
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe caffeic acid moiety plays an essential role in attenuating lipid accumulation by chlorogenic acid and its analogues†
Xiaoxue Cao‡
 ,
Chongming Wu‡,
Yu Tian* and
Peng Guo*
,
Chongming Wu‡,
Yu Tian* and
Peng Guo*
Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100193, China. E-mail: ytian@implad.ac.cn; pguo@implad.ac.cn; Tel: +86-10-5783-3235
First published on 17th April 2019
Abstract
Chlorogenic acid (5-caffeoylquinic, CA) possesses distinct hypolipidemic properties in vivo and in vitro, yet the structure–activity relationship (SAR) of CA on lipid metabolism remains unknown. To achieve this aim, we designed and synthesized two sets of CA analogues and evaluated their efficacies to prevent oleic acid (OA)-elicited lipid accumulation in HepG2 cells. Blockage of all hydroxyl and carboxyl groups on the quinic acid moiety did not deteriorate the hypolipidemic effect of CA while blockage of all phenolic hydroxyl groups on the caffeic acid moiety abolished the activity of CA. Further replacement of the quinic acid moiety with cyclohexane and modification of individual phenolic hydroxyl groups on the caffeic acid moiety showed that the phenolic-hydroxyl-reserved analogues displayed a more potent hypolipidemic effect than CA, whereas the analogue with no phenolic hydroxyl displayed little effect on the OA-elicited lipid accumulation. In accordance, the modulating effects of CA on the transcription of the lipogenic gene sterol-regulatory element binding protein (SREBP)1c/1a, acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS) and peroxisome proliferator-activated receptor α (PPARα) were also abolished when the phenolic hydroxyl groups on the caffeic acid moiety were blocked. Our results suggest that the phenolic hydroxyl on the caffeic acid moiety is vital for the lipid-lowering activity of CA.
1. Introduction
Hyperlipidemia, a disorder of lipid metabolism, involves an imbalance of cholesterol level. Other forms include hypertriglyceridemia and mixed hyperlipidemia in which both cholesterol and triglyceride levels are elevated. It is known as a major cause of cardiovascular disease (CVD) that currently contributes to half of the world's mortality.1–3 Patients with hyperlipidemia are at approximately twice the risk of developing CVD compared to those with normal total cholesterol level.4 In current clinical prescription, statins are the main treatment for hyperlipidemia. However, they are associated with numerous adverse events and limitations.5,6 Thus developing novel drugs originating from natural products of appreciable action to combat lipid metabolic disorders would have significant medical and economic impacts.A general approach to drug discovery, which has been embraced by the pharmaceutical industry, includes developing lead compounds and structure modification of templates. It lies in structure–activity relationship (SAR)-oriented synthesis.7,8 Formulation of SARs provides information of essential functional groups to guide practical structure optimization of parental compounds,9 which further benefits lowering side effects and improving efficacy. Therefore, the study of SARs of lead compounds is considerably vital in drug discovery.
Chlorogenic acid (5-CQA, CA), the ester of caffeic acid with quinic acid (shown in Fig. 1), is distributed in a wide range of plants and human diet, such as Lonicera japonica Thunb,10 coffee and tea.11 It possesses various biological properties.12–15 Recent studies demonstrated a distinct potency of CA on improving dyslipidemia.16–19 Our previous works have indicated CA dramatically attenuated lipid accumulation in vivo and in vitro.20–23 Existing studies have reported structure–activity relationships on certain effects of CA. For instance, caffeic acid moiety was important for suppression of hepatic gluconeogenesis and hyperglycemia.24 The 3,4-position hydroxyl groups were responsible for its antibacterial activity.25 However, the functional groups in structure that are responsible for anti-hyperlipidemia activity have not been characterized yet.
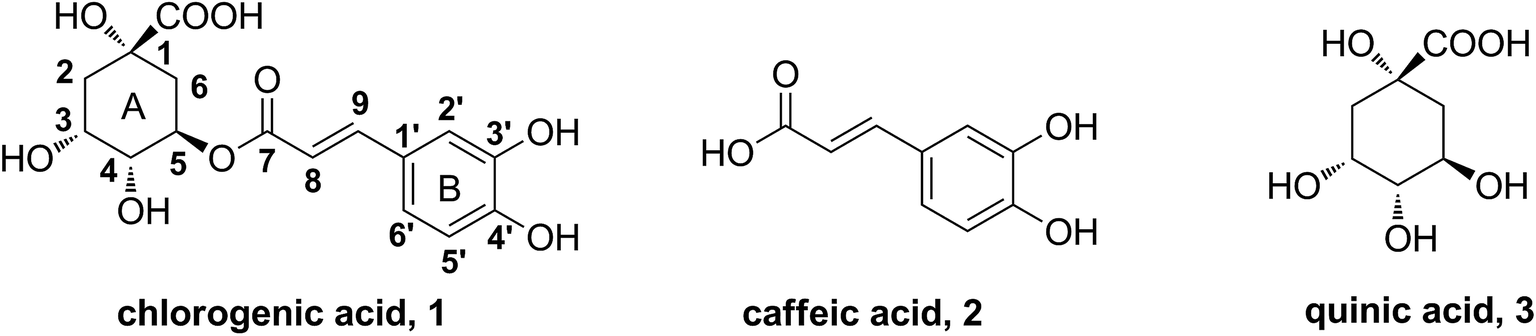 | ||
| Fig. 1 The structures of chlorogenic acid, caffeic acid and quinic acid. | ||
In this study, in order to investigate the primary structure–hypolipidemic activity relationship of chlorogenic acid and clarify the role of the caffeic acid or quinic acid moiety on lipid-lowering effect, an implementation was designed as shown in Fig. 2. We first modified the active groups such as the hydroxyl and carboxyl groups on quinic acid moiety in the A ring and the phenolic hydroxyl groups on caffeic acid moiety in the B ring. Moreover, a series of cinnamic acid cyclohexanol esters were synthesized to explore the significance of the phenolic hydroxyl groups in chlorogenic acid and caffeic acid. By the approach of a cell-based bioassay,26 the regulation of above compounds on oleic acid-elicited lipid accumulation in HepG2 cells and the expression of lipogenic, lipid oxidation-related genes are reported in this paper.
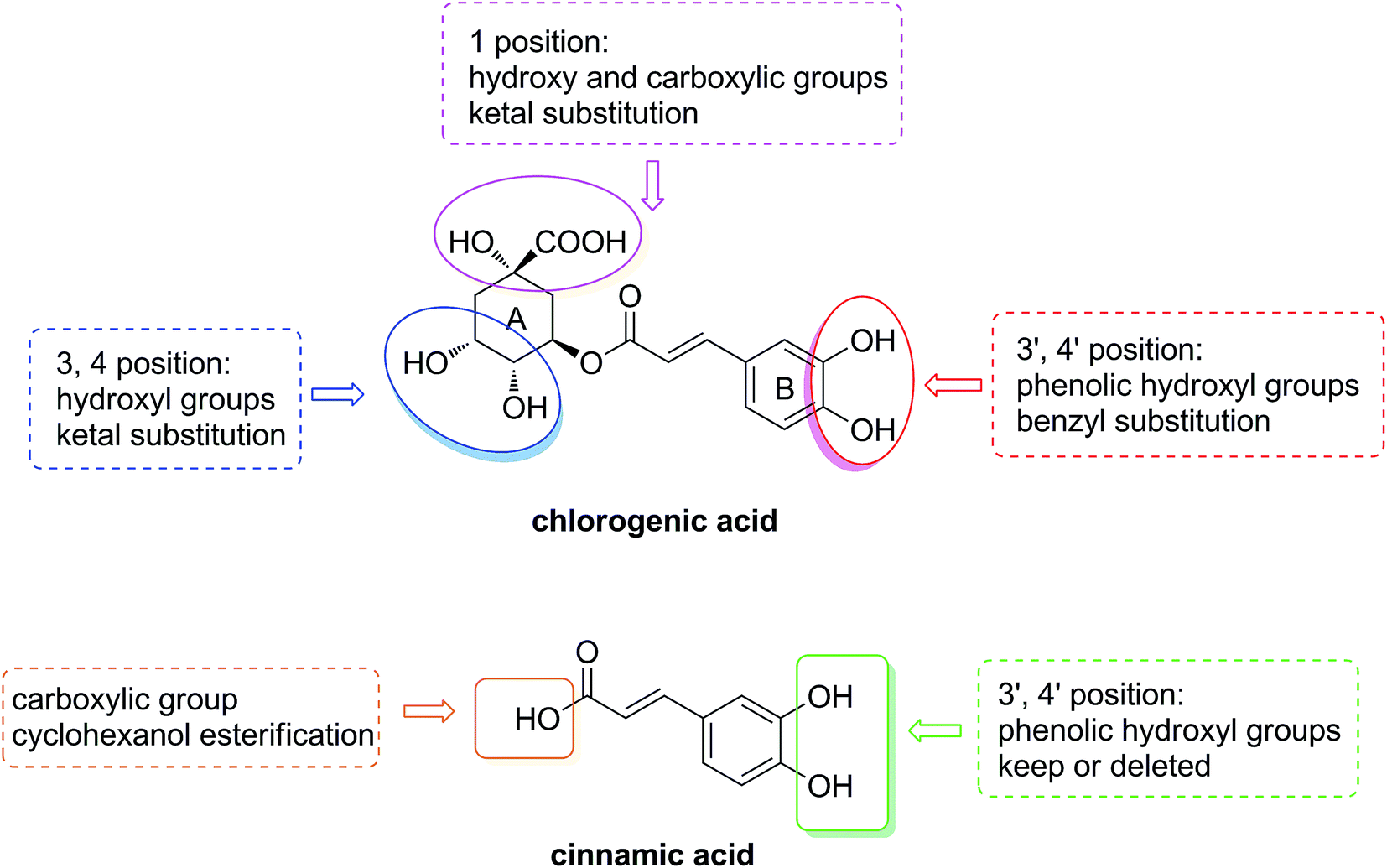 | ||
| Fig. 2 The design of structure–activity relationship research. | ||
2. Experimental section
2.1. General information
All the reagents were used without further purification unless otherwise specified. Solvents were dried and redistilled prior to use in the usual manner. Analytical TLC was performed using silica gel HF254. Preparative column chromatography was performed with silica gel H. 1H and 13C NMR spectra were recorded on a Bruker Advance III 600 MHz spectrometer. HRMS were obtained on a Thermofisher LTQ-Obitrap XL.2.2. Reagents and materials
Dulbecco's modified Eagle medium (DMEM), fetal bovine serum were purchased from Corning Inc. (CA, USA). Penicillin and streptomycin were procured from Hyclone (Logan, Utah, USA). Simvastatin, oil-red O, oleic acid and dimethyl Sulphoxide DMSO were purchased from Sigma-Aldrich (St. Louis, MO, USA). Chlorogenic acid was purchased from the Energy Chemical Company. The kit for Triglyceride (TG) was purchased from Jian Cheng Biotechnology Company (Nanjing, China). Total RNA extraction reagent Trizol was purchased from invitrogen (Carlsbad, CA, USA), PrimeScript RT reagent kit and SYBR-Green PCR kit were purchased from Transgene Biotech, Inc. (Beijing, China).2.3. Chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to offer pure light yellow powder, 90% yield; 1H-NMR (600 MHz, DMSO-d6) δ: 9.58 (1H, s, Ph′–OH), 9.14 (1H, s, Ph′–OH), 7.48 (1H, d, J = 15.8 Hz, H-9), 7.05 (1H, d, J = 1.5 Hz, H-2′), 7.00 (1H, dd, J = 1.4, 8.2 Hz, H-6′), 6.77 (1H, d, J = 8.1 Hz, H-5′), 6.23 (1H, d, J = 15.9 Hz, H-8), 5.34–5.30 (1H, m, H-5), 4.39–4.38 (1H, m, H-4), 4.11–4.09 (1H, m, H-3), 2.20–2.16 (1H, m, H-2, 6), 2.03–2.01 (1H, m, H-2, 6), 1.92–1.89 (1H, m, H-2, 6), 1.83–1.79 (1H, m, H-2, 6), 1.40 (3H, s, –CH3), 1.25 (3H, s, –CH3); 13C-NMR (150 MHz, DMSO-d6) δ: 176.2, 165.8, 148.5, 145.6, 145.4, 125.6, 121.5, 115.8, 114.9, 114.1, 108.1, 76.5, 73.2, 72.7, 70.4, 36.4, 34.8, 28.1, 26.0; HRMS (ESI): calcd for [M + Na]+C19H22NaO9: 417.1162, found 417.1167.:1) to gain pure light yellow powder, 65% yield; 1H-NMR (600 MHz, DMSO-d6) δ: 9.60 (1H, s, Ph'–OH), 9.16 (1H, s, Ph'–OH), 7.51 (1H, d, J = 15.8 Hz, H-9), 7.11–6.95 (2H, m, H-2′, 6′), 6.77 (1H, d, J = 7.7 Hz, H-5′), 6.25 (1H, d, J = 15.8 Hz, H-8), 5.24–5.10 (1H, m, H-5), 4.50–4.36 (1H, m, H-4), 4.26–4.14 (1H, m, H-3), 2.29–2.18 (2H, m, H-2, 6), 2.14–2.04 (1H, m, H-2, 6), 1.90–1.79 (1H, m, H-2, 6), 1.57 (6H, s, 2× –CH3), 1.41 (3H, s, –CH3), 1.27 (3H, s, –CH3); 13C-NMR (150 MHz, DMSO-d6) δ: 173.7, 165.9, 148.4, 145.7, 145.6, 125.5, 121.4, 115.8, 114.9, 113.7, 110.4, 108.2, 78.2, 75.6, 72.5, 69.4, 35.7, 34.0, 28.1, 27.9, 27.8, 25.7; HRMS (ESI): calcd for [M + Na]+ C22H26NaO9: 457.1475, found 457.1472.:1) to offer pure white solid, 67% yield; 1H-NMR (600 MHz, DMSO-d6) δ: 7.55 (1H, d, J = 15.7 Hz, H-9), 7.50 (1H, d, J = 1.7 Hz, Bn′–H), 7.47–7.44 (4H, m, Bn′–H), 7.40–7.36 (8H, m, Bn′–H), 7.33–7.31 (3H, m, Bn′–H, H-2′), 7.22 (1H, dd, J = 2.0 Hz, 8.3 Hz, H-6′), 7.08 (1H, d, J = 8.3 Hz, H-5′), 6.50 (1H, d, J = 15.9 Hz, H-8), 5.37–5.33 (1H, m, H-5), 5.19 (4H, s, Bn′–CH2), 5.15–5.09 (2H, m, Bn′–CH2), 4.41–4.39 (1H, m, H-4), 4.12–4.11 (1H, m, H-3), 2.24–2.20 (1H, m, H-2, 6), 2.10–2.07 (1H, m, H-2, 6), 1.99–1.96 (1H, m, H-2, 6), 1.88–1.84 (1H, m, H-2, 6), 1.41 (3H, s, –CH3), 1.26 (3H, s, –CH3); 13C-NMR (150 MHz, DMSO-d6) δ: 174.2, 166.3, 151.2, 149.0, 145.1, 137.0, 136.8, 135.2, 128.8, 128.7, 128.7, 128.3, 128.1, 128.0, 127.4, 127.3, 123.1, 115.9, 114.3, 113.9, 109.7, 77.4, 77.0, 74.0, 71.4, 71.1, 70.7, 67.9, 37.0, 34.5, 28.1, 26.0; HRMS (ESI): calcd for [M + Na]+ C40H40NaO9: 687.2570, found 687.2573.
:1) to offer pure light yellow powder, 90% yield; 1H-NMR (600 MHz, DMSO-d6) δ: 9.58 (1H, s, Ph′–OH), 9.14 (1H, s, Ph′–OH), 7.48 (1H, d, J = 15.8 Hz, H-9), 7.05 (1H, d, J = 1.5 Hz, H-2′), 7.00 (1H, dd, J = 1.4, 8.2 Hz, H-6′), 6.77 (1H, d, J = 8.1 Hz, H-5′), 6.23 (1H, d, J = 15.9 Hz, H-8), 5.34–5.30 (1H, m, H-5), 4.39–4.38 (1H, m, H-4), 4.11–4.09 (1H, m, H-3), 2.20–2.16 (1H, m, H-2, 6), 2.03–2.01 (1H, m, H-2, 6), 1.92–1.89 (1H, m, H-2, 6), 1.83–1.79 (1H, m, H-2, 6), 1.40 (3H, s, –CH3), 1.25 (3H, s, –CH3); 13C-NMR (150 MHz, DMSO-d6) δ: 176.2, 165.8, 148.5, 145.6, 145.4, 125.6, 121.5, 115.8, 114.9, 114.1, 108.1, 76.5, 73.2, 72.7, 70.4, 36.4, 34.8, 28.1, 26.0; HRMS (ESI): calcd for [M + Na]+C19H22NaO9: 417.1162, found 417.1167.:1) to gain pure light yellow powder, 65% yield; 1H-NMR (600 MHz, DMSO-d6) δ: 9.60 (1H, s, Ph'–OH), 9.16 (1H, s, Ph'–OH), 7.51 (1H, d, J = 15.8 Hz, H-9), 7.11–6.95 (2H, m, H-2′, 6′), 6.77 (1H, d, J = 7.7 Hz, H-5′), 6.25 (1H, d, J = 15.8 Hz, H-8), 5.24–5.10 (1H, m, H-5), 4.50–4.36 (1H, m, H-4), 4.26–4.14 (1H, m, H-3), 2.29–2.18 (2H, m, H-2, 6), 2.14–2.04 (1H, m, H-2, 6), 1.90–1.79 (1H, m, H-2, 6), 1.57 (6H, s, 2× –CH3), 1.41 (3H, s, –CH3), 1.27 (3H, s, –CH3); 13C-NMR (150 MHz, DMSO-d6) δ: 173.7, 165.9, 148.4, 145.7, 145.6, 125.5, 121.4, 115.8, 114.9, 113.7, 110.4, 108.2, 78.2, 75.6, 72.5, 69.4, 35.7, 34.0, 28.1, 27.9, 27.8, 25.7; HRMS (ESI): calcd for [M + Na]+ C22H26NaO9: 457.1475, found 457.1472.:1) to offer pure white solid, 67% yield; 1H-NMR (600 MHz, DMSO-d6) δ: 7.55 (1H, d, J = 15.7 Hz, H-9), 7.50 (1H, d, J = 1.7 Hz, Bn′–H), 7.47–7.44 (4H, m, Bn′–H), 7.40–7.36 (8H, m, Bn′–H), 7.33–7.31 (3H, m, Bn′–H, H-2′), 7.22 (1H, dd, J = 2.0 Hz, 8.3 Hz, H-6′), 7.08 (1H, d, J = 8.3 Hz, H-5′), 6.50 (1H, d, J = 15.9 Hz, H-8), 5.37–5.33 (1H, m, H-5), 5.19 (4H, s, Bn′–CH2), 5.15–5.09 (2H, m, Bn′–CH2), 4.41–4.39 (1H, m, H-4), 4.12–4.11 (1H, m, H-3), 2.24–2.20 (1H, m, H-2, 6), 2.10–2.07 (1H, m, H-2, 6), 1.99–1.96 (1H, m, H-2, 6), 1.88–1.84 (1H, m, H-2, 6), 1.41 (3H, s, –CH3), 1.26 (3H, s, –CH3); 13C-NMR (150 MHz, DMSO-d6) δ: 174.2, 166.3, 151.2, 149.0, 145.1, 137.0, 136.8, 135.2, 128.8, 128.7, 128.7, 128.3, 128.1, 128.0, 127.4, 127.3, 123.1, 115.9, 114.3, 113.9, 109.7, 77.4, 77.0, 74.0, 71.4, 71.1, 70.7, 67.9, 37.0, 34.5, 28.1, 26.0; HRMS (ESI): calcd for [M + Na]+ C40H40NaO9: 687.2570, found 687.2573.3,4-Dihydroxycinnamic acid cyclohexanol ester (11). Column chromatography (eluent: DCM–CH3OH, 10
:1), white power, 68% yield; 1H-NMR (600 MHz, CDCl3) δ: 7.57 (d, J = 15.9 Hz, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.11 (d, J = 2.0 Hz, 1H, H-2), 7.02 (dd, J = 8.2 Hz, 2.0 Hz, 1H, H-6), 6.88 (d, J = 2.0 Hz, 1H, H-5), 6.27 (d, J = 15.9 Hz, 1H, CHCH), 4.90–4.86 (m, 1H, O–CH), 1.93–1.90 (m, 2H, O–CH–CH2), 1.79–1.74 (m, 2H, O–CH–CH2), 1.58–1.25 (m, 6H, –(CH2)3–); 13C-NMR (150 MHz, CDCl3) δ: 167.7, 146.5, 144.9, 143.9, 127.6, 122.4, 116.2, 115.6, 114.6, 73.3, 31.8, 25.5, 23.9.
CH), 7.11 (d, J = 2.0 Hz, 1H, H-2), 7.02 (dd, J = 8.2 Hz, 2.0 Hz, 1H, H-6), 6.88 (d, J = 2.0 Hz, 1H, H-5), 6.27 (d, J = 15.9 Hz, 1H, CHCH), 4.90–4.86 (m, 1H, O–CH), 1.93–1.90 (m, 2H, O–CH–CH2), 1.79–1.74 (m, 2H, O–CH–CH2), 1.58–1.25 (m, 6H, –(CH2)3–); 13C-NMR (150 MHz, CDCl3) δ: 167.7, 146.5, 144.9, 143.9, 127.6, 122.4, 116.2, 115.6, 114.6, 73.3, 31.8, 25.5, 23.9.
Cinnamic acid cyclohexanol ester (12). Column chromatography (eluent: PE–EA, 30
:1), colourless oil, 53% yield; 1H-NMR (600 MHz, pyridine-d5) δ: 7.67 (d, J = 16.1 Hz, 1H, CHCH), 7.54–7.52 (m, 2H, H-2, 6), 7.38–7.37 (m, 1H, H-3, 4, 5), 6.44 (d, J = 16.0 Hz, 1H, CHCH), 4.91–4.87 (m, 1H, O–CH), 1.94–1.91 (m, 2H, O–CH–CH2), 1.79–1.75 (m, 2H, O–CH–CH2), 1.61–1.25 (m, 6H, –(CH2)3–); 13C-NMR (150 MHz, CDCl3) δ: 166.5, 144.4, 134.6, 130.2, 129.0, 128.1, 119.0, 72.9, 31.9, 25.5, 23.9.
3-Hydroxycinnamic acid cyclohexanol ester (13). Column chromatography (eluent: PE–EA, 20
:1), white solid, 62% yield; 1H-NMR (600 MHz, pyridine-d5) δ: 7.60 (d, J = 16.0 Hz, 1H, CHCH), 7.22–7.19 (m, 1H, H-5), 7.05–7.02 (m, 1H, H-4, 6), 6.91–6.89 (m, 1H, H-2), 6.38 (d, J = 16.0 Hz, 1H, CHCH), 4.90–4.86 (m, 1H, O–CH), 1.92–1.91 (m, 2H, O–CH–CH2), 1.78–1.72 (m, 2H, O–CH–CH2), 1.57–1.23 (m, 6H, –(CH2)3–); 13C-NMR (150 MHz, CDCl3) δ: 167.0, 156.9, 144.7, 135.9, 130.0, 120.3, 118.8, 117.7, 114.7, 73.2, 31.8, 25.5, 23.9.
4-Hydroxycinnamic acid cyclohexanol ester (14). Column chromatography (eluent: PE–EA, 20
:1), white solid, 64% yield; 1H-NMR (600 MHz, pyridine-d5) δ: 7.61 (d, J = 15.9 Hz, 1H, CHCH), 7.38–7.37 (m, 2H, H-2, 6), 6.87–6.86 (m, 2H, H-3, 5), 6.26 (d, J = 15.9 Hz, 1H, CHCH), 4.89–4.86 (m, 1H, O–CH), 1.91–1.90 (m, 2H, O–CH–CH2), 1.79–1.72 (m, 2H, O–CH–CH2), 1.55–1.22 (m, 6H, –(CH2)3–); 13C-NMR (150 MHz, CDCl3) δ: 168.0, 158.5, 145.0, 130.1, 126.8, 116.1, 115.6, 73.3, 31.8, 25.5, 23.9.
2.4. Evaluation of the biological activity
| Name | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| SREBP1c | CCATGGATGCACTTTCGAA | CCAGCATAGGGTGGGTCAA |
| ACC | TGATGTCAATCTCCCCGCAGC | TTGCTTCTTCTCTGTTTTCTCCCC |
| FAS | CGGTACGCGACGGCTGCCTG | GCTGCTCCACGAACTCAAACACCG |
| SREBP1a | TGCTGACCGACATCGAAGAC | CCAGCATAGGGTGGGTCAA |
| PPARα | AAAAGCCTAAGGAAACCGTTCTG | TATCGTCCGGGTGGTTGCT |
| β-Actin | CCTGGCACCCCAGCACAAT | GCCGATCCACACACGGAGTACT |
3. Results and discussion
3.1. Chemistry
The synthesis of analogues 4–6 is outlined in Fig. 3. The naturally abundant chlorogenic acid (1) was treated with 2,2-dimethoxypropane (DMP) and p-toluenesulfonic acid (TsOH) in dry acetone to obtain compound 4. Moreover, compound 1 reacted with dry acetone in Lewis acidic conditions trimethylsilyl trifluoromethanesulfonate (TMSOTf) to provided compound 5. Compound 6 was attained via alkylation of compound 4 with benzyl bromide (BnBr) and potassium carbonate (K2CO3) in acetonitrile.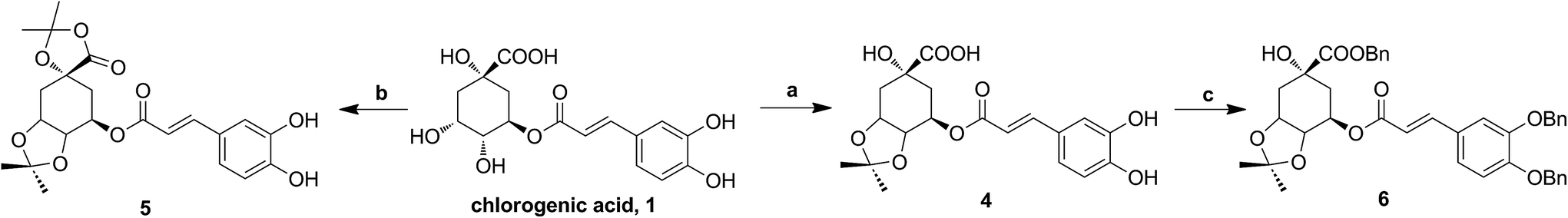 | ||
| Fig. 3 The synthesis of chlorogenic acid analogues. Reagents and conditions: (a) DMP, TsOH; (b) TMSOTf, acetone; (c) BnBr, K2CO3. | ||
The synthesis of cinnamic acid ester analogues 11–14 is showed in Fig. 4. Compounds 7–10 were reacted respectively with cyclohexanol in the presence of sulfoxide chloride (SOCl2), and finally gained the target compounds 11–14.
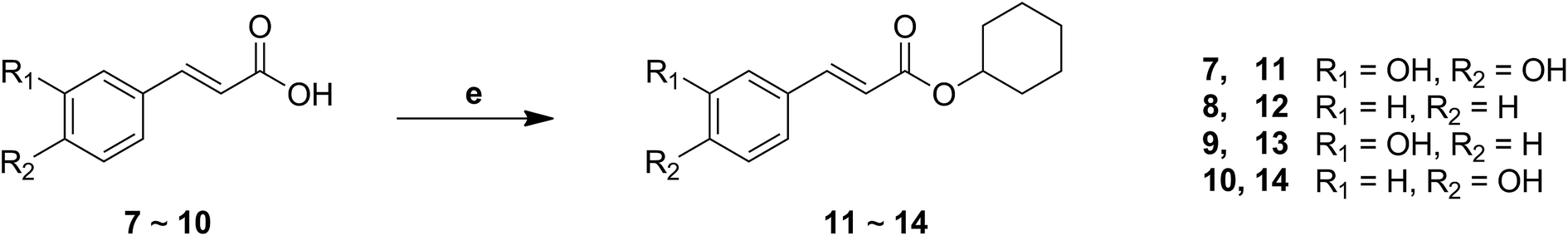 | ||
| Fig. 4 The synthesis of a series of cinnamic acid ester derivatives. Reagents and conditions: (e) SOCl2, cyclohexanol, reflux. | ||
3.2. Biological assessment
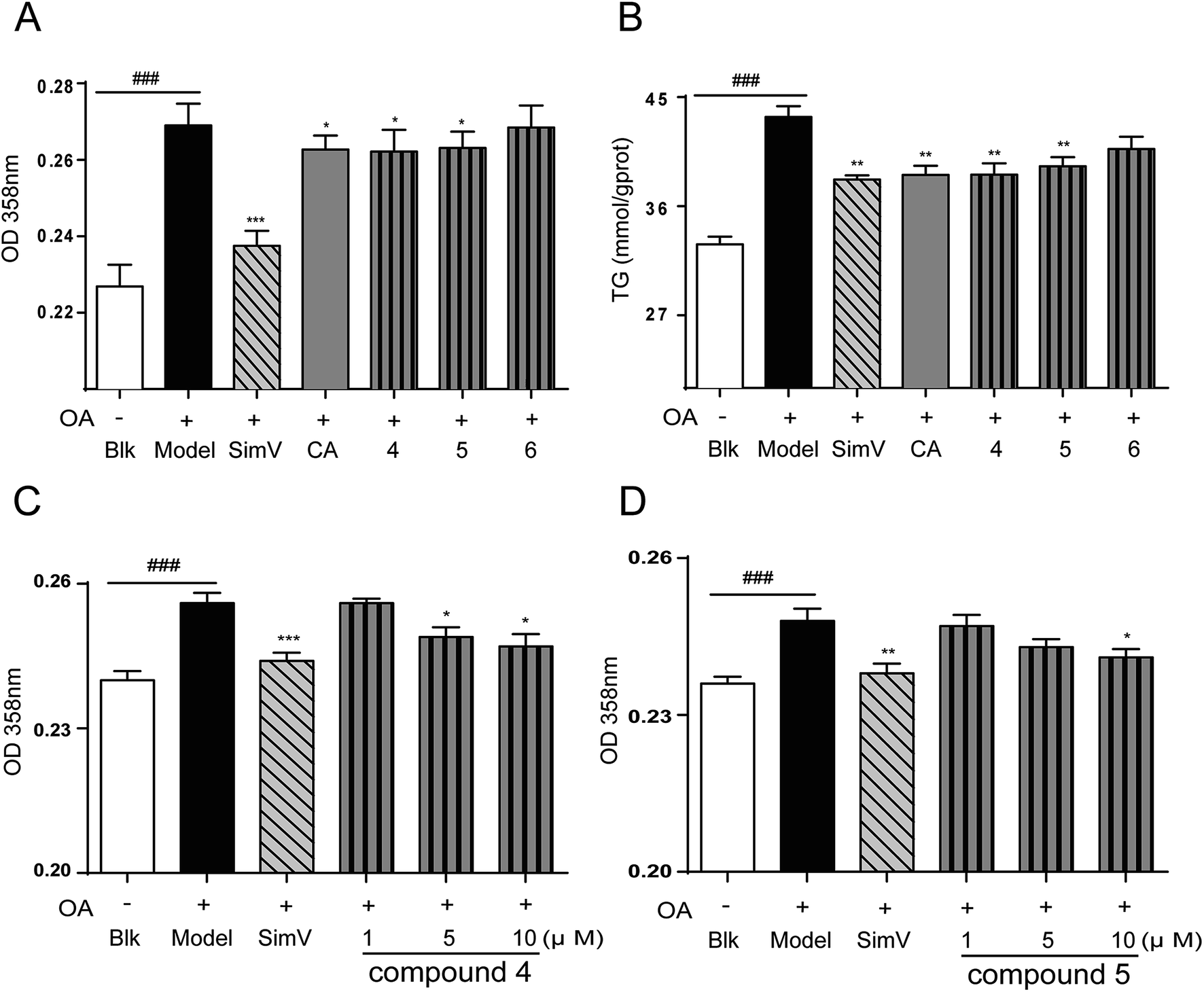 | ||
| Fig. 5 Effects of CA analogues 4–6 (10 μmol L−1) on OA-elicited lipid accumulation in HepG2 cells. (A) 358 nm measurement after oil-red O staining (n = 8); (B) intracellular TG levels (n = 6); (C) and (D) compounds 4, 5 inhibited OA-elicited lipid accumulation in a dose-dependent way. Data are depicted as means ± SEM. ###p < 0.001, model vs. blank; *p < 0.05, **p < 0.01, test groups vs. model group. Blk: blank; SimV: simvastatin, used as a positive control; CA: chlorogenic acid; compound 4: 3,4-di-O-isopropylidene-5-O-(3′,4′-dihydroxycinnamoyl)-quinic acid; compound 5: 3,4-di-O-isopropylidene-5-(3′,4′-dihydroxycinnamoyl)-1-O-quinic acid isopropylidene ester; compound 6: 3,4-di-O-isopropylidene-5-O-(3′,4′-dibenzyloxycinnamoyl)-quinic acid benzyl ester. | ||
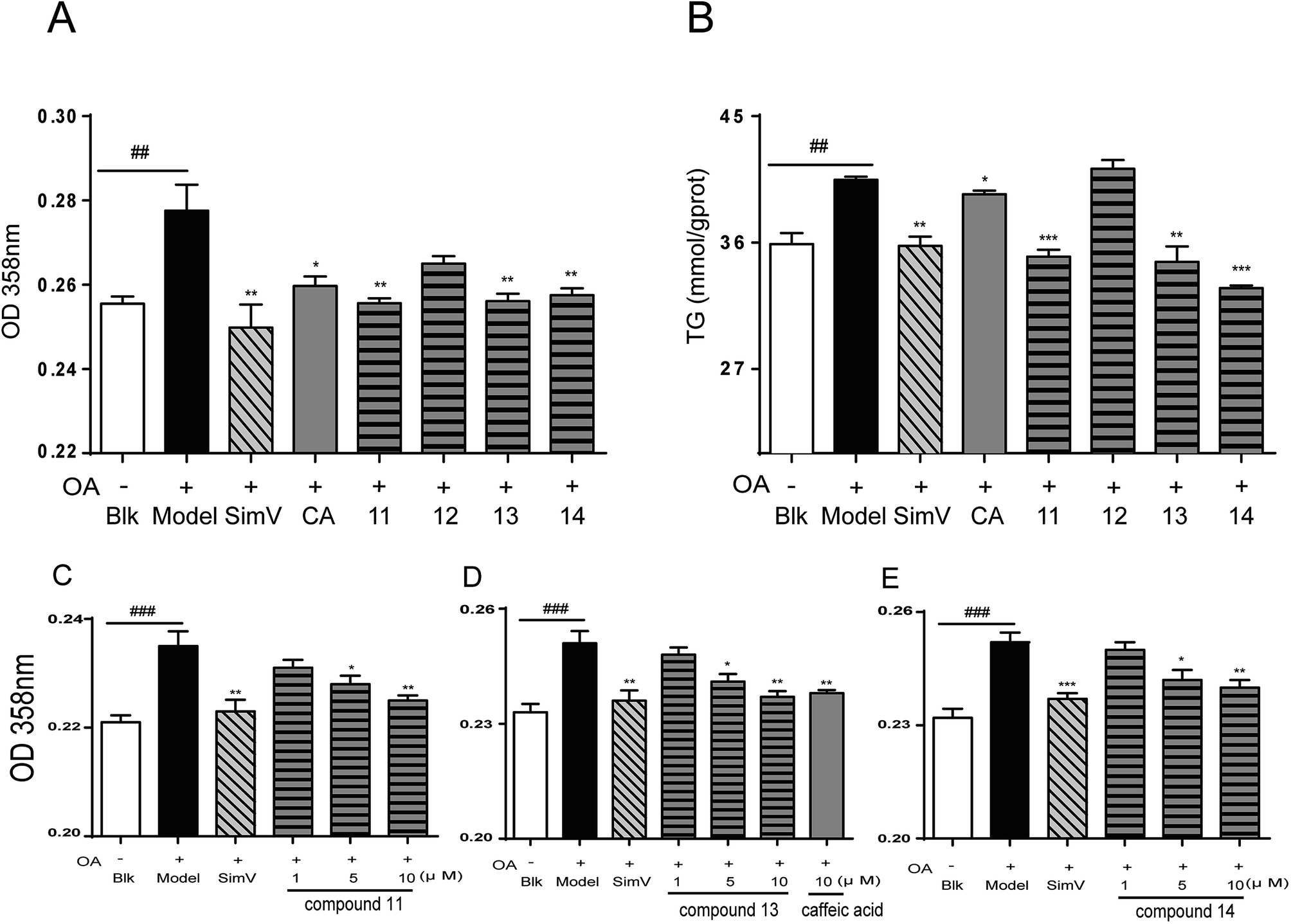 | ||
| Fig. 6 Effects of CA analogues 11–14 (10 μmol L−1) on OA-elicited lipid accumulation in HepG2 cells. (A) 358 nm measurement after oil-red O staining (n = 8); (B) intracellular TG levels (n = 6); (C–E) compounds 11, 13, 14 inhibited OA-elicited lipid accumulation in a dose-dependent way. Data are depicted as means ± SEM. ##p < 0.01, model vs. blank; *p < 0.05, **p < 0.01, ***p < 0.001, test groups vs. model group. Blk: blank; SimV: simvastatin, used as a positive control; CA: chlorogenic acid; compound 11: 3,4-dihydroxycinnamic acid cyclohexanol ester; compound 12: cinnamic acid cyclohexanol ester; compound 13: 3-hydroxycinnamic acid cyclohexanol ester; compound 14: 4-hydroxycinnamic acid cyclohexanol ester. | ||
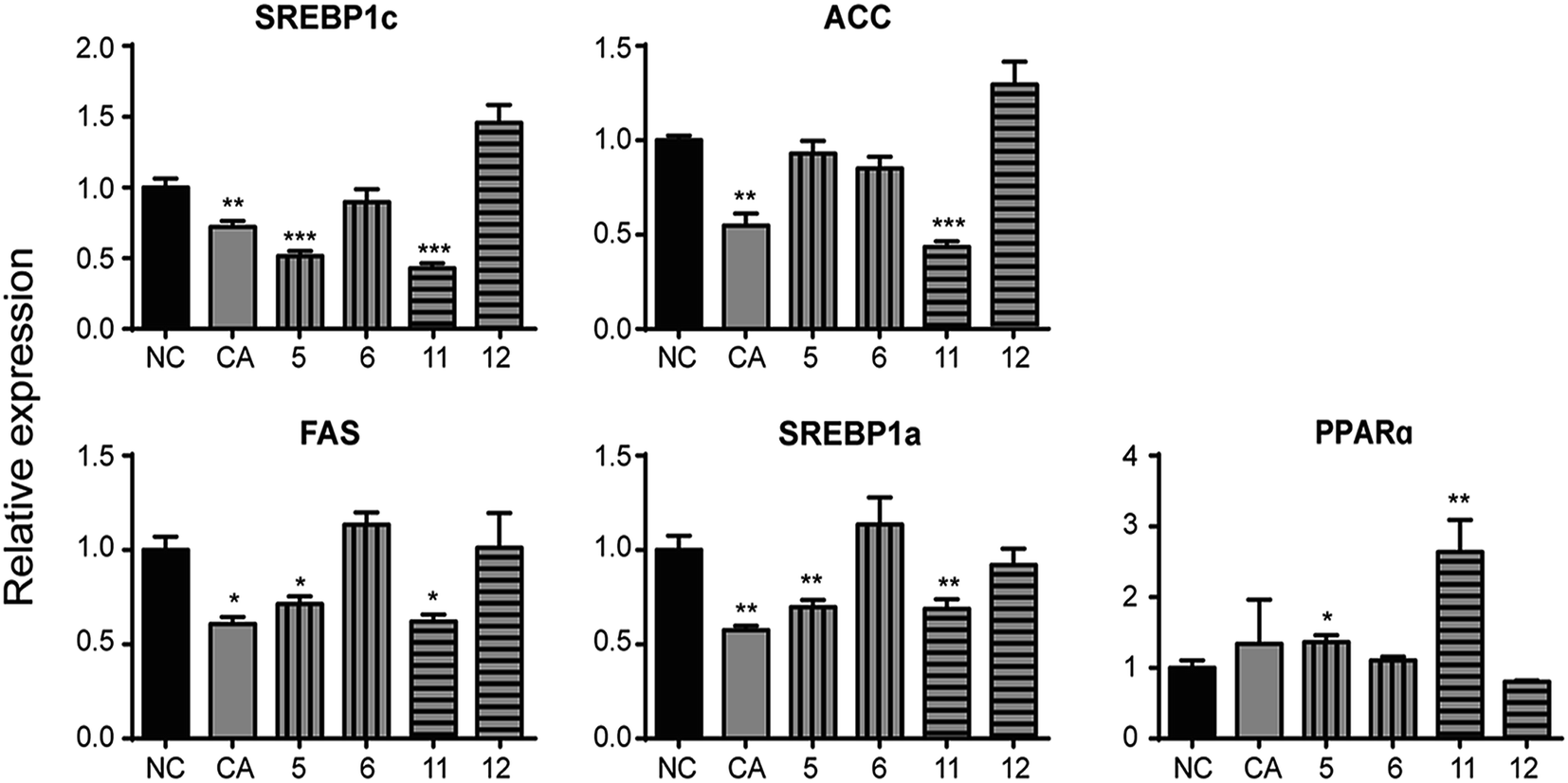 | ||
| Fig. 7 Effects of CA and its analogues (10 μmol L−1) on the mRNA levels of lipogenic genes SREBP1c, ACC, FAS, SREBP1a and lipid-oxidation related gene PPARα in OA-elicited HepG2 cells. Real-time quantitative PCR was conducted with gene-specific oligonucleotide primers. The amplification of β-actin served as an internal control (n = 6). Data are depicted as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, test groups vs. negative control group. NC: negative control; CA: chlorogenic acid; compound 5: 3,4-di-O-isopropylidene-5-(3′,4′-dihydroxycinnamoyl)-1-O-quinic acid isopropylidene ester; compound 6: 3,4-di-O-isopropylidene-5-O-(3′,4′-dibenzyloxycinnamoyl)-quinic acid benzyl ester. compound 11: 3,4-dihydroxycinnamic acid cyclohexanol ester; compound 12: cinnamic acid cyclohexanol ester; SREBP1c/1a: sterol response element binding protein 1c/1a; ACC: acetyl-CoA carboxylase; FAS: fatty acid synthase; PPARα: peroxisome proliferator-activated receptor α. | ||
4. Conclusions
The study of SAR demonstrated caffeic acid moiety with phenolic hydroxyl is essential for hypolipidemic activity instead of quinic acid moiety. Additionally, the amount and the position of phenolic hydroxyl groups have little influence on lipid-lowering CA. Caffeic acid moiety with phenolic hydroxyl directly affects the modulation of CA on altering mRNA levels of lipometabolic-modulating genes. This study introduced a series of chemical agents with improving hypolipdemic activity in vitro, thus are worthy for further study and the report lied foundation for developing novel drugs of promising anti-hyperlipidemic effect.Conflicts of interest
We declare there are no financial or other contractual agreements that might cause conflicts of interest or be perceived as causing conflicts of interest.Acknowledgements
This work was supported by the National Natural Sciences Foundation of China (Grant No. 81302656), Beijing Natural Sciences Foundation (Grant No. 7192129), the CAMS Innovation Fund for Medical Science (CIFMS) (Grant No. 2016-I2M-1-012, 2016-I2M-3-015).References
- E. J. Lee, H. Oh, B. G. Kang, M. K. Kang, D. Y. Kim, Y. H. Kim, J. Y. Lee, J. G. Ji, S. S. Lim and Y. H. Kang, J. Agric. Food Chem., 2018, 66, 10447–10457 CrossRef CAS PubMed.
- K. K. Poh, B. Ambegaonkar, C. A. Baxter, P. Brudi, W. Buddhari, F. T. Chiang, M. Horack, Y. Jang, B. Johnson, D. Lautsch, J. Sawhney, A. Vyas, B. P. Yan and A. K. Gitt, Eur. J. Prev. Cardiol., 2018, 25(18), 1950–1963 CrossRef PubMed.
- C. Wu, Y. Guo, Y. Su, X. Zhang, H. Luan, X. Zhang, H. Zhu, H. He, X. Wang, G. Sun, X. Sun, P. Guo and P. Zhu, J. Cell. Mol. Med., 2014, 18, 293–304 CrossRef CAS PubMed.
- S. Karr, Am. J. Manag. Care, 2017, 23, S139–S148 Search PubMed.
- M. Apostolopoulou, A. Corsini and M. Roden, Eur. J. Clin. Invest., 2015, 45, 745–754 CrossRef CAS PubMed.
- N. Sattar, D. Preiss, H. M. Murray, P. Welsh, B. M. Buckley, A. J. d. Craen, J. J. McMurray, D. J. Freeman and J. W. Jukema, Lancet, 2010, 375, 735–742 CrossRef CAS.
- J. Chen, W. Li, H. Yao and J. Xu, Fitoterapia, 2015, 103, 231–241 CrossRef CAS PubMed.
- H. Yao, J. Liu, S. Xu, Z. Zhu and J. Xu, Expert Opin. Drug Discovery, 2017, 12, 121–140 CrossRef CAS PubMed.
- M. Lahlou, Pharmacology & Pharmacy, 2013, vol. 04, no. 03, p. 15 Search PubMed.
- L. Zhang, J. Liu, P. Zhang, S. Yan, X. He and F. Chen, Chromatographia, 2011, 73, 129–133 CrossRef CAS.
- D. D. Rio, A. Stalmach, L. Calani and A. Crozier, Nutrients, 2010, 2, 820–833 CrossRef PubMed.
- A. Karunanidhi, R. Thomas, B. A. Van and V. Neela, BioMed Res. Int., 2013, 2013, 392058 Search PubMed.
- N. Liang and D. D. Kitts, Nutrients, 2016, 8, 16 CrossRef PubMed.
- W. Li, X. Liu, G. Zhang and L. Zhang, Zhongguo Feiai Zazhi, 2017, 20, 555–561 Search PubMed.
- H. Y. Ye, J. Jin, L. W. Jin, Y. Chen, Z. H. Zhou and Z. Y. Li, Inflammation, 2017, 40, 523–529 CrossRef CAS PubMed.
- S. Meng, J. Cao, Q. Feng, J. Peng and Y. Hu, Evid.-Based Complementary Altern. Med., 2013, 2013(4), 801457 Search PubMed.
- K. W. Ong, A. Hsu and B. K. Tan, Biochem. Pharmacol., 2013, 85, 1341–1351 CrossRef CAS PubMed.
- A. S. Cho, S. M. Jeon, M. J. Kim, J. Yeo, K. I. Seo, M. S. Choi and M. K. Lee, Food Cosmet. Toxicol., 2010, 48, 937–943 CrossRef CAS PubMed.
- L. T. Zhang, C. Q. Chang, Y. Liu and Z. M. Chen, Acta Acad. Med. Sin., 2011, 33, 281 CAS.
- C. Wu, X. Zhang, X. Zhang, H. Luan, G. Sun, X. Sun, X. Wang, P. Guo and X. Xu, J. Nutr. Biochem., 2014, 25, 412–419 CrossRef CAS PubMed.
- X. Zhang, C. Wu, H. Wu, L. Sheng, Y. Su, X. Zhang, H. Luan, G. Sun, X. Sun and Y. Tian, PLoS One, 2013, 8, e61922 CrossRef CAS PubMed.
- C. Wu, H. Luan, X. Zhang, S. Wang, X. Zhang, X. Sun and P. Guo, PLoS One, 2014, 9, e95452 CrossRef PubMed.
- C. M. Wu, H. Luan, S. Wang, X. P. Zhang, H. T. Liu and P. Guo, Acta Pharm. Sin., 2015, 50, 278 CAS.
- K. M. Jackson, T. Rathinasabapathy, D. Esposito and S. Komarnytsky, Mol. Nutr. Food Res., 2017, 61(9), 1601118 CrossRef PubMed.
- M. Zhang, W. X. Liu, M. F. Zheng, Q. L. Xu, F. H. Wan, J. Wang, T. Lei, Z. Y. Zhou and J. W. Tan, Molecules, 2013, 18, 14096–14104 CrossRef CAS PubMed.
- S. Wang, C. Wu, X. Li, Y. Zhou, Q. Zhang, F. Ma, J. Wei, X. Zhang and P. Guo, Acta Pharm. Sin. B, 2017, 7, 453–460 CrossRef PubMed.
- S. Chan, R. Q. Sun and X. Y. Zeng, Diabetes, 2013, 62, 2095–2105 CrossRef CAS PubMed.
- G. Musso, R. Gambino and M. Cassader, Obes. Rev., 2010, 11, 430–445 CrossRef CAS PubMed.
- Y. L. Liu, G. L. Patman, J. B. Leathart, A. C. Piguet, A. D. Burt, J. F. Dufour, C. P. Day, A. K. Daly, H. L. Reeves and Q. M. Anstee, J. Hepatol., 2014, 61, 75–81 CrossRef CAS PubMed.
- T. Utsunomiya and M. Shimada, Hepatol. Res., 2011, 41, 711–721 CrossRef PubMed.
- W. Cui, S. L. Chen and K. Q. Hu, Am. J. Transl. Res., 2010, 2, 95–104 CAS.
- M. Szewczyk, M. Morawiak, A. Narczyk, Z. Pakulski and Z. Urbanczyk-Lipkowska, Journal of Chemistry and Biochemistry, 2018, 6, 28–37 Search PubMed.
- M. B. Dantas, T. L. Sampaio, R. Roacute, J. Magalhatilde, E. Ferreira, T. S. Melo, H. G. Rodrigues and D. B. Lima, Afr. J. Pharm. Pharmacol., 2018, 12(20), 263–268 CrossRef CAS.
- K. Karthikesan and L. Pari, Fundam. Clin. Pharmacol., 2010, 22, 523–527 CrossRef PubMed.
- J. H. Kim, S. I. Kang, H. S. Shin, S. A. Yoon, S. W. Kang, H. C. Ko and S. J. Kim, Journal of the Agricultural Chemical Society of Japan, 2013, 77, 1595–1598 CAS.
- T. Uemura, T. Goto, M. S. Kang, N. Mizoguchi, S. Hirai, J. Y. Lee, Y. Nakano, J. Shono, S. Hoshino, K. Taketani, N. Tsuge, T. Narukami, M. Makishima, N. Takahashi and T. Kawada, J. Nutr., 2011, 141, 17–23 CrossRef CAS PubMed.
- L. P. Bechmann, R. A. Hannivoort, G. Guido, G. K. S. Hotamisligil, T. Michael and C. Ali, J. Hepatol., 2012, 56, 952–964 CrossRef CAS PubMed.
- Q. Liu, B. Yuan, K. A. Lo, H. C. Patterson, Y. Sun and H. F. Lodish, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14568–14573 CrossRef CAS PubMed.
- B. Kim, M. J. Woo, C. S. Park, S. H. Lee, J. S. Kim, B. Kim, S. An and S. H. Kim, Phytother. Res., 2017, 31, 132–139 CrossRef CAS PubMed.
- C. Wang, R. Batey, J. Yamahara and Y. Li, J. Funct. Foods, 2017, 36, 43–51 CrossRef CAS.
- C. W. Wan, C. N. Wong, W. K. Pin, M. H. Wong, C. Y. Kwok, R. Y. Chan, P. H. Yu and S. W. Chan, Phytother. Res., 2013, 27, 545–551 CrossRef CAS PubMed.
- T. Murase, K. Misawa, Y. Minegishi, M. Aoki, H. Ominami, Y. Suzuki, Y. Shibuya and T. Hase, Am. J. Physiol.: Endocrinol. Metab., 2011, 300, 122–133 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09395d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |