
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced control of plasmonic properties of silver–gold hollow nanoparticles via a reduction-assisted galvanic replacement approach†
Josée R. Daniela,
Lauren A. McCarthyb,
Emilie Ringe*bcd and
Denis Boudreau *a
*a
aDépartement de Chimie et Centre D'optique, Photonique et Laser (COPL), Université Laval, Québec (QC) G1V 0A6, Canada. E-mail: denis.boudreau@chm.ulaval.ca
bDepartment of Chemistry, Rice University, Houston, Texas 77005, USA
cDepartment of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge, CB3 0FS, UK. E-mail: er407@cam.ac.uk
dDepartment of Earth Sciences, University of Cambridge, Downing Street, Cambridge, CB2 3EQ, UK
First published on 2nd January 2019
Abstract
Hollow noble metal nanoparticles are of growing interest due to their localized surface plasmon resonance (LSPR) tunability. A popular synthetic approach is galvanic replacement which can be coupled with a co-reducer. Here, we describe the control over morphology, and therefore over plasmonic properties including energy, bandwidth, extinction and scattering intensity, offered by co-reduction galvanic replacement. This study indicates that whereas the variation of atomic stoichiometry using the co-reduction method described in this work offers a rather modest tuning range of LSPR energy when compared to traditional galvanic replacement, it nevertheless has a profound effect on shell thickness, which imparts a degree of control over scattering intensity and sensitivity to changes in the dielectric constant of the surrounding environment. Therefore, in this context particle size and gold content become two design parameters that can be used to independently tune LSPR energy and intensity.
Introduction
Localized surface plasmon resonance (LSPR) is defined as the light-driven collective oscillations of the conduction electrons of a metallic nanostructure, leading to enhanced photon absorption and scattering cross sections (far-field effects) as well as enhancement of the local electromagnetic field around the nanostructure (near-field effects).1 These phenomena are being harnessed in applications spanning numerous fields, including biomedicine,2 biophysics,3 energy conversion4 and catalysis.5The near- and far-field optical properties of a plasmonic nanoparticle are dictated by its size and shape, the dielectric function of the constituent metal as well as that of the surrounding environment. Therefore, controlling shape and size is key to tailoring properties, a realization that fostered a rich set of synthetic approaches leading to cubes,6 triangles,7 decahedra,8 bars,9 and stars,10 to name a few. An interesting addition to the numerous chemical synthesis tools developed to control nanocrystal geometry (and therefore properties) is the ability to further manipulate shape via the synthesis of hollow structures. In such nanoparticles, not only can the shape be modulated but also the cavity's radius-to-shell thickness ratio, thereby providing an extra degree of freedom for LSPR tuning.11 An analytical solution to the plasmonic behaviour of shells based on Mie theory was first described by Aden and Kerker.12 This has later been augmented by a semiclassical approach13 and the plasmon hybridization model14–16 as well as numerical approaches.17–19 Together, these works contributed to the current understanding of the phenomenon, which can be summarized as follows: the non-isotropic electron oscillation in hollow structures leads to a shape-dependent depolarization and LSPR energy splitting into lower-energy symmetric (bonding) and higher-energy anti-symmetric (anti-bonding) plasmon modes. The size of the cavity and the wall thickness dictate the extent of interaction between the inner and outer surface resonances. The LSPR of these shell structures are therefore related to the nanoshell's inner and outer radii, r1 and r2, where the aspect ratio of the radii is equal to r1/(r2 − r1) (Fig. 1A). For weaker interaction between these surfaces (thick wall, small r) the symmetric plasmon resonance approaches the solid particle value, while a stronger interaction (thin wall, large r) leads to a larger energy splitting and a lower energy symmetric LSPR, when compared to a solid particle of the same size. Along with the LSPR energy, the proportion of the scattering and absorption is modulated by shape: a particle with a thin wall scatters less than a particle with a thick wall.20
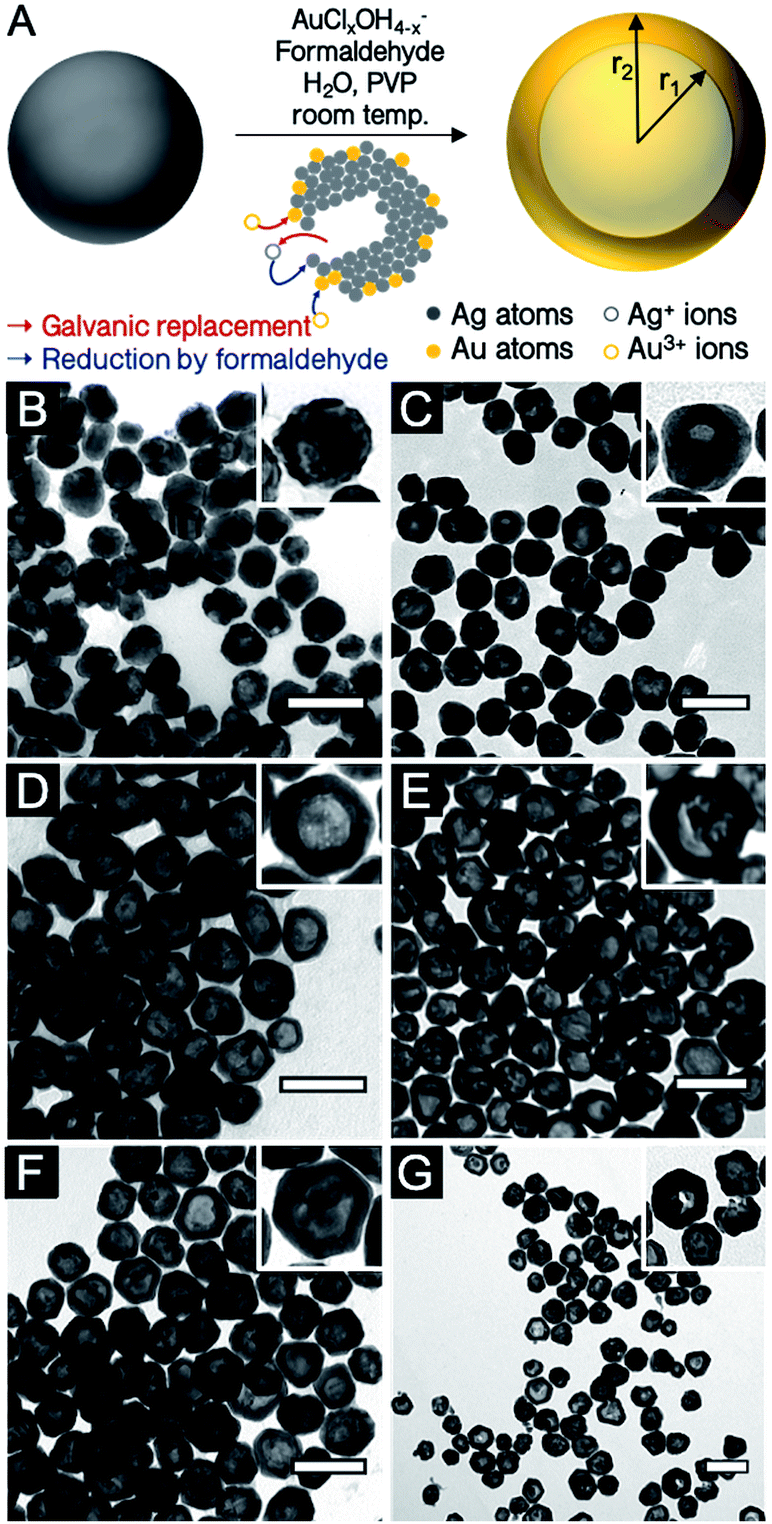 | ||
| Fig. 1 (A) Schematic illustration of the co-reduction galvanic replacement procedure used. (B–G) TEM images of reaction products for different Au:Ag stoichiometries (B) 0.1Au:Ag, (C) 0.25Au:Ag, (D) 0.50Au:Ag, (E) 0.75Au:Ag, (F) Au:Ag, and (G) 2Au:Ag. Scale bars, 100 nm. | ||
Galvanic replacement has emerged as a popular synthesis route to prepare hollow metallic structures with control over composition and morphology.21,22 Nanoscale galvanic replacement occurs when a metal ion encounters a nanoparticle made of a zerovalent metal atom with a lower redox potential (often referred as a sacrificial template), leading to an exchange of electrons, where the template metal will be oxidized and dissolved in solution while the oxidant metal will be reduced onto the surface of the particle. Owing to their plasmonic properties, a commonly studied metal pair is Ag/Au; galvanic replacement operates because Au has a more positive redox potential and can gradually replace the Ag template. Taking advantage of the well-known shape control mechanisms for the Ag template, the Ag/Au pair has been used extensively as a synthetic approach to generate simple to exotic hollow structures, including spheres,23,24 rods,25 wires,26 rings,27 octahedrons,28,29 multilayer spheres,30 as well as boxes and cages.31
By altering the resulting shell characteristics, including composition, the relative amount of oxidant metal added to the nanoparticle template suspension controls the optical properties of the final solution. The shape and composition of the final product mainly depends on alloying and dealloying steps occurring during the galvanic replacement reaction. Early on, the oxidant metal alloys by deposition at the surface of the reductant template while the template's interior is dissolved through pinholes and cavities. As the galvanic exchange proceeds further, dealloying may occur and lead to a cage/frame-like structure or fragmentation of the nanoparticle shell.21,22 The extend of alloying to dealloying is controlled by the stoichiometry of the metals, which thereby dictates the thickness, porosity, and structural integrity of the walls.
Augmenting traditional galvanic replacement with the addition of a secondary reducing agent acting upon both metallic species (often termed “co-reducer” when referring to the oxidant metal) provides the ability to tune the alloying process. With co-reduction, shape and composition can therefore be manipulated beyond what is achievable with simple galvanic replacement.32–35 The secondary reducing agent in the solution introduces new reaction pathways and products owing to the competition between the reduction of oxidant ions by the metal template and this reducing agent. In addition, the newly oxidized atoms emerging from the template can be reduced back by the reducing agent, providing further composition control. Together, galvanic replacement and reduction strategies provide rich possibilities for shape and composition control for enhanced LSPR tuning.
The dependence of LSPR energy and bandwidth on AgAu hollow nanoparticle morphology has been probed experimentally, for instance by Yang et al., who investigated the optical properties of heterogeneous hollow particles as a function of their inner and outer diameters,36 and by Hu and co-workers, who unravelled the effect of edge length in nanoboxes and nanocages on their optical properties.37–39 Despite the promise of remarkable LSPR tuning in such systems, an extensive study of the possibilities and limitations of modulating optical properties using co-reduction galvanic replacement has yet to appear in the literature. Herein, we present a systematic investigation of the extent of control over morphology and optical properties offered by co-reduction galvanic replacement, including energy, bandwidth, extinction and scattering intensity. We obtained these results using bulk extinction spectroscopy as well as single particle (SP) scattering experiments on a systematic variation of the aspect ratio of the radii (r1/(r2 − r1)) by changing either the concentration of Au or the particle size. These optical measurements were correlated with the average size and wall thickness of the particles obtained with transmission electron microscopy. This study indicates that while the variation of atomic stoichiometry using the co-reduction method described in this work offers a rather modest tuning range of LSPR energy when compared to traditional galvanic replacement, it nevertheless has a profound effect on shell thickness, which imparts a degree of control over scattering intensity and sensitivity to changes in the dielectric constant of the surrounding environment. Since particle size can be used to adjust LSPR energy, particle size and gold content become two design parameters that can be used to independently tune LSPR energy and intensity.
Experimental section
Materials
Tannic acid (C76H52O46, ACS Reagent), sodium citrate tribasic (Na3C6H5O7, 99.0% ACS Reagent), polyvinylpyrrolidone (PVP10, MW ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and gold(III) chloride trihydrate (HAuCl4·3H2O, ≥ 99.9%, trace metal grade) were purchased from Sigma Aldrich. Silver nitrate (AgNO3, 99.9995%) was obtained from Strem Chemicals. Aqueous formaldehyde solution stabilized with methanol, (CH2O, 37% ACS) was obtained from Fisher Scientific. Aqueous hydrochloric acid (HCl, 36.5–38.0%, ACS reagent) and aqueous nitric acid (HNO3, 68.0–70.0%, ACS reagent) were purchased from VWR International. Potassium carbonate anhydrous (K2CO3, ACS ISO reagent) was purchased from EMD Millipore. All chemicals and reagents were used without further purification. Ultrapure water (18 MΩ) was used in all experiments. All glassware used for synthesis was washed with concentrated acid (nitric acid or aqua regia) and then rinsed thoroughly with ultrapure water.
000) and gold(III) chloride trihydrate (HAuCl4·3H2O, ≥ 99.9%, trace metal grade) were purchased from Sigma Aldrich. Silver nitrate (AgNO3, 99.9995%) was obtained from Strem Chemicals. Aqueous formaldehyde solution stabilized with methanol, (CH2O, 37% ACS) was obtained from Fisher Scientific. Aqueous hydrochloric acid (HCl, 36.5–38.0%, ACS reagent) and aqueous nitric acid (HNO3, 68.0–70.0%, ACS reagent) were purchased from VWR International. Potassium carbonate anhydrous (K2CO3, ACS ISO reagent) was purchased from EMD Millipore. All chemicals and reagents were used without further purification. Ultrapure water (18 MΩ) was used in all experiments. All glassware used for synthesis was washed with concentrated acid (nitric acid or aqua regia) and then rinsed thoroughly with ultrapure water.
Synthesis procedures
Characterization techniques
:H2O2) for one hour. Coverslips were then rinsed with water followed by ethanol and dried in an oven. 10 μL of NP solution in water was diluted in 1000 μL of ethanol, and 100 μL of this solution was drop cast on the coverslip. Correlated TEM and optical spectroscopy was performed to validate the technique (Fig. S4†). When performing correlated LSPR/TEM experiments, the NPs were instead drop cast on a 200-mesh carbon/formvar copper grids (Ted Pella Inc.).Results and discussion
Hollow AgAu nanoparticles were obtained with a co-reduction galvanic replacement procedure, with formaldehyde as the secondary reducing agent, where reaction kinetics are controlled by the pH of the Au solution (Fig. 1A).24 The reaction was performed with several oxidant:reductant atomic ratios, from 0.1 to 2 equivalents of Au3+ with respect to Ag0 (i.e., 0.1Au:Ag to 2Au:Ag). Throughout this manuscript, Ag and Au concentrations are described as atomic ratios between Ag and Au; their galvanic replacement reaction being Au3+ + 3Ag0 → Au0 + 3Ag+, 1Au:Ag (or 1 equivalent of Au) therefore contains three times more Au than is required to replace all the Ag present in the absence of a secondary reducing agent (here formaldehyde). Transmission electron microscopy (TEM) images of hollow nanoparticles (Fig. 1B–G) show a progressive enlargement of the cavity with increasing Au (dimensions listed in Table 1). At 0.1Au:Ag, the majority of the nanoparticles remain solid spheres with a rough surface, a prelude to pinhole formation. At 0.25Au:Ag, nanoparticles are mostly semi-hollow and the voids non-concentric. At 0.5Au:Ag, the voids are significantly larger and regular, resulting in a homogenous suspension of hollow nanoparticles with a shell thickness of about 12 nm. As more Au is added, particle size and wall thickness increases (0.75Au:Ag, Fig. 1E). At equimolar stoichiometry (Au:Ag, Fig. 2F), the galvanic replacement reaction by the oxidant overcomes the co-reduction process such that smaller nanoparticles with thinner shells are produced. An even larger excess of Au eventually leads to the loss of structural integrity of the shells and to homogeneous nucleation of small Au nanoparticles (2Au:Ag, Fig. 1G).| Atomic stoichiometry | Particle size (nm) | Cavity (nm) | Shell thick. (nm) | r1/(r2 − r1) | LSPR (nm) | Spectral Shift (nm (eV)) | LSPR (nm) | Spectral shift (nm (eV)) | Norm. scatt. intensity | N | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bulk (A) | Single particle | ||||||||||
| a The 0.25Au:Ag reaction products were found too heterogeneous, including mainly non-concentric semi-hollow structures or multiple small cavities such that an average shell thickness could not be determined. | |||||||||||
| Ag | 82 ± 8 | 460 | (B) | 457 ± 13 | 1.0 ± 0.3 | 86 | |||||
| 025Au:Ag | 87 ± 9 | a | a | a | 594 | 134 (−0.44) | (C) | 545 ± 18 | 88 (−0.44) | 1.2 ± 0.2 | 107 |
| 0.5Au:Ag | 94 ± 9 | 70 ± 9 | 12 ± 3 | 2.9 | 603 | 143 (−0.64) | (D) | 548 ± 18 | 91 (−0.45) | 1.5 ± 0.2 | 143 |
| 0.75Au:Ag | 102 ± 10 | 71 ± 8 | 16 ± 3 | 2.4 | 604 | 144 (−0.64) | (E) | 549 ± 17 | 92 (−0.45) | 1.7 ± 0.2 | 229 |
| Au:Ag | 98 ± 11 | 78 ± 8 | 10 ± 2 | 3.9 | 651 | 191 (−0.79) | (F) | 570 ± 20 | 113 (−0.54) | 1.2 ± 0.2 | 111 |
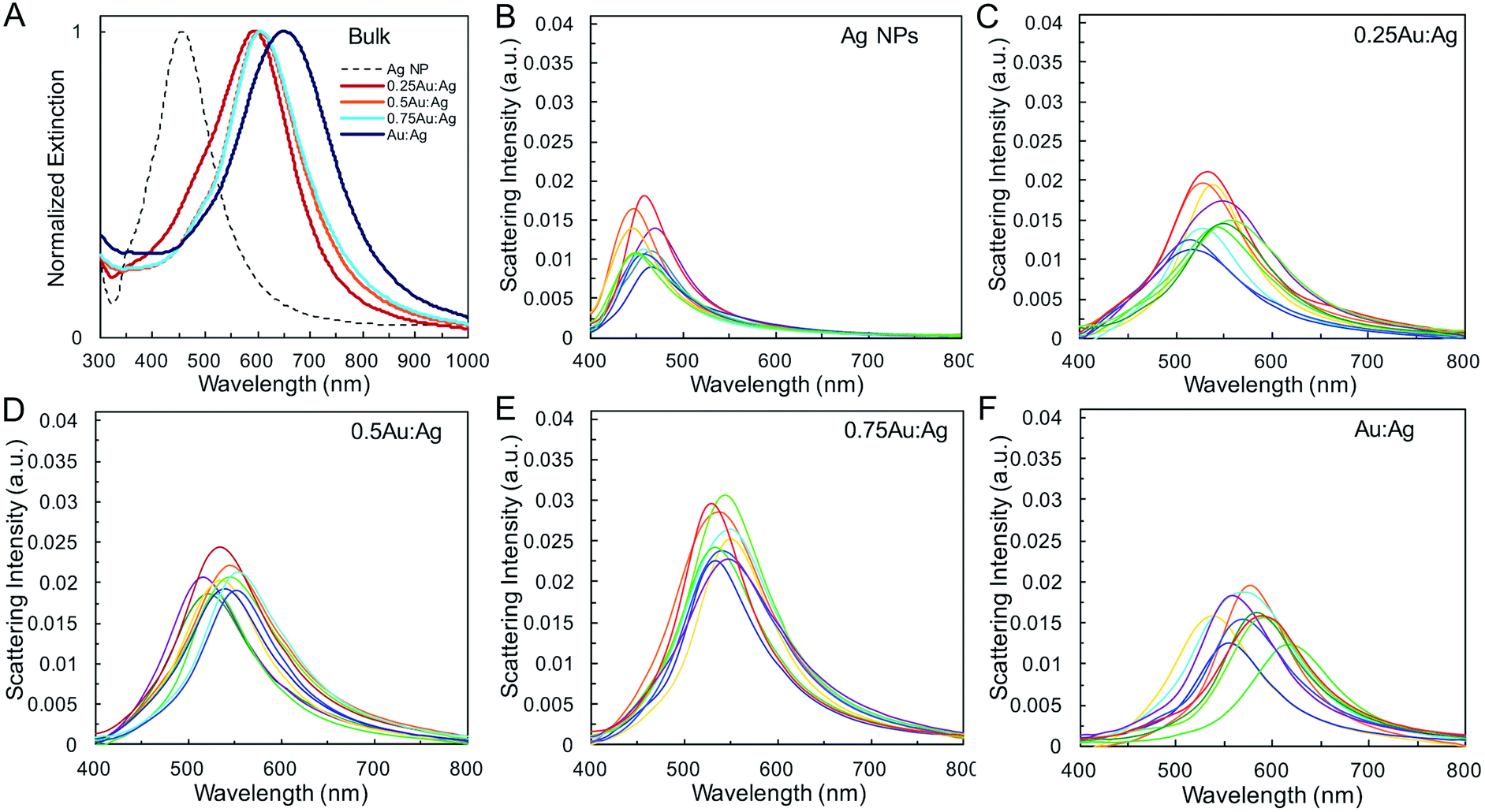 | ||
| Fig. 2 Optical properties of AgAu shells. (A) Normalized bulk UV-vis spectra and representative single nanoparticle scattering spectra for (B) Ag, (C) 0.25Au:Ag, (D) 0.5Au:Ag, (E) 0.75Au:Ag, and (F) Au:Ag. | ||
Single-particle measurements from hyperspectral experiments in an optical dark field scattering microscope41 (Table 1 and Fig. 2) reveal an increase in the average scattering intensity from Ag spheres to AgAu shells, attributed to the increase in overall nanoparticle diameter. Variations in average shell thickness are observed with the amount of Au added to the Ag nanoparticle suspension. As expected, stronger scattering is measured for larger hollow nanoparticles with thicker walls (Table 1 and Fig. 2E). A relationship is also present between the bandwidth and shell thickness, with narrower LSPR bands observed for thicker shells. This is observed in bulk (Fig. 2A), and, more importantly, in single-particle (Fig. 2B–F) LSPR spectra, which are not affected by heterogeneous broadening (Table S2†). These single-particle LSPR spectra indeed display a large distribution arising from particle-to-particle variations in, primarily, diameter and wall thickness, but also in void concentricity,16,19,42 surface roughness and the presence of pinholes,43,44 all of which alter optical properties. Interestingly, although composition may play a role in the observed scattering intensity it is not the dominant effect (Fig. S1†). Calculations by Wu and Liu using Mie theory have suggested that while the LSPR properties of Ag/Au alloy nanoshells are both sensitive to shell thickness and composition, the effect of geometry on near-field and far-field enhancement dominates, whereas the gold content plays a more significant role in the modulation of energy frequency, as we observe here.45
The geometric change from solid sphere to hollow shell structure and the alloying with Au lead to a marked redshift of the LSPR from 460 to 594 nm (bulk LSPR, Table 1). The slight redshifts (i.e., from 594 to 651 nm) linked to changes in the reaction's Au stoichiometry indicate that this parameter offers only mediocre LSPR energy control (Fig. 2 and Table 1). Here, the shell thickness control offered by galvanic replacement provides further means to manipulate the LSPR, until the point at which shells become thin and/or porous and the extinction intensity dramatically decreases, hindering applications needing strong light–matter interactions such as enhanced spectroscopies46–48 and photothermal cancer therapy.48–50 An alternative approach to further tune the LSPR energy is to manipulate the radius-to-shell thickness ratio by changing the initial size of the nanoparticle templates. Fig. 3C–F shows the galvanic replacement product of Ag nanoparticle templates with initial diameters ranging from 58 to 109 nm. When using the same atomic stoichiometry (i.e., same atomic concentration, meaning the Ag nanoparticle concentration decrease with increasing particle size), the average shell thickness remains relatively constant at about 11–12 nm (Table 2). The observed redshift in LSPR frequency associated with hollowing is more significant as the initial nanoparticle size increases, with shifts of 130, 140, 155, and 190 nm (dipole mode) for 58 ± 8, 76 ± 8, 87 ± 9, and 109 ± 12 nm diameter nanoparticles, respectively. This is attributed to both the overall nanoparticle size (from electromagnetic retardation effect) and the increasing cavity-shell aspect ratio from 2.1 (for 66 nm diameter) to 4.6 (for 118 nm diameter), as reported in Table 2, Fig. 3A and S2.† The LSPR full width at half maximum (FWHM) also broadens with increasing nanoparticle size, mainly due to increased contributions of electron surface scattering and radiation damping.39,51 Note that for the larger particles (i.e., 118 nm), only the dipole mode at lower energy is considered here. The experimental extinction coefficient increases with particle size (Fig. 3B, calculated from extinction optical density and particle concentration in molarity obtained from nanoparticle tracking analysis (NTA)), and agrees well with predictions from Mie theory calculations (Fig. S3†) and reported values.52
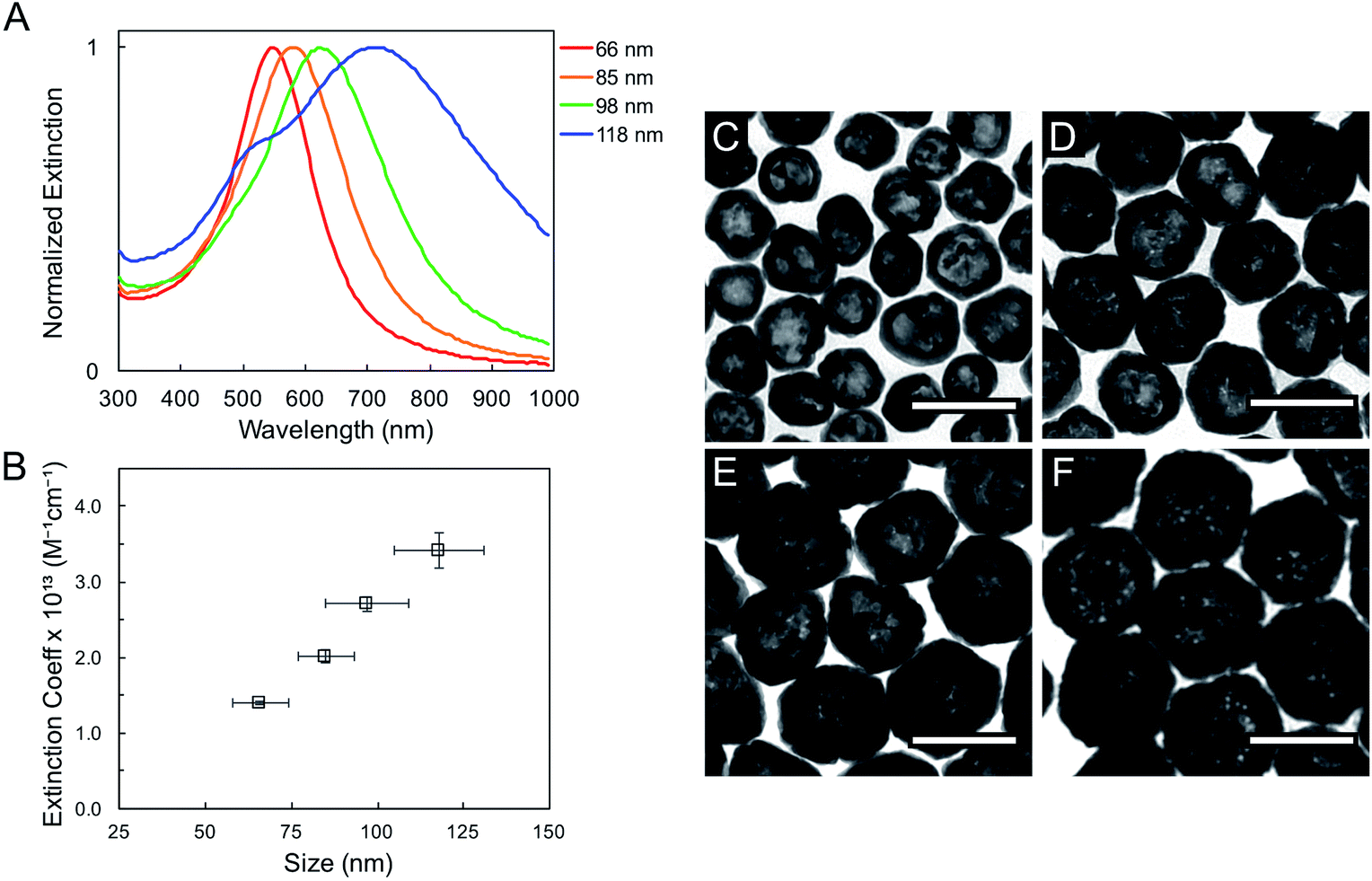 | ||
| Fig. 3 Bulk UV-vis extinction spectra (A) and extinction coefficient (B) for hollow AgAu nanoparticles of four different outer diameters: (C) 66 ± 8 nm (D) 85 ± 8 nm (E) 98 ± 12 nm and (F) 118 ± 13 nm, obtained from Ag nanoparticles of 58 ± 8, 76 ± 8, 87 ± 9, and 109 ± 12 nm diameter, respectively. Scale bars: 100 nm. | ||
| Size Ag (nm) | Size AgAu (nm) | Cavity size (nm) | Shell thickness (nm) | r1/(r2 − r1) | LSPR Ag (nm) | LSPR AgAu (nm) | Spectral shift (nm (eV)) | FWHM Ag (nm) | FWHM AgAu (nm) |
|---|---|---|---|---|---|---|---|---|---|
| 58 ± 8 | 66 ± 8 | 45 ± 7 | 11 ± 2 | 2.1 | 430 | 560 | 130 (−0.67) | 90 | 150 |
| 76 ± 8 | 85 ± 8 | 62 ± 6 | 12 ± 3 | 2.7 | 450 | 590 | 140 (−0.65) | 105 | 190 |
| 87 ± 9 | 98 ± 12 | 76 ± 8 | 11 ± 2 | 3.5 | 475 | 630 | 155 (−0.64) | 155 | 245 |
| 109 ± 12 | 118 ± 13 | 97 ± 10 | 11 ± 2 | 4.6 | 520 | 710 | 190 (−0.63) | 245 | 440 |
The LSPR energy depends on the dielectric properties of the surrounding environment including solvent, substrate and adsorbates, enabling refractive index-based sensing applications. A manifestation of this phenomenon can be readily seen in the difference between the LSPR energies measured in water (refractive index (RI) = 1.33) and those measured for particles supported on a silica coverslip (RI ∼ 1.5) and surrounded by air (RI ∼ 1), where the average medium RI is lower than that of water (Table 1). The latter are blue-shifted with respect to the former, and the shift appears to increase with an increase in LSPR wavelength.
Systematic investigations of the bulk RI sensitivity (RIS) of nanoshell solutions of the same shell thickness but different sizes, (Fig. 3) performed with various water/glycerol mixtures, reveal an exponential increase in RIS with particle size (Fig. 4A, blue markers). However, experimental RIS data from nanoparticles prepared by manipulating the Au:Ag stoichiometry (Fig. 1C, E and F) do not follow an obvious trend with respect to their sizes (Fig. 4A). This is attributed to the variation in both size and shell thickness in these samples (Table 1). RIS is best described as a function of the cavity radius-to-shell thickness ratio, as seen in Fig. 4B. This is consistent with theoretical reports from Jain and El-Sayed,53 where sensitivity was predicted to increase near-exponentially with the ratio of the core radius-to-shell thickness when overall size was kept constant. This observation can be explained by the enhanced polarizability of thin nanoshells owing to the stronger coupling between the inner and outer surfaces, leading to a more responsive nanoparticle. Analogously, and as observed, an increase in RIS is expected with increasing particle size for a given shell thickness, as this results in a larger r1/(r2 − r1) value.54 Another effect to consider is that an increase in the medium's dielectric permittivity reduces the wavelength of the incident light, leading to an additional redshift for larger nanoparticles due to phase retardation effects.53 Together, these RIS measurements suggest that larger hollow nanoparticles with thin walls (higher r1/(r2 − r1) value) might perform better than their thick, small counterparts in sensing applications. However, one must consider that increasing nanoparticle size induces a broadening of the LSPR bandwidth, which can be detrimental to the sensing Figure of Merit (FOM).55 FOMs as a function of core–shell ratio for particles with same shell thickness are shown in Fig. 4B. Additional discussion and data are available in the SI, Table S3.†
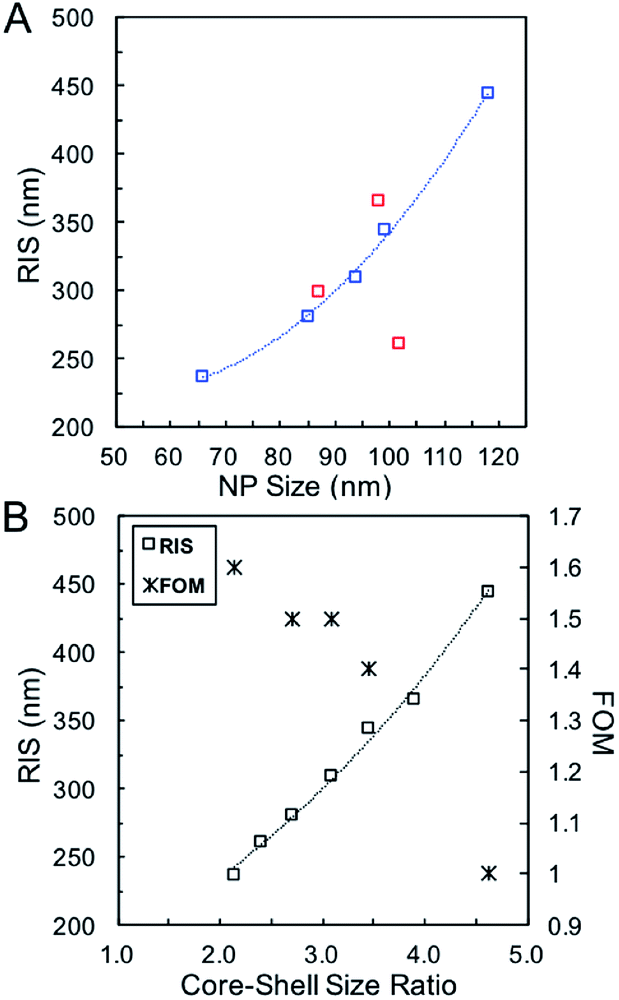 | ||
| Fig. 4 (A) RIS as function of particle size using Ag nanoparticles of different sizes (blue, Table 2) and for various Ag:Au stoichiometries (red, Table 1). (B) RIS and FOM as function of the core–shell ratio. | ||
Conclusion
The optical properties of AgAu hollow nanoparticles prepared through co-reduction galvanic replacement were characterized to quantify the plasmonic behaviour of the resulting structures. Two key parameters were investigated, namely the atomic stoichiometry of the metals and the size of the initial nanoparticle template used. LSPR energy, extinction/scattering intensity and bandwidth were studied using bulk extinction spectroscopy as well as single particle scattering experiments. Experimental data shows that the plasmonic properties depend on the final shape and composition, the latter being controllable directly by varying the Au:Ag atomic ratio, which also affects the final particle shape. We demonstrated that the combination of both approaches, variation of the atomic stoichiometry and sacrificial template size, gives more possibilities for LSPR tunability in terms of peak position as well as control on scattering/absorption intensity and RIS sensitivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the American Chemical Society Petroleum Research Fund under grant number 56256 DNI5 (E. R.) and a 3M Nontenured Faculty Award (E. R.). L. A. M. wishes to acknowledge financial support from a National Science Foundation Graduate Research Fellowship #1450681. D. B. and J. R. D. acknowledge funding from the Natural Sciences and Engineering Research Council of Canada, the Canadian Foundation for Innovation and the Fonds de la Recherche du Québec – Nature et Technologies as well as technical support from Julie-Christine LevesqueReferences
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- X. Yang, M. Yang, B. Pang, M. Vara and Y. Xia, Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- H. Shen, L. J. Tauzin, R. Baiyasi, W. Wang, N. Moringo, B. Shuang and C. F. Landes, Chem. Rev., 2017, 117, 7331–7376 CrossRef CAS PubMed.
- G. V. Hartland, L. V. Besteiro, P. Johns and A. O. Govorov, ACS Energy Lett., 2017, 2, 1641–1653 CrossRef CAS.
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed.
- D. Yu and V. W.-W. Yam, J. Am. Chem. Soc., 2004, 126, 13200–13201 CrossRef CAS PubMed.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzán, ACS Nano, 2014, 8, 5833–5842 CrossRef CAS PubMed.
- A. Sánchez-Iglesias, N. Winckelmans, T. Altantzis, S. Bals, M. Grzelczak and L. M. Liz-Marzán, J. Am. Chem. Soc., 2017, 139, 107–110 CrossRef PubMed.
- B. J. Wiley, Y. Chen, J. M. McLellan, Y. Xiong, Z.-Y. Li, D. Ginger and Y. Xia, Nano Lett., 2007, 7, 1032–1036 CrossRef CAS PubMed.
- P. Senthil Kumar, I. Pastoriza-Santos, B. Rodríguez-González, F. Javier García de Abajo and L. M. Liz-Marzán, Nanotechnology, 2008, 19, 015606 CrossRef PubMed.
- N. Halas, MRS Bull., 2005, 30, 362–367 CrossRef CAS.
- A. L. Aden and M. Kerker, J. Appl. Phys., 1951, 22, 1242–1246 CrossRef.
- G. Mukhopadhyay and S. Lundqvist, Nuovo Cimento B 11, 1975, 27, 1–18 CrossRef.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419–422 CrossRef CAS PubMed.
- T. H. Park and P. Nordlander, Chem. Phys. Lett., 2009, 472, 228–231 CrossRef CAS.
- Y. Wu and P. Nordlander, J. Chem. Phys., 2006, 125, 124708 CrossRef PubMed.
- E. Prodan and P. Nordlander, Nano Lett., 2003, 3, 543–547 CrossRef CAS.
- E. Prodan, P. Nordlander and N. J. Halas, Nano Lett., 2003, 3, 1411–1415 CrossRef CAS.
- M. W. Knight and N. J. Halas, New J. Phys., 2008, 10, 105006 CrossRef.
- S. Schelm and G. B. Smith, J. Opt. Soc. Am. A, 2005, 22, 1288–1292 CrossRef PubMed.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6332 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- Y. Choi, S. Hong, L. Liu, S. K. Kim and S. Park, Langmuir, 2012, 28, 6670–6676 CrossRef CAS PubMed.
- J. R. Daniel, L. A. McCarthy, S. Yazdi, M. Chagnot, E. Ringe and D. Boudreau, J. Phys. Chem. C, 2018, 122, 18168–18176 CrossRef.
- D. Seo and H. Song, J. Am. Chem. Soc., 2009, 131, 18210–18211 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- T. J. A. Slater, A. Macedo, S. L. M. Schroeder, M. G. Burke, P. O'Brien, P. H. C. Camargo and S. J. Haigh, Nano Lett., 2014, 14, 1921–1926 CrossRef CAS PubMed.
- Y. Yin, C. Erdonmez, S. Aloni and A. P. Alivisatos, J. Am. Chem. Soc., 2006, 128, 12671–12673 CrossRef CAS PubMed.
- X. Hong, D. Wang, S. Cai, H. Rong and Y. Li, J. Am. Chem. Soc., 2012, 134, 18165–18168 CrossRef CAS PubMed.
- J. A. I. Acapulco, S. Hong, S. K. Kim and S. Park, J. Colloid Interface Sci., 2016, 461, 376–382 CrossRef CAS PubMed.
- J. Chen, J. M. Mclellan, A. Siekkinen, Y. Xiong and Z. Li, J. Am. Chem. Soc., 2006, 128, 14776–14777 CrossRef CAS PubMed.
- E. Gonzalez, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377–1380 CrossRef CAS PubMed.
- L. Polavarapu, D. Zanaga, T. Altantzis, S. Rodal-Cedeira, I. Pastoriza-Santos, J. Pérez-Juste, S. Bals and L. M. Liz-Marzán, J. Am. Chem. Soc., 2016, 138, 11453–11456 CrossRef CAS PubMed.
- J.-E. Park, S. Kim, J. Son, Y. Lee and J.-M. Nam, Nano Lett., 2016, 16, 7962–7967 CrossRef CAS PubMed.
- H. Jing and H. Wang, Chem. Mater., 2015, 27, 2172–2180 CrossRef CAS.
- L. Yang, B. Yan and B. M. Reinhard, J. Phys. Chem. C, 2008, 112, 15989–15996 CrossRef CAS PubMed.
- M. Hu, H. Petrova, A. R. Sekkinen, J. Chen, J. M. McLellan, Z.-Y. Li, M. Marquez, X. Li, Y. Xia and G. V Hartland, J. Phys. Chem. B, 2006, 110, 19923–19928 CrossRef CAS PubMed.
- M. Hu, J. Chen, M. Marquez, Y. Xia and G. V Hartland, J. Phys. Chem. C, 2007, 111, 12558–12565 CrossRef CAS PubMed.
- M. Hu, C. Novo, A. Funston, H. Wang, H. Staleva, S. Zou, P. Mulvaney, Y. Xia and G. V. Hartland, J. Mater. Chem., 2008, 18, 1949–1960 RSC.
- L. Rainville, M. Dorais and D. Boudreau, RSC Adv., 2013, 3, 13953 RSC.
- A. Kumar, E. Villarreal, X. Zhang and E. Ringe, Adv. Struct. Chem. Imaging, 2018, 4, 8 CrossRef.
- O. Peña-Rodríguez, P. Díaz-Núñez, V. Rodríguez-Iglesias, L. Montaño-Priede, A. Rivera and U. Pal, Nanoscale Res. Lett., 2017, 12, 16 CrossRef.
- E. Hao, S. Li, R. C. Bailey, S. Zou, G. C. Schatz and J. T. Hupp, J. Phys. Chem. B, 2004, 108, 1224–1229 CrossRef CAS.
- A. Grubisic, S. Mukherjee, N. Halas and D. J. Nesbitt, J. Phys. Chem. C, 2013, 117, 22545–22559 CrossRef CAS.
- D. Wu and X. Liu, Appl. Phys. Lett., 2010, 97, 061904 CrossRef.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS.
- A. M. Gobin, M. H. Lee, N. J. Halas, W. D. James, R. A. Drezek and J. L. West, Nano Lett., 2007, 7, 1929–1934 CrossRef CAS.
- X. Huang and M. A. El-Sayed, Alexandria J. Med., 2011, 47, 1–9 CrossRef.
- G. V Hartland, Chem. Rev., 2011, 111, 3858–3887 CrossRef PubMed.
- S. E. Skrabalak, J. Chen, L. Au, X. Lu, X. Li and Y. Xia, Adv. Mater., 2007, 19, 3177–3184 CrossRef CAS PubMed.
- P. K. Jain and M. A. El-Sayed, J. Phys. Chem. C, 2007, 111, 17451–17454 CrossRef CAS.
- F. Tam, C. Moran and N. Halas, J. Phys. Chem. B, 2004, 108, 17290–17294 CrossRef CAS.
- L. J. Sherry, R. Jin, C. A. Mirkin, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2006, 6, 2060–2065 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09364d |
| This journal is © The Royal Society of Chemistry 2019 |