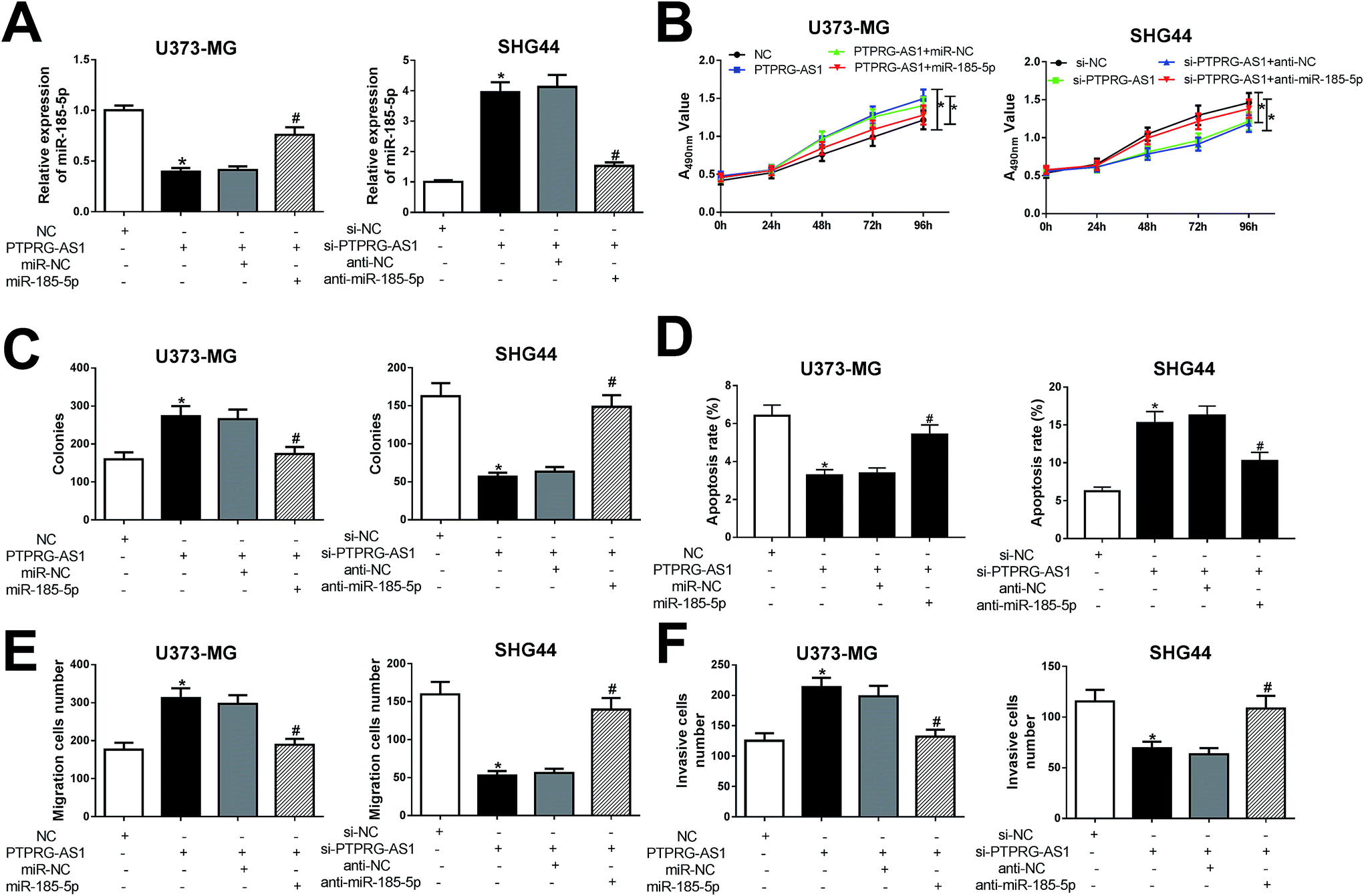 | ||
| Fig. 6 LncRNA PTPRG-AS1 regulated cell proliferation, migration, invasion, and apoptosis by sponging miR-185-5p in U373-MG and SHG44 cells. (A) U373-MG cells were transfected with pcDNA3.0 vector, pcDNA PTPRG-AS1, negative control mimic + pcDNA PTPRG-AS1, or miR-185-5p mimic + pcDNA PTPRG-AS1. SHG44 cells were transfected with small interfering RNA negative control, si-PTPRG-AS1, si-PTPRG-AS1 + negative control inhibitor, or si-PTPRG-AS1 + miR-185-5p inhibitor. (B and C) Cell proliferation ability was evaluated by CCK8 and colony formation assays. (D) Cell apoptotic rate was evaluated by flow cytometry. (E and F) Cell migration and invasion abilities were evaluated by transwell assay. *P < 0.05, #P < 0.05. | ||
LncRNA PTPRG-AS1 affected autophagy by sponging miR-185-5p in U373-MG and SHG44 cells
To determine whether PTPRG-AS1 affects autophagy through interacting with miR-185-5p, U373-MG cells and SHG44 cells were treated as Fig. 6. Our results showed that upregulation of PTPRG-AS1 increased the GFP-LC3 positive cells in U373-MG cells, which was significantly attenuated by miR-185-5p mimic transfection (Fig. 7A). However, downregulation of PTPRG-AS1 decreased the GFP-LC3 positive cells in SHG44 cells, which was markedly abated by miR-185-5p inhibitor (Fig. 7B). In addition, miR-185-5p overexpression undermined the effect of upregulation of PTPRG-AS1 on the expressions of LC3 and p62 (Fig. 7C). By contrast, loss of miR-185-5p also receded the effect of PTPRG-AS1 depletion on the expressions of LC3 and p62 (Fig. 7D).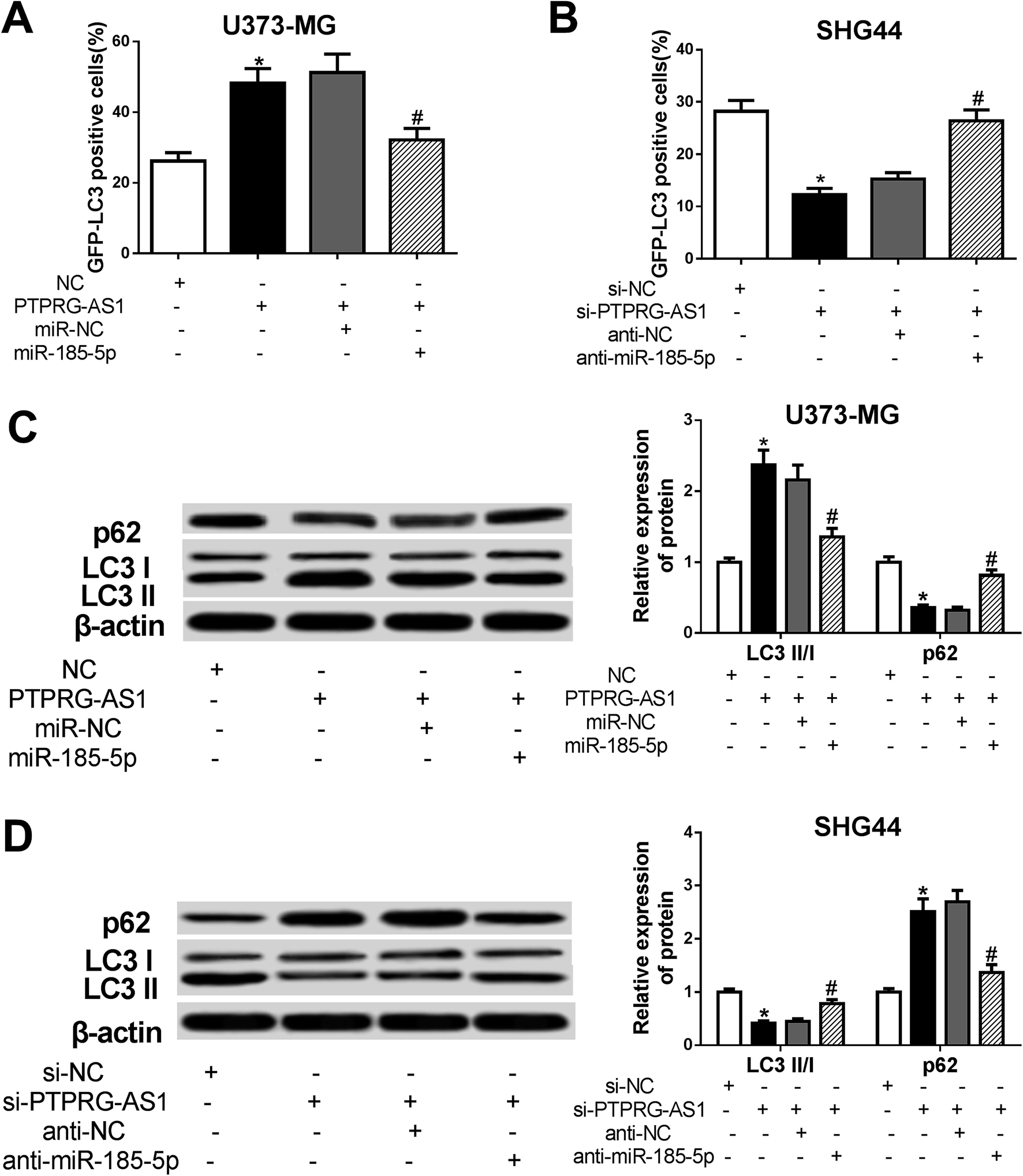 | ||
| Fig. 7 LncRNA PTPRG-AS1 affected autophagy by sponging miR-185-5p in U373-MG and SHG44 cells. U373-MG cells and SHG44 cells were treated as Fig. 6. (A and B) Autophagy formation was analyzed by transfection of GFP-LC3 into U373-MG cells and SHG44 cells. (C and D) Western blot was performed to detect the protein expressions of p62 and LC3 in U373-MG cells and SHG44 cells. *P < 0.05, #P < 0.05. | ||
Discussion
In the current study, we found that lncRNA PTPRG-AS1 was upregulated in glioma tissues, suggesting that PTPRG-AS1 may contribute to glioma progression. Moreover, it was observed that high level of PTPRG-AS1 was correlated with a high survival rate of glioma patients. Similarly, the level of PTPRG-AS1 was increased in cell lines U373-MG, U87-MG, U251, and SHG44 compared with that of human astroglia cell. Meanwhile, no obvious correlation between the expression of PTPRG-AS1 and p53 phenotype was observed. To further explore the role of PTPRG-AS1 in glioma, we knocked down the expression of PTPRG-AS1 in SHG44 cells while upregulated it in U373-MG cells. The results showed that PTPRG-AS1 knockdown inhibited the proliferation, invasion, and migration, while promoted apoptosis in SHG44 cells. Moreover, upregulation of PTPRG-AS1 promoted cell growth in U373-MG cells. Interestingly, the effect of PTPRG-AS1 on apoptosis in U373-MG is consistent with that of SHG44 cells, suggesting that p53 phenotype is not responsible for PTPRG-AS1-mediated apoptosis. Those observations demonstrated that PTPRG-AS1 may play oncogene role in glioma progression.Autophagy is an essential and conserved lysosomal degradation pathway, which controls the homeostasis of the cytoplasm by bulk degradation of unnecessary or dysfunctional cellular organelles and protein aggregates.24,25 Autophagy is considered as a double-edged sword. It can prolong the survival of cancer cells and enhance its resistance to apoptosis, and paradoxically, defective autophagy has been linked to increased of tumorigenesis, but the mechanisms behind this phenomenon are unclear. Polymerization of the polyubiquitin-binding protein p62/SQSTM1 (hereafter referred to as p62) yields protein bodies that either reside free in the cytosol and nucleus or occur within autophagosomes and lysosomal structures.26,27 Inhibition of autophagy lead to an increase of p62 protein levels.28 Furthermore, increasing evidence have shown that disruption of autophagy genes led to the decrease of proliferation and survival in oncogene transformed tumor cells and reduced tumor growth in vivo,29,30 suggesting that the inhibition of autophagy may contribute to cancer treatment. In the following experiments, GFP-LC3 puncta assay also demonstrated that GFP-LC3 positive cells were also decreased in PTPRG-AS1-knockdown cells, while they were increased in pcDNA PTPRG-AS1 cells. In addition, our study also found that PTPRG-AS1 could regulate the ratio of LC3II/I and the expression of p62, suggesting PTPRG-AS1 may be implicated in autophagy.
Mounting evidence has shown that lncRNAs serve as competing endogenous RNAs (ceRNAs) in the regulation of oncogenesis by binding to miRNAs. miR-185-5p was abnormally expressed in variety cancers and associated with tumorigenesis and metastasis by interacting with lncRNAs. Recent study has shown that knockdown of lncRNA UCA1 inhibited tumor growth and invasion via sponging miR-185-5p through Wnt/β-catenin signaling pathway.31 Retinal non-coding RNA3 (RNCR3 also known as LINC00599) knockdown suppressed cell proliferation, colony formation, and invasion in prostate cancer cell by increasing the expression of miR-185-5p.32 Nevertheless, still relatively little is known about the molecular mechanism of miR-185-5p and tumor aggressiveness in glioma. We found that miR-185-5p was predicted to have binding sites with PTPRG-AS1 through bioinformatic software. Furthermore, the relationship between PTPRG-AS1 and miR-185-5p was authenticated by luciferase reporter and RIP assays. We thought that miR-185-5p may be required for PTPRG-AS1 in regulating biological process in the development of glioma. In this report, miR-185-5p expression was downregulated in glioma tissues and a negative correlation between miR-185-5p and PTPRG-AS1 in glioma tissues was also observed. Moreover, upregulation of miR-185-5p suppressed proliferation, invasion, migration, and autophagy, while promoted apoptosis in SHG44 cells. Additionally, miR-185-5p depletion has inverse effect on proliferation, apoptosis, autophagy, invasion, and migration in U373-MG cells. We further demonstrated that upregulation of PTPRG-AS1 inhibited miR-185-5p expression, whereas downregulation of PTPRG-AS1 promoted miR-185-5p expression. Moreover, our study disclosed that miR-185-5p inhibitor weakened the tumor-suppressive effect of PTPRG-AS1 knockdown on SHG44 cells, and miR-185-5p mimic attenuated the oncogene effect of upregulation of PTPRG-AS1 on U373-MG cells. These results showed that PTPRG-AS1 regulated proliferation, apoptosis, invasion, and migration by sponging miR-185-5p. Our study further demonstrated that PTPRG-AS1 also regulated the ratio of LC3II/I and the expression of p62 through interacting with miR-185-5p. Therefore, our study manifested that lncRNA PTPRG-AS1 may regulate the proliferation, apoptosis, migration, invasion and autophagy in glioma cells by sponging miR-185-5p.
There are some limitations in this report. For example, we only explored the roles of PTPRG-AS1/miR-185-5p axis in the proliferation, invasion, migration, apoptosis and autophagy of glioma cells (U373-MG and SHG44). Moreover, the influence of PTPRG-AS1 and miR-185-5p, alone or in combination, on the development of glioma needs to be further investigated in other glioma cells and xenograft models. Although our results showed no correlation between the expression and function of PTPRG-AS1 and p53 phenotype, whether lncRNA PTPRG-AS1 is associated with the other phenotypic and genomic features of those cells should be investigated in future.
Taken together, our study first found that lncRNA PTPRG-AS1 was upregulated in glioma cell lines (U373-MG, U87-MG, U251, and SHG44). PTPRG-AS1 regulated glioma growth by affecting cell invasion, migration, apoptosis and autophagy in vitro. A new signaling pathway PTPRG-AS1/miR-185-5p was first observed.
Fundings
This study was approved by the Science and Technology Research Project of Henan Province (Grant no. 182102311180).Conflicts of interest
The authors have no conflict of interest to declare.References
- D. N. Louis, H. Ohgaki, O. D. Wiestler, W. K. Cavenee, P. C. Burger, A. Jouvet, B. W. Scheithauer and P. Kleihues, Acta Neuropathol., 2007, 114, 97–109 CrossRef PubMed.
- E. A. Maher, F. B. Furnari, R. M. Bachoo, D. H. Rowitch, D. N. Louis, W. K. Cavenee and R. A. DePinho, Genes Dev., 2001, 15, 1311–1333 CrossRef CAS PubMed.
- L. L. Morgan, Neuro-Oncology, 2015, 17, 623–624 CrossRef PubMed.
- D. N. Louis, A. Perry, G. Reifenberger, A. von Deimling, D. Figarella-Branger, W. K. Cavenee, H. Ohgaki, O. D. Wiestler, P. Kleihues and D. W. Ellison, Acta Neuropathol., 2016, 131, 803–820 CrossRef PubMed.
- Y. Qin, W. Chen, B. Liu, L. Zhou, L. Deng, W. Niu, D. Bao, C. Cheng, D. Li, S. Liu and C. Niu, Theranostics, 2017, 7, 1663–1673 CrossRef CAS PubMed.
- M. L. Goodenberger and R. B. Jenkins, Cancer Genet., 2012, 205, 613–621 CrossRef CAS PubMed.
- B. K. Rasmussen, S. Hansen, R. J. Laursen, M. Kosteljanetz, H. Schultz, B. M. Nørgård, R. Guldberg and K. O. Gradel, J. Neuro-Oncol., 2017, 135, 571–579 CrossRef PubMed.
- M. Huarte, Nature Medicine, 2015, 21, 1253–1261 CrossRef CAS PubMed.
- P. F. Li, S. C. Chen, T. Xia, X. M. Jiang, Y. F. Shao, B. X. Xiao and J. M. Guo, World J. Gastroenterol., 2014, 20, 5411–5419 CrossRef CAS PubMed.
- Y. Liu, S. Sharma and K. Watabe, Front. Biosci., Scholar Ed., 2015, 7, 94–108 CrossRef.
- E. S. Martens-Uzunova, R. Bottcher, C. M. Croce, G. Jenster, T. Visakorpi and G. A. Calin, Eur. Urol., 2014, 65, 1140–1151 CrossRef CAS PubMed.
- N. Ye, B. Wang, Z. F. Quan, S. J. Cao, X. T. Wen, Y. Huang, X. B. Huang, R. Wu, X. P. Ma and Q. G. Yan, Asian Pac. J. Cancer Prev., 2014, 15, 5993–5997 CrossRef PubMed.
- X.-Q. Zhang and G. K.-K. Leung, Neurochem. Int., 2014, 77, 78–85 CrossRef CAS PubMed.
- Y. Sun, J.-G. Jin, W.-Y. Mi, W. Hao, S.-R. Zhang, Q. Meng and S.-T. Zhang, Oncol. Res., 2018, 26, 103–110 CrossRef PubMed.
- Y. Wang, Y. Wang, J. Li, Y. Zhang, H. Yin and B. Han, Cancer Lett., 2015, 367, 122–128 CrossRef CAS PubMed.
- Y. Kondo, T. Kanzawa, R. Sawaya and S. Kondo, Nat. Rev. Cancer, 2005, 5, 726–734 CrossRef CAS PubMed.
- L. Li, H. Chen, Y. Gao, Y. W. Wang, G. Q. Zhang, S. H. Pan, L. Ji, R. Kong, G. Wang, Y. H. Jia, X. W. Bai and B. Sun, Mol. Cancer Ther., 2016, 15, 2232–2243 CrossRef CAS PubMed.
- L. Yang, X. Zhang, H. Li and J. Liu, Mol. BioSyst., 2016, 12, 2605–2612 RSC.
- J.-F. Huo and X.-B. Chen, J. Cell. Biochem., 2019, 120, 6127–6136 CrossRef CAS PubMed.
- M. Iranpour, M. Soudyab, L. Geranpayeh, R. Mirfakhraie, E. Azargashb, A. Movafagh and S. Ghafouri-Fard, Tumor Biol., 2016, 37, 2933–2940 CrossRef CAS PubMed.
- M. Soudyab, M. Iranpour and S. Ghafouri-Fard, Arch. Iran. Med., 2016, 19(7), 508–517 Search PubMed.
- Z. Xu, Y. Yan, L. Qian and Z. Gong, Oncol. Rep., 2017, 37, 1359–1366 CrossRef CAS PubMed.
- K. Wang, C. Y. Liu, L. Y. Zhou, J. X. Wang, M. Wang, B. Zhao, W. K. Zhao, S. J. Xu, L. H. Fan, X. J. Zhang, C. Feng, C. Q. Wang, Y. F. Zhao and P. F. Li, Nat. Commun., 2015, 6, 6779 CrossRef CAS PubMed.
- E. White, J. M. Mehnert and C. S. Chan, Clin. Cancer Res., 2015, 21, 5037–5046 CrossRef CAS PubMed.
- K. R. Parzych and D. J. Klionsky, Antioxid. Redox Signaling, 2014, 20, 460–473 CrossRef CAS PubMed.
- H. Wei, C. Wang, C. M. Croce and J. L. Guan, Genes Dev., 2014, 28, 1204–1216 CrossRef CAS PubMed.
- Y. Katsuragi, Y. Ichimura and M. Komatsu, FEBS J., 2015, 282, 4672–4678 CrossRef CAS PubMed.
- G. Bjorkoy, T. Lamark, A. Brech, H. Outzen, M. Perander, A. Overvatn, H. Stenmark and T. Johansen, J. Cell Biol., 2005, 171, 603–614 CrossRef PubMed.
- Y. F. Peng, Y. H. Shi, Z. B. Ding, A. W. Ke, C. Y. Gu, B. Hui, J. Zhou, S. J. Qiu, Z. Dai and J. Fan, Autophagy, 2013, 9, 2056–2068 CrossRef CAS PubMed.
- T. F. Fan, L. L. Bu, W. M. Wang, S. R. Ma, J. F. Liu, W. W. Deng, L. Mao, G. T. Yu, C. F. Huang, B. Liu, W. F. Zhang and Z. J. Sun, Oncotarget, 2015, 6, 43581–43593 Search PubMed.
- X. Chen, J. Gao, Y. Yu, Z. Zhao and Y. Pan, Gene, 2018, 676, 298–305 CrossRef CAS PubMed.
- C. Tian, Y. Deng, Y. Jin, S. Shi and H. Bi, Am. J. Transl. Res., 2018, 10, 1562–1570 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |