
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSNP discovery and functional annotation in the Panax japonicus var. major transcriptome
Jian Lia,
Ding-Ping Baia and
Xi-Feng Zhang *b
*b
aFujian Key Laboratory of Traditional Chinese Veterinary Medicine and Animal Health, Fujian Agriculture and Forestry University, Fuzhou 350002, China
bCollege of Biological and Pharmaceutical Engineering, Wuhan Polytechnic University, Wuhan 430023, China. E-mail: zhangxf9465@163.com
First published on 10th July 2019
Abstract
Due to the lack of a Panax japonicus var. major reference genome, we assembled a reference transcriptome from P. japonicus C. A. Mey transcriptome sequencing data, and 203![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 283 unigenes were obtained. In this study, with the assistance from the Trinity, Bowtie2 and SAMtools softwares, 218465 single nucleotide polymorphisms (SNPs) were identified by mapping the Illumina sequences to the reference transcriptome. The SNP forms included 126262 transformations and 92203 transversions. A large number of SNP loci were associated with triterpenoid saponin synthesis: 54 SNPs were associated with cytochrome P450, one with glycosyl transferase and 94 with the biosynthesis of the triterpenoid saponin backbone.
283 unigenes were obtained. In this study, with the assistance from the Trinity, Bowtie2 and SAMtools softwares, 218465 single nucleotide polymorphisms (SNPs) were identified by mapping the Illumina sequences to the reference transcriptome. The SNP forms included 126262 transformations and 92203 transversions. A large number of SNP loci were associated with triterpenoid saponin synthesis: 54 SNPs were associated with cytochrome P450, one with glycosyl transferase and 94 with the biosynthesis of the triterpenoid saponin backbone.
Introduction
Transcriptome sequencing (RNA-seq) is an efficient method to reveal the specific period and global gene expression of individual genes in a particular organism.1 This applies especially in the cases of limited genome sequence information and turn group sequenced gene coding region of lay particular stress on, rich in GC stitching is relatively easy. With the development of high-throughput sequencing, many species have been investigated at the molecular level. The discovery of single nucleotide polymorphisms (SNPs) plays key roles in the studies of disease treatment, genetics and evolution in animal and plant breeding.2The availability of high-throughput sequencing methods has led to the discovery of thousands to millions of SNPs in diverse organisms, particularly humans, model experimental organisms and agriculturally important plants and animals. Since SNPs provide a powerful tool for the discovery of high-risk groups, identification of disease genes, design and testing of drugs and basic biological research, they have become important in the application of the Human Genome Project.3
Panax japonicus var. major, a perennial herb in the Araliaceae family, is mainly distributed in Shaanxi, Gansu, Anhui, Zhejiang, Jiangxi, Fujian, Hunan, Hubei, Guangxi, Tibet and other places. It is a traditional medicine widely used in China. It promotes blood circulation and has anti-inflammatory and anti-oxidant activities; it is also responsible for hemostasis. Moreover, it is used to treat a variety of diseases, as documented in the Pharmacopoeia of the People's Republic of China.4 Panax japonicus var. major, P. ginseng C. A. Mey, P. quinquefolius and P. pseudoginseng are closely related, and all share similar chemical compositions: mainly, saponins, polysaccharides, volatile oils, amino acids, trace elements and many types of active components.5
Our main purpose was to develop and examine the SNP markers of P. japonicus var. major to enhance and accelerate its breeding via genomic selection. To date, the reference sequence to P. japonicus var. major, which is needed for SNP loci identification of the interested gene, has not been completed. Thus, we used the transcriptome data of the traditional Chinese medicinal plant P. japonicus, which is closely related, as a reference sequence for mapping P. japonicus var. major.6
Results
Illumina sequencing and de novo assembly
In this study, we prepared three biological replications of P. japonicus for sequencing with the Illumina platform. Illumina Hiseq2000 high-throughput sequencing resulted in a total of 155862844 effective reads with an average length of 94.74 bp (NCBI SRA accession: SRP062943) and about a total number of 15.6 Gbp nucleotides. All the clean reads were assembled with the Trinity software, and 188914 unigenes with total length of 117.1 Mb were obtained. The average length of the unigenes was 620 bp and the N50 length was 941 bp. There were 62240 and 29425 unigenes with lengths of over 500 bp and 1000 bp, respectively. Also, the length of the unigenes was in the range from 201 bp to 16000 bp. The frequency distribution of the GC content had an optimum of 35.54% (Fig. 1). The length of the unigenes was evenly distributed. From the analysis of the length distribution characteristics of all the unigenes, we found that the largest proportion of unigenes was 200–400 bp in length, representing 57.41%. Compared to the sequencing data of P. ginseng C. A. Meyer transcriptome assembled in 2013 (which afforded 2423076 reads and 45846 unigenes),7 we had deeper sequencing and recovered more unigenes (Table 1).
 | ||
| Fig. 1 GC content frequency distribution. | ||
| All (≥200 bp) | ≥500 bp | ≥1000 bp | N50 | N90 | Total length | Max length | Min length | Average length | |
|---|---|---|---|---|---|---|---|---|---|
| Transcript | 531296 |
296826 |
173132 |
1472 | 380 | 488565172 |
16000 |
201 | 919.57 |
| Unigene | 188914 |
62315 |
29425 |
941 | 258 | 117148035 |
16000 |
201 | 620.11 |
Unigene functional annotation
A genetic similarity comparison was performed with the BLAST (Basic Local Alignment Search Tool) algorithm. Of the total 62240 unigenes, the homologous matching information was 32003 (51.42%) in the Nr database, 18455 in the Swiss-Prot database, 10381 in the KOG (EuKaryotic Orthologous Group) database, 21278 in the GO (Gene Ontology) database, and 8369 in the KEGG (Kyoto Encyclopedia of Genes and Genomes) database; 30237 unigenes lacked functional annotation (Table 2).
| Database | Match number | Match rate (%) |
|---|---|---|
| Total unigenes | 62240 |
100.00 |
| Nr | 32003 |
51.42 |
| Swiss-Prot | 18455 |
29.65 |
| KOG | 10381 |
32.44 |
| GO | 21278 |
34.19 |
| KEGG | 8369 | 13.45 |
| Unknown | 30237 |
48.58 |
To eliminate the influence of the differences in the lengths of the genes and sequencing depths, RPKM was used to calculate the level of gene expression. In measuring the amount of gene expression, if calculating the amount by mapping to the read number, statistics is not a director. This is because in random sampling, longer gene sequences are more likely to be extracted than shorter gene sequences. Thus, sequencing would falsely indicate a high expression of longer genes. The RPKM value corrected this for the P. japonicus transcriptome expression quantity. Thus, the RPKM value was applied as an index to select the validation of SNP loci with an RPKM threshold value ≥ 3.8 Based on the Qual value in the VCF file of SNP calling and RPKM value, 10 variant loci were selected for verification. Due to the lack of reference genome information, the construction of the transcriptome model was very difficult. Especially, the stitching accuracy and splicing length of the sequencing technology greatly depend on the sequencing depth. At present, China has launched a medicinal plant transcriptome study; however, depth of sequencing and the number of genes detected are required for improvement. In this study, we sequenced rhizomes of P. japonicus var. major considering the limitation of Panax genus transcriptome data and that there is no public reference sequence for SNP. Panax japonicus var. major is a variation of the traditional Chinese medicine plant P. japonicus; therefore, we used P. japonicus unigenes as the reference sequences for P. japonicus var. major. This is because the difference between different transcripts can be detected.9,10 The Bowtie2 software was used to compare the P. japonicus var. major and P. japonicus unigenes, and the result indicated an average matching rate of around 70%. As the congener plant, the evolution type of P. japonicus var. major was in ancient groups, and the evolution relationship with the species in this group such as P. ginseng, American ginseng, and P. notoginseng plants was established. Although the biosynthesis of saponins is unclear, the key functions in the level of the genes in sequence are quite similar. Squalene epoxidase (comp164607_c0_seq3), dammarenediol-II-synthase (comp159106_c2_seq21) and beta-amyrin synthase (comp158446_c0_seq8) genes are important genes of P. japonicus var. major. Their respective DNA sequences showed 99, 99 and 98% identities with those of P. ginseng; 99, 99 and 94% with those of American ginseng; and 95, 95 and 95% with those of P. notoginseng. However, cytochrome P450 and glycosyl transferase with modifying functions, both in the form of a gene family, exist in plants, and there is no strict sequence consistency between different species. For example, the 18 known glycosyl transferase genes showed only 63% identity of their genetic sequences among ginseng, American ginseng and P. notoginseng. Saponins in different plant metabolic regulations are generally visible. Thus, the matching rate at 70% was available.
With the application of the SAMtools software, there were 371358 SNPs in the transcriptome sequencing of P. japonicus var. major. To ensure the accuracy of SNP loci, screening SNPs should ensure that the coverage of two transcripts is greater than the sum of 20 contigs and candidate SNP loci have at least 5 bp of conserved sequences on both sides. The large-scale high-throughput sequencing resulted in a total of 371358 variant loci. According to the above conditions, the screening of candidate SNPs determined a total of 218465 SNP loci, including 126262 transitions and 92203 transversions.11 In the transition loci, the T/C and A/G transitions represented 63165 and 63097 loci, respectively. In the transversion loci, the A/T, G/T, A/C, and G/C transversions represented 29574, 22910, 22750 and 16969 loci, respectively (Fig. 2). The functional annotations of unigenes with 10 SNP loci are summarized in Table 3. In the Nr database, six of the 10 SNP loci were functionally annotated as cytochrome P450, which is a terminal oxygenase and participates in the biological internal sterol hormone synthesis.12 Cytochrome P450 can affect metabolism and pharmacodynamics, and the SNPs can cause an alteration (loss or gain) of the functions.13 The SNPs of cytochrome P450 will be a key factor in influencing the function of P. japonicus var. major in therapy processing.
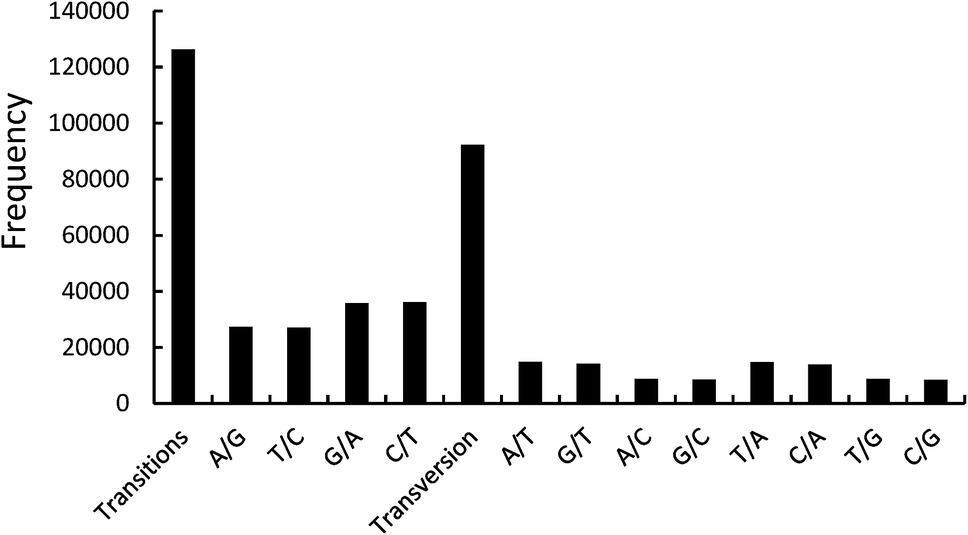 | ||
| Fig. 2 Distribution of SNP variants. | ||
| Query name | SNP form | Annotation | RPKM |
|---|---|---|---|
| Comp179123_c0_seq1_zzs | T/G | Cytochrome P450 (Panax notoginseng) | 27.4 |
| Comp179123_c0_seq1_zzs | C/A | Cytochrome P450 (Panax notoginseng) | 27.4 |
| Comp171079_c0_seq17_zzs | T/G | Cytochrome P450 (Panax ginseng) | 38.2 |
| Comp171079_c0_seq17_zzs | A/G | Cytochrome P450 (Panax ginseng) | 38.2 |
| Comp170147_c0_seq6_zzs | C/A | Cytochrome P450 (Panax notoginseng) | 4.65 |
| Comp167639_c0_seq17_zzs | A/G | HMG-CoA (Eleutherococcus senticosus) | 12.17 |
| Comp167465_c1_seq6_zzs | T/A | Squalene synthase (Panax notoginseng) | 19.44 |
| Comp178663_c2_seq2_zzs | T/A | Acetyl-CoA,C-acetyltransferase protein (Camellia oleifera) | 77.47 |
| Comp178663_c2_seq2_zzs | G/A | Acetyl-CoA,C-acetyltransferase protein (Camellia oleifera) | 77.47 |
| Comp160382_c0_seq3_zzs | A/C | Cytochrome P450 (Panax notoginseng) | 6.49 |
Squalene synthase had one annotation in the Nr database. The syntheses of triterpenoid saponins, sterols, cholesterol and other terpenes in Panax are all through squalene synthase catalysis.
Conclusion
Traditional Chinese medicine is the creation and accumulation of Chinese civilization for thousands of years, and is the wisdom crystallization of the Chinese nation. Traditional Chinese medicine and Chinese herbology are complementary to each other. In recent years, the demand for Panax has been increasing, and plant ecosystems are being destroyed by excessive excavation. Due to the rapid development of molecular biology, research on the molecular markers of the Panax genus and the use of important economic characteristics for molecular genetic markers to select high-quality P. japonicus var. major are particularly important. The development of P. japonicus var. major SNP molecular markers has great potential in promoting its genetics and breeding, and a large number of SNP loci can also allow large-scale tag scanning. In the future, we will further expand the sample size to validate the accuracy of other sites.The gene chip method is also currently popular, in which the gene chip is a probe to sample for mRNA sequence information using known sequence mRNA hybridization. To date, the mRNA of P. japonicus var. major has not been reported, and new mRNA cannot be detected without the corresponding gene chip probe sequences. Also, Panax japonicus var. major has no reference sequence; thus, we compared its gene sequence with P. japonicus. This not only resulted in the identification of a large number of SNP loci, but also provided evidence of the genetic and evolutionary relationship between the two species.
At present, there are lots of SNP calling softwares, which include GATK and SAMtools. Although the description of the SNP results of VCF files in GATK gives the best support, the function of SAMtools is more powerful. For SNP calling, SAMtools and GATK could be used together to finish the work accurately.
Irrespective of variation calling or ChIP-seq, the first step of data analysis is to compare the reads to the genome. The premise of the work is reads being mapped successfully. However, some reads could not be identified in the reference genome and were termed ‘unmapped reads’ because of the individual differences, differences between reference genomes, and the quality of the reference genome itself. Normally, these types of data would be removed, but they also contain a large amount of sequence information and are worthy of analysis. Individual data was compared with reference genome sequencing by the assembly software to Unmapped reads splicing locally. Comparing the contigs and database, reads reached the purpose of gene structure prediction and gene functional annotation. The analysis process is exhibited in Fig. 3. Mace et al. identified new genes in sorghum through the assembly of unmapped reads and the achieved results showed that their new breed guinea-margaritiferums is a sorghum variety containing mostly new genes; its genetic diversity is unique and it has great research value.14 Our database contained many unmapped reads and further analysis will be conducted to verify more valuable information from them.
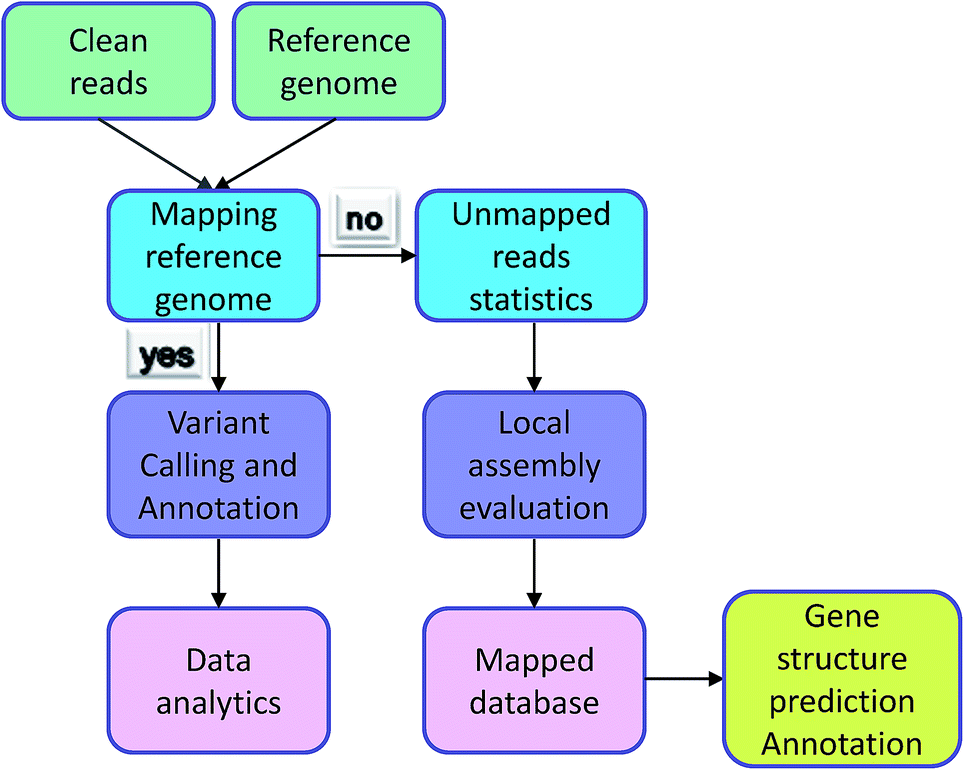 | ||
| Fig. 3 Unmapped reads analysis. | ||
Methods
Illumina sequencing
Illumina sequencing is a useful tool to uncover the character of an organism. With its development and improvement, the bioinformatics is promoted. It mainly includes the following processes: extracting nucleotides (DNA or RNA), quality evaluation of the extracted nucleotides, quantifying the library, sequence template amplification and fixed sequencing primers.Preparation of plant samples
We collected plant materials of P. japonicus var. major from 6 year-old plants in Enshi, Hubei Province of China. The rhizomes were harvested from more than ten plants with three biological replications. The rhizomes of each sample were separated, cleaned with sterile water and then stored promptly in liquid nitrogen.RNA isolation and library construction
Total RNA was extracted from Panax japonicus var. major rhizomes using the RNeasy Mini Kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's protocol. The integrity and quantity of the total RNA were evaluated with 1% agarose gel electrophoresis and ultraviolet spectrophotometry (DU800, Beckman Coulter, USA). The mRNAs with a poly(A) tail were fragmented into short fragments (about 200 bps), which were used as the template to synthesize the first strand of cDNA. The library was prepared with the TruSeq RNA Sample Preparation kit v2 (Illumina, USA).De novo assembly
The Illumina Hiseq 2000 platform (Illumina, California, USA) was used for sequencing of the cDNA library.15 Paired-ended reads with an average length of 94.74 bp were obtained. Before assembly, we filtered out the reads containing N's, unpaired reads and other low-quality reads. After quality control, the clean reads were assembled with the Trinity software (trinityrnaseq_r2013_08_14). Clustering analysis of differentially expressed genes was performed in Trinity using analyze_diff_expr.pl. Following the assembly, further sequence splicing and redundant sequences were removed using the sequence clustering software TGICL.16,17 The sequence number and length are important evaluations of assembly quality.Short reads mapped by Bowtie2
The Bowtie2 and Bwa softwares were used for sequence alignment. The level of gene expression was calculated by RPKM (reads per kilobase transcriptome per million reads) to eliminate the effect of gene length and differences in sequencing.18 Bowtie2 mainly gives the length of 50–1000 bp reads mapped to the genome and generates the SAM file format of the comparison results. Mapping reads to the genome is the first step in many analyses, such as variation calling, ChlP-seq (Chromatin Immunoprecipitation-sequencing), RNA-seq (RNA-sequencing) and BS-seq (Bisulfite-sequencing). Bowtie2 was used for sequence alignment and the first step was to generate the reference sequence index database.19 Bowtie2 produced the results of alignment files in the SAM format and at the end of the alignment, in standard error output.20There are many variants of calling software, and SAMtools and GATK (Genome Analysis Toolkit) are currently the most popular to discover SNPs and INDELs (insertion and deletions), respectively.21,22 GATK was developed in the One-Thousand Genome project for genome analysis. The SAMtools software has two parts, namely, SAMtools and BCFtools; the former has a subcommand ‘mpileup analysis’ that compares the results of reference sequence base sites, produces a BCF file and uses BCFtools for SNP/INDEL calling.7 After using mpileup, the parameter ‘-g/-v’ generates a BCF file (a VCF (variant call format) binary file). If this parameter is not used, it generates a text file, which statistically compares information of each base site in the reference sequence. BCFtools was used to filter the results of variants and obtain a reliable result. According to the quality of the sixth column value in the VCF file, we could also write a new program for filtering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (31502110, 81274023) and the Nature Science Foundation of Fujian Province of China (2019J01377). The authors have no financial interest or other potential conflicts of interest.References
- Ż. Agnieszka, J. Paulina and F. Marek, Transcriptome sequencing:next generation approach to RNA functional analysis, J. Clin. Monit. Comput., 2011, 25(3), 155–161 CrossRef
.
- K. K. Fugate, D. Fajardo, B. Schlautman, J. P. Ferrareze, M. D. Bolton, L. G. Campbell, E. Wiesman and J. Zalapa, Generation and Characterization of a Sugarbeet Transcriptome and Transcript-Based SSR Makers, Plant Genome, 2014, 7(2), 9–22 CrossRef
- D. Altshuler, V. J. Pollara, C. R. Cowles, W. J. Van Etten, J. Baldwin, L. Linton and E. S. Lander, An SNP map of the human genome generated by reduced representation shotgun sequencing, Nature, 2000, 407(6803), 513–516 CrossRef CAS
- T. Yang, S. Zhang, R. Wang, D. Li, Y. Hu, J. Nie, X. Zhao, Q. Wang, Y. Chen, Y. Zheng and P. Chen, Polysaccharides from Rhizoma Panacis Majoris and its anti-oxidant activity, Int. J. Biol. Macromol., 2016, 86, 756–763 CrossRef CAS
- T. Morita, Y. Kasai, O. Tanaka, J. Zhou, T. R. Yang and J. Shoji, Saponins of Zu-Tziseng, rhizomes of Panax japonicus C.A. Meyer var. Major C.Y. Wu et K.M.Feng, collected in Yunnan, China, Chem. Pharm. Bull., 1982, 30(12), 4341–4346 CrossRef CAS
- Q. Hua and L. R. Song, Zhao Shen” Interpretation of “Ben Cao Gang Mu Shi Yi”, J. Nanjing Univ. Tradit. Chin. Med., 1991, 7, 53 Search PubMed
- C. F. Li, Y. J. Zhu, X. Guo, C. Sun, H. Luo, J. Song, Y. Li, L. Wang, J. Qian and S. Chen, Transcriptome analysis reveals ginsenosides biosynthetic genes, microRNAs and simple sequence repeats in Panax ginseng C. A. Meyer, BMC Genomics, 2013, 14, 245 CrossRef CAS
- S. Wickramasinghe, G. Rincon, A. Islas-Trejo and J. F. Medrano, Transcriptional profiling of bovine milk using RNA sequencing, BMC Genomics, 2012, 13, 45 CrossRef CAS
- F. M. You, N. Huo, K. R. Deal, Y. Q. Gu, M. C. Luo, P. E. McGuire, J. Dvorak and O. D. Anderson, Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence, BMC Genomics, 2011, 12, 59 CrossRef CAS
- F. M. You, K. R. Deal, J. Wang, M. T. Britton, J. N. Fass, D. Lin, A. M. Dandekar, C. A. Leslie, M. Aradhya, M. C. Luo and J. Dvorak, Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms, BMC Genomics, 2012, 13, 354 CrossRef CAS
- D. Wu, S. C. Daugherty, S. E. Van Aken, G. H. Pai, K. L. Watkins, H. Khouri, L. J. Tallon, J. M. Zaborsky, H. E. Dunbar, P. L. Tran, N. A. Moran and J. A. Eisen, Metabolic complementarity and genomics of the dual bacterial symbiosis of sharpshooters, PLoS Biol., 2006, 4, 1079–1092 CrossRef CAS
- R. A. Kahn and F. Durst, Function and evolution of plant cytochrome P450, Recent Adv. Phytochem., 2000, 34, 151–189 CAS
- T. Ahmad, M. A. Valentovic and G. O. Rankin, Effects of cytochrome P450 single nucleotide polymorphisms on methadone metabolism and pharmacodynamics, Biochem. Pharmacol., 2018, 153, 196–204 CrossRef CAS
- E. S. Mace, S. Tai, E. K. Gilding, Y. Li, P. J. Prentis, L. Bian, B. C. Campbell, W. Hu, D. J. Innes, X. Han, A. Cruickshank, C. Dai, C. Frère, H. Zhang, C. H. Hunt, X. Wang, T. Shatte, M. Wang, Z. Su, J. Li, X. Lin, I. D. Godwin, D. R. Jordan and J. Wang, Whole-genome sequencing reveals untapped genetic potential in Africa's indigenous cereal crop sorghum, Nat. Commun., 2013, 4, 2320 CrossRef
- S. Anders, P. T. Pyl and W. Huber, HTSeq--a Python framework to work with high-throughput sequencing data, Bioinformatics, 2015, 31(2), 166–169 CrossRef CAS
- M. G. Grabherr, B. J. Haas, M. Yassour, J. Z. Levin, D. A. Thompson, I. Amit, X. Adiconis, L. Fan, R. Raychowdhury, Q. Zeng, Z. Chen, E. Mauceli, N. Hacohen, A. Gnirke, N. Rhind, F. di Palma, B. W. Birren, C. Nusbaum, K. Lindblad-Toh, N. Friedman and A. Regev, Fulllength transcriptome assembly from RNA-Seq data without a reference genome, Nat. Biotechnol., 2011, 29(7), 644–652 CrossRef CAS
- G. Pertea, X. Huang, F. Liang, V. Antonescu, R. Sultana, S. Karamycheva, Y. Lee, J. White, F. Cheung, B. Parvizi, J. Tsai and J. Quackenbush, TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets, Bioinformatics, 2003, 9, 651–652 CrossRef PubMed
- K. J. Vining, K. R. Pomraning, L. J. Wilhelm, H. D. Priest, M. Pellegrini, T. C. Mockler, M. Freitag and S. H. Strauss, Dynamic DNA cytosine methylation in the Populus trichocarpa genome: tissue-level variation and relationship to gene expression, BMC Genomics, 2012, 13, 27 CrossRef CAS
- W. Guo, P. Fiziev, W. Yan, S. Cokus, X. Sun, M. Q. Zhang, P. Y. Chen and M. Pellegrini, BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data, BMC Genomics, 2013, 14(1), 1–8 CrossRef
- R. Ahmad, D. E. Parfitt, J. Fass, E. Ogundiwin, A. Dhingra, T. M. Gradziel, D. Lin, N. A. Joshi, P. J. Martinez-Garcia and C. H. Crisosto, Whole genome sequencing of peach (Prunus persica L.) for SNP identification and selection, BMC Genomics, 2011, 12, 569 CrossRef CAS
- H. Li, Mathematical notes on samtools algorithms, 2010 Search PubMed
- Genome Sequencing and Analysis Group, Unified genotyper - gsa, 2011 Search PubMed
| This journal is © The Royal Society of Chemistry 2019 |