
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-Myrcene/isobornyl methacrylate SG1 nitroxide-mediated controlled radical polymerization: synthesis and characterization of gradient, diblock and triblock copolymers†
Adrien Métafiot *acd,
Lysandre Gagnona,
Sébastien Pruvostc,
Pascal Hubertb,
Jean-François Gérardc,
Brigitte Defoortd and
Milan Marić*a
*acd,
Lysandre Gagnona,
Sébastien Pruvostc,
Pascal Hubertb,
Jean-François Gérardc,
Brigitte Defoortd and
Milan Marić*a
aDepartment of Chemical Engineering, McGill University, 3610 University St., Montreal, H3A 0C5 Quebec, Canada. E-mail: milan.maric@mcgill.ca; adrien.metafiot2@mcgill.ca
bDepartment of Mechanical Engineering, McGill University, 817 Sherbrooke St. W., Montreal, H3A 0C3 Quebec, Canada
cIngénierie des Matériaux Polymères (IMP), CNRS UMR5223, INSA Lyon, 17 Jean Capelle Avenue, 69621 Villeurbanne, France
dArianeGroup, Avenue du Général Niox, 33160 Saint-Médard-en-Jalles, France
First published on 25th January 2019
Abstract
β-Myrcene (My), a natural 1,3-diene, and isobornyl methacrylate (IBOMA), from partially bio-based raw materials sources, were copolymerized by nitroxide-mediated polymerization (NMP) in bulk using the SG1-based BlocBuilder™ alkoxyamine functionalized with an N-succinimidyl ester group, NHS-BlocBuilder, at T = 100 °C with initial IBOMA molar feed compositions fIBOMA,0 = 0.10–0.90. Copolymer reactivity ratios were rMy = 1.90–2.16 and rIBOMA = 0.02–0.07 using Fineman–Ross, Kelen–Tudos and non-linear least-squares fitting to the Mayo–Lewis terminal model and indicated the possibility of gradient My/IBOMA copolymers. A linear increase in molecular weight versus conversion and a low dispersity (Đ ≤ 1.41) were exhibited by My/IBOMA copolymerization with fIBOMA,0 ≤ 0.80. My-rich and IBOMA-rich copolymers were shown to have a high degree of chain-end fidelity by performing subsequent chain-extensions with IBOMA and/or My, and by 31P NMR analysis. The preparation by NMP of My/IBOMA thermoplastic elastomers (TPEs), mostly bio-sourced, was then attempted. IBOMA-My-IBOMA triblock copolymers containing a minor fraction of My or styrene (S) units in the outer hard segments (Mn = 51–95 kg mol−1, Đ = 1.91–2.23 and FIBOMA = 0.28–0.36) were synthesized using SG1-terminated poly(ethylene-stat-butylene) dialkoxyamine. The micro-phase separation was suggested by the detection of two distinct Tgs at about −60 °C and +180 °C and confirmed by atomic force microscopy (AFM). A plastic stress–strain behavior (stress at break σB = 3.90 ± 0.22 MPa, elongation at break εB = 490 ± 31%) associated to an upper service temperature of about 140 °C were also highlighted for these triblock polymers.
Introduction
The community of polymer researchers has long desired to elaborate on polymerization techniques that couple the control of microstructure offered by living ionic polymerization1–3 with the simplicity of industrial implementation made possible by other processes such as free radical polymerization.4 Since the early 1980s, an easy-to-use synthetic technique now termed reversible-deactivation radical polymerization (RDRP)5–9 has been developed, encompassing several novel controlled/pseudo-living radical polymerizations and allowing the preparation of tailor-made macromolecules with precise and predetermined molar masses, compositions, topologies, and functionalities. Among them, nitroxide-mediated polymerization (NMP)7,8,10 is conceptually and practically the simplest of these systems. Based on a reversible termination mechanism between the propagating free radical and the nitroxide used as a controlling agent, NMP establishes a thermal dynamic equilibration between dormant macro-alkoxyamines capped by the nitroxide moiety and actively propagating macroradicals.11 At a minimum, only an alkoxyamine, available from commercial sources and easily functionalizable, and the monomer(s) are necessary to implement an NMP process initiated via a mono-component pathway (i.e. unimolecular initiator). NMP relies on the elimination of oxygen prior to polymerization as the main experimental constraint.6,12The opportunity to prepare high-performance polymers by taking advantage of NMP's simplicity appears to be highly desirable from both practical and economical points of view.13 The synthesis of polymers having desirable mechanical properties such as a high tensile strength, a remarkable extensibility or a combination of both seems achievable by NMP. In particular, the development of styrenic-diene block copolymers, generally composed of a dispersed poly(styrene) (PS) or PS derivative phase in a continuous elastomer domain,14,15 looks specifically suited to the NMP process. Indeed, the NMP of conjugated dienes such as butadiene (B)16,17 and especially isoprene (I)18 was explored from the onset of the development of NMP, using 2,2,6,6-tetramethylpiperidinyl-1-oxy (TEMPO),7 the original first-generation stable free nitroxide. Well-tailored homopolymers and copolymers based on either B or I were produced as well,19–23 mediated by the 2,2,5-trimethyl-4-phenyl-3-azahexane-3-oxyl nitroxide (TIPNO),19,24 a more labile second-generation initiator. N-tert-Butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)]nitroxide (SG1)25 showed also its effectiveness to control the polymerization of I.26–28 Recently, we studied the well-controlled polymerization of β-myrcene,29 an acyclic monoterpene, initiated by the unimolecular SG1-based succinimidyl ester-functionalized BlocBuilder™ alkoxyamine called NHS-BlocBuilder,30 demonstrating once again the facility of the NMP process to synthesize elastomers based on 1,3-dienes with low dispersity (Đ) and substantial chain-end fidelity.
Well-tailored styrene (S)/I or B statistical, diblock and triblock polymers were thereby made by NMP.19,31,32 Surprisingly enough and to the best of our knowledge, no NMP-based styrenic-diene block copolymers were reported in the literature as possible alternative plastics with valuable mechanical properties or as thermoplastic elastomers (TPEs). This observation prompted us to synthesize NMP-based block copolymers based on an unusual but promising monomer coupling, namely β-myrcene (My)/isobornyl methacrylate (IBOMA).
A naturally occurring monoterpene with a highly active diene structure, β-myrcene (My, 7-methyl-3-methylene-octa-1,6-diene), typically exhibits chemistry similar to unsaturated hydrocarbons.33–35 Its polymerization leads to a bio-sourced P(My) elastomer exhibiting an expected sub-zero glass transition temperature (Tg) of about −75 °C and a pseudo-plastic behavior.36 My-based TPEs were notably produced by anionic polymerization, making P(My) a possible elastomer substitute to well-known poly(butadiene) (PB) and poly(isoprene) (PI) rubbers. In 1983, S-My-S triblock copolymers, initiated by sec-butyllithium at 30 °C in benzene solution, were made by sequential monomer addition (number-average molecular weight Mn = 165–194 kg mol−1, final S molar fraction FS = 0.23–0.58) and exhibited tensile strengths at break σB = 4.3–12.8 MPa and elongations at break εB = 670–1290%.37,38 Such triblocks exhibited the classical self-assembled morphologies in this case. More recently, Bolton et al. produced AMMS-My-AMMS (AMMS = α-methyl-p-methylstyrene) triblock polymers by sequential anionic polymerization,39 which showed ultimate tensile stress values ranging from 0.5 to 10.8 MPa and εB ranging from 525 to 1340%. Interestingly, the insertion of high Tg P(AMMS)s as outer hard blocks (Tg,P(AMMS) ∼ 180 °C) allowed the extension of the upper service temperature of these triblocks compared to that of traditional PS-based TPEs, which are limited to 90–100 °C.
Similarly, P(IBOMA), displaying a high Tg ∼ 190 °C,40 can be used as the rigid domain of block copolymer TPEs to provide strength, hardness and an improved performance at high temperature. This was illustrated by Yu and coworkers, who prepared IBOMA-B-IBOMA triblock copolymers initiated by the m-diisopropenylbenzene/tert-butyllithium diadduct,41 which exhibited excellent stress–strain properties and an upper service temperature of 160 °C. With the development of alkoxyamine unimolecular initiators and SG1 nitroxide in the late 1990s, the polymerization of IBOMA, a bulky hydrophobic methacrylate, is no longer a barrier by NMP. Control of methacrylate polymerizations was accomplished by Charleux and coworkers via SG1-based initiators using a small fraction of co-monomer such as S42,43 or acrylonitrile44 (<10 mol%). A comprehensive review was released by Nicolas et al. about the most successful strategies directed toward the control of the NMP of methacrylic esters.45 The development of dedicated nitroxides and alkoxyamines has led to improving the NMP of methacrylic esters. Guillaneuf, Gigmes and coworkers focused their attention on the indolynoxyl radical 2,2-diphenyl-3-phenylimino-2,3-dihydroindol-1-yloxyl (DPAIO) nitroxide,46 and the design of indolinic nitroxides derived from DPAIO.47 The bulk methyl methacrylate (MMA) polymerization initiated by the corresponding DPAIO-based alkoxyamines at 85–100 °C showed a good control. Recently, a new series of alkoxyamines were designed by Asua et al. and allowed the well-controlled synthesis of poly(methacrylate)s at moderate temperatures.48
In the present study, we report first the copolymerization of My and IBOMA in bulk at 100 °C, initiated by the succinimidyl ester-terminated NHS-BlocBuilder alkoxyamine. The active feature of these NMP-based My/IBOMA copolymers, ideally capped by a SG1 nitroxide moiety, was then assessed quantitatively via 31P NMR spectroscopy and qualitatively by chain-extensions with a fresh batch of IBOMA and/or My. After guaranteeing the efficient control of the SG1-mediated copolymerization of My with IBOMA, the second part of this investigation was focused on the preparation of P(My)-(SG1)2 macroinitiators using telechelic poly(ethylene-stat-butylene) initiator terminated with SG1 nitroxide groups49 and their subsequent chain-extension with IBOMA-rich/My or S mixtures. The phase behavior, the thermal behavior, notably along with viscoelastic properties, and the stress–strain properties of the resulting triblock copolymers were elucidated to emphasize the possibility to produce valuable diene-based block copolymers from largely sustainable feedstocks, in a simple way by NMP.
Experimental
Materials & methods
β-Myrcene (My, ≥ 90%), basic alumina (Al2O3, Brockmann, Type I, 150 mesh), calcium hydride (CaH2, 90–95% reagent grade), thiophenol (97%), and benzene (≥99%, ACS reagent) were purchased from Sigma-Aldrich and used as received. Toluene (≥99%), methanol (MeOH, ≥99%), methylene chloride (CH2Cl2, 99.9% certified ACS), chloroform (CHCl3, 99.8%), and tetrahydrofuran (THF, 99.9% HPLC grade) were obtained from Fisher Scientific and used as received. 2-Methyl-2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)-aminoxy]-N-propionyloxysuccinimide, also known as NHS-BlocBuilder (NHS-BB), was prepared according to a published method30 from 2-(tert-butyl[1-(diethoxyphosphoryl)-2,2-dimethylpropyl]aminooxy)-2-methylpropionic acid, also known as MAMA SG1 (BlocBuilder-MA™, (BB), 99%, provided by Arkema and used without further purification), N-hydroxysuccinimide (NHS, 98%, purchased from Aldrich and used as received), and N,N′-dicyclohexylcarbodiimide (DCC, 99%, purchased from Aldrich and used as received). Telechelic poly(ethylene-stat-butylene) difunctional initiator PEB-(SG1)2 terminated with N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)]nitroxide (SG1) was produced relying on a published synthesis route,49 from hydroxyl terminated poly(ethylene-stat-butylene) (PEB-(OH)2, Kraton™ D2205) obtained from Kraton, acryloyl chloride (98%) acquired from Sigma-Aldrich and BlocBuilder™. Styrene (S, 99%) was obtained from Fisher Scientific and isobornyl methacrylate (IBOMA, Visiomer® Terra) was obtained from Evonik, and both S and IBOMA were purified by passing through a column of basic alumina mixed with 5 wt% calcium hydride and then stored in a sealed flask under a head of nitrogen in a refrigerator until needed. The deuterated chloroform (CDCl3, 99.8%) used as a solvent for nuclear magnetic resonance (NMR) was obtained from Cambridge Isotopes Laboratory. Diethyl phosphite (98%) was purchased from Sigma-Aldrich.My/IBOMA copolymerization by NMP
The copolymerizations, depicted in Scheme 1a, were done in a 10 mL three-necked round-bottom glass flask equipped with a vertical reflux condenser (inserted in the middle neck), a thermal well, and a magnetic stir bar. Table 1 gives the formulations for the various My/IBOMA copolymerizations studied. For example, for the experiment My/IBOMA-50 (molar fraction of IBOMA in the initial feed fIBOMA,0 = 0.50), the reactor was sealed with rubber septa after the addition of NHS-BB (0.102 g, 0.213 mmol) and the magnetic stir bar. My (2.381 g, 17.478 mmol) and previously purified IBOMA (3.897 g, 17.529 mmol) were then injected into the reactor. The initial molar ratio of monomers and NHS-BB was calculated to give theoretically a copolymer sample with target number-average molecular weight Mn,theo = (MMyfMy,0 + MIBOMAfIBOMA,0) DP ∼ 30 kg mol−1 at complete overall conversion with DP = ([My]0 + [IBOMA]0)/[NHS-BB]0 = 164, the number average degree of polymerization. A mixture of ethylene glycol/distilled water (20/80 vol%) at a temperature of 3 °C was circulated (Fisher Scientific Isotemp 3016D digital refrigerated bath) through the condenser connected to one of the necks of the reactor to prevent any evaporative loss of the monomers. A purge of ultrapure nitrogen was then introduced to the reactor for 30 min to deoxygenate at room temperature the reactants prior to polymerization. After purging, the reactor was heated at a rate of about 10 °C min−1 to 100 °C with continuous nitrogen purge. The time at which the reactor temperature reached 90 °C was taken arbitrarily as the commencement of the reaction (t = 0 min). Samples were then taken from the reactor periodically by a syringe until the end of the experiments or until the samples became too viscous to withdraw. Reactions were then stopped by removing the reactor from the heating mantle and letting the contents cool down to room temperature, while under continuous nitrogen purge. For each sample withdrawn during the polymerization, the crude polymer was precipitated with excess methanol. After filtration and recovery, the precipitated polymer was dried at 50 °C under vacuum in the oven overnight to remove unreacted monomers. Samples were analyzed by nuclear magnetic resonance (NMR) and gel permeation chromatography (GPC). At the end of the experiment (t = 340 min), the overall conversion for My/IBOMA-50 was 52.7% (individual conversions: XMy = 75.2% and XIBOMA = 30.2%) as determined by 1H NMR, with a number-average molecular-weight Mn,MHS = 9.4 kg mol−1 and a dispersity Đ = 1.28, as determined by GPC. The molar composition of IBOMA in the final copolymer was FIBOMA = 0.43 according to 1H NMR spectroscopy. The exact same procedure was followed for all My/IBOMA copolymerizations. My/IBOMA copolymer characteristics can be found in Table 2.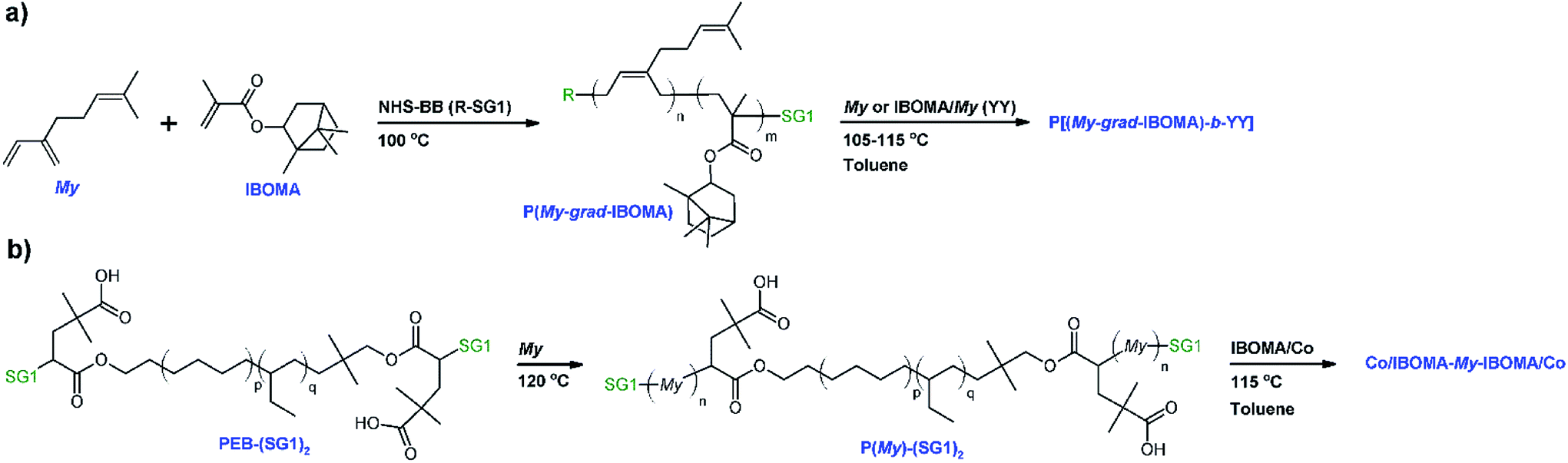 | ||
| Scheme 1 (a) My/IBOMA gradient copolymerization in bulk initiated by NHS-BB and subsequent My or IBOMA/My (∼93/7 mol%) chain-extension in toluene and (b) synthesis of P(My)-(SG1)2 from PEB-(SG1)2 difunctional initiator (n ≫ p, q) and subsequent IBOMA/Co chain-extension in toluene (Co = My or S co-monomer, 8–9 mol%). | ||
| IDa | [NHS-BB]0 (M) | [My]0 (M) | [IBOMA]0 (M) | fMy,0 | T (°C) | Solvent | t (min) |
|---|---|---|---|---|---|---|---|
| a Experimental identification given by My/IBOMA-XX where XX refers to the rounded % initial molar fraction of My in the mixture (fMy,0).b 50 wt% of toluene in the initial feed.c Highly viscous reaction medium after relatively short times for experiments having IBOMA-rich starting mixture. | |||||||
| My/IBOMA-50-T120 | 0.030 | 2.49 | 2.51 | 0.50 | 120 | — | 240 |
| My/IBOMA-70-T120 | 0.029 | 3.76 | 1.57 | 0.71 | 120 | — | 200 |
| My/IBOMA-0-Tol | 0.015 | 0 | 2.08 | 0 | 100 | Tolueneb | 140 |
| My/IBOMA-0 | 0.033 | 0 | 4.40 | 0 | 100 | — | 4c |
| My/IBOMA-10 | 0.032 | 0.45 | 4.04 | 0.10 | 100 | — | 108 |
| My/IBOMA-20 | 0.032 | 0.91 | 3.73 | 0.20 | 100 | — | 240 |
| My/IBOMA-30 | 0.031 | 1.45 | 3.33 | 0.30 | 100 | — | 300 |
| My/IBOMA-40 | 0.031 | 1.94 | 2.92 | 0.40 | 100 | — | 280 |
| My/IBOMA-50 | 0.031 | 2.51 | 2.52 | 0.50 | 100 | — | 340 |
| My/IBOMA-60 | 0.028 | 3.07 | 2.10 | 0.59 | 100 | — | 380 |
| My/IBOMA-70 | 0.029 | 3.70 | 1.61 | 0.70 | 100 | — | 480 |
| My/IBOMA-80 | 0.028 | 4.46 | 1.08 | 0.81 | 100 | — | 380 |
| My/IBOMA-90 | 0.027 | 5.09 | 0.55 | 0.90 | 100 | — | 400 |
| My/IBOMA-100 | 0.026 | 5.80 | 0 | 1 | 100 | — | 500 |
| ID | FMya | XMyb (%) | XIBOMAb (%) | Xb (%) | Mn,MHSc (kg mol−1) | Đc | 〈kp〉〈K〉d (105 s−1) | 1,4-e (%) | 1,2-e (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Molar fraction of My in the copolymer (FMy), as determined by 1H NMR in CDCl3 of the final dry sample (Fig. S5 in ESI for the spectral assignments).b Individual monomer conversions XMy and XIBOMA, determined by 1H NMR in CDCl3. Overall conversion X = XMyfMy,0 + XIBOMAfIBOMA,0.c Mn,GPC and Mw,GPC determined by GPC calibrated with PMMA standards in THF at 40 °C. Mn,MHS obtained from Mn,GPC and corrected using the Mark–Houwink relationship (further details in the Experimental section).d 〈kp〉〈K〉 derived from the slopes 〈kp〉[P˙] taken from the semilogarithmic kinetic plots of ln((1 − X)−1) versus time in the linear region generally from 0 to 60 min (0 to 20 min for My/IBOMA-0-Tol and 0 to 120 min for My/IBOMA-80, My/IBOMA-90 and My/IBOMA-100; squared linear regression coefficient = R2 ≥ 0.91 for every experiment. The linear fits to the experimental data during the initial stages of the polymerizations are provided in the ESI, Fig. S3). 〈kp〉〈K〉's estimated from 〈kp〉[P˙] and r = [SG1]0/[NHS-BB]0 (eqn (5)). Error bars derived from the standard errors in the slope from the linear fits of ln((1 − X)−1) versus time.e My regioselectivity determined by 1H NMR in CDCl3. 3,4-content% = 100 − 1,4-content% − 1,2-content% (Fig. S5 in ESI for further details).f No kinetic study led due to the early “caking” (high viscosity) of the reaction medium.g 1H NMR peaks could not be detected. | |||||||||
| My/IBOMA-50-T120 | 0.52 | 97.1 | 73.5 | 85.3 | 16.1 | 1.48 | 6.4 ± 0.4 | 58.8 | 37.5 |
| My/IBOMA-70-T120 | 0.70 | 98.8 | 82.9 | 94.2 | 13.7 | 1.71 | 12.7 ± 0.2 | 63.6 | 30.1 |
| My/IBOMA-0-Tol | 0 | 0 | 14.1 | 14.1 | 12.3 | 1.72 | 2.3 ± 0.8 | — | — |
| My/IBOMA-0 | 0 | 0 | 38.4 | 38.4 | 27.1 | 1.75 | —f | — | — |
| My/IBOMA-10 | 0.14 | 86.5 | 36.8 | 41.8 | 18.3 | 1.57 | 3.5 ± 0.2 | —g | —g |
| My/IBOMA-20 | 0.51 | 81.5 | 11.1 | 25.2 | 8.6 | 1.38 | 0.9 ± 0.4 | 68.2 | 29.5 |
| My/IBOMA-30 | 0.45 | 83.6 | 31.6 | 47.2 | 10.9 | 1.35 | 3.0 ± 0.2 | 61.9 | 36.0 |
| My/IBOMA-40 | 0.64 | 66.2 | 16.6 | 36.4 | 6.5 | 1.36 | 0.9 ± 0.5 | 70.1 | 26.2 |
| My/IBOMA-50 | 0.57 | 75.2 | 30.2 | 52.7 | 9.4 | 1.28 | 2.5 ± 0.3 | 65.9 | 30.5 |
| My/IBOMA-60 | 0.83 | 77.5 | 15.3 | 52.0 | 7.3 | 1.26 | 2.7 ± 0.2 | 67.4 | 29.9 |
| My/IBOMA-70 | 0.77 | 77.9 | 35.0 | 65.0 | 7.5 | 1.29 | 1.7 ± 0.6 | 71.3 | 24.9 |
| My/IBOMA-80 | 0.81 | 57.8 | 56.0 | 57.5 | 7.4 | 1.41 | 1.8 ± 0.5 | 85.6 | 10.5 |
| My/IBOMA-90 | 0.86 | 29.7 | 67.4 | 33.5 | 5.9 | 1.34 | 1.3 ± 0.7 | 86.4 | 8.5 |
| My/IBOMA-100 | 1.0 | 39.4 | 0 | 39.4 | 5.0 | 1.32 | 1.4 ± 0.4 | 86.6 | 7.0 |
Chain-extension of My/IBOMA copolymers with My, IBOMA and S
All chain-extensions from P(My-grad-IBOMA) macroinitiators were performed in a very similar setup to that used for the My/IBOMA copolymerizations with the use of a 10 mL reactor. The formulations can be found in Table 3B. As a brief illustration, My/IBOMA-82-IBOMA/My was synthesized using a gradient P(My-grad-IBOMA) copolymer (My/IBOMA-82, FMy = 0.82, Mn,MHS = 13.6 kg mol−1, Đ = 1.51, 0.367 g, 0.027 mmol) that was added to the reactor along with toluene (3.24 g), My (0.135 g, 0.991 mmol) and previously purified IBOMA (2.787 g, 12.530 mmol). The reaction medium was mixed and bubbled with ultrapure nitrogen for at least 30 min, then heated to 105 °C and allowed to react for 210 min while maintaining a N2 purge. t = 0 min was selected once the reaction medium reached 90 °C. Samples were drawn periodically via syringe at 0, 60, 120 and 210 min. The samples and the final diblock copolymer were precipitated in excess methanol and allowed to dry overnight in a vacuum oven at 50 °C. My/IBOMA-82-IBOMA/My exhibited Mn,MHS = 23.2 kg mol−1, Đ = 1.61 (GPC) and FMy = 0.49 (1H NMR). The results of the chain-extensions from P(My-grad-IBOMA)s are given in Table 3C.| (A) P(My-grad-IBOMA) macroinitiatora | |||||||
|---|---|---|---|---|---|---|---|
| ID | fMy,0 | FMy,1 | LFb (%) | X1 (%) | Mn,MHS,1 (kg mol−1) | Mn,theo,X1c (kg mol−1) | Đ1 |
| a The index “1” is associated to the characteristics of the P(My-grad-IBOMA) macroinitiators (MI) whereas the index “2” refers to the features of the whole chain-extended diblock copolymer (MI + P(My) or P(IBOMA-co-My) segment added).b Living molar fraction of MI chains capped by a SG1 group, measured by 31P NMR (Fig. S6 in ESI for the spectra). Standard deviation derived from the difference in macroinitiator Mn value between Mn,GPC and Mn,MHS.c Predicted Mn,MHS,1 at X1 measured experimentally and calculated as follows: Mn,theo,X1 = (X1/100)Mn,theo,1 with Mn,theo,1 = 30 kg mol−1 at X = 100%.d Targeted number-average molecular weight of the whole chain-extended diblock copolymer (MI block + extended block) at X = 100%.e Predicted Mn,MHS,2 of the whole chain-extended diblock copolymer (MI block + second block added) at X2, measured experimentally, and calculated as follows: Mn,theo,X2 = (X2/100)(Mn,theo,2 − Mn,MHS,1) + Mn,MHS,1 (=predicted Mn of the second block added at X2 + experimental Mn,MHS of MI). | |||||||
| My/IBOMA-44 | 0.30 | 0.44 | 74 ± 9 | 39.1 | 8.8 | 11.7 | 1.32 |
| My/IBOMA-82 | 0.79 | 0.82 | 69 ± 3 | 64.2 | 13.6 | 19.3 | 1.51 |
| (B) Formulation of chain-extension | |||||||
|---|---|---|---|---|---|---|---|
| ID | [MI]0 (M) | [My]0 (M) | [IBOMA]0 (M) | [Toluene]0 (M) | Mn,theod (kg mol−1) | T (°C) | t (min) |
| My/IBOMA-44-My | 0.004 | 2.414 | 0 | 3.859 | 91.0 | 115 | 390 |
| My/IBOMA-82-IBOMA/My | 0.004 | 0.147 | 1.857 | 5.216 | 122.0 | 105 | 210 |
Synthesis of Co/IBOMA-My-IBOMA/Co (Co = My or S co-monomer) triblock copolymers by NMP
A two-step process was implemented in a similar reactor apparatus used for the above-mentioned copolymerizations and chain-extensions: (1) My homopolymerization initiated by PEB-(SG1)2 dialkoxyamine; (2) IBOMA/Co chain-extension from P(My)-(SG1)2 macroinitiator. The entire set of formulations is given in Table 4. For example, the synthesis of S/IBOMA-My-IBOMA/S triblock polymer (Co = S), characterized then mechanically and rheologically, can be detailed. To the 50 mL reactor was added PEB-(SG1)2 difunctional initiator (Mn,GPC = 5.7 kg mol−1, Đ = 1.17, 0.912 g, 0.140 mmol) and My (21.005 g, 143.671 mmol) with magnetic stirring (experiment My-52, Table 4A). A purge of nitrogen was applied for 30 min. The reactor was then heated to 120 °C to commence polymerization. t = 0 min was selected once the reaction medium reached 100 °C. At t = 360 min, the polymerization was stopped, the polymer was precipitated in excess methanol and dried overnight in a vacuum oven at 50 °C. 1H NMR was performed to ensure the complete elimination of methanol and unreacted My in the dry polymer, and to determine the conversion (49.1%). P(My) synthesized exhibited Mn,MHS = 51.7 kg mol−1 and Đ = 1.53 as determined by GPC. The chain-extension was then done by reacting P(My)-(SG1)2 (1.50 g, 0.029 mmol), purified IBOMA (3.32 g, 14.94 mmol) and purified S (0.15 g, 1.42 mmol) in toluene (∼4.3 g) under continuous nitrogen flow and magnetic stirring for 3 h at 115 °C (experiment My-52-IBOMA/S, Table 4B). The resulting polymer sample (Mn,MHS = 78.1 g mol−1, Đ = 2.41) was precipitated in excess methanol and dried under vacuum at 50 °C overnight. Lastly, fractionation was performed using a methanol (non-solvent)/benzene (solvent) pair (2 cycles). The final fractionated triblock polymer exhibited Mn,MHS = 94.7 kg mol−1, Đ = 2.23, FIBOMA = 0.34 and FS = 0.07. Final Co/IBOMA-My-IBOMA/Co copolymer characteristics can be found in Table 4C.| (A) P(My)-(SG1)2a | |||||||
|---|---|---|---|---|---|---|---|
| ID | t (min) | XMy (%) | Mn,MHS,My (kg mol−1) | Mn,theo,XMyb (kg mol−1) | ĐMy | 1,4-c (%) | 1,2-c (%) |
| a The indexes “My” and “CE” refer, respectively, to the final characteristics of P(My)-(SG1)2 and the whole chain-extended triblock copolymer.b Predicted Mn,MHS,My at XMy measured experimentally and calculated as follows: Mn,theo,XMy = (XMy/100)Mn,theo,My (Mn,theo,My = 143.4 and 139.8 kg mol−1 for experiments My-35 and My-52, respectively).c See footnote “e” in Table 2.d Targeted number-average molecular weight of the whole chain-extended triblock copolymer at XCE = 100%.e Predicted Mn,MHS,CE of the whole chain-extended triblock copolymer at XCE, measured experimentally, and calculated as follows: Mn,theo,XCE = (XCE/100)(Mn,theo,CE − Mn,MHS,My) + Mn,MHS,My. | |||||||
| My-35 | 290 | 33.4 | 35.2 | 47.9 | 1.45 | 91.7 | 3.2 |
| My-52 | 360 | 49.1 | 51.7 | 68.6 | 1.53 | 88.2 | 5.0 |
| (B) Formulation of chain-extension | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | [P(My)-(SG1)2]0 (M) | [IBOMA]0 (M) | [My]0 (M) | [S]0 (M) | [Toluene]0 (M) | Mn,theo,CEd (kg mol−1) | T (°C) | t (min) |
| My-35-IBOMA/My | 0.004 | 1.681 | 0.142 | 0 | 4.929 | 133.4 | 115 | 90 |
| My-52-IBOMA/S | 0.003 | 1.546 | 0 | 0.147 | 4.848 | 171.2 | 115 | 180 |
Characterization
Monomer conversion was determined with a Varian NMR Mercury spectrometer (1H NMR, 300 MHz, 32 scans) using CDCl3. In the case of a copolymerization between two monomers A and B, the overall monomer conversion X was calculated from formula (1):| X = XAfA,0 + XBfB,0 | (1) |
Phosphorus nuclear magnetic resonance (31P NMR) was performed using a 5 mm diameter up NMR tube with 840 scans being processed in a 200 MHz Varian Gemini 2000 spectrometer operating at 81 MHz. 0.0352 g of My-rich My/IBOMA-82 (Mn,MHS = 13.6 kg mol−1) and 0.0676 g of IBOMA-rich My/IBOMA-44 (Mn,MHS = 8.8 kg mol−1) were characterized in CDCl3 with the addition of diethylphosphite as internal reference (0.0027 g and 0.0072 g respectively). To confirm similar relaxation rates between diethylphosphite and the copolymer samples, My/IBOMA-82 and My/IBOMA-44 were run under the exact same conditions with only one scan and no dummy scans (ss = 0). Slight differences (<3.2%) in integral values were measured between these spectra and the standard ones with multiple scans. Moderate Mn (∼10–20 kg mol−1) IBOMA/My copolymers were thus assumed to relax at the same rate compared to that of diethylphosphite.
The number-average molecular weights (Mn,GPC) and the dispersities (Đ = Mw,GPC/Mn,GPC) were measured using gel permeation chromatography (GPC, Water Breeze, differential refractive index RI 2414 detector, 40 °C) with HPLC grade THF as the mobile phase (flow rate of 0.3 mL min−1). The GPC was equipped with 3 Waters Styragel® HR columns (HR1 with a molecular weight measurement range of 102 to 5 × 103 g mol−1, HR2 with a molecular weight measurement range of 5 × 102 to 2 × 104 g mol−1 and HR4 with a molecular weight measurement range of 5 × 103 to 6 × 105 g mol−1) and a guard column was used. Mn,GPC values were determined by calibration with 10 linear narrow molecular weight distribution P(MMA) standards (Varian Polymer Standards, molecular weights ranging from 875 to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 677000 g mol−1). The P(My) and P(IBOMA) contributions to Mn,GPC were converted using Mark–Houwink–Sakurada (MHS) coefficients (MHS parameters determined at 35 °C with THF eluent for P(MMA):51 KP(MMA) = 12.2 × 10−5 dL g−1 and αP(MMA) = 0.690; MHS parameters determined at 25 °C with THF eluent for P(IBOMA):52 KP(IBOMA) = 3.8 × 10−5 dL g−1 and αP(IBOMA) = 0.748; MHS parameters determined at 30 °C with THF eluent for P(My) containing 90 and 10 mol% of 1,4- and 3.4-content respectively:53 KP(My) = 7.5 × 10−5 dL g−1 and αP(My) = 0.772). Therefore, the converted Mn values were calculated according to the Mark–Houwink relationship54 and the molar composition of the copolymer samples (determined by 1H NMR) as given in eqn (2) in the case of My/IBOMA copolymers:
677000 g mol−1). The P(My) and P(IBOMA) contributions to Mn,GPC were converted using Mark–Houwink–Sakurada (MHS) coefficients (MHS parameters determined at 35 °C with THF eluent for P(MMA):51 KP(MMA) = 12.2 × 10−5 dL g−1 and αP(MMA) = 0.690; MHS parameters determined at 25 °C with THF eluent for P(IBOMA):52 KP(IBOMA) = 3.8 × 10−5 dL g−1 and αP(IBOMA) = 0.748; MHS parameters determined at 30 °C with THF eluent for P(My) containing 90 and 10 mol% of 1,4- and 3.4-content respectively:53 KP(My) = 7.5 × 10−5 dL g−1 and αP(My) = 0.772). Therefore, the converted Mn values were calculated according to the Mark–Houwink relationship54 and the molar composition of the copolymer samples (determined by 1H NMR) as given in eqn (2) in the case of My/IBOMA copolymers:
| Mn,MHS = FMy[(KP(MMA)/KP(My))Mn,GPCαP(MMA)+1]1/(αP(My)+1) + (1 − FMy)[(KP(MMA)/KP(IBOMA))Mn,GPCαP(MMA)+1]1/(αP(IBOMA)+1) | (2) |
Thermogravimetric analysis (TGA) was carried out using a Q500™ from TA Instruments under nitrogen flow at a ramp rate of 15 °C min−1. Samples were heated in aluminium pans. Differential scanning calorimetry (DSC, Q2000™ from TA Instruments) was used under N2 atmosphere. Indium was used as a standard to calibrate temperature while heat flow was calibrated via a benzoic acid standard. Three scans per cycle (heat/cool/heat) at a rate of 10 °C min−1 was set with a temperature range ranging for instance from −90 °C to +220 °C for the characterization of the triblock copolymers. Only the second heating run was considered to eliminate the thermal history. The reported Tgs were calculated using the inflection method from the change in slope observed in the DSC traces.
The stress–strain features of the Co/IBOMA-My-IBOMA/Co triblock polymer (Co = S) were determined using a MTS Insight material testing system with a 5 kN load cell at room temperature and a cross-head speed of 10 mm min−1. Dog-bone style tensile specimens (ASTM D638 type V for reference, overall length = 63.5 mm, overall width = 9.53 mm) were prepared by solvent casting. To cast the film, the polymer sample was fully dissolved in dichloromethane (∼80 wt%) for an hour with continuous stirring using a magnetic stir bar. The solution was cast into a level Teflon Petri dish at room temperature for approximately two days. Then, the drying was completed after placing the film in the oven at 50 °C under vacuum until constant weight was measured. The film samples were cut into typical dog-bone shapes using a sharp blade. Specimens with defects (nicked sides, cracks, air bubbles) on their surface were discarded and were not used in mechanical testing. For each specimen, the thickness and the width were measured at five different points along the small centre portion with a digital caliper (Marathon, CO 030150F128) and the average film thickness and width were used for all calculations. The samples tested exhibited a mean thickness from 0.55 to 0.69 mm and a mean width from 3.11 to 3.32 mm. Each test was considered finished after the complete breakup of the specimen at the narrow section (films that broke near the grips were discarded). 6 specimens were tested, and averaged results were reported using TestWorks 4 software. The Young's modulus was determined as the slope of the stress–strain curve at strains of 0–0.5%.
The mechanical response of Co/IBOMA-My-IBOMA/Co (Co = S) to torsional oscillation at 0.15 Hz and 1% strain over a temperature range of 25 to 230 °C was measured at a rate of 5 °C min−1 under N2 using the CDT 450 convection heated measuring chamber mounted on the Anton Paar Modular Compact Rheometer MCR302. The solid rectangular fixture (SFR) was used, consisting of an upper and a lower holder with insets. Rectangular bars (thickness = 1.05 ± 0.04 mm; width = 8.91 ± 0.13 mm; length = 46.13 ± 0.24 mm) were made by solvent casting, following the above protocol used for preparing the tensile bars. Prior to the torsion tests, dynamic amplitude sweeps at 0.15 Hz and varying strain from 0.01 to 10% were conducted at various temperatures to ensure the material was kept within the linear viscoelastic regime (Fig. S12 in ESI†). Please note that, before the rheological characterization, the removal of the SG1 groups of Co/IBOMA-My-IBOMA/Co chain-ends was performed as described elsewhere.44,55
The micro-phase behaviour of Co/IBOMA-My-IBOMA/Co (Co = My) triblock polymer was studied by Atomic Force Microscopy (AFM). AFM images were collected in tapping mode using an AFM Bruker Multimode 8 equipped with Nanoscope V controller. The scanning speed for image acquisition was 0.5 Hz. The used cantilevers are Bruker TAP 300A with a radius of curvature of the tip of 8 nm. Data analyses were processed using the Nanoscope Analysis software (version 1.5). The samples were prepared as follows: the polymer (∼0.5 g) was fully dissolved in chloroform (∼10 mL) and one or two drops was spin coated on a silicon wafer, previously rinsed with acetone and cleaned by UV–O3 for 30 min. Lastly, the coated film was dried at 50 °C under vacuum overnight.
Results and discussion
Two main parts are introduced thereafter and consist of the synthesis by nitroxide-mediated polymerization (NMP), via the SG1 nitroxide rate moderator, and the characterization of β-myrcene-rich (My-rich) and isobornyl methacrylate-rich (IBOMA-rich) copolymers. First, the nature of My/IBOMA copolymerization initiated by NHS-BlocBuilder (NHS-BB) was examined as well as its kinetics, its level of control and the ability of the resulting copolymers to re-initiate a second batch of monomer(s). Glass transition temperature (Tg) and decomposition temperature (Tdec) of the My/IBOMA copolymers were also investigated. Secondly, the preparation of relatively high Mn P(My)-(SG1)2 was explored by using the PEB-(SG1)2 dialkoxyamine. The possibility to synthesize triblock polymers by IBOMA/Co chain-extensions (Co = My or S co-monomer ∼ 8–9 mol%, relative to IBOMA) from P(My)-(SG1)2 was then presented. Scheme 1 depicts the various polymerizations done in this study.My/IBOMA copolymerization by NMP
The equations used for the calculation of the reactivity ratios can be found in the ESI (page S4).† By using the Fineman–Ross (FR) method (Fig. 1a),65 the reactivity ratios were determined: rMy = 2.16 ± 0.34 and rIBOMA = 0.07 ± 0.04 (rMyrIBOMA = 0.15 ± 0.19). Similar values were calculated via the Kelen–Tudos (KT) approach (Fig. 1b):66 rMy = 1.90 ± 0.18 and rIBOMA = 0.02 ± 0.17 (rMyrIBOMA = 0.04 ± 0.13). The errors associated were derived from the standard errors of the slopes from FR and KT plots. Even though the KT equation refines the linearization method by introducing an arbitrary positive constant to spread the data more evenly, it remains a rearrangement of the copolymer composition equation and a non-linear least-squares (NLLS) fit to the Mayo–Lewis equation67 is likely the soundest method to determine rMy and rIBOMA. Consequently, the reactivity ratios determined by the KT method were used as the initial guesses for a NLLS fitting of the data (page S4 in ESI†). At 95% confidence level and with a regression coefficient R2 = 0.91, the statistical fit to the data yielded reactivity ratios rMy = 2.07 ± 0.58 and rIBOMA = 0.05 ± 0.07. These latter results are similar to those obtained by FR and KT approaches, confirming the general trends with respect to My/IBOMA NMP at 100 °C in bulk and initiated by NHS-BB: rMy > 1.50, rIBOMA < 0.30 and rMyrIBOMA < 0.50. The large difference in reactivity between My and IBOMA can be first noted. Whatever the terminal unit of the propagating species (⋯My˙ or ⋯IBOMA˙), the macro-radical showed a high preference for My. The insertion of IBOMA units at low conversions, either by self-propagation or cross-propagation, is not favoured: IBOMA-terminated macro-radical has a strong tendency to alternate. Accordingly, it can be assumed that My/IBOMA copolymerization under these conditions produces copolymers that possess a gradient in composition consisting of My enrichment at the start of the polymer chain followed by the incremental IBOMA enrichment at the end of the chain due to My depletion. However, the gradient nature of this copolymerization was moderate as indicated by the presence of methacrylate units at the start of the copolymer chain synthesized (X = 3–12% and FIBOMA = 13–18% at t = 0–60 min for experiment My/IBOMA-50 for instance). Even though samples exhibiting relatively low conversions were used, it can be assumed that the composition drift in the monomer feed was non-negligible, especially taking the difference between rMy and rIBOMA into account and was the main source of error by relying on the instantaneous copolymer composition model.68 To the best of our knowledge, no reactivity ratios were reported in the literature for diene/IBOMA systems, limiting thus the discussion. Nonetheless, for the My/GMA pair copolymerized under similar conditions (120 °C in bulk initiated by NHS-BB),56 statistical copolymers were synthesized with rMy = 0.48–0.80 and rGMA = 0.50–0.71. Such a difference compared to the My/IBOMA system highlights the probable influence of the bulky and rigid bicyclic isobornyl groups over the reactivity of the methacrylate monomer (steric hindrance, hydrophobicity, electronic interactions/stabilization), as suggested by Trumbo as well.69
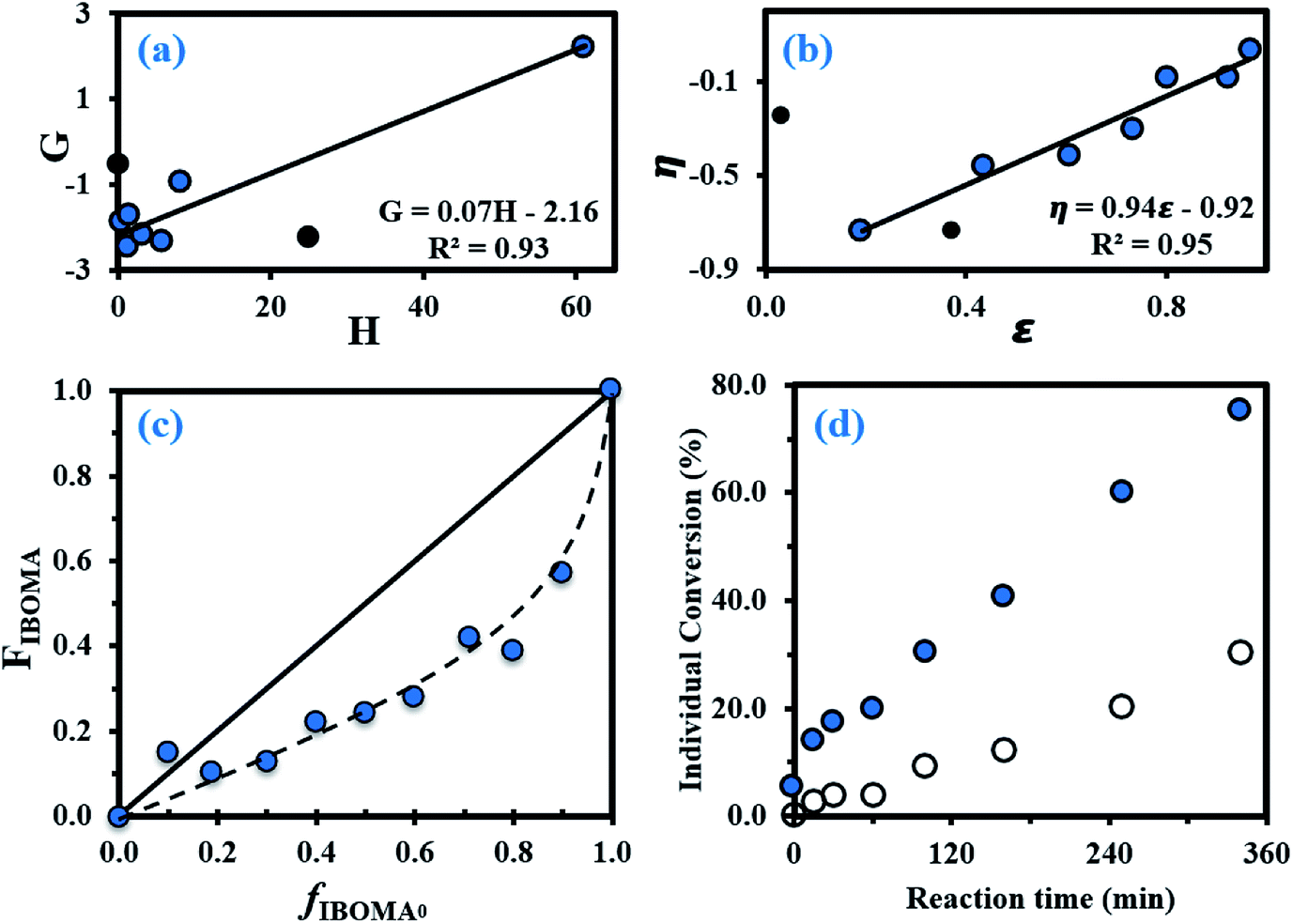 | ||
Fig. 1 (a) Fineman–Ross (FR) and (b) Kelen–Tudos (KT) plots (solid black circles (●) corresponding to experimental outliers not taken into account for the calculations while solid lines refer to the linear trend lines) to determine the binary reactivity ratios for My and IBOMA for copolymerizations done in bulk at 100 °C initiated by NHS-BB (parameters G, H, η and ε defined in the ESI, page S4†). (c) Mayo–Lewis plot of My/IBOMA copolymerizations with respect to IBOMA, using the final molar composition FIBOMA, and the initial monomer feed composition fIBOMA,0. The solid straight line indicates the azeotropic composition where fIBOMA,0 = FIBOMA while the dashed line is the associated trend line of the experimental data (solid blue circles ( )). Table S1 in the ESI† lists the samples used for these plots. (d) Individual My ( )). Table S1 in the ESI† lists the samples used for these plots. (d) Individual My ( ) and IBOMA (○) conversions, determined by 1H NMR in CDCl3, versus reaction time t for the gradient copolymerization My/IBOMA-50 exhibiting fMy,0 = 0.50. ) and IBOMA (○) conversions, determined by 1H NMR in CDCl3, versus reaction time t for the gradient copolymerization My/IBOMA-50 exhibiting fMy,0 = 0.50. | ||
The Mayo–Lewis plot with respect to IBOMA is shown in Fig. 1c. Except for fIBOMA,0 = 0.10, the copolymer compositions are always below the diagonal line where the copolymer composition equals the feed composition (FIBOMA = fIBOMA,0). Thus, the polymer formed at the early stages of the reaction was always richer in My than in IBOMA. The composition gradient of the My/IBOMA copolymers can be highlighted by determining the individual My and IBOMA conversions as a function of polymerization time, as illustrated in Fig. 1d with equimolar amounts of each monomer in the initial feed (fMy,0 = 0.50). Throughout the first hour, essentially only My was reacting (XMy = 19.8% and XIBOMA = 3.7% at t = 60 min) leading to essentially quasi P(My) propagating macro-radicals. Afterwards, while the consumption rate of My remained constant, IBOMA monomer reacted more significantly (XIBOMA = 30.2% at t = 340 min) from a My-poor feed, resulting in an increase of the composition of IBOMA in the chain. Although this experiment was stopped at XMy = 75.2% and XIBOMA = 30.2%, the last stages of the copolymerization can be easily deduced with the gradual IBOMA enrichment of the chain due to the depletion of My monomer.
Before exploring the kinetics of My/IBOMA copolymerization at various initial feed compositions, it is useful to examine the homopolymerization of My and IBOMA by NMP under the same conditions (experiments My/IBOMA-100 and My/IBOMA-0, Table 1). My NMP at 100 °C in bulk mediated by NHS-BB showed a satisfactory control with Mn,MHS increasing linearly with conversion and Đ ≤ 1.36 (Fig. S4, ESI†), resulting in 1,4-rich P(My) (Table 2), even though non-negligible deviations of Mn,MHS values from theoretical ones were measured. The NMP of IBOMA without neither co-monomer nor additional free nitroxide was also attempted under these conditions. Controlled polymerization of methacrylates is still challenging for NMP because of their very high equilibrium constant K resulting from slow recombination of nitroxides and sterically-hindered poly(methacrylate) radicals.71,72 This long-time obstacle was confirmed once again by the inability to implement the SG1-based NMP of IBOMA in bulk at 100 °C in a controlled manner: the reaction medium polymerized spontaneously, giving P(IBOMA) chains exhibiting Mn,MHS = 27.1 kg mol−1 and Đ = 1.75 (My/IBOMA-0, Table 2). Due to the early high viscosity of the medium after only a few minutes, it was decided to repeat the experiment in 50 wt% toluene (experiment My/IBOMA-0-Tol, Table 1) in vain. The polymerization stagnated at X ∼ 10% with Mn,MHS = 11.4–12.3 kg mol−1 and Đ = 1.63–1.76 (Fig. S4, ESI†), illustrating presumably the disproportionation between P(IBOMA) macro-radicals and SG1 nitroxide.
In order to produce IBOMA-rich polymers by SG1-mediated NMP, the controlling co-monomer approach, developed by Charleux and co-workers,42–44 was tried where the addition of a small concentration of a co-monomer exhibiting a much lower activation–deactivation equilibrium constant K with respect to the methacrylate enhances the control of the polymerization. It was herein decided to use My as a potential controlling co-monomer although its individual K has not been reported. 10–20 mol% of My had a marked impact on the outcome of My/GMA NMP initiated by NHS-BB in bulk. It gave a linear evolution of Mn with monomer conversion together with Đ ≤ 1.52 up to almost quantitative overall conversion.56 Previously, 10–20 mol% of isoprene, another conjugated 1,3-diene, allowed the well-controlled bulk NMP of tert-butyl acrylate and GMA.19 In the present study, the addition of 10 mol% of My (experiment My/IBOMA-10, Table 1) into the IBOMA feed undoubtedly slowed down the reaction even though the features of a controlled system were only partially observed due to the upward curvature of the semi-logarithmic plot and Đ = 1.51–1.60 at X = 4–42% (Fig. 2a and b). Expectations were fully met by performing the NMP of IBOMA in the presence of 20 mol% of My as a co-monomer (My/IBOMA-20, Table 1) with Đ ≤ 1.39, Mn increasing linearly with X and a quasi-linearity of ln((1 − X)−1) versus t during the reaction with R2 = 0.92 (Fig. 2a–c). It can be assumed that the addition of My in the IBOMA-rich feed induced a significant decrease of the average 〈K〉 and therefore enhanced the control of the polymerization.
 | ||
| Fig. 2 (a) Semi-logarithmic kinetic plots of ln((1 − X)−1) (X = overall conversion) versus polymerization time t, (b) Đ versus overall conversion X and (c) Mn determined by GPC relative to PMMA standards in THF at 40 °C, and corrected using the Mark–Houwink relationship, versus overall conversion X for the various My/IBOMA copolymerizations in bulk at 100 °C initiated by NHS-BB. The dashed line indicates the theoretical Mn versus overall conversion based on the monomer to initiator ratio (Mn,theo ∼ 30 kg mol−1 at X = 100% for every experiment). All experimental ID and characterization of experiments are listed in Tables 1 and 2. The same legend at the bottom right of the figure is used for each of the three plots. | ||
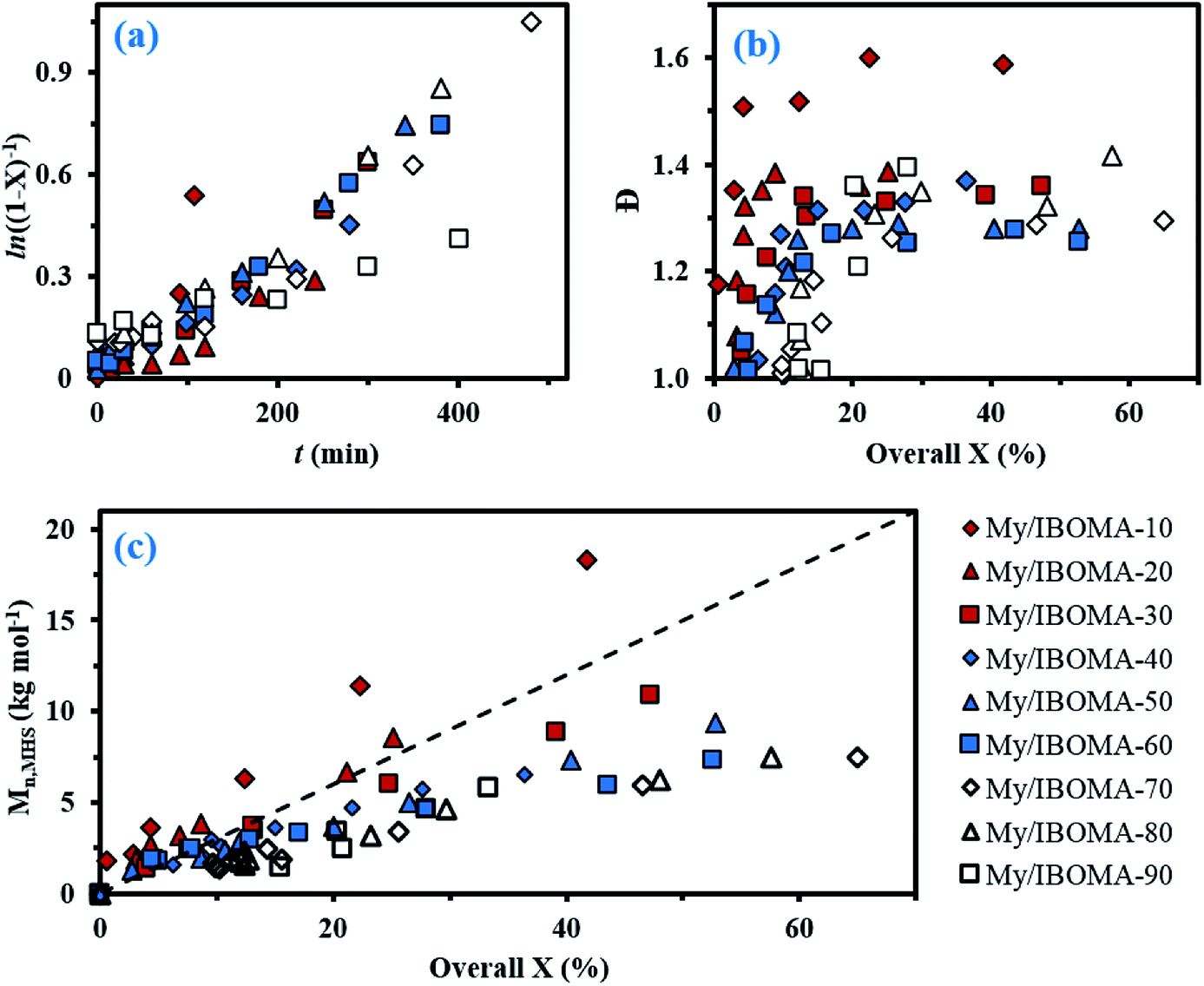
For fMy,0 = 0.30–0.90, linear Mn,MHS increase with overall X and narrow molecular weight distributions (Đ ≤ 1.41) of the P(My-grad-IBOMA)s were observed (Fig. 2b and c). These results presumably illustrate the effective mediation of the stable SG1 free nitroxide, capping the propagating chains and thus shifting the equilibrium toward deactivation.12 Although termination and transfer side reactions were still present, they were likely minimized due to the high number of reversibly terminated chains (dormant P(My-grad-IBOMA)-SG1 chains). Nonetheless, as fMy,0 increased, the deviations of Mn,MHS from Mn,theo became more significant. Even though the principle of universal calibration was applied, the MHS coefficients did not match exactly the GPC conditions used (THF, 40 °C). The likely large differences in hydrodynamic volumes of the P(My-grad-IBOMA)s and that of the PMMA standards that was used to calibrate the GPC may partly explain the deviations. The copolymerization with fMy,0 = 59–90% had a flatter slope compared to the others (Fig. 2c), where the unreliability of the GPC calibration cannot be the sole reason. Chain transfer side reactions to the monomers, creating new chains and resulting in shorter average chain length, may also explain the loss of control.73 The GPC chromatograms at different polymerization times are shown in Fig. 3 for the experiment My/IBOMA-50. The monomodal shift of the curves to lower elution times with no major tails confirmed the well-controlled NMP of this monomer pair. As reported previously with regard to the homopolymerization of My initiated by NHS-BB in bulk at 120 °C,29 a minor peak, corresponding to high Mn polymers, was detectable mostly at the commencement of the copolymerization (20–25 min, Fig. 3), presumably due to My auto-initiation. Indeed, the spontaneously free radical polymerization of My was reported at elevated temperatures as well as at room temperature.74 Under this assumption, My auto-initiation during the NMP of the My/IBOMA mixture would result in the presence of extra active species, not necessarily mediated by the nitroxide radical.
 | ||
| Fig. 3 Normalized GPC traces of P(My-grad-IBOMA) with fMy,0 = 0.50, initiated by NHS-BB at 100 °C in bulk targeting Mn,theo = 30 kg mol−1 at X = 100% (experiment My/IBOMA-50). | ||
Attention can also be paid to the kinetic plots of ln((1 − X)−1) versus reaction time t (Fig. 2a). For fMy,0 = 0.10–0.81, all copolymerizations obeyed first order kinetics as described by eqn (3) and generated good fits to linear kinetic plots (squared linear regression coefficient R2 ≥ 0.94 for the whole experiments).
| ln([M]0/[M]t) = ln([M]0/([M]0(1 − X))) = ln((1 − X)−1) = 〈kp〉[P˙] time | (3) |
In eqn (3), [M]0 and [M]t are the concentrations of monomer at time zero and subsequent later time t, respectively, 〈kp〉 is the average propagation rate constant and [P˙] is the propagating radical concentration. Marked upward curvatures (Fig. 2a) can be seen for experiments My/IBOMA-10 and My/IBOMA-20 having an initial feed rich in IBOMA (fIBOMA,0 = 0.80–0.90). It suggests an increase in [P˙], which can be explained by the contribution of the likely high individual K of IBOMA, resulting from slow recombination of nitroxides and poly(methacrylate) radicals (decrease in the concentration of dormant chains) and leading to a high [P˙]. 〈kp〉[P˙] values were determined from the slopes of the semi-logarithmic plots of conversion versus time (Fig. 2a) in the linear regions where [P˙] is expected to remain relatively constant (see Table 2, footnote d). These slopes can be related to the equilibrium between the dormant and the active chains as shown in eqn (4).
| K = ([P˙][SG1˙])/[P-SG1] | (4) |
| 〈kp〉〈K〉 ≈ (〈kp〉[P˙][SG1]0)/[NHS-BB]0 = 〈kp〉[P˙]r | (5) |
Initially, we expected significant differences in 〈kp〉〈K〉 values between My-rich and IBOMA-rich initial feeds. Indeed, even though no kinetic constants for My are reported yet, kp,I = 125 ± 30 L mol−1 s−1 for the radical polymerization of isoprene, another similar conjugated 1,3-diene, initiated by di-tert-butyl peroxide at 5 °C, was reported.75 Comparatively, kp,IBOMA ∼ 280 L mol−1 s−1 at 5 °C can be estimated from the study led by Beuermann and co-workers using the pulsed-laser polymerization-GPC technique.76 IBOMA and My homopolymerizations performed herein (My/IBOMA-0 and My/IBOMA-100, Table 1) confirmed the kinetic singularity of these two monomers polymerized by NMP. While a quasi-spontaneous polymerization was performed when an IBOMA solution was heated at 100 °C in the presence of NHS-BB, My polymerized slowly under identical experimental conditions and this reaction exhibited kpK = (1.4 ± 0.4)10−5 s−1 (Table 2) consistent with the kinetic values reported for the NMP of dienes.27,29 Very interestingly, 〈kp〉〈K〉 was on average (2.03 ± 0.94)10−5 s−1 for the NMP of My with IBOMA with fIBOMA,0 = 0.10–0.90 (Table 2), which was substantially close to the 〈kp〉〈K〉 value of the NMP of My. Accordingly, it can be assumed that the copolymerization kinetics was largely governed by the My kinetics, even when the initial mixture was rich in IBOMA. This behaviour is illustrated in Fig. 2a where all curves almost overlap, regardless of fIBOMA,0.
The molar fractions of 1,4-, 1,2- and 3,4-P(My) additions in the My/IBOMA copolymers were determined by 1H NMR (Fig. S5 in ESI† for the assignments). Interestingly, while the experiment My/IBOMA-100 led to a P(My) homopolymer rich in 1,4-content (86.6 mol%, Table 2), the fraction of 1,2-motif in the copolymers markedly increased when IBOMA-rich feeds were used (∼30 mol% of 1,2-content for fIBOMA,0 = 0.41–0.80). A very similar trend was observed for the systems My/GMA and My/tert-butyl acrylate copolymerized under the same conditions at 120 °C and 115 °C respectively.56 Therefore, it must be caused by the presence of a methacrylate radical as a terminal unit, which might have altered the steric and/or the electronic environment of the active end of the propagating species.
The glass transition behaviour of P(My-grad-IBOMA) copolymer with cumulative IBOMA mole fractions ranging from 0.10 to 0.96 was also characterized using DSC heat curves (Fig. S7, ESI†). Tg versus IBOMA composition plot is given in Fig. 4. A trend curve was included, which was obtained by fitting the Gordon–Taylor equation78 (eqn (6)) to the experimental data.
| Tg = (Tg,P(My)wMy + KTg,P(IBOMA)wIBOMA)/(wMy + KwIBOMA) | (6) |
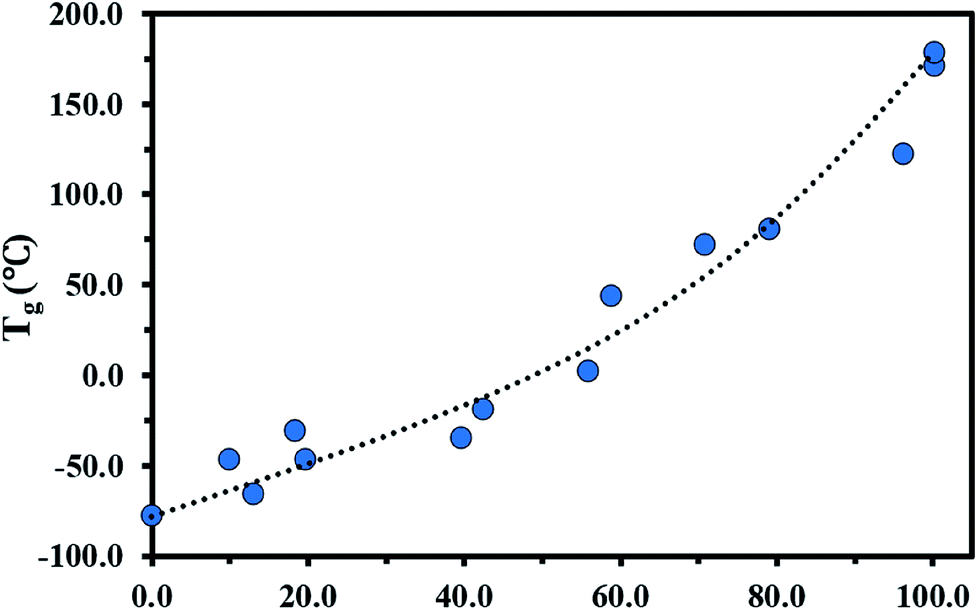 | ||
| Fig. 4 FIBOMA effects on Tg in P(My-grad-IBOMA) gradient copolymers. The DSC traces can be found in the ESI, Fig. S7.† Tg = −77.0 °C for FIBOMA = 0 was determined previously.29 The black dotted line represents the experimental data fitted to the Gordon–Taylor equation. | ||
Thermogravimetric analysis (TGA, Experimental section) has been applied to decomposition study on the final dry samples taken from experiments My/IBOMA-20 and My/IBOMA-80, exhibiting 51 and 81 mol% of My units respectively (Table 2). Fig. 5 represents the thermal decomposition curve of both gradient copolymers. Interestingly, the synthesized polymers exhibited markedly a two-step degradation pattern. A first weight loss was observed from 145 to 305 °C with a Tdec,max of 242 °C (temperature at which the weight loss is most apparent, maximum of the derivative curve) for the sample richer in My (My/IBOMA-80, Fig. 5b) and from 205 to 315 °C with Tdec,max = 307 °C for My/IBOMA-20 sample (Fig. 5a). This early degradation peak was likely due to the loss of the side chains by cleavage of the O-isobornyl bond, releasing camphene as a main product, as reported by Matsumoto and coworkers.40 This first weight loss was proportional to the IBOMA fraction possessed by the samples: while about 40% of mass loss was measured for My/IBOMA-20 with FIBOMA = 0.49, only about 22 wt% of My/IBOMA-80, exhibiting FIBOMA = 0.19, was lost (Fig. 5). This observation supports the contribution of the IBOMA units to the premature P(My-grad-IBOMA) thermal decomposition. The second degradation step is similar for both samples, starting at 305–315 °C with Tdec,max = 370–380 °C and decomposing totally the copolymers at about 450 °C (Fig. 5), which could be mostly caused by the scission reactions of the main chain (depolymerization) resulting in the formation of monomer and oligomer radicals. We previously reported the decomposition behaviour of SG1-mediated P(My), from 290 to 485 °C (Tdec,max = 385 °C),29 matching closely the second degradation step of the two P(My-grad-IBOMA)s characterized.
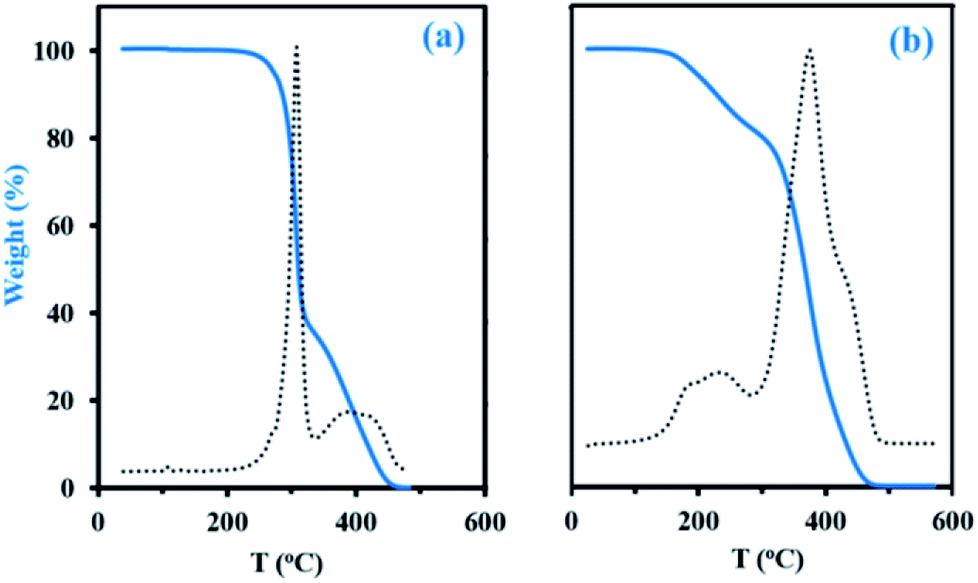 | ||
| Fig. 5 TGA traces (N2 atmosphere, 10 °C min−1) of final gradient copolymers (a) My/IBOMA-20 and (b) My/IBOMA-80, previously precipitated in excess methanol and dried under vacuum at 50 °C. Sample weight versus temperature is represented by the solid blue line whereas the dotted line represents the derivative of weight relative to the temperature versus temperature in order to determine precisely the temperature at which weight loss is most apparent (Tdec,max). | ||
31P NMR was first performed to evaluate the living fraction (LF) of these two gradient copolymers initiated by the NHS-BB alkoxyamine (Fig. S6 in ESI for the spectra). The proportion of chains containing a SG1 end group was 74 ± 9 mol% and 69 ± 3 mol% for My/IBOMA-44 and My/IBOMA-82 copolymers, respectively (Table 3A). It is interesting to note that about two thirds of the My-rich My/IBOMA-82 chains remained SG1-capped despite a relatively high overall conversion (X1 = 64.2%). It can thus be deduced that about 30% of these chains underwent irreversible terminations, becoming more prevalent at high conversion, resulting notably in a relatively broad molecular weight distribution (Đ1 = 1.51, Table 3A). A similar LF was measured for the IBOMA-rich gradient copolymer exhibiting a lower overall conversion (X1 = 39.1%). A moderate conversion was targeted for the synthesis of this macro-initiator since an IBOMA-rich (fIBOMA,0 = 0.70) feed was polymerized, where disproportionation side reactions likely occur. It can be assumed that the synthesis of a high conversion P(My-grad-IBOMA) rich in IBOMA would have a relatively low LF, caused by the occurrence of terminations by H-transfer notably.
To further examine the capacity of these samples to re-initiate and produce diblock copolymers, two chain-extension experiments were done using My/IBOMA-44 and My/IBOMA-82 as macroinitiators (Table 3A). The reaction conditions and results of the chain-extensions are given in Tables 3B and C respectively. My/IBOMA-44 macroinitiator (FIBOMA,1 = 0.56, Mn,MHS,1 = 8.8 kg mol−1) was cleanly chain-extended at 115 °C in toluene with My to obtain a P[(My-grad-IBOMA)-b-My] diblock copolymer (FIBOMA,2 = 0.07, Mn,MHS,2 = 30.1 kg mol−1, Table 3C) containing predominantly My units. The dispersity of this chain-extended product (Đ2 = 1.65) was higher than that of the parent macro-initiator (Đ1 = 1.32), which suggests that termination occurred during the reaction and/or some macroinitiator was not initiated. This latter assumption can be supported by the previous quantitative spectroscopic analysis, indicating that about 25 mol% of My/IBOMA-44 was not terminated by SG1 (Table 3A), as well as the GPC traces of the chain-extension experiment (Fig. 6b) showing that the tails of the final chain-extended copolymer overlapped to some degree with the associated macroinitiator trace. The asymmetrical final GPC trace at 390 min may also indicate the occurrence of irreversible terminations throughout the chain-extension, which generate “dead” species having lower Mn (∼15–25 kg mol−1). Generally, this successful My chain-extension from IBOMA-rich P(My-grad-IBOMA) copolymer prompted us to investigate the reversed situation where a My-rich macroinitiator can be re-activated to add an IBOMA-rich segment. With a fresh IBOMA/My mixture (93 mol% of purified IBOMA), My/IBOMA-82 (FMy,1 = 0.82, Mn,MHS,1 = 13.6 kg mol−1, Đ1 = 1.51, Table 3A) was extended at 105 °C in 50 wt% toluene (Table 3B). Due to the beneficial kinetic effects observed of the addition of 10–20 mol% of My over the control of the SG1-based NMP of IBOMA (Fig. 2), 7 mol% of the acyclic monoterpene was initially introduced in the feed. After 210 min, the resulting chain-extended diblock exhibited Mn,MHS,2 = 23.2 kg mol−1, Đ2 = 1.61 and the overall My molar composition was decreased from 82 to 49 mol% (Table 3C). Similar to the first chain-extension, the GPC trace of the chain-extended product retained generally its mono-modal nature and had a clear shift to the left (Fig. 6a), indicating a high-level of chain-end fidelity of the original macroinitiator.
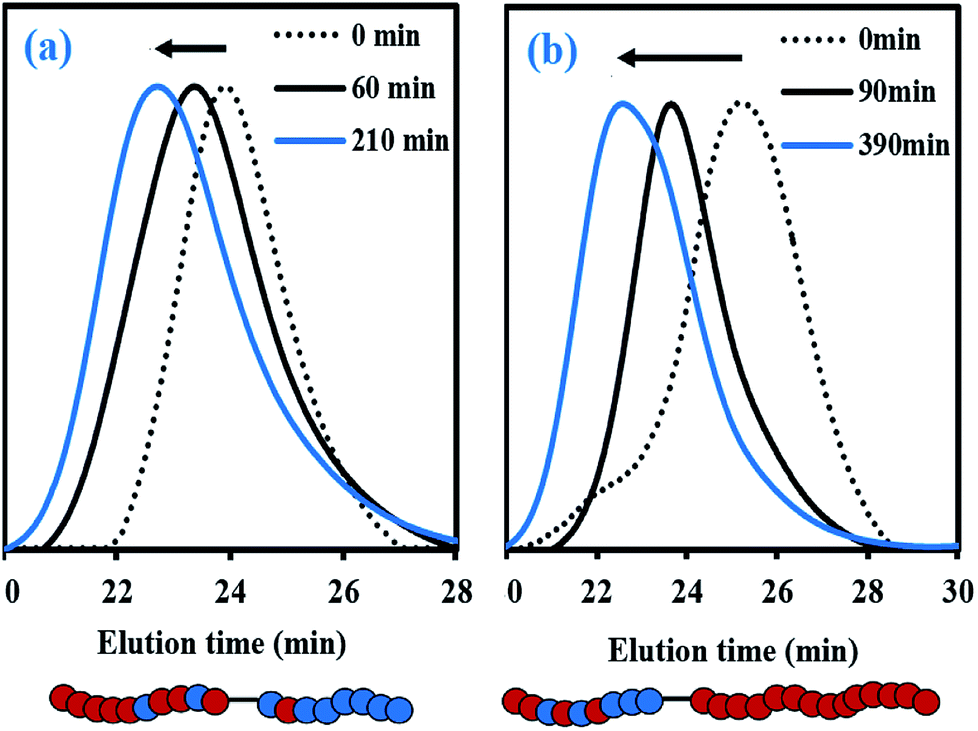 | ||
| Fig. 6 Normalized GPC traces for the chain-extensions of (a) My/IBOMA-82 with a IBOMA/My (93/7 mol%) mixture (experiment My/IBOMA-82-IBOMA/My) and (b) My/IBOMA-44 with My (experiment My/IBOMA-44-My) at T = 105–115 °C in 50 wt% toluene. Schematic representations of the final chain-extended copolymers, where solid red and blue circles represent My and IBOMA units respectively, are given below the GPC chromatograms. | ||
These satisfactory results naturally paved the way to the preparation by NMP of well-defined 1,3-diene-based triblock copolymers composed of a flexible and bio-sourced P(My) mid-segment33 and two rigid P(IBOMA) outer segments, the latter coming from partially bio-based raw material source.81,82
Co/IBOMA-My-IBOMA/Co triblock copolymers by NMP
The synthesis by SG1-mediated NMP of triblock copolymers consisting of a soft P(My) middle block and two hard blocks, containing mostly IBOMA units, was aimed in this second part. A two-step approach was applied for preparing these triblock copolymers: (1) NMP of My initiated by the PEB-(SG1)2 difunctional initiator (telechelic poly(ethylene-stat-butylene) copolymer terminated with SG1 groups);49 (2) chain-extension of the P(My)-(SG1)2 macroinitiator with a IBOMA/Co mixture (Co = My or S co-monomer, ∼8–9 mol% relative to IBOMA). The synthesis route is depicted in Scheme 1b. A triblock molecular architecture was targeted so that both ends of the rubbery mid-segment are theoretically anchored to a glassy domain,83 mimicking thus the traditional micro-structure of styrene block copolymers such as SIS or SBS (I = isoprene, B = butadiene).PEB-(SG1)2 dialkoxyamine macroinitiator (Mn,MHS = 5.7 kg mol−1, Đ = 1.17) was first produced, according to the method reported by Lessard et al.49 1H NMR of the final product revealed that about 84 mol% of the chain ends were capped by a SG1 unit. It can thus be deduced that a minor fraction of the synthesized macroinitiator consisted of monofunctional alkoxyamine terminated by SG1 only at one chain end and “dead” PEB chains. To know the optimal conditions for making high average chain length P(My) in a controlled manner, NMP of My initiated by various concentrations of PEB-(SG1)2 at 120 °C in bulk was then pursued. Four different theoretical Mn were thus targeted at quantitative conversion, namely 79.1, 105.0, 137.2 and 174.0 kg mol−1 (Table S2, ESI†), and kinetic studies were led (Fig. S8, ESI†) to assess the level of control. As Mn,theo increased, higher dispersity values and greater deviations of Mn,MHS from the theoretical molar masses were observed (Fig. S7b and S7c†), confirming likely the greater presence of irreversible terminations and chain transfer reactions to My and P(My) with [My]0. Nonetheless, lowering the dialkoxyamine concentration resulted in the synthesis of higher Mn,MHS P(My)s. Particularly, when targeting Mn,theo = 137.2 kg mol−1 at XMy = 100% (experiment My-137), a polyterpene exhibiting Mn,MHS = 49.5 kg mol−1 and Đ = 1.52 was obtained at 51% conversion (Fig. S7b and S7c†), which consisted of the most satisfactory balance between a relatively high Mn and a moderate dispersity. It was then attempted to produce longer P(My) chains with a relatively narrow molecular weight distribution by using a low concentration of the dialkoxyamine initiator [PEB-(SG1)2] (Mn,theo = 169–173 kg mol−1 at X = 1.0) combined with additional free SG1 nitroxide (9–18 mol% with respect to the initiator, experiments My-173SG1,9 and My-169SG1,18, Table S2†). An improved control of the NMP when using a slight excess of this potent nitroxide could have been expected since the dormant state should be favoured (lower concentration of propagating macro-radicals, reduction of the overall polymerization rate).86–88 Despite apparent rate constants somewhat lower as [SG1˙]0 increased (Fig. S9a in ESI†), similar Đ versus X and Mn,MHS versus X trends were determined (Fig. S8b and S8c†) indicating the ineffectiveness of adding SG1 free radical to the reaction mixture. This observation echoes a previous study showing that the addition of 4.9–11.7 mol% of the SG1 mediator with respect to the initiator did not enhance the NMP of My at 120 °C in bulk initiated by NHS-BB.29 A last experiment in 50 wt% toluene, without extra SG1, was led, predicting Mn,theo ∼ 170 kg mol−1 again (experiment My-170Tol, Table S2†). The use of a solvent was thought to help, as seen by the increase in viscosity at XMy > 30%, when performing this reaction in bulk, which might account for the occurrence of diffusional limitations,89 bringing about a loss of control. Interestingly, while the polymerization in toluene was slower (Fig. S8a†), similar molecular characteristics of the growing P(My) were obtained compared to the previous mass polymerizations (Fig. S8b and S8c†). Higher Đ values were even observed (Fig. S8b†), which may be assigned to chain-transfer side reactions to toluene.90,91
Consequently, the P(My) segment was prepared via the PEB-(SG1)2-initiated NMP of My at 120 °C in bulk for 280–400 min (XMy ∼ 40–50%) targeting Mn,theo ∼ 140 kg mol−1 and without any additional free nitroxide. Two 1,4-rich P(My)s (experiments My-35 and My-52, Table 4A), exhibiting Mn,MHS = 35–52 kg mol−1 and acceptable Đ = 1.45–1.53, were synthesized this way. Attention was now shifted to the capacity of these P(My) segments, ideally capped by two SG1 units, to form triblock copolymer with a mixture of IBOMA containing a minor fraction of co-monomer.
Afterwards, a second triblock copolymer, with a higher overall Mn as well as harder outer blocks, was targeted. To that end, My-52 macroinitiator (Mn,MHS,My = 51.7 kg mol−1, Đ = 1.53, Table 4A) was reacted with a IBOMA/S mixture (8.7 mol% of S used as a controlling co-monomer, as previously reported24,42,92) for 3 h in 50 wt% toluene at 115 °C (Table 4B). The resulting product My-52-IBOMA/S had Mn,MHS,CE = 78.1 kg mol−1 and Đ = 2.41. Likewise, the final chain-extended product shifted to lower elution time compared to the macroinitiator (Fig. S9b†). To increase My-52-IBOMA/S Mn, a fractional precipitation using the non-solvent addition method was implemented, relying on the benzene (solvent)/methanol (non-solvent) pair. Eventually, the fractionated triblock copolymer had Mn,MHS = 94.7 kg mol−1 and Đ = 2.23 (Table 4C), reflecting the removal of a fraction of relatively short chains. The rigid segments were constituted of about 88 mol% of methacrylate units and 12 mol% of S, as determined by 1H NMR.
A high Đ = 1.91–2.23 was measured for these final triblock copolymers, which may be mainly caused by the difficulty to control the polymerization of a methacrylic ester-rich feed (fIBOMA,0 ≥ 0.91), although a small amount of My or S were added to possibly decrease the average activation–deactivation equilibrium constant 〈K〉. The inactivity of a fraction of PEB-(SG1)2 and P(My)-(SG1)2 macroinitiators may also contribute to the broadening of the molecular weight distribution. Besides, the very likely presence of PEB-SG1 monofunctional chains may have resulted in the synthesis of short diblock copolymer chains, assuming a similar activity between PEB-SG1 and PEB-(SG1)2. All these hypotheses would lead to a non-negligible population of shorter than expected chains, as observed in Fig. S10 (ESI†) by the tails on the low molecular weight edge of the GPC chromatograms (elution time ∼ 23–25 min).
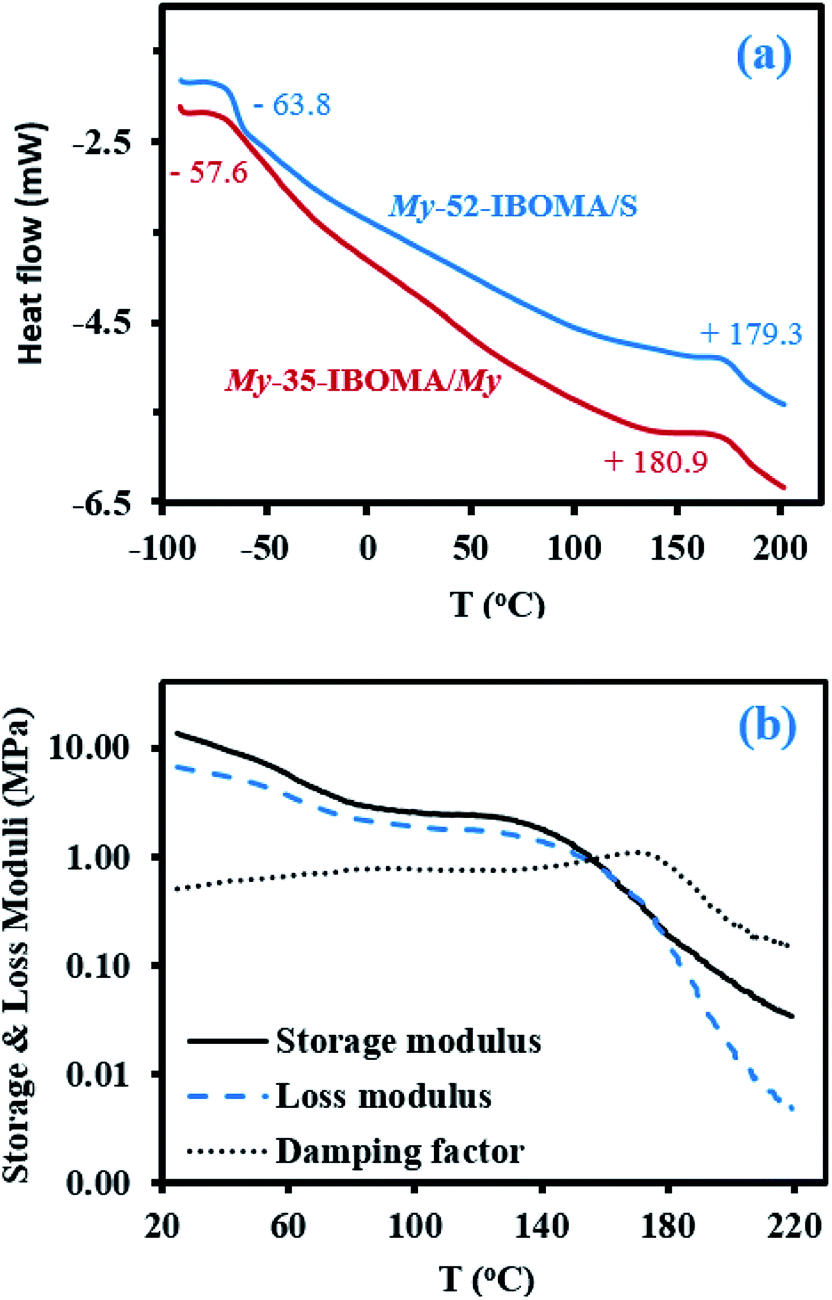 | ||
| Fig. 7 (a) DSC traces (second heating run) of the triblock copolymers My-52-IBOMA/S (blue) and My-35-IBOMA/My (red). The numbers near the changes in slope correspond to the Tgs determined via the inflection method. (b) Dynamic mechanical analysis of the sample My-52-IBOMA/S by torsional oscillation, yielding the storage modulus, the loss modulus and the damping factor versus temperature (0.15 Hz, 1% strain, 5 °C min−1, N2 atmosphere). | ||
These DSC results suggest a two-phase system, due to the immiscibility between P(My) and IBOMA-rich segments. To further examine immiscibility, the solubility parameters δ were compared to estimate the tendency towards micro-phase separation. Using the group contribution method developed by van Krevelen and coworkers,94 theoretical solubility parameters for P(IBOMA) and 1,4-P(My) (favoured regioselectivity in our case) were determined (calculations can be found in ESI, page S16†) and δP(My) = 16.7 MPa1/2 and δP(IBOMA) = 21.8 MPa1/2 were calculated. Such a δ difference indicates a relatively high immiscibility between P(My) and P(IBOMA) segments and would suggest a strong bulk phase separation of P(My-b-IBOMA) block copolymer. Atomic force microscopy (AFM)95 was applied to the study of the surface morphology of My-35-IBOMA/My prepared by spin-coating a 3–4 wt% solution in chloroform. Fig. 8 shows a 2 μm × 2 μm scan of the polymeric surface (two other AFM phase images of My-35-IBOMA/My in different regions are given in ESI, Fig. S11†). The nature of the light and dark domains was determined by quantifying the relative contribution of the light component (ImageJ software application, binary mode). This image contained 23–24% of light areas which is consistent with the molar fraction of P(IBOMA) in the characterized triblock (FIBOMA = 0.28, Table 4). The light areas can thus be attributed to P(IBOMA) and the dark ones to the rubbery P(My) midblock material. The phase separation of the two component blocks appeared clear with the embedding of glassy P(IBOMA) aggregates (disperse phase) in the soft P(My) (continuous phase). A spherical or cylindrical morphology might be obtained. An in-depth analysis, using for instance small-angle X-ray scattering (SAXS), would be required to ascertain the block copolymer morphology. It is of importance to note that the My-35-IBOMA/My self-assembly was disordered with a random location of the rigid aggregates, which exhibited various sizes (27% of large aggregates exhibiting a surface area of 2650 ± 740 nm2 and 73% of smaller aggregates having a surface area of 930 ± 480 nm2, quantification via Nanoscope Analysis software). By assuming that cylinders were observed (FIBOMA = 0.28, possibly elevated for sphere formation), the radius of the cylinders R can be estimated according to the following formula (7):
| R = 1.0αKM1/2 | (7) |
 | ||
| Fig. 8 Atomic force microscopy (AFM, Experimental section) phase image (2 μm × 2 μm) under tapping mode of operation of the surface morphology of the triblock copolymer My-35-IBOMA/My cast film. The dark domain represents the My component (color-coded height scale given to the right of the image). | ||
Fig. 7b shows the temperature dependence of the shear storage and loss moduli (G′ and G′′ respectively) as well as the damping factor (tanδ = G′′/G′) from room temperature to 220 °C for the triblock copolymer My-52-IBOMA/S (Table 4C). The significant decrease of G′ from about 13 to 5 MPa with temperature until 70 °C can be first noted. A quasi constant G′ could have been expected in this temperature range due to the glassy state of the IBOMA units embedded in the soft rubbery P(My) phase. The presence of 12 mol% of S in the extended blocks can partly explain this behaviour. Compared to P(IBOMA), the solubility parameter of PS, δPS = 18.4 MPa1/2 (Hoftyzer and Van Krevelen approach), is closer to that of P(My), which may result in the partial homogeneity of the triblock copolymer. Moreover, short PS sequences, softening at relatively low temperature, were likely contained by the outer blocks. It was notably reported by DSC that Tg,PS = 32–69 °C for low Mn,PS = 0.9–3.0 kg mol−1.99 The rubbery plateau was then observed at T = 70–140 °C where G′ slightly decreased from 3.6 to 2.2 MPa. At T > 140 °C, the modulus dropped due to the softening of the P(IBOMA) phase, the flexible P(My) chains being no longer held together by the rigid domains. The maximum tanδ peak appeared at 171 °C, which is consistent with the P(IBOMA)'s Tg measured previously (Tg,P(IBOMA) = 171–177 °C, Fig. 4). Generally, this dynamic mechanical analysis (DMA) suggests that My-52-IBOMA/S may have an upper service temperature T ∼ 140 °C with G′ ≥ 2.2 MPa at T ≤ 140 °C.
Lastly, the mechanical properties at room temperature of My-52-IBOMA/S were determined by uniaxial tensile testing. The stress–strain curves are given in Fig. 9 along with the mean Young's modulus E, yield stress σY, tensile strength at break σB and tensile elongation at break εB values. The linear elastic region was apparent until 13–19% elongation, at which point the yield strength was observed. This low-strain region allowed the determination of E = 2.32 ± 0.28 MPa and σY = 5.02 ± 0.23 MPa (Fig. 9). Beyond the proportionality limit, the tensile strength decreased (23% on average) until about 130% elongation. This can be presumably explained by the onset of the plastic deformation, not allowing the total recovery of the strains (permanent deformation of the material). The plastic region was then markedly observed with a plateau, characteristic of the drawing of the My-52-IBOMA/S chains, ending at the fracture point (εB = 490 ± 31%). It has to be noted that the strain hardening phenomenon, consisting of the orientation and alignment of polymer chains in the direction of the load which increases the strength and stiffness of the material, was not observed. Consequently, no increase in stress was measured at the fracture (σB = 3.90 ± 0.22 MPa). A key-factor allowing the understanding of this behaviour is presumably the morphology of the triblock copolymer. The possible disorganized structure of My-52-IBOMA/S, not containing necessarily P(IBOMA) glassy spheres can be detrimental. In this case, the expected physical crosslinking structure does not exist, bringing about a decrease of the entropic contribution to strain hardening. By assuming that the strain hardening process is associated with the debonding of the entanglement network (chain disentanglement), it may be also caused by the P(My) continuous phase, not being sufficiently entangled (Mn,MHS,My = 51.7 kg mol−1 < 3Me,P(My)),85 resulting in an unconstrained uncoiling of the chains. Generally, a flexible plastic was identified, having moderate elastic modulus and tensile strength at break combined with a good irreversible extensibility.
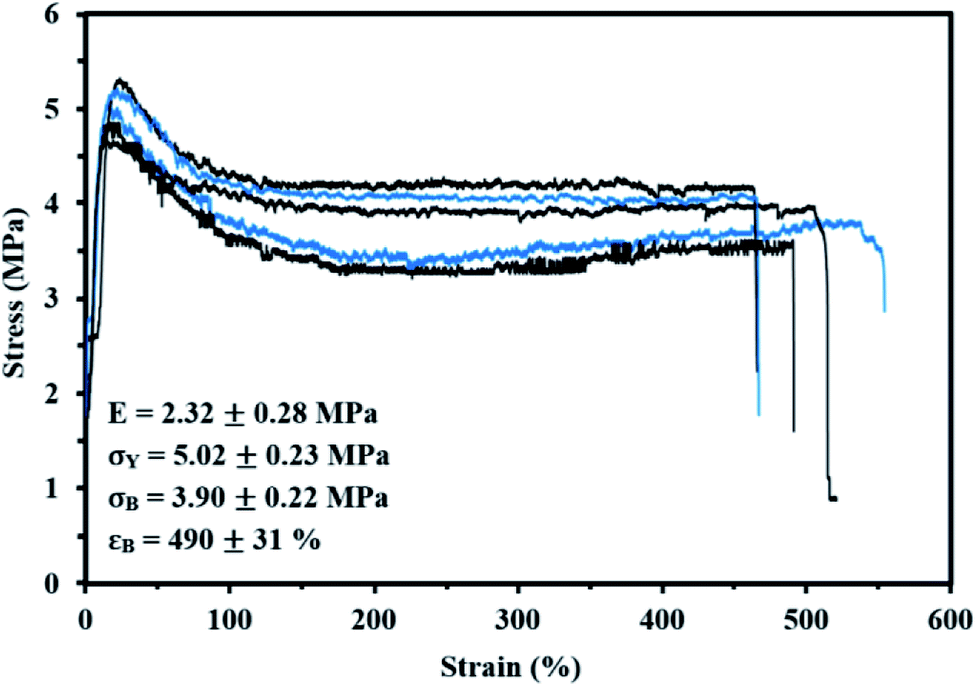 | ||
| Fig. 9 Tensile stress–strain curves of five My-52-IBOMA/S samples (same batch, color only used for differentiation) at room temperature and at a cross-head speed of 10 mm min−1. The average Young's modulus E, yield stress σY, tensile strength at break σB and tensile elongation at break εB, obtained from these curves are also given. | ||
Conclusions
My/IBOMA NMP in bulk at 100 °C initiated by NHS-BB highlighted the gradient nature of this copolymerization with the higher reactivity of the monoterpene (rMy = 1.90–2.16) toward both propagating species (⋯My˙ and ⋯IBOMA˙) compared of that of the bulky methacrylate (rIBOMA = 0.02–0.07). While the homopolymerization of IBOMA was not controlled, likely due to the high equilibrium constant of this methacrylate, the addition of 20 mol% of My as a co-monomer allowed the synthesis of a well-defined P(My-grad-IBOMA) copolymer with Đ ≤ 1.39 and Mn,MHS close to the predicted one. More generally, well-controlled copolymerizations were performed for fIBOMA,0 ≤ 0.80 and the resulting P(My-grad-IBOMA)s exhibited Đ ≤ 1.41, Mn,MHS = 5.9–10.9 kg mol−1 and monomodal GPC distributions at overall conversion X = 25–65%. The deviations of experimental Mn,MHS values from theoretical ones were greater with fMy,0, which may be caused by chain transfer side reactions to the monomers. Regardless of the initial feed composition, these copolymerizations had 〈kp〉〈K〉 = 2.0 ± 0.9 × 10−5 s−1 relatively close to that of My homopolymerization (1.4 ± 0.4 × 10−5 s−1), stressing the strong influence of My kinetics over the copolymerization kinetics. P(My-grad-IBOMA) displayed a glass transition temperature between −65 and +123 °C depending on the molar composition of IBOMA. A two-step thermal decomposition was observed by TGA, consisting of an early decomposition peak at Tdec,max = 242–307 °C, which results likely from the release of the isobornyl group, and a final decomposition peak at Tdec,max = 370–380 °C, due to the scission of the copolymer backbone. The chain-end fidelity of My-rich and IBOMA-rich P(My-grad-IBOMA)s was quantitatively deemed by 31P NMR (69–74% of SG1-terminated chains) and confirmed via successful chain-extensions in toluene with IBOMA and/or My (monomodal shift of the GPC traces).The satisfactory mediation of the My/IBOMA system by the SG1 nitroxide allowed subsequently the preparation of triblock copolymers, composed of a soft P(My) middle block and two outer IBOMA-rich segments. 1,4-P(My)-(SG1)2 macro-initiator (Mn,MHS = 35–52 kg mol−1, Đ = 1.45–1.53) was first synthesized using the PEB-(SG1)2 dialkoxyamine initiator in bulk at 120 °C, followed by the IBOMA/Co chain-extension (Co = My or S co-monomer, <9 mol%) at 115 °C in toluene. Displaying two distinct Tgs at −58 and +181 °C, My/IBOMA-My-IBOMA/My triblock copolymer (Mn,MHS = 51 kg mol−1, Đ = 1.91, FIBOMA = 0.28) was studied by AFM, which revealed the micro-phase separation of the continuous P(My) domain and the disperse IBOMA-rich aggregates, despite an apparent disorganization. Lastly, rheological analysis as well as stress–strain tests were performed for the NMP-based S/IBOMA-My-IBOMA/S triblock copolymer (Mn,MHS = 95 kg mol−1, Đ = 2.23, FIBOMA = 0.36, FS = 0.05). In comparison to traditional styrenic block copolymers, an extended upper service temperature at around 140 °C was observed by DMA. Furthermore, this triblock copolymer exhibited a tensile strength at break σB ∼ 4 MPa and an elongation at break εB ∼ 500%, despite the absence of strain hardening.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
ArianeGroup and the French Defense Procurement Agency (Direction Générale de l'Armement, DGA) supported this work financially. We would like to thank the Association Nationale de la Recherche et de la Technologie (ANRT) for its financial contribution. We also thank Mary Sullivan and Noah Macy of Arkema for their aid in obtaining the BlocBuilder™ alkoxyamine initiator and SG1 used in this work.Notes and references
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- R. P. Quirk, Q. Zhuo, S. H. Jang, Y. Lee and G. Lizarraga, in Applications of Anionic Polymerization Research, American Chemical Society, 1998, ch. 1, pp. 2–27 Search PubMed.
- H.-G. Elias in Macromolecules: Synthesis, Materials, and Technology, Springer, 2nd edn, 1984, ch. 20, pp. 681–736 Search PubMed.
- J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. Le, R. Mayadunne, G. Meijs, C. Moad, G. Moad, E. Rizzardo and S. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32(1), 93–146 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26(11), 2987–2988 CrossRef CAS.
- R. P. N. Veregin, M. K. Georges, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26(20), 5316–5320 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117(20), 5614–5615 CrossRef CAS.
- D. H. Solomon, E. Rizzardo and P. Cacioli, Eur. Pat. Appl. 0135280A3, Commonwealth Scientific and Industrial Research Organization, 1986.
- G. Moad and E. Rizzardo, in Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science, The Royal Society of Chemistry, 2016, ch. 1, pp. 1–44 Search PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38(1), 63–235 CrossRef CAS.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- J. G. Drobny, in Handbook of Thermoplastic Elastomers, Plastics Design Library, 2007, ch. 5, pp. 161–178 Search PubMed.
- R. C. Bening, W. H. Korcz and D. L. Handlin, in Modern Styrenic Polymers: Polystyrenes and Styrenic Copolymers, John Wiley & Sons, 2003, ch. 21, pp. 463–500 Search PubMed.
- J. L. Pradel, B. Ameduri and B. Boutevin, Macromol. Chem. Phys., 1999, 200, 2304–2308 CrossRef CAS.
- J. L. Pradel, B. Boutevin and B. Ameduri, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3293–3302 CrossRef CAS.
- B. Keoshkerian, M. Georges, M. Quinlan, R. Veregin and B. Goodbrand, Macromolecules, 1998, 31, 7559–7561 CrossRef CAS.
- D. Benoit, E. Harth, P. Fox, R. M. Waymouth and C. J. Hawker, Macromolecules, 2000, 33(2), 363–370 CrossRef CAS.
- C. Cheng, K. Qi, E. Khoshdel and K. L. Wooley, J. Am. Chem. Soc., 2006, 128, 6808–6809 CrossRef CAS PubMed.
- J. K. Wegrzyn, T. Stephan, R. Lau and R. B. Grubbs, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2977–2984 CrossRef CAS.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130(37), 12264–12265 CrossRef CAS PubMed.
- Y. Cai, K. B. Aubrecht and R. B. Grubbs, J. Am. Chem. Soc., 2010, 133, 1058–1065 CrossRef PubMed.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- D. Benoit, S. Grimaldi, S. Robin, J.-P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122(25), 5929–5939 CrossRef CAS.
- H. Matsuoka, Y. Suetomi, P. Kaewsaiha and K. Matsumoto, Langmuir, 2009, 25(24), 13752–13762 CrossRef CAS PubMed.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromolecules, 2011, 44(23), 9230–9238 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromol. Rapid Commun., 2012, 33(9), 805–810 CrossRef CAS PubMed.
- A. Métafiot, Y. Kanawati, J. -F. Gérard, B. Defoort and M. Maric, Macromolecules, 2017, 50(8), 3101–3120 CrossRef.
- J. Vinas, N. Chagneux, D. Gigmes, T. Trimaille, A. Favier and D. Bertin, Polymer, 2008, 49, 3639–3647 CrossRef CAS.
- M. K. Georges, G. K. Hamer and N. A. Listigovers, Macromolecules, 1998, 31(25), 9087–9089 CrossRef CAS.
- C. Detrembleur, V. Sciannamea, C. Koulic, M. Claes, M. Hoebeke and R. Jérome, Macromolecules, 2002, 35(19), 7214–7223 CrossRef CAS.
- A. Behr and L. Johnen, ChemSusChem, 2009, 2(12), 1072–1095 CrossRef CAS PubMed.
- P. Sarkar and A. K. Bhowmick, Ind. Eng. Chem. Res., 2018, 57, 5197–5206 CrossRef CAS.
- N. Bauer, J. Brunke and G. Kali, ACS Sustainable Chem. Eng., 2017, 5(11), 10084–10092 CrossRef CAS.
- P. Sarkar and A. K. Bhowmick, RSC Adv., 2014, 4, 61343–61354 RSC.
- R. P. Quirk and T. -L. Huang, in New Monomers and Polymers, Springer, 1984, ch. 19, pp. 329–355 Search PubMed.
- R. P. Quirk, US Pat. 4 374 957, Michigan Molecular Institute, 1983.
- J. M. Bolton, M. A. Hillmyer and T. R. Hoye, ACS Macro Lett., 2014, 3(8), 717–720 CrossRef CAS.
- A. Matsumoto, K. Mizuta and T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 1993, 31(10), 2531–2539 CrossRef CAS.
- J. M. Yu, P. Dubois and R. JérÔme, Macromolecules, 1996, 29(26), 8362–8370 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38(13), 5485–5492 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39(24), 8274–8282 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(1), 34–47 CrossRef CAS.
- E. Guégain, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2015, 36(13), 1227–1247 CrossRef PubMed.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Astolfi, L. Greci, P. Tordo and D. Bertin, Macromolecules, 2007, 40, 3108–3114 CrossRef CAS.
- P. Astolfi, L. Greci, P. Stipa, C. Rizzoli, C. Ysacco, M. Rollet, L. Autissier, A. Tardy, Y. Guillaneuf and D. Gigmes, Polym. Chem., 2013, 4, 3694–3704 RSC.
- N. Ballard, M. Aguirre, A. Simula, A. Agirre, J. R. Leiza, J. M. Asua and S. van Es, ACS Macro Lett., 2016, 5, 1019–1022 CrossRef CAS.
- B. Lessard, C. Aumand-Bourque, R. Chaudury, D. Gomez, A. Haroon, N. Ibrahimian, S. Mackay, M.-C. Noel, R. Patel, S. Sitaram, S. Valla, B. White and M. Maric, Int. Polym. Process., 2011, 26(2), 197–204 CrossRef CAS.
- S. Georges, M. Bria, P. Zinck and M. Visseaux, Polymer, 2014, 55, 3869–3878 CrossRef CAS.
- H. K. Mahabadi, J. Appl. Polym. Sci., 1985, 30(4), 1535–1544 CrossRef CAS.
- X. Q. Zhang and C. H. Wang, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 1951–1956 CrossRef CAS.
- P. Hattam, S. Gauntlett, J. W. Mays, N. Hadjichristidis, R. N. Young and L. J. Fetters, Macromolecules, 1991, 24(23), 6199–6209 CrossRef CAS.
- H. Pasch and B. Trathnigg, in Multidimensional HPLC of Polymers, Springer, 2013, ch. 4 and 5, pp. 37–90 Search PubMed.
- C. Petit, B. Luneau, E. Beaudoin, D. Gigmes and D. Bertin, J. Chromatogr. A, 2007, 1163, 128–137 CrossRef CAS PubMed.
- A. Métafiot, J. -F. Gérard, B. Defoort and M. Maric, J. Polym. Sci., Part A: Polym. Chem., 2018, 56(8), 860–878 CrossRef.
- G. S. Ananchenko, M. Souaille, H. Fischer, C. LeMercier and P. Tordo, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3264–3283 CrossRef CAS.
- S. Beuermann and M. Buback, Prog. Polym. Sci., 2002, 27(2), 191–254 CrossRef CAS.
- C. Burguiere, M. Dourges, B. Charleux and J. P. Vairon, Macromolecules, 1999, 32(12), 3883–3890 CrossRef CAS.
- J. Nicolas, E. Guégain and Y. Guillaneuf, in Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science, The Royal Society of Chemistry, 2016, ch. 7, pp. 305–348 Search PubMed.
- C. Dire, B. Charleux, S. Magnet and L. Couvreur, Macromolecules, 2007, 40(6), 1897–1903 CrossRef CAS.
- B. Lessard and M. Maric, J. Polym. Sci., Part A: Polym. Chem., 2009, 47(10), 2574–2588 CrossRef CAS.
- A. Moayeri, B. Lessard and M. Maric, Polym. Chem., 2011, 2(9), 2084–2092 RSC.
- M. Berger and I. Kuntz, J. Polym. Sci., Part A: Polym. Chem., 1964, 2, 1687–1698 Search PubMed.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5(2), 259–262 CrossRef CAS.
- T. Kelen and F. Tudos, J. Macromol. Sci., Chem., 1975, 9(1), 1–27 CrossRef.
- P. W. Tidwell and G. A. Mortimer, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 369–387 CrossRef CAS.
- M. A. Dubé, E. Saldivar-Guerra and I. Zapata-Gonzales, in Handbook of Polymer: Synthesis, Characterization, and Processing, 1st edn, John Wiley & Sons, 2013, ch. 6, pp. 105–125 Search PubMed.
- D. L. Trumbo, Polym. Bull., 1993, 31, 629–636 CrossRef CAS.
- H. Benoit, Z. Grubisic, P. Rempp, P. Decker and J. Zilliox, J. Chim. Phys., 1966, 63, 1507–1514 CrossRef CAS.
- R. B. Grubbs, Polym. Rev., 2011, 51, 104–137 CrossRef CAS.
- G. S. Ananchenko, M. Souaille, H. Fischer, C. LeMercier and P. Tordo, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3264–3283 CrossRef CAS.
- G. Gryn'ova, C. Y. Lin and M. L. Coote, Polym. Chem., 2013, 4(13), 3744–3754 RSC.
- W. J. Runckel and L. A. Goldblatt, Ind. Eng. Chem., 1946, 38(7), 749–751 CrossRef CAS.
- N. Kamachi and A. Kajiwara, Macromolecules, 1996, 29(7), 2378–2385 CrossRef.
- S. Beuermann, M. Buback, T. P. Davis, N. Garcia, R. G. Gilbert, R. A. Hutchinson, A. Kajiwara, M. Kamachi, I. Lacik and G. T. Russel, Macromol. Chem. Phys., 2003, 204(10), 1338–1350 CrossRef CAS.
- N. Hadjichristidis, J. Mays, W. Ferry and L. J. Fetters, J. Polym. Sci., Polym. Phys. Ed., 1984, 22(10), 1745–1751 CrossRef CAS.
- M. Gordon and J. S. Taylor, J. Chem. Technol. Biotechnol., 1952, 2(9), 493–500 CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39(24), 8274–8282 CrossRef CAS.
- C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromol. Rapid Commun., 2004, 25, 1215–1220 CrossRef CAS.
- J. W. Reed and W. L. Jolly, J. Org. Chem., 1977, 42, 3963–3965 CrossRef CAS.
- R. N. Cavalcanti, T. Forster-Carneiro, M. T. M. Gomes, M. A. Rostagno, J. M. Prado and M. A. A. Meireles, in Natural Product Extraction: Principles and Applications, The Royal Society of Chemistry, 2013, ch. 1, pp. 1–57 Search PubMed.
- M. D. Gehlsen, K. Almdal and F. S. Bates, Macromolecules, 1992, 25(2), 939–943 CrossRef CAS.
- S. Schlogl, M. -L. Trutschel, W. Chassé, G. Riess and K. Saalwachter, Macromolecules, 2014, 47(9), 2759–2773 CrossRef.
- L. J. Fetters, D. J. Lohse and R. H. Colby, in Physical Properties of Polymers Handbook, 2nd edn, Springer, 2007, ch. 25, pp. 447–454 Search PubMed.
- S. Grimaldi, J. -P. Finet, F. Le Moigne, A. Zeghdaoui, P. Tordo, D. Benoit, M. Fontanille and Y. Gnanou, Macromolecules, 2000, 33(4), 1141–1147 CrossRef CAS.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- S. Robin, O. Guerret, J. -L. Couturier, R. Pirri and Y. Gnanou, Macromolecules, 2002, 35(10), 3844–3848 CrossRef CAS.
- D. R. D'Hooge, M. -F. Reyniers and G. B. Marin, Macromol. React. Eng., 2013, 7(8), 362–379 CrossRef.
- D. Gigmes, D. Bertin, C. Lefay and Y. Guillaneuf, Macromol. Theory Simul., 2009, 18, 402–419 CrossRef CAS.
- B. Lessard, C. Tervo and M. Maric, Macromol. React. Eng., 2009, 3, 245–256 CrossRef CAS.
- C. J. Hawker, E. Elce, J. L. Dao, W. Volksen, T. P. Russel and G. G. Barclay, Macromolecules, 1996, 29, 2686–2688 CrossRef CAS.
- B. Zhang, Y. Ma, D. Chen, J. Xu and W. Yang, J. Appl. Polym. Sci., 2013, 129(1), 113–120 CrossRef CAS.
- D. W. van Krevelen and K. Te Nijenhuis, in Properties of Polymers, 4th edn, Elsevier, 2009, ch. 7, pp. 189–227 Search PubMed.
- M. Pinna, S. Hiltl, X. Guo, A. Böker and A. V. Zvelindovsky, ACS Nano, 2010, 4(5), 2845–2855 CrossRef CAS PubMed.
- D. J. Meier, J. Polym. Sci., 1969, 26C, 81–98 Search PubMed.
- D. J. Meier, Polym. Prepr., 1970, 11, 400–404 CAS.
- F. S. Bates, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- L. -P. Blanchard, J. Hesse and S. L. Malhotra, Can. J. Chem., 1974, 52, 3170–3175 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and calculations; NMR spectra; GPC and DSC traces; kinetic plots; AFM images. See DOI: 10.1039/c8ra09192g |
| This journal is © The Royal Society of Chemistry 2019 |