
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-nanoparticle-dispersed amorphous BaTiO3 thin films as hole-trapping centers: enhanced photocatalytic activity and stability†
Su-Wei Zhang‡
ab,
Shun Li‡ cd,
Bo-Ping Zhang*a,
Dongfang Yuc,
Zuotai Zhangc and
Jing-Feng Lib
cd,
Bo-Ping Zhang*a,
Dongfang Yuc,
Zuotai Zhangc and
Jing-Feng Lib
aThe Beijing Municipal Key Laboratory of New Energy Materials and Technologies, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: bpzhang@ustb.edu.cn; Tel: +86-010-62334195
bState Key Laboratory of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
cSchool of Environmental Science and Engineering, Guangdong Provincial Key Laboratory of Soil and Groundwater Pollution Control, Southern University of Science and Technology, Shenzhen 518055, China
dSUSTech Academy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen 518055, China
First published on 11th February 2019
Abstract
Nobel metal (Au and Ag) nanoparticles are often used in semiconductor photocatalysis to enhance the photocatalytic activity, while inexpensive Cu attracts less attention due to its easy oxidization. Herein, an elaborate study was conducted using Cu-nanoparticle-dispersed amorphous BaTiO3 films as photocatalysts. Photocatalytic and photoelectrochemical measurements demonstrated that the degradation efficiency and photocurrent density of the nanocomposite films are approximately 3.5 and 10 times as high as the pristine BaTiO3 film, respectively, which can be ascribed to a synergetic effect of the surface plasmon resonance and interband excitation. In addition, a good stability was also demonstrated by cyclic tests for the degradation of rhodamine B, which may be due to the amorphous nature of the BaTiO3 matrix providing hole-trapping centers. The high photocatalytic stability suggests that Cu is a promising alternative metal to replace Au and Ag for the development of cost-effective photocatalysts. Our work demonstrates a simple and promising strategy for improving the photostability of Cu nanomaterials and may provide a useful guideline for designing Cu-based composite materials toward various photocatalytic applications such as water pollution treatment.
1. Introduction
Photocatalysis technology is considered as one of the most promising strategies for harnessing sufficient and renewable solar energy for clean energy production and environmental remediation.1 However, the photocatalytic efficiency of most semiconductors with large bandgaps (e.g., TiO2, ZnO and SrTiO3) is seriously hindered because they can only respond to UV light which occupies merely approximately 4% of the whole solar spectrum.1,2 In addition, possessing a highly stable structure during the photocatalytic process is highly desirable for photocatalytic materials. Therefore, it is very important to develop suitable photocatalytic materials, which can utilize a wider range of visible light and maintain stability under light irradiation. To this end, many strategies have been carried out such as metal-ion and nonmetal doping,3 metal–oxide complexes,4–6 surface modification,7,8 and dispersal of noble metals.9,10Over the past few decades, dispersing noble metal nanoparticles (NPs) such as Pt, Au and Ag onto semiconductors has emerged as an efficient method to improve the photocatalytic activity of semiconductors.11–14 As an important low-cost metal, Cu NPs can provide active centers with great promise in catalysis that are expected to be an alternative material for the replacement of noble metals.15–20 A few reports have elaborated that the addition of Cu NPs can promote photocatalytic activity of semiconductors due to the localized surface plasmon resonance (LSPR) effect.21–26 Furthermore, the interband transition energy for Cu is very low (1.9 eV), suggesting that dispersing Cu into semiconductor photocatalysts could potentially enhance the photocatalytic property. However, up until now, Cu has drawn less attention in the field of photocatalysis compared to Au and Ag due to its easy oxidization under ambient conditions. For example, Sato et al. reported that the hydrogen production efficiency using Cu–graphene was much stronger than that of the pure graphene, whereas Cu NPs were oxidized into Cu2O after being irradiated for 8 h.22 Very recently, Qiao et al. have demonstrated that hybrid Cu/CQDs when used as a photocatalyst manifest an outstanding photoactivity. However, the Cu/CQD hybrids exhibited a CuO impurity after photocatalytic reaction.27 These phenomena could be attributed to the fact that excess photogenerated holes (h+) can accumulate on the Cu surface and subsequently induce rapid oxidation of Cu to form Cu2+ or Cu+.
Therefore, enhancing photoinduced stability (or preventing oxidation) of Cu is an important and meaningful subject for practical applications. One possible strategy is to modify the surface by loading with an h+-trapping cocatalyst to prevent oxidation of the photocatalyst via the rapid transfer of photogenerated h+.28,29 Considering the advantages of the low-cost and stable properties of BaTiO3 (BTO) for using as a photocatalyst,30,31 we have investigated for the first time whether its amorphous form can act as h+-trapping centers to improve the photoinduced stability of Cu, with the aim of improving its photocatalytic activity. Herein, we conducted an elaborate study for an amorphous BTO and Cu NP composite thin film system, in which both photocatalytic activity and photoinduced stability of Cu are enhanced during the photocatalytic process.
2. Experimental
2.1 Material preparation
Cu/BTO films with varying Cux/(Cu + BTO) ratios (x = 0, 10, 15, 20, 25, 30, 35 and 40, where x is mol% of Cu, named as Cux/BTO) were fabricated by using a sol–gel and spin-coating method, starting with barium acetate (Ba(CH3COO)2, 99.0%), tetrabutyl titanate (Ti(CH9O4)4, 98.0%) and copper nitrate (Cu(NO3)2·3H2O, 99.8%) as raw materials, as well as acetic acid (CH3COOH, 99.5%), deionized water (H2O) and glycerol (C3H8O3, 99.0%) as solvents. First, 1 mol of Ba(CH3COO)2 was dissolved into the blend solution of CH3COOH (5 ml), H2O (5 ml) and C3H8O3 (5 ml). After stirring for 2 h, 1 mol of Ti(C4H9O4)4 was added to the solution, followed by stirring for another 2 h. Several drops of nitric acid for hydrolysis catalysis were added to adjust the pH value of the solution between 2 and 3. Then, a resultant solution of C3H8O3 and Cu(NO3)2·3H2O was added into the above precursor solution containing varying chemical compositions of Cux/BTO. To form the Cux/BTO films, the obtained solution was spin coated onto a glass substrate with a size of 2 × 2 cm at 500 rpm for 5 s and subsequently 3000 rpm for 15 s. Finally, the films were dried at 150 °C for 30 s and subsequently heated at 500 °C for 1 h in a protection atmosphere of 5% H2 + 95% Ar gas to prevent the oxidization of Cu NPs. These sequences for coating and pyrolysis treatments were repeated for 10 times to form the final Cux/BTO films.2.2 Characterization
The crystal structures for all the samples were investigated by XRD (X-ray diffraction: DMAX-RB, Rigaku Inc.) with Cu Kα radiation (λ = 1.5406 Å) filtered using a Ni foil. TG (Thermogravimetry) and DTA (differential thermal analysis) were carried out for the precursor powders in a protection atmosphere of N2 gas to prevent the oxidization of Cu NPs (HCT-1, Beijing Optical Instrument Inc.). The powders were obtained by drying the precursor solution in an oven at 60 °C. Characterization of the morphology and chemical composition were performed by FESEM (field-emission scanning electron microscopy, JEOL JSM7401F). The TEM (transmission electron microscopy) images and SAED (selected area electron diffraction) patterns were obtained using an FEI Tecnai G2 F20 microscope. The chemical binding state for the elements composing the prepared films was characterized by XPS (X-ray photoelectron spectroscopy: PHI-5300, PHI) using Al Kα radiation (hν = 1486.6 eV) as an X-ray source. The energies for all spectra were adjusted by normalizing with respect to the C 1s peak at 284.8 eV. The Cu 2p, Ba 3d and Ti 2p spectra were fitted using the XPS data, which were determined by deconvolution with the program XPS Peak 4.0. The absorption spectra were obtained by using an UV-2800 (UV-Visible spectrophotometer, UNICO Instruments Co., Ltd.). Stable-state PL (photoluminescence) spectra for the films were measured using an F-4500 FL spectrophotometer with an excitation wavelength of 335 nm at room temperature. The ESR (electron spin resonance) spectra were measured by a Bruker model ESR 300E spectrometer equipped with a quanta-Ray Nd:YAG laser system as the irradiation source (λ = 355 nm, 10 Hz). The corresponding parameters for the spectrometer, such as sweep width, center field, microwave frequency, modulation frequency and power, were set as 3486.70 G, 100 G, 9.82 GHz, 100 kHz and 5.05 mW, respectively.2.3 Photocatalytic and photoelectrochemical tests
The photocatalytic activity was estimated from the degradation of rhodamine B (RhB) under UV-Vis light with a 500 W xenon lamp (Beijing institute of electrical light sources, China). A 20 mM RhB solution was spin coated onto the surface of the films at a rate of 3000 rpm for 30 s. The reaction temperature was kept at room temperature to prevent any thermal catalytic effect. Films with the RhB layer were kept in the dark for 30 min to reach the adsorption/desorption equilibrium between the photocatalyst and RhB before illumination, and were 15 cm away from the light source. The degradation rate was monitored by monitoring the maximal absorbance peak value of RhB (550–580 nm) relative to its initial intensity under various illumination times using a UV-Vis spectrophotometer on the basis of the following formula: η = (A0 − At)/A0 × 100%, η is the decolorization ratio of the reaction, A0 is the absorbance of the RhB solution before the reaction, and At is the absorbance of the RhB solution after t min.The photoelectrochemical measurements were performed using a CHI660E electrochemical station in 1 M Na2SO4 solution by a three-electrode cell, where the FTO glass substrates coated with a Cu/BTO film were used as the working electrode, a platinum wire as the counter electrode and Ag/AgCl as the reference electrode. The EIS (electrochemical impedance spectra) were recorded by applying an AC voltage of 5 mV amplitude in the frequency range of 0.01 Hz to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz with the initial potential referring to the open-circuit voltage. The IPCE (incident photon-to-current conversion efficiency) was measured at a series of wavelengths by inserting various bandpass filters in front of the reaction cell to obtain the desired incident wavelength. The light intensities were measured using a spectroradiometer (PL-MW2000 Photoradiometer; PerfectLight, China).
000 Hz with the initial potential referring to the open-circuit voltage. The IPCE (incident photon-to-current conversion efficiency) was measured at a series of wavelengths by inserting various bandpass filters in front of the reaction cell to obtain the desired incident wavelength. The light intensities were measured using a spectroradiometer (PL-MW2000 Photoradiometer; PerfectLight, China).
3. Results and discussion
Fig. 1a shows the XRD results obtained for the Cux/BTO films. The standard diffraction peaks for Cu (PDF#04-0836) are shown with vertical lines for comparison. A broad diffraction peak for all the films was detected at approximately 25°, which corresponds to the BTO matrix,31 indicating that the matrix is amorphous. From a typical TG-DTA curve for the Cu25/BTO precursor powders in Fig. S1,† we can see that no further weight loss was observed above 600 °C, which demonstrates that the starting crystallization temperature for the BTO matrix is ca. or higher than 600 °C. In addition, the diffraction peaks at 2θ values of 43.3°, 50.4° and 74.1° belong to the (111), (200) and (220) crystal planes of metallic Cu, respectively. The strengthened diffraction intensity for Cu with increasing x indicates an enhanced crystallinity and grain growth, which is in accordance with the TEM results obtained for the Cux/BTO films with x = 10, 15 and 35 (Fig. S2a–c†). The in-plane TEM image for the Cu25/BTO films in Fig. 1b reveals that most of the Cu NPs (as indicated by the red imaginary line) are almost spherical in shape and homogeneously distributed in the amorphous BTO matrix. The SAED pattern in Fig. 1c shows characteristic diffraction rings for metallic Cu (111), (200), (220) and (311) from inner to outer, which match well with the XRD results (Fig. 1a). From the HAADF-STEM characterization for the Cu35/BTO sample (Fig. S2d†), bright spots due to Cu can be clearly discerned from the background. Elemental mapping results (Fig. S2e and f†) further confirm the composition of the bright spots to be Cu elements with a uniform distribution in the film.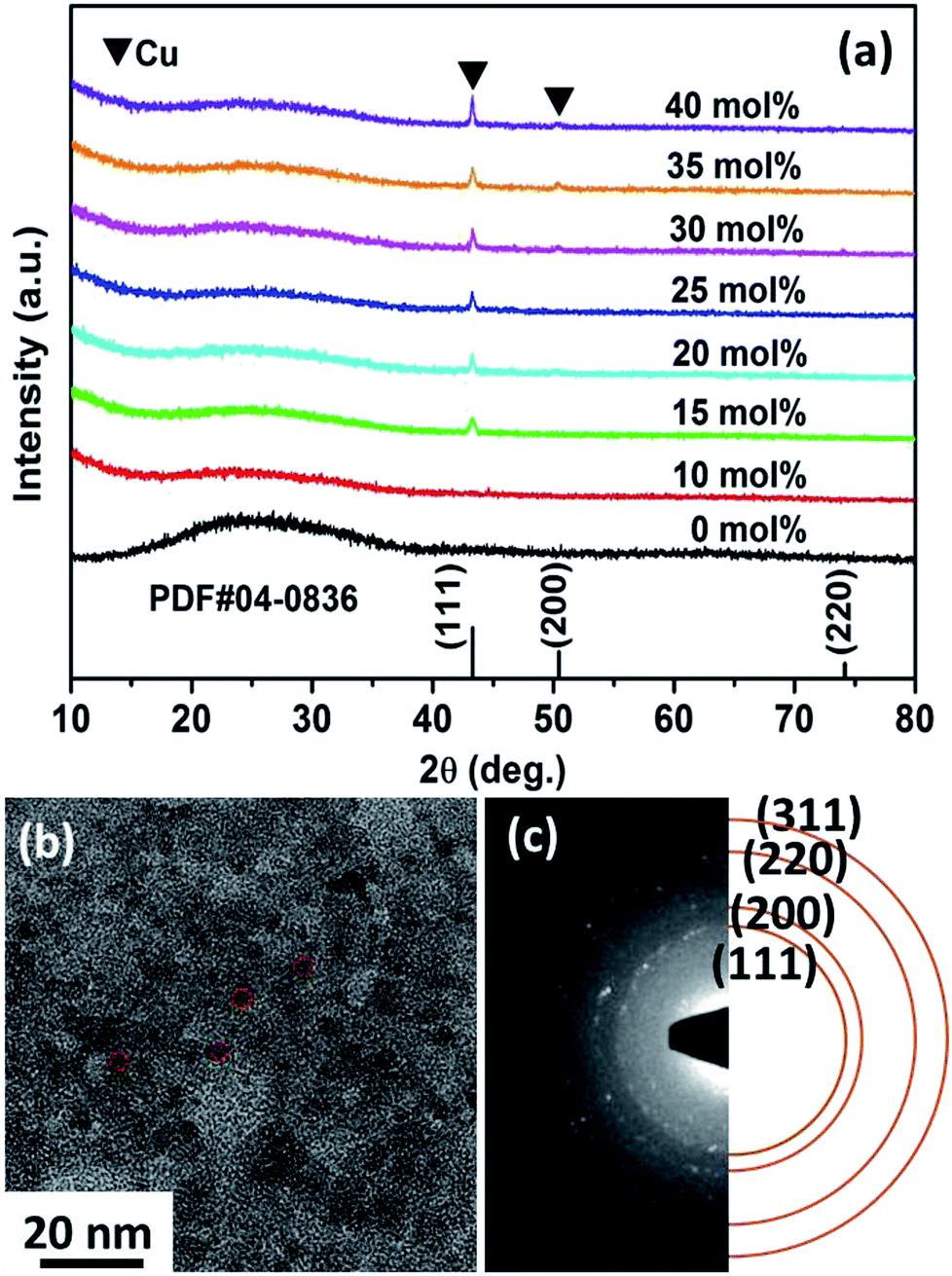 | ||
| Fig. 1 (a) XRD patterns for the Cux/BTO thin films. (b) TEM micrograph and (c) SAED patterns of the Cu25/BTO sample. | ||
The XPS spectra for the Cu25/BTO composite films (Fig. S3a†) confirm the existence of elemental Cu, Ba, Ti, and O. Peaks at 932.5 and 952.3 eV in Fig. S3b† refer to Cu0 2p3/2 and Cu0 2p1/2, respectively, which are slightly shifted to lower binding energies compared to the corresponding values 932.7 and 952.5 eV for characteristic metallic Cu.32 The Ba2+ 3d5/2 and Ba2+ 3d3/2 peaks in Fig. S3c† centered at 780.2 and 795.5 eV are slightly lower than those in BTO,33 respectively. The two peaks at 458.3 and 464.1 eV in Fig. S3d† are in accordance with the Ti4+ 2p3/2 and Ti4+ 2p1/2 peaks at 458.1 and 463.9 eV.32 The latter pair of peaks arise from Ti3+ 2p3/2 and Ti3+ 2p1/2, which are located at lower binding energies of 457.2 and 463.0 eV, approximately similar to the reported values of 457.1 and 463.1 eV.34 This may be due to the deficient oxygen supply in an air environment at 500 °C, which is lower than the crystallization temperature of BTO at 600 °C, as demonstrated by the TG-DTA results (Fig. S1†). As a result, the oxygen vacancies (named as  ) can be generated resulting in the partial reduction of Ti4+ to Ti3+, as depicted by the following defect equations:
) can be generated resulting in the partial reduction of Ti4+ to Ti3+, as depicted by the following defect equations:
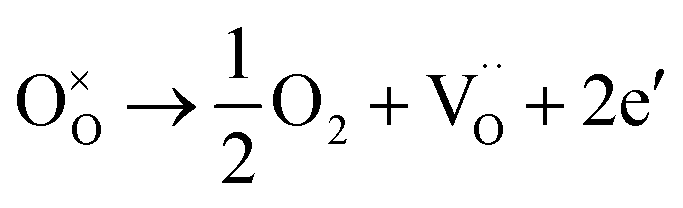 | (1) |
| Ti4+ + e− → Ti3+ | (2) |
The reduction of tetravalent cations to a lower valence state in B-sites has been found in a BTO ceramic system.34
To understand the optical properties of the Cux/BTO films, the theoretical SPR absorption spectra were calculated first based on Mie theory,35–38 as shown in Fig. S4a.† An SPR peak located at 625 nm was found for all samples, while an obvious enhancement of the interband transition (ca. 300 to 652 nm) was observed with increasing x value. On the other hand, the experimentally measured optical absorption spectra for Cux/BTO in Fig. S4b† exhibit an absorption peak in the range of 560–650 nm due to the SPR effect in the Cu NPs under visible light. The intensity of the SPR peak strengthens, while an obvious enhancement of the interband transition was also observed with increased x, along with an overlap of the SPR band and interband transition absorption edge, which agrees well with the theoretical simulation.
The photocatalytic and photoelectrochemical activity of Cux/BTO films were evaluated. The absorption spectra of RhB after photocatalytic degradation by various samples are shown in Fig. S5a.† The photodegradation results in Fig. 2a manifest that the self-degradation for RhB is very weak under UV-Vis light without a catalyst, with only 2% of RhB degraded after 5 h of irradiation, while ca. 12% of RhB can be degraded by the pristine BTO. All the Cux/BTO samples exhibited an improved photocatalytic activity, with the highest photodegradation rate for RhB of 42% reached when x = 25. The RhB degradation rate over Cu25/BTO is approximately 3.5 times as high as the pristine BTO. The corresponding photodegradation rate kinetics are shown in Fig. S5b,† and the inset of (b) shows the histogram of the photodegradation rates (k is derived as the slope of the ln(C0/C) − t plot according to the first-order kinetics). Cycling measurements for RhB degradation over Cu25/BTO were carried out to evaluate the photostability of the Cu. After three cycles, the RhB degradation rate in Fig. 2b slightly decreased from 42% to 40%, indicating a high durability and a good stability for the Cu25/BTO catalyst. Fig. 2c shows the photocurrent profiles for these photoanodes recorded under zero-bias. Obviously, the Cu25/BTO can produce a photocurrent density that is approximately 10 times higher than that of the pristine BTO. The EIS curves, which can be used to evaluate the charge transfer (CT) resistance (RCT), were measured,39 as shown Fig. 2d. It is well known that the smaller the radius of the impedance-related arc, the faster the CT between the electrode and the electrolyte.40 Due to the smaller radius of the arc for the Cu25/BTO sample compared to the pristine BTO, Cu25/BTO has a smaller RCT and stronger CT ability.
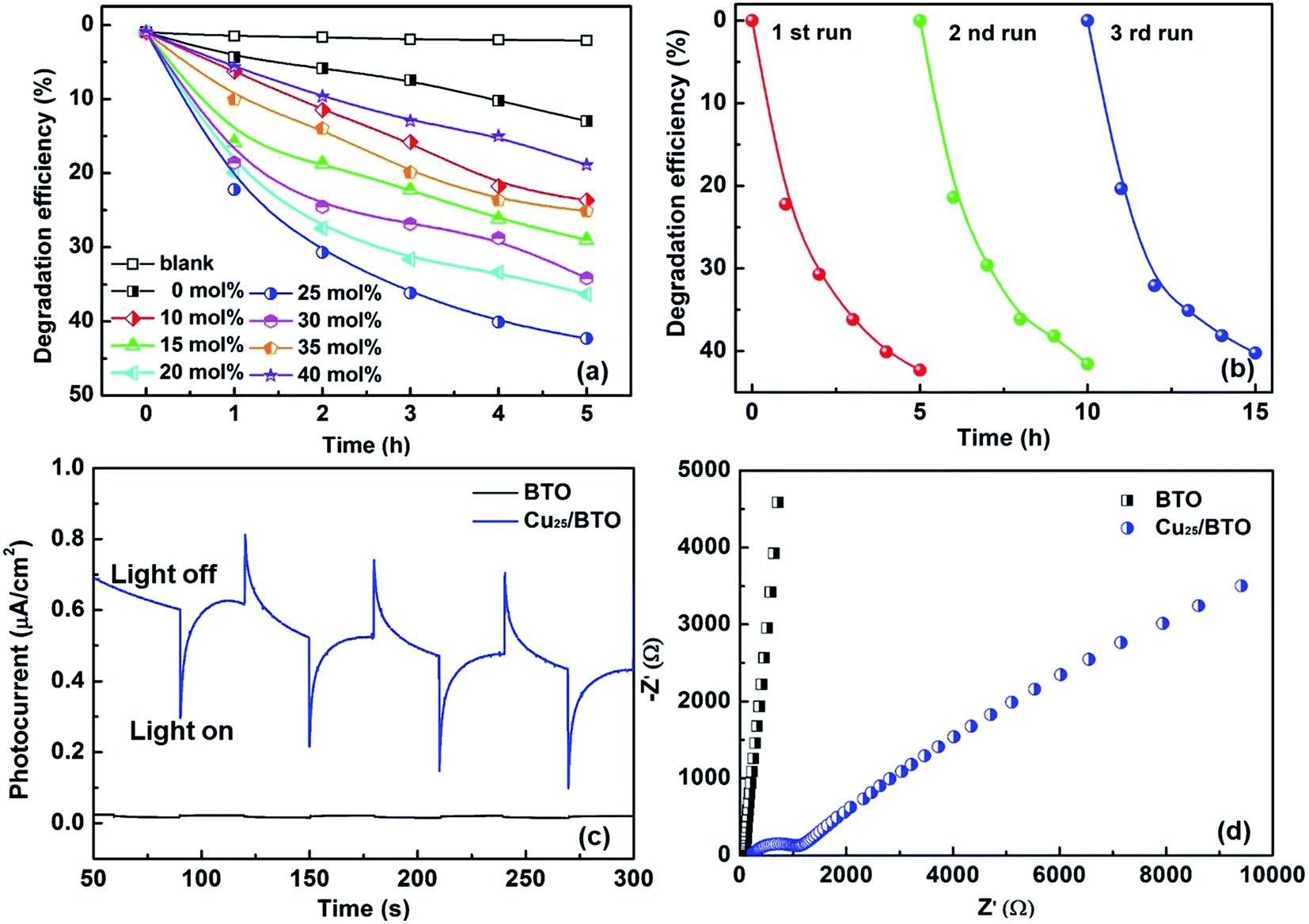 | ||
| Fig. 2 (a) Degradation rate of RhB using the Cux/BTO thin films under UV-Vis light irradiation. (b) Cycling measurements for RhB degradation efficiency. (c) Photocurrent density vs. time and (d) Nyquist plots for pristine BTO and Cu25/BTO thin-film photoelectrodes. | ||
Photoluminescence (PL) spectroscopy is widely used to follow the irradiative recombination of photogenerated charge carriers.41 Therefore, we carried out PL measurements to analysis the CT behavior in the Cu/BTO system using the Cu25/BTO sample showing the highest photocatalytic efficiency, as well as pristine BTO for comparison. In Fig. 3a, both the spectra possess two emission peaks at 387 and 424 nm, which can be ascribed to recombination between e− in the conduction band (CB) and h+ in the valence band (VB) and recombination between e− in a defect level and h+ in the VB, respectively. The defect level may result from the 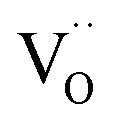 (as demonstrated by the XPS results in Fig. S3d†), which can usually be found in amorphous BTO.42 Notably, the PL intensity of the BTO obviously decreased after the dispersion of Cu NPs, suggesting that the recombination of the photogenerated e−–h+ pairs is greatly inhibited in the Cu25/BTO. Similar behavior has also been observed in other material systems.43
(as demonstrated by the XPS results in Fig. S3d†), which can usually be found in amorphous BTO.42 Notably, the PL intensity of the BTO obviously decreased after the dispersion of Cu NPs, suggesting that the recombination of the photogenerated e−–h+ pairs is greatly inhibited in the Cu25/BTO. Similar behavior has also been observed in other material systems.43
 | ||
| Fig. 3 (a) PL spectra for pristine BTO and Cu25/BTO thin films with an excitation wavelength of 335 nm at room temperature. (b) IPCE curves, simulated and measured absorption spectra for the Cu25/BTO thin film. (c) Optical absorption spectra and (d) degradation rate of RhB over pristine BTO and (Au35,31 Ag25,45 Cu25)/BTO thin films under UV-Vis light. | ||
Fig. 3b shows the IPCE curve for the Cu25/BTO sample. Noteworthy, the IPCE curve for the Cu25/BTO shows a strong similarity to the interband transition band at shorter wavelength (400–550 nm) and is also consistent to some extent with the SPR spectrum measured at longer wavelengths (550–700 nm). This result gives clear proof that the hot e− transferred from Cu to BTO for RhB degradation over the Cu25/BTO arises from the synergetic effect of the interband transition and SPR effect of Cu NPs, which is unlike that reported for the Au–Cu/SrTiO3 system.44 For further evaluating the photocatalytic performance, we compared the photodegradation efficiency of RhB over pristine BTO, Au35/BTO,31 Ag25/BTO45 and Cu25/BTO (this work) samples (the optical absorption spectra are shown in Fig. 3c) under UV-Vis light, as shown in Fig. 3d. We can see that the photodegradation efficiency over Cu25/BTO is comparable to that of Au35/BTO and Ag25/BTO, which shows that Cu is a highly promising alternative to noble metals.
To test the photostability of Cu in the composite film, XPS measurements were performed. Fig. 4a shows the high-resolution XPS spectra for Cu 2p before reaction and after the 1st, 2nd and 3rd run, respectively. It can be clearly seen that the binding energy peaks for Cu 2p located at 952.3 and 932.5 eV remain unchanged after the photocatalytic reactions, suggesting good photostability for the metallic Cu during the photocatalysis process. The oxidization of Cu was successfully restrained by the dispersion in amorphous BTO, which is possibly because amorphous BTO can act as h+-trapping centers to avoid photoinduced oxidation of Cu. To prove our hypothesis, an ESR test was used to detect the reactive h+ species both in the dark and under UV-Vis light illumination on the Cu25/BTO sample. As shown in Fig. 4b, the ESR signal (g = 2.004) occurs in the dark. Previous work suggested that the g = 2.004 signal is associated with titanium vacancies (ionized  ) contained in the surface of BTO.46,47 It is, therefore, reasonable to speculate that the surface of the amorphous BTO prepared in this study also contains such
) contained in the surface of BTO.46,47 It is, therefore, reasonable to speculate that the surface of the amorphous BTO prepared in this study also contains such 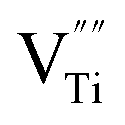 defects, which could be ascribed to the lack of Ti4+ due to the amorphous structure of BTO:
defects, which could be ascribed to the lack of Ti4+ due to the amorphous structure of BTO:
 | (3) |
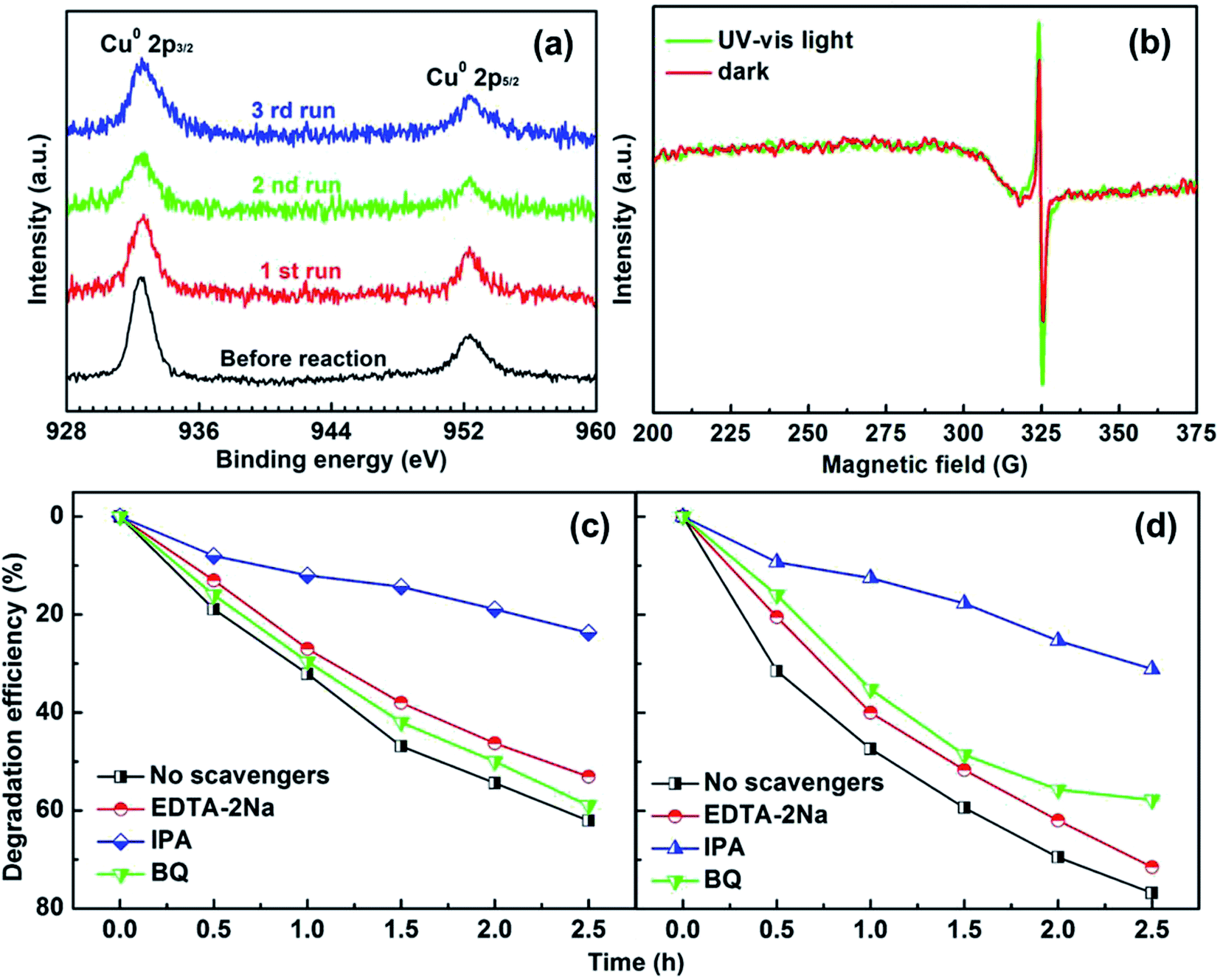 | ||
| Fig. 4 (a) High-resolution XPS spectra for Cu 2p before and after the 1st, 2nd and 3rd run reaction. (b) ESR signals for the Cu25/BTO thin film measured in the dark and under UV-Vis light irradiation for 40 min. Photocatalytic trapping experiments for the degradation of RhB solution over (c) pristine BTO and (d) Cu25/BTO photocatalysts with the addition of EDTA-2Na, IPA, and BQ as scavengers. | ||
The intensity obviously increases after irradiation by UV-Vis light, indicating an increase of h+ accumulated in the BTO surface region. While no ESR signal for ionic Cu was detected both in the dark and under UV-Vis light irradiation,48,49 indicating that Cu remains in the metallic state without oxidation. According to a previous report, amorphous TiO2 can act as h+-trapping centers to improve the stability of CdS via the effective capture of photogenerated h+.29,50 In the present work, we believe that the amorphous BTO can also work as h+-trapping centers to avoid the photoinduced oxidation of Cu NPs in the photocatalytic process.
To understand in depth the role of active species in the photocatalytic process of RhB over the pristine BTO and Cu25/BTO photocatalysts, various scavengers including disodium ethylenediaminetetraacetate (EDTA-2Na), iso-propanol (IPA) and benzoquinone (BQ) were employed as quenchers of superoxide radicals (·O2), holes (h+) and hydroxyl radicals (·OH), respectively.51–53 Typically, films were immersed in 20 ml of 10 mg L−1 RhB aqueous solution, which was irradiated with a 500 W high pressure Xe lamp light source (positioned 8 cm away from the catalytic reactor). From Fig. 4c, the addition of IPA has the most significant negative effect on RhB degradation, with only 23% RhB degraded, which suggests the pivotal role of ·OH in the process of photocatalytic reaction over pristine BTO. However, when BQ and EDTA-2Na were added, degradation efficiencies are merely 53% and 59% respectively, implying that ·O2 and h+ have no obvious effect on degradation of RhB. On the other hand, for the Cu25/BTO sample in Fig. 4d, the degradation efficiencies of RhB increase after adding IPA and EDTA-2Na, compared to that of the pristine BTO. These results clearly indicate that the role of h+ weakened in the photocatalytic process after dispersing Cu NPs into the amorphous BTO matrix. Therefore, we can conclude that the amorphous BTO can work as h+-trapping centers in the photocatalytic process, thus avoiding the photoinduced oxidation of Cu NPs.
A possible band configuration for amorphous BTO and Cu is proposed to explain the potential mechanism, as illustrated in Fig. 5. Under UV-light illumination, e− are produced in the CB and defect level, leaving h+ on the VB in BTO. Meanwhile, under UV and visible light, the SPR effect and interband transition promotes efficient CT from Cu NPs to BTO (CTSPR and CTinter), as indicated by the blue and plum-purple arrows. Such CTSPR and CTinter can promote the generation of active ·O2 radicals on the BTO surface, thus leading to an enhanced photocatalytic activity. On the other hand, the h+ generated in both the BTO and Cu can be trapped in the amorphous BTO with many 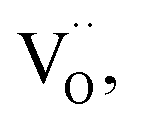 and, then, rapidly consumed by the h+-trapping centers, thus hindering the oxidation of Cu. A similar behavior was reported in amorphous TiO2 systems.29,50 As a result, the photoinduced stability of Cu is enhanced by the amorphous BTO matrix acting as h+ trapping centers.
and, then, rapidly consumed by the h+-trapping centers, thus hindering the oxidation of Cu. A similar behavior was reported in amorphous TiO2 systems.29,50 As a result, the photoinduced stability of Cu is enhanced by the amorphous BTO matrix acting as h+ trapping centers.
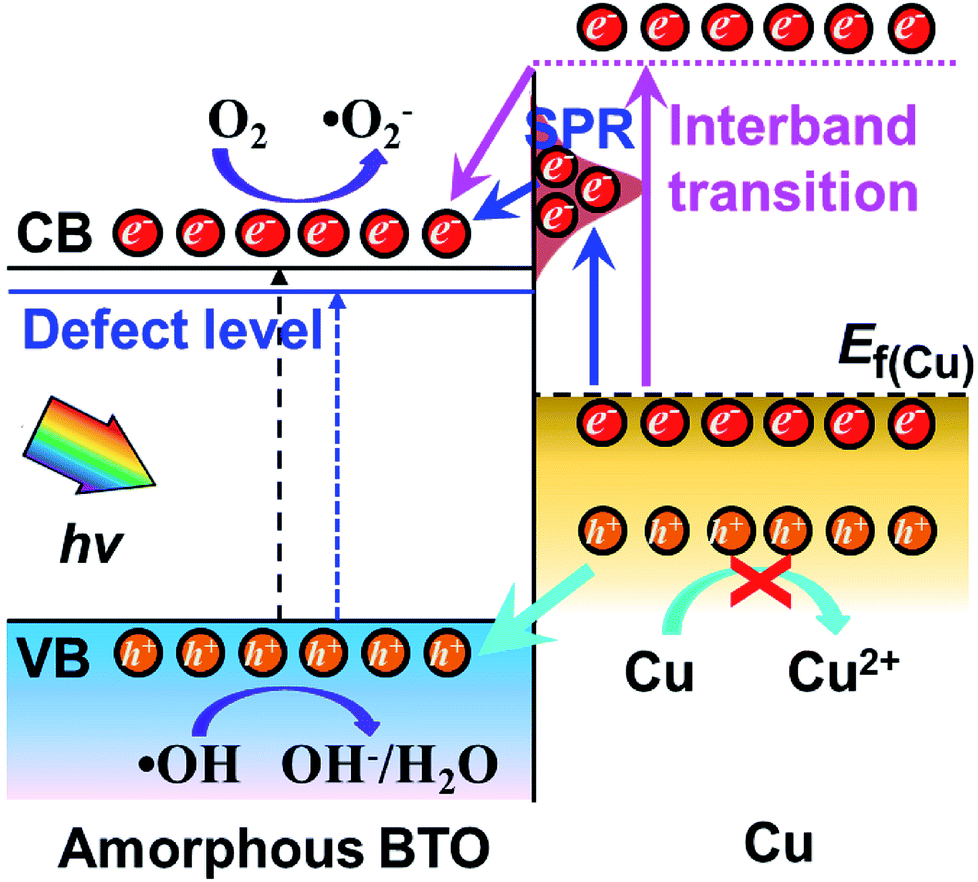 | ||
| Fig. 5 Schematic diagram for the band structure and possible charge transfer mechanism for amorphous BTO dispersed with Cu NPs. | ||
4. Conclusions
In summary, amorphous BTO films dispersed with Cu NPs were fabricated using a sol–gel and spin-coating method. Both the photocatalytic degradation activity and photoelectrochemical performance of the amorphous BTO films were improved after introducing Cu NPs. Such enhancements could be related to both surface plasmon resonance and interband excitation effects. Interestingly, the photocatalytic activity of Cux/BTO is comparable to that of Au/BTO and Ag/BTO counterparts with good stability observed after three cycles. We also found that the oxidation of Cu is strongly hindered when dispersing in amorphous BTO films that serve as h+-trapping centers. The strategy described here may benefit the application of highly stable and nonprecious Cu in amorphous semiconductor photocatalysis and open up new opportunities for designing and preparing UV-Vis responsive photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51472026) and Specialized Research Fund for the Doctoral Program of Higher Education (Grant No. 20130006110006).References
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- M. M. Hasan and N. K. Allam, RSC Adv., 2018, 8, 37219–37228 RSC.
- H. Yin, K. Yu, C. Song, R. Huang and Z. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 14851–14860 CrossRef CAS PubMed.
- K. Xiong, K. Wang, L. Chen, X. Wang, Q. Fan, J. Courtois, Y. Liu, X. Tuo and M. Yan, Nano-Micro Lett., 2017, 10, 17 CrossRef PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Zhang, Y. Su, H. Wei, J. Wang, C. Zhang, J. Zhao, Z. Yang, M. Xu, L. Zhang and Y. Zhang, Mater. Lett., 2013, 107, 251–254 CrossRef CAS.
- S. Demirci, T. Dikici, M. Yurddaskal, S. Gultekin, M. Toparli and E. Celik, Appl. Surf. Sci., 2016, 390, 591–601 CrossRef CAS.
- J. Manna, S. Goswami, N. Shilpa, N. Sahu and R. K. Rana, ACS Appl. Mater. Interfaces, 2015, 7, 8076–8082 CrossRef CAS PubMed.
- E. Jiang, X. Liu, H. Che, C. Liu, H. Dong and G. Che, RSC Adv., 2018, 8, 37200–37207 RSC.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133–5146 RSC.
- Y. Wang, Q. Lai, F. Zhang, X. Shen, M. Fan, Y. He and S. Ren, RSC Adv., 2014, 4, 44442–44451 RSC.
- U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. da Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinacé and E. L. Pires, Appl. Catal., A, 2001, 211, 1–17 CrossRef CAS.
- H. Yu, F. Peng, J. Tan, X. Hu, H. Wang, J. Yang and W. Zheng, Angew. Chem., 2011, 123, 4064–4068 CrossRef.
- B. P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270, 16–25 CrossRef CAS.
- A. M. Kirillov, M. N. Kopylovich, M. V. Kirillova, M. Haukka, M. F. C. G. da Silva and A. J. L. Pombeiro, Angew. Chem., Int. Ed., 2005, 44, 4345–4349 CrossRef CAS PubMed.
- N. Niklas, F. W. Heinemann, F. Hampel, T. Clark and R. Alsfasser, Inorg. Chem., 2004, 43, 4663–4673 CrossRef CAS PubMed.
- W.-Z. Shen, L. Yi, P. Cheng, S.-P. Yan, D.-Z. Liao and Z.-H. Jiang, Inorg. Chem. Commun., 2004, 7, 819–822 CrossRef CAS.
- J. Dong, J. Ye, D. Ariyanti, Y. Wang, S. Wei and W. Gao, Chemosphere, 2018, 204, 193–201 CrossRef CAS PubMed.
- Y. Sato, S.-I. Naya and H. Tada, APL Mater., 2015, 3, 104502 CrossRef.
- A. Marimuthu, J. Zhang and S. Linic, Science, 2013, 339, 1590–1593 CrossRef CAS PubMed.
- M. S. Rodrigues, D. Costa, R. P. Domingues, M. Apreutesei, P. Pedrosa, N. Martin, V. M. Correlo, R. L. Reis, E. Alves, N. P. Barradas, P. Sampaio, J. Borges and F. Vaz, Appl. Surf. Sci., 2018, 438, 74–83 CrossRef CAS.
- G. V. Hartland, Chem. Rev., 2011, 111, 3858–3887 CrossRef CAS PubMed.
- X.-J. Lv, S.-X. Zhou, C. Zhang, H.-X. Chang, Y. Chen and W.-F. Fu, J. Mater. Chem., 2012, 22, 18542–18549 RSC.
- S. Qiao, B. Fan, Y. Yang, N. Liu, H. Huang and Y. Liu, RSC Adv., 2015, 5, 43058–43064 RSC.
- M. K. I. Senevirathna, P. K. D. D. P. Pitigala and K. Tennakone, J. Photochem. Photobiol., A, 2005, 171, 257–259 CrossRef CAS.
- H. Yu, X. Huang, P. Wang and J. Yu, J. Phys. Chem. C, 2016, 120, 3722–3730 CrossRef CAS.
- W.-J. Liang, L. Ma, H. Liu and J. Li, Chemosphere, 2013, 92, 1390–1395 CrossRef CAS PubMed.
- S.-W. Zhang, B.-P. Zhang, S. Li, X. Y. Li and Z. C. Huang, J. Alloys Compd., 2016, 654, 112–119 CrossRef CAS.
- G. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corporation, 1979, vol. 64.
- M. Wegmann, L. Watson and A. Hendry, J. Am. Ceram. Soc., 2004, 87, 371–377 CrossRef CAS.
- L. Zhao, B.-P. Zhang, P.-F. Zhou, L.-F. Zhu and N. Wang, Ceram. Int., 2016, 42, 1086–1093 CrossRef CAS.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Condens. Matter Mater. Phys., 1972, 6, 4370–4379 CrossRef CAS.
- H. Hövel, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 18178 CrossRef.
- J. Lermé, B. Palpant, B. Prével, M. Pellarin, M. Treilleux, J. L. Vialle, A. Perez and M. Broyer, Phys. Rev. Lett., 1998, 80, 5105–5108 CrossRef.
- S. Wang and B. Zhang, Appl. Catal., A, 2013, 467, 585–592 CrossRef CAS.
- M. Ye, X. Wen, N. Zhang, W. Guo, X. Liu and C. Lin, J. Mater. Chem. A, 2015, 3, 9595–9600 RSC.
- M. A. Mohamed, J. Jaafar, M. F. M. Zain, L. J. Minggu, M. B. Kassim, M. S. Rosmi, N. H. Alias, N. A. Mohamad Nor, W. N. W. Salleh and M. H. D. Othman, Appl. Surf. Sci., 2018, 436, 302–318 CrossRef CAS.
- X. Yuan, Z. Wu, G. Zeng, L. Jiang, J. Zhang, T. Xiong, H. Wang and D. Mo, Appl. Surf. Sci., 2018, 454, 293–304 CrossRef CAS.
- L. V. Maneeshya, V. S. Anitha, S. S. Lekshmy, I. John Berlin, P. B. Nair, G. P. Daniel, P. V. Thomas and K. Joy, J. Mater. Sci.: Mater. Electron., 2013, 24, 848–854 CrossRef CAS.
- Y. Li, B.-P. Zhang, J.-X. Zhao, Z.-H. Ge, X.-K. Zhao and L. Zou, Appl. Surf. Sci., 2013, 279, 367–373 CrossRef CAS.
- M. Liu, W. Zhou, T. Wang, D. Wang, L. Liu and J. Ye, Chem. Commun., 2016, 52, 4694–4697 RSC.
- S.-W. Zhang, B.-P. Zhang, S. Li, Z. Huang, C. Yang and H. Wang, J. Adv. Ceram., 2017, 6, 1–10 CrossRef CAS.
- L. Da-Yong, T. Masayuki, O. Takeaki and S. Xiu-Yun, Jpn. J. Appl. Phys., 2009, 48, 021401 CrossRef.
- G. Dong, D. L. Jacobs, L. Zang and C. Wang, Appl. Catal., B, 2017, 218, 515–524 CrossRef CAS.
- P. H. Kasai, E. B. Whipple and W. Weltner Jr, J. Chem. Phys., 1966, 44, 2581–2591 CrossRef CAS.
- P. H. Kasai and D. McLeod Jr, J. Chem. Phys., 1971, 55, 1566–1575 CrossRef CAS.
- M. Liu, R. Inde, M. Nishikawa, X. Qiu, D. Atarashi, E. Sakai, Y. Nosaka, K. Hashimoto and M. Miyauchi, ACS Nano, 2014, 8, 7229–7238 CrossRef CAS PubMed.
- X. Bai, L. Wang and Y. Zhu, ACS Catal., 2012, 2, 2769–2778 CrossRef CAS.
- B. Luo, D. Xu, D. Li, G. Wu, M. Wu, W. Shi and M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 17061–17069 CrossRef CAS PubMed.
- Y. Xiang, X. Zhang, X. Wang, X. Ding, D. Huang and H. Chen, J. Catal., 2018, 357, 188–194 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09204d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |