
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica cubosomes templated by a star polymer†
Congcong Cui a,
Lu Han*a and
Shunai Cheab
a,
Lu Han*a and
Shunai Cheab
aSchool of Chemical Science and Engineering, Tongji University, 1239 Siping Road, Shanghai, 200092, China. E-mail: luhan@tongji.edu.cn
bSchool of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, 200240, P. R. China
First published on 19th February 2019
Abstract
The organization of amphiphilic molecules into well-defined geometries and morphologies is an area of fundamental and practical importance. Herein, we report silica cubosomes synthesized by the cooperative self-assembly of the amphiphilic star polymer poly(ethylene glycol)-(polystyrene)2. The silica cubosomes exhibit a spherical shape and a highly ordered bicontinuous diamond-surface structure. A new synthesis-field diagram was constructed based on the mass ratio of THF, HCl (2 M) and the polymer. Moreover, the mechanism of the formation of silica cubosomes has been revealed. Due to the inorganic framework, the silica cubosomes show enhanced stability and a wide scope of applications.
Introduction
Due to the versatile design of block copolymers (BCPs), the solution self-assembly of amphiphilic BCPs leads to a variety of materials with a well-defined morphology, and nanostructures with desirable functions are obtained due to the arrangement and physicochemical properties of the building blocks.1 The structure of the BCP system can be manipulated by the incompatibility and volume fractions of the different blocks and is associated with the packing parameter g = V/a0l, where V is the volume of the hydrophobic segment, a0 is the molecular area of the hydrophilic head group, and l represents the dynamic chain length of the hydrophobic part.2 By increasing the value of the packing parameter, spherical, cylindrical, bicontinuous, lamellar, and their corresponding inverted phases can be formed.Among these materials, colloidal particles with bicontinuous structures (cubosomes) are the focus of an increasing number of studies due to their controllable structures and extensive applications.3–8 These bicontinuous structures consist of two interwoven labyrinths separated by a single continuous surface that follows the triply periodic minimal surface.9 The attraction of these materials lies not only in their complex and highly symmetrical structures, but also in their structural analogies in both natural and artificial systems e.g. cubic membranes,10 the lyotropic liquid crystal phases of amphiphilic molecules,11,12 the self-assembly of block copolymers,13 etc.
The cubosomes show high water-dispersibility, chemical stability, and oral bioavailability.14–17 The interior of the cubosomes consists of regular interpenetrating networks with a large surface area. Therefore, polymer cubosomes are widely used in the fields of drug delivery, catalysis and so on.8,16–19 For the formation of polymer cubosomes, both top-down and bottom-up methods have been applied. The examples include self-organized precipitation by slow evaporation of a good solvent from a polymer solution containing both poor and good solvents,20–23 a spray drying technique for the synthesis of cubosomes from dry powder,24,25 flash nanoprecipitation, spontaneous emulsification, etc.; however, cubosomes primarily consist of self-assembled lipid molecules or amphiphilic block polymers; this limits their use under extreme conditions such as in strong acids and alkalis, at high temperatures and in complex solution environments. Moreover, several mesoporous silica microspheres have been reported,26–28 with pore sizes in the mesoporous range.
Note that an evaporation-induced self-assembly method has been employed in the synthesis of mesoporous solids. However, it is still difficult to control the structure and morphology of the ordered macroporous scaffolds with a large unit cell parameter due to the complex interactions between inorganic precursors and polymers, and bulk structures are often formed in this case. Herein, we report the synthesis via the self-assembly of a star polymer with an inorganic precursor in the evaporation process, and it has been demonstrated that this method can be utilized to form silica cubosomes with a spherical morphology and controllable bicontinuous structures. Our strategy was to use the microphase-templating route through the self-assembly of the amphiphilic star polymer poly(ethylene glycol)-(polystyrene)2 (sGS2) with one hydrophilic block (PEG), two hydrophobic blocks (PS) and a silica source, tetraethyl orthosilicate (TEOS), in a mixture of tetrahydrofuran (THF) and water. At a large THF/water ratio, the hydrophobic PS block was present in the THF-rich phase; therefore, water was driven out of the hydrophobic phase and mainly appeared in the hydrophilic PEG region.29 With the evaporation of solvents and the hydrolysis and condensation of TEOS, the sample formed two interwoven diamond labyrinths through cooperative self-assembly. As THF evaporated, the packing parameter decreased, causing the self-assembled structure to transform from an inverted bicontinuous double-diamond to a lamellar structure and eventually to silica cubosomes (Scheme 1). Finally, the template molecules could be removed by calcination.
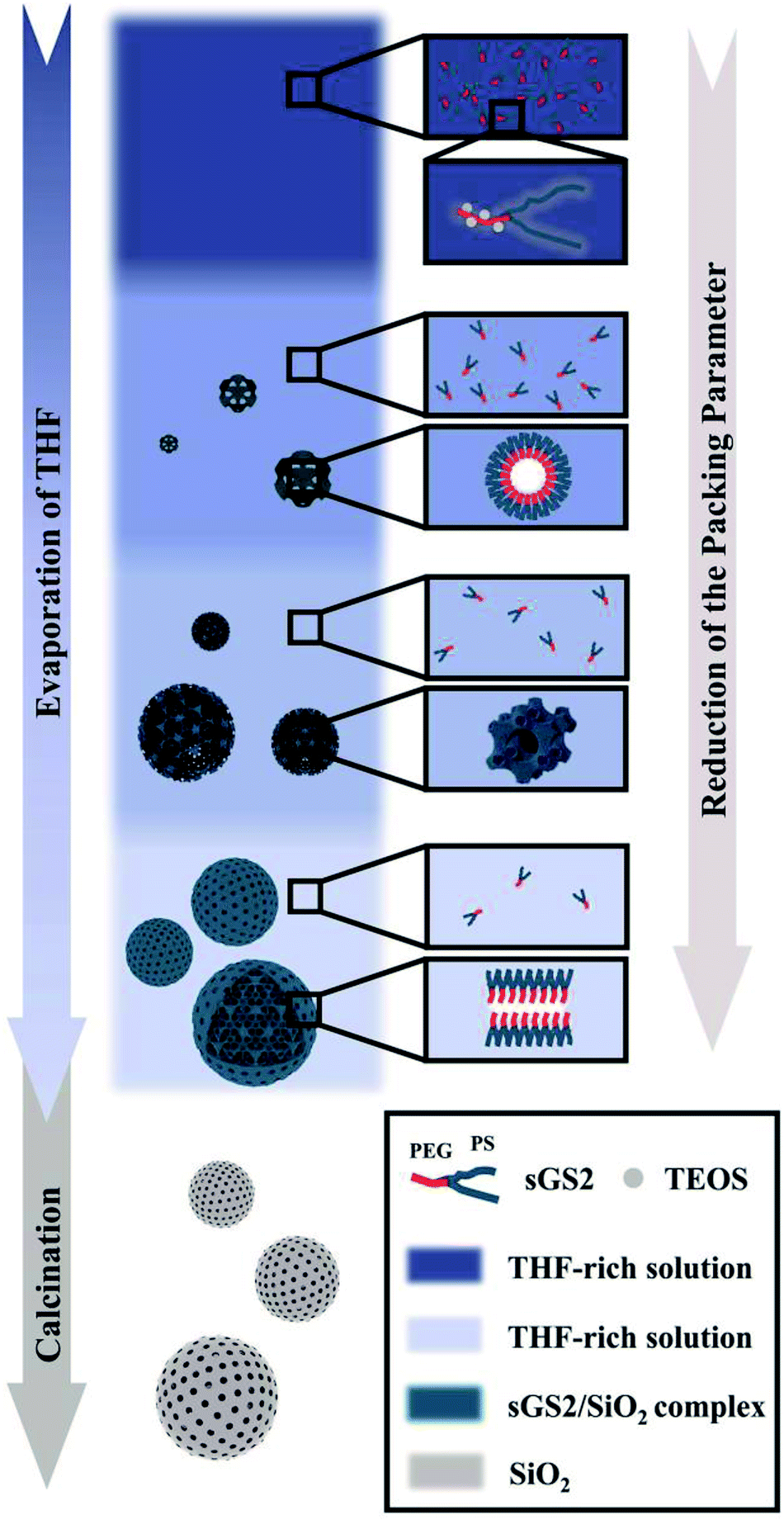 | ||
| Scheme 1 Synthesis of silica cubosomes. | ||
Experimental
Materials
Diethanolamine (99%, Sinopharm Chemical Reagent Co., Ltd), p-toluenesulfonyl chloride (99%, J&K), triethylamine (99.5%, Macklin), dimethyl pyridine, (99%, Transoole Reagent Co., Ltd), tetraethyl orthosilicate (TEOS, 98%, J&K), 2-bromoisobutyryl bromide (98%, TCI), 1,1,4,7,7-pentamethyldiethylenetriamine (PMEDTA, 98%, TCI), copper bromide (CuBr, 97%, Sinopharm Chemical Reagent Co., Ltd), stannous octoate (Sn(Oct)2, 95%, Adamas-beta), diethyl ether (anhydrous) (99%, Sinopharm Chemical Reagent Co. Ltd), hydrochloric acid (HCl, 36%, Sinopharm Chemical Reagent Co., Ltd), methanol (99%, Sinopharm Chemical Reagent Co., Ltd), petroleum ether (60–90, Sinopharm Chemical Reagent Co., Ltd), N,N-dimethylformamide (DMF, 99.5%, General-Reagent), dichloromethane (99.5%, General-Reagent), tetrahydrofuran (THF, 99%, Sinopharm Chemical Reagent Co., Ltd), ε-caprolactone (99%, Adamas-beta), methylene blue trihydrate (82%, General-Reagent), aminopropyltrimethoxysilane (APS, 96%, TCI) and deionized water (Milli-Q, 18.2 MΩ cm) were used without further purification. The polymerization inhibitor of styrene (99%, Sinopharm Chemical Reagent Co., Ltd) was removed by 20% sodium hydroxide solutions, dried with anhydrous magnesium sulfate, and finally distilled under reduced pressure before use. Methyl poly(ethylene glycol) with a hydroxyl terminal group (PEG–OH 5000) at one end was purchased from Aldrich (Mn, GPC = 5.17 kg mol−1, PDI = 1.12, labelled as PEG). The DNA sodium salt from herring testes was purchased from Sigma, USA, listed as Type XIV and processed by the methods reported in our previous study.30Synthesis of PEG-OTs
Poly(ethylene glycol) (20 g), which was previously dried in vacuo at 80 °C for 4 h, was dissolved in methylene chloride (55 mL). Then, p-toluenesulfonyl chloride (4 g) and triethylamine (20 mL) were added to this solution, and the mixture was stirred overnight under a nitrogen atmosphere. The reaction mixture was concentrated to about 30% of its original volume and then stirred for another 2 h at room temperature. The polymer was precipitated from this solution by the dropwise addition of ether (300 mL) under rapid stirring. Stirring was continued for another 15 min, and the reaction mixture was placed in an ice bath. The precipitate was filtered, washed with ether, and dried in vacuo to obtain PEG-OTs.Synthesis of PEG–N–OH2
PEG-OTs (5 g), triethylamine (6.95 mL) and diethanolamine (4 mL) in DMF (40 mL) were heated at 80 °C for 12 h. After cooling down the abovementioned solution to room temperature, the product was precipitated by the dropwise addition of ether to the solution. The precipitated PEG–N–OH2 was filtered, washed, and dried in vacuo.Synthesis of PEG–N–Br2
PEG–N–OH2 (4 g) was dissolved in dry tetrahydrofuran (20 mL). Then, 4 mL of triethylamine was added to the abovementioned solution. After the solution was stirred for 30 min while being cooled down in an ice-water bath, 2-bromoisobutyryl bromide (0.99 mL) was added to the solution over 1 h under N2 protection. Finally, the solution was stirred at room temperature for 12 h and then filtered to obtain a homogeneous solution. Subsequently, 500 mL of cold ether was added to the abovementioned solution to obtain a white precipitate. The precipitate was filtered and washed several times with cold ether. After the precipitate was dried under vacuum, the macroinitiator PEG–N–Br2 was obtained.Synthesis of PEG-PS2 by atom transfer radical polymerization (ATRP)
The PEG-PS2 star polymers were synthesized using the well-established ATRP of styrene at 110 °C with PEG–N–Br2 as the macroinitiator and CuBr/PMDETA as the catalyst. Typically, PEG–N–Br2 (1.00 g) was dissolved in styrene (10.00 g) in a 100 mL Schlenk flask, and then, PMDETA (0.17 mL) and CuBr (0.12 g) were added to the abovementioned solution. The reaction system was fully degassed via more than three freeze–pump–thaw cycles and sealed under vacuum. The bottle was then placed in an oil bath at 110 °C to allow the polymerization to occur. After the polymerization was terminated by exposing the reaction mixture to air, the catalyst was removed by filtration through an Al2O3 column using methylene chloride as the eluent. Cold methanol (500 mL) was poured into the clear filtrate to precipitate the PEG-PS2 (sGS2) star polymer, which was obtained by filtration and dried under vacuum at room temperature.Synthesis of macroporous silica
The macroporous silica was prepared via solvent-evaporation-induced aggregation assembly, and the miktoarm star polymer was used as the template. Then, 0.10 g of sGS2 was added to 7.00 g of THF with 5.00 g of HCL (2 M) for the hydrolysis of TEOS. After the mixture was stirred for approximately 2 h at 50 °C, 0.70 g of TEOS was finally added to the solution, and then, the mixture was allowed to stir for an additional 2 h. The solvent was then allowed to completely evaporate at 50 °C. The as-made samples were calcined at 550 °C in air for 6 h to remove the template.Surface amination of the silica cubosomes
Typically, 0.125 mg silica cubosomes were added to 12.5 mL of 10% (vol%) aminopropyltrimethoxysilane in toluene. The solution was reacted at 80 °C for 12 h. The aminated silica cubosomes were then centrifuged and washed with water and ethanol.Adsorption experiment
Herein, 20 mg silica cubosomes were added to 20 mL of a 10 mg L−1 methylene blue solution. Alternatively, 20 mg aminated silica cubosomes were added to 20 mL of a 20 mg L−1 DNA solution. After a certain period of time, 3 mL of the solution was taken out, and the adsorbent was centrifuged. The supernatant was analyzed by UV-visible absorption spectroscopy.Characterizations
The nuclear magnetic resonance (NMR) spectra were obtained via the Bruker Avance 600 MHz NMR spectrometer using tetramethylsilane (TMS) as the internal reference. The polymers were dissolved in deuterated chloroform (CDCl3). The molecular weights and molecular weight distributions of the polymers were determined via the HLC-8320GPC (TOSOH Corp.) gel permeation chromatography (GPC) apparatus, and the measurements were conducted using DMF as the eluent at 40 °C at the elution rate of 0.6 mL min−1. The apparent molecular weights were determined via the Tskgel AWM-H (6.0 ID × 150 mm) columns using linear polystyrene standards. The SAXS experiments were conducted by beamline BL16B1 at the Shanghai Synchrotron Radiation Facility. The microscopic features of the samples were observed using SEM, which was performed using JEOL JSM-7401F, JSM-7800F Prime and JSM-7900F; the samples were observed with a landing energy of 1 kV without any metal coating. TEM observations were performed using the JEOL JEM-2100 microscope equipped with a LaB6 gun operated at 200 kV (Cs = 1.0 mm, point resolution 2.3 Å). Images were obtained using the TENGRA CCD camera (with a resolution of 2304 × 2304 pixels, a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 fibre-optical taper and an effective pixel size of 8 μm2). The zeta potential was obtained using Malvern Zetasizer Nano ZS90. UV-Vis spectra were obtained by Persee TU-1810. Finally, the nitrogen adsorption–desorption experiment was performed using Quantachrome Instruments Autosorb iQ.
:1 fibre-optical taper and an effective pixel size of 8 μm2). The zeta potential was obtained using Malvern Zetasizer Nano ZS90. UV-Vis spectra were obtained by Persee TU-1810. Finally, the nitrogen adsorption–desorption experiment was performed using Quantachrome Instruments Autosorb iQ.
Results and discussion
Properties of the miktoarm star polymer (sGS2)
We simulated the structure of the phospholipid molecules to synthesize a star polymer with one hydrophilic block and two hydrophobic blocks (Fig. S1†). The amphiphilic star polymer can cause microphase separation due to the incompatibility between two blocks in the solution; therefore, it undergoes self-assembly to form a variety of complex structures.The Mn of sGS2 star polymers is 51.40 kg mol−1 with a volume fraction of 10.19% for the PEG segment (see Table 1). The Flory–Huggins interaction parameter for the two blocks has been calculated as 139.02 by
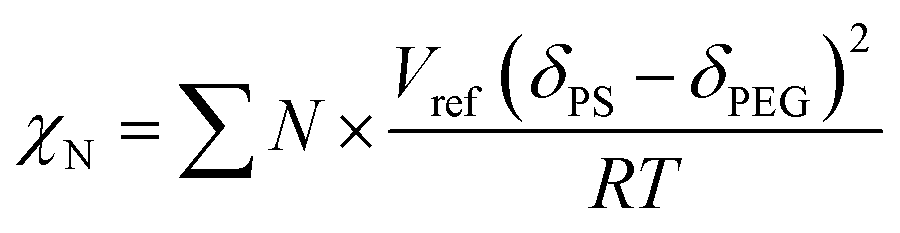
| Polymers | Mn(NMR) (kg mol−1) | Mn(GPC) (kg mol−1) | PDI | VPEG % | VPS % |
|---|---|---|---|---|---|
| sGS2 | 51.40 | 40.00 | 1.84 | 10.19 | 89.81 |
The nuclear magnetic resonance (NMR) spectra of the polymers are shown in Fig. 1. The polymers were dissolved in deuterated chloroform. The resonance signals of the methoxy protons and PEG backbone protons appear at 3.40 and 3.80–3.50 ppm, respectively. The resonance signals of the PS backbone protons and benzene ring protons appear at 1.98–1.12 and 7.23–6.26 ppm, respectively, which confirm the presence of PS. Moreover, the a/l signal ratio is 3:2, which proves that the star polymers have one PEG backbone and two PS backbones.
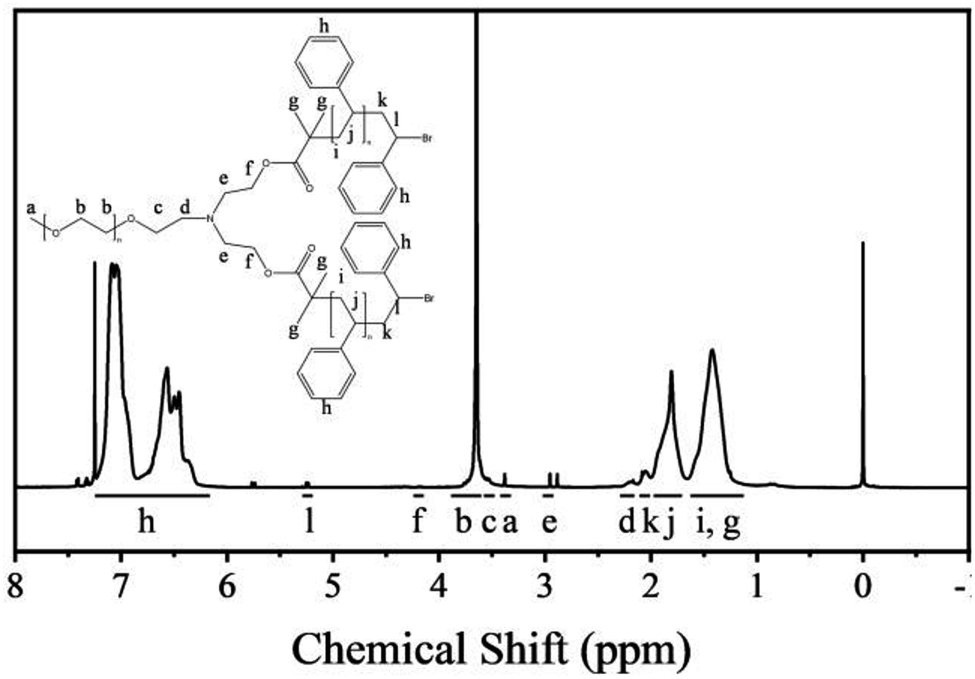 | ||
| Fig. 1 1H NMR spectra of the polymers. | ||
The Mn(NMR) is calculated by the degree of polymerization (DP).31,32 The ratio of the peak areas between 3.80 and 3.50 ppm and 7.23 and 6.26 ppm is 4.77. Therefore, the total DP for the PS block is 446. This can be used as a basis for estimating the Mn(NMR) (that is, Mn(NMR) = MPEG + DP × Mmonomer) of star polymers, and the Mn(NMR) has been estimated to be 51.40 kg mol−1.
Characterization of the cubosome structures
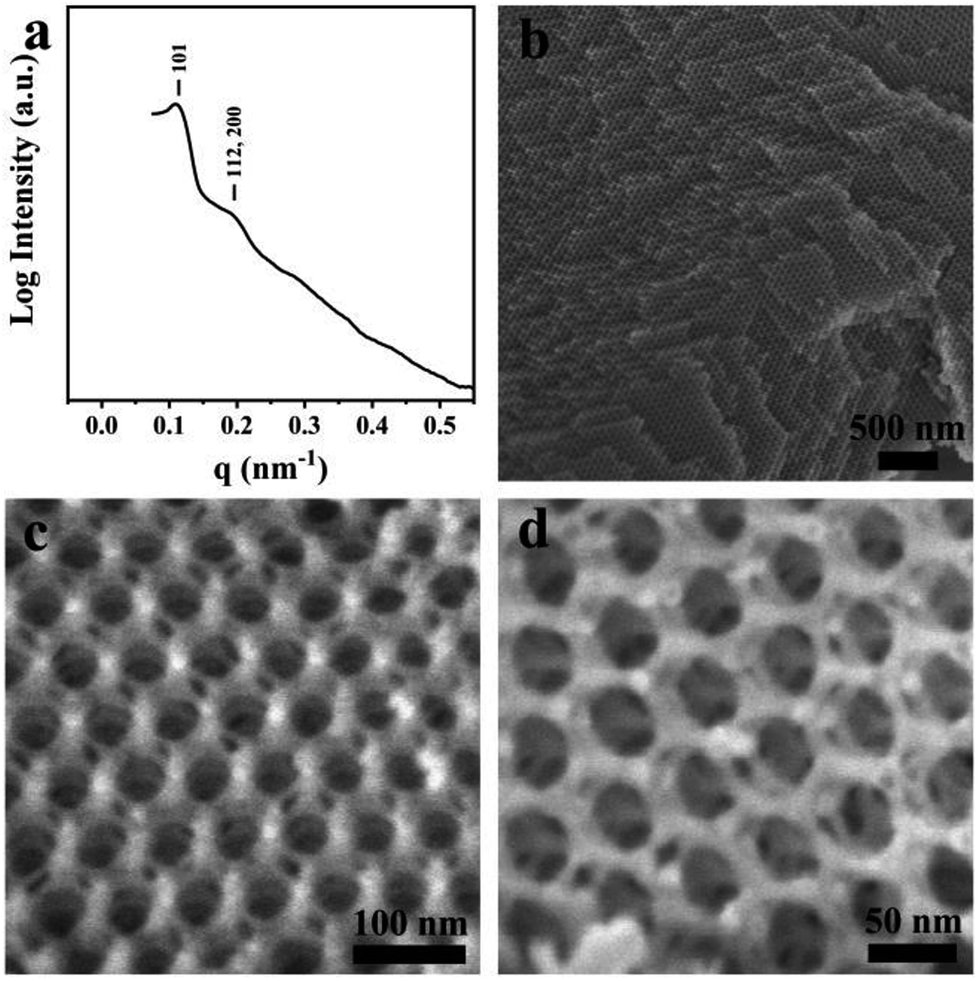
Macroporous silica was synthesized by evaporation-induced self-assembly with sGS2 as the templating agent. As the evaporation process proceeds, the PEG and PS blocks experience microphase separation and thus form different structures in the solution. Moreover, TEOS gradually hydrolyzes and bonds with PEG through hydrogen bonding interactions. After the completion of evaporation, the organic templating agents were removed by calcination to obtain silica with an ordered structure.Fig. 2a shows the low-magnification scanning electron microscopy (SEM) image of the sample, presenting a spherical morphology with 2–15 μm diameter. The surfaces of these spherical particles show ordered pores (Fig. 2b) with hexagonal perforation and ∼70 nm diameter, which are formed by the change in structure from bicontinuous to lamellar (Scheme 1). After crushing the particles into pieces, the inner structure can be revealed. As shown in Fig. 2c, the cross sections of the particles show that the silica frameworks contain smooth surfaces with tetrahedral arrangements of the nodes, which can be identified as a diamond structure. The structure consists of two sets of diamond networks with an inner diameter of ∼26 nm and a wall thickness of ∼18 nm. The unit cell parameter obtained from the SEM image is a ≈ 250 nm with the original cubic symmetry (space group Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Note that one channel of the diamond networks is connected to the hexagonal array of openings on the surface of the spheres (Fig. 2d). The TEM images (Fig. 2e and S3†) of the thin section of the sample after slicing show ordered hollow network structures with the contrast of the double diamond structure. Note that the structure transforms into a layered structure approaching the surface. However, due to the extremely large unit cell parameter, it is very difficult to measure the small angle X-ray scattering profile of this sample because the first reflection exceeds the limit of the instrument (Fig. 2f).
m). Note that one channel of the diamond networks is connected to the hexagonal array of openings on the surface of the spheres (Fig. 2d). The TEM images (Fig. 2e and S3†) of the thin section of the sample after slicing show ordered hollow network structures with the contrast of the double diamond structure. Note that the structure transforms into a layered structure approaching the surface. However, due to the extremely large unit cell parameter, it is very difficult to measure the small angle X-ray scattering profile of this sample because the first reflection exceeds the limit of the instrument (Fig. 2f).
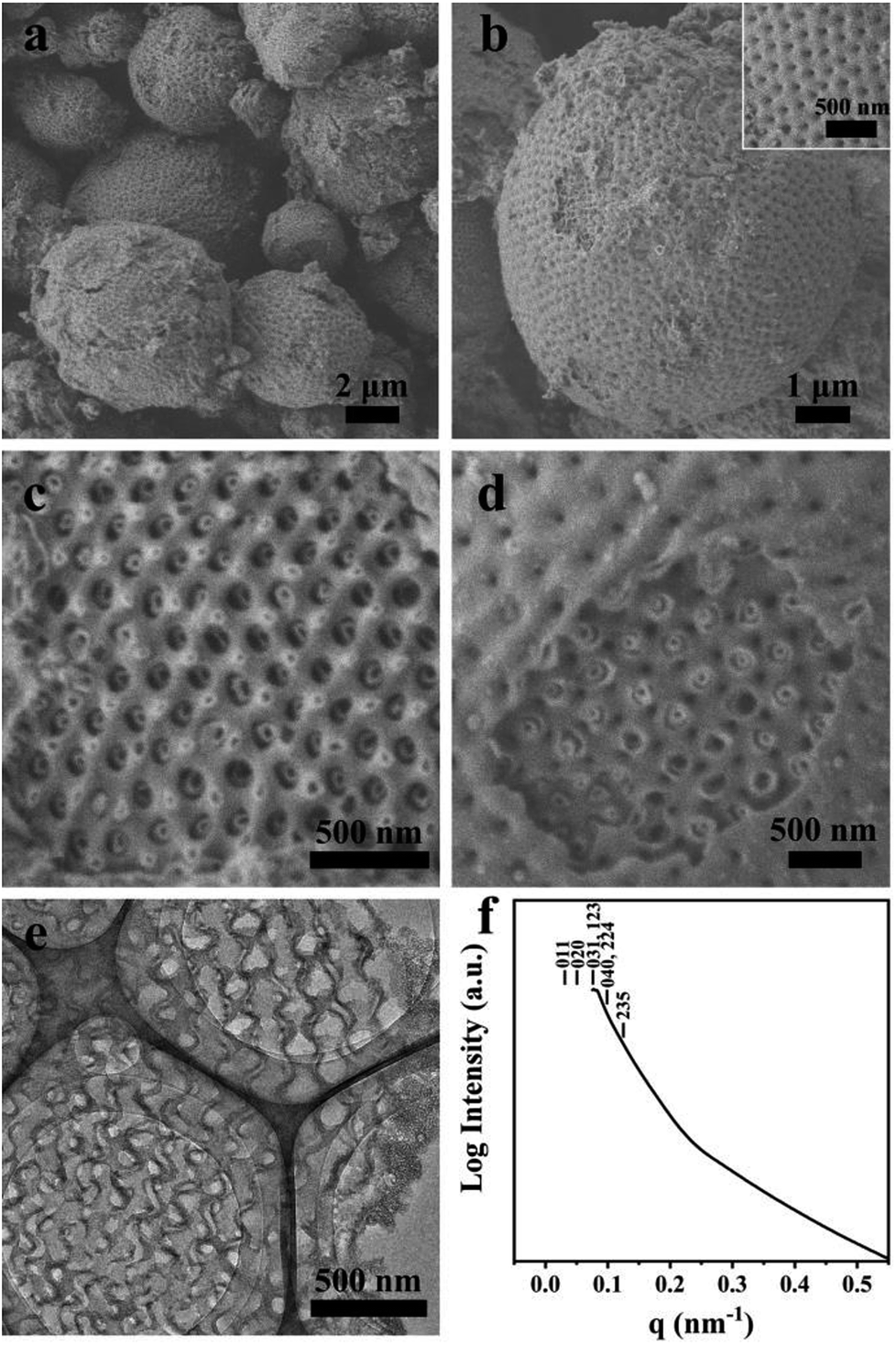 | ||
| Fig. 2 SEM (a–d), TEM (e) images and SAXS pattern (f) of the synthesized sample. The mass ratio of sGS2:THF:HCl (2 M):TEOS is 1:70:50:7. | ||
The nitrogen adsorption–desorption isotherm of the silica cubosomes exhibits a complex behaviour due to the hierarchical porous structure (Fig. S4a†). It shows a type II isotherm with an H4 type hysteresis loop, and a sharp increase in the relative pressure range of 0.90–0.99 indicates the presence of macropores. The surface area is calculated as 693 m2 g−1 by the NLDFT method. The relatively high surface area is mainly contributed by the mesopores. The pore size distribution obtained from the NLDFT method is broad (Fig. S4b†). This is mainly due to the existence of multi-level pores, whereas the large macropores cannot be accurately predicted.
The surface potentials of the as-made sample and the calcined sample in the aqueous medium are −18.87 mV and −11.18 mV, respectively. The as-made sample can stably disperse in THF for a long time and in water for 12 hours (Fig. S5†). However, the calcined sample in the aqueous medium precipitates within 3 hours. This is due to the fact that the as-made sample consists of silica coated with a large number of stretched PS blocks (the PEG block is embedded inside the silica). Therefore, it can stably disperse in THF; however, it cannot stably disperse in an aqueous medium for a long time. The calcined sample is completely composed of silica, and the Si–OH groups have been removed during the calcination step; therefore, the sample cannot form a stable dispersion in an aqueous medium. However, this dispersity can be improved by further surface modification.
Moreover, the adsorption of silica cubosomes on methylene blue and DNA was explored (Fig. S7†). The maximum adsorption ratio for methylene blue was 79.0% for 2 hours. Since the silicon hydroxy group on the surface of the silica has a negative charge, it has no adsorption effect on the negatively charged DNA molecules. By aminating the surface of silica cubosomes, the silicon hydroxy group can adsorb DNA molecules in a neutral aqueous medium. The relationship between the adsorption rate and time was calculated by measuring the absorbance change at 258 nm, which reached a maximum of 95.2% at 90 min. The adsorption of small molecules on porous materials is mainly related to the surface area of these materials. As the surface area of silica cubosomes is not advantageous as compared to that of mesoporous materials, their adsorption on methylene blue is limited. However, the large pore size and accessibility of the porous structure play an important role for the diffusion and adsorption of macromolecules into the material.
Formation mechanism of cubosomes
To further understand the formation mechanism of cubosomes, a series of samples with an ordered structure have been synthesized by changing the amounts of THF, HCl and the template. The three-component synthesis-field diagram based on the mass fraction of THF, HCl (2 M) and sGS2 × 100 (the mass of sGS2 in the diagram is 100 times that of the actual quantity) is shown in Fig. 3. The three-component synthesis-field diagram drawn without the 100 times relationship is shown in Fig. S6.† Inverse 2D hexagonal, bulk double diamond, cubosomes and disordered structures have been obtained.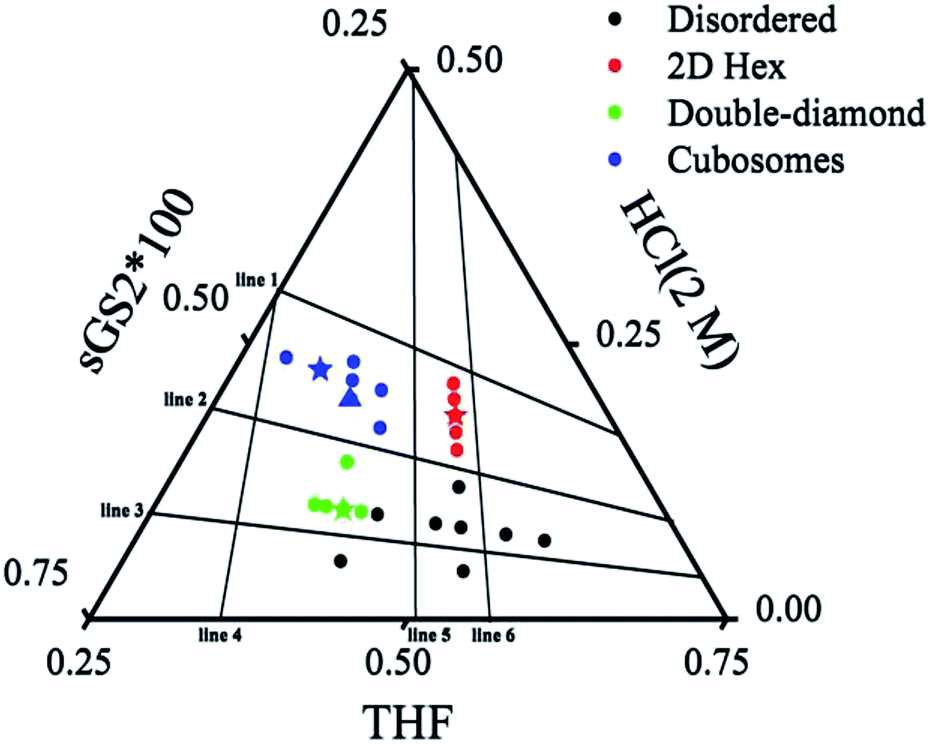 | ||
| Fig. 3 Three-component synthesis-field diagram with structures derived from various compositions (in mass fractions) of the sGS2 polymer. The mass of sGS2 in the diagram is 100 times that of the actual quantity. The blue, red, green stars and blue triangle are the samples shown in Fig. 2, 4, 5, and 6, respectively. | ||
Fig. 4 presents the SEM images of the calcined sample with an inverse 2D hexagonal synthesized with a mass ratio of sGS2:THF:HCl (2 M) = 1:120:50. The sample consisted of stacked tubes with an average diameter of ∼20 nm and a wall thickness of ∼5 nm. The length ranges from several to tens of microns.
 | ||
| Fig. 4 SEM (a and b) images of the inverse 2D hexagonal structure. | ||
The bulk double diamond (Fig. 5) can be synthesized with a mass ratio of sGS2:THF:HCl (2 M) = 1:80:20. The structure is similar to the shifted double-diamond structure reported in our previous study.29 Moreover, two reflections can be observed in the SAXS pattern, which can be indexed to 101 and 112 (200) reflections with the unit cell parameters of a ≈ 54 nm and c ≈ 77 nm based on the tetragonal lattice. From the SEM images, two sets of silica frameworks can be observed, which show a tetrahedral connection at the nodes and can be considered as a shifted double diamond structure.
 | ||
| Fig. 5 SAXS (a) pattern and SEM (b–d) images of the double-diamond structure. | ||
To illustrate the formation of different structures, several lines originating from zero points divided the diagram into several regions, which indicated different mass fractions of THF, HCl (2 M) and sGS2 × 100, respectively. The three lines (line 1–3) derived from the point of THF indicate that with a change in the mass fraction of THF, mHCl(2 M)/msGS2×100 remains fixed. Among them, line 2 separates hexagonal and cubosomes from double-diamond in two areas. In addition, three lines (line 4–6) derived from the point of HCl (2 M) indicate that mTHF/msGS2×100 remains fixed as the mass fraction of HCl (2 M) changes. Cubosomes and bulk double-diamond are formed in the area between line 4 and line 5. As the mass fraction of HCl (2 M) increases, the structure of the resulting silica is transformed from bulk double-diamond to double-diamond structure with a perforated layered coverage and then is completely converted to cubosomes.
Based on the abovementioned results, the structural change from a bulk double-diamond structure to the cubosomes can be explained by the decrease in the THF content. Because the star polymer sGS2 has two hydrophobic tails that are similar to those of phospholipid molecules, it is easier to form bicontinuous structures. At the beginning of the reaction, the corporative self-assembly of the template molecules with an inorganic source leads to the formation of a nucleus with a double-diamond structure. Upon solvent evaporation, the structure gradually grows with the self organization of the template and the hydrolysis and condensation of TEOS. Due to the faster evaporation of THF, the packing parameters are reduced, leading to a decrease in the hydrophobic volume fraction. Therefore, the packing parameters are decreased, and the structure is changed from an inverted bicontinuous double-diamond to a lamellar structure. The double-diamond core is covered by layers of lamellar surface, and the cubosomes can be obtained. This process can be demonstrated by cubosomes with multiple layers of lamellar on the surface (Fig. 6). Moreover, the water content for the synthesis of cubosomes is relatively high; this leads to significantly enlarged unit cell parameters due to the expanded channels in the diamond framework.29 When the water content in the system is very high, the polymer will precipitate out of the solution due to the large hydrophobic tails of the star polymer. Therefore, it is difficult to form a normal micelle structure (oil-in-water) in our system.
 | ||
| Fig. 6 An SEM image of the cubosomes with multiple layer lamellar on the surface. The sample was synthesized with a mass ratio of sGS2:THF:HCl (2 M):TEOS = 1:80:45:7. | ||
Conclusions
We have showed the synthesis of silica cubosomes using an amphiphilic star polymer as a template, TEOS as a silicon source, and a volatile self-assembly method for preparing microporous silica in a THF and water system. Compared with mesoporous materials, our materials have larger pore diameters, which facilitate the diffusion of macromolecules inside the materials, and have high mass transfer efficiency. Due to the inorganic skeleton, the materials show enhanced stability. We hope that our strategy will provide a deeper understanding of the block copolymer systems and widen the synthesis and applications of new porous materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China, (21533002, 21471099, 21571128), the National Key R&D Program of China (2016YFC0205900), the National Excellent Doctoral Dissertation of PR China (201454), and the Shanghai Rising-Star Program (17QA1401700).Notes and references
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- Z. Lin, S. Liu, W. Mao, H. Tian, N. Wang, N. Zhang, F. Tian, L. Han, X. Feng and Y. Mai, Angew. Chem., Int. Ed., 2017, 56, 7135–7140 CrossRef CAS PubMed.
- M. G. Jeong and K. T. Kim, Macromolecules, 2016, 50, 223–234 CrossRef.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed.
- Q. Xiao, Z. Wang, D. Williams, P. Leowanawat, M. Peterca, S. E. Sherman, S. Zhang, D. A. Hammer, P. A. Heiney, S. R. King, D. M. Markovitz, S. André, H.-J. Gabius, M. L. Klein and V. Percec, ACS Cent. Sci., 2016, 2, 943–953 CrossRef CAS PubMed.
- C. Park, Y. La, T. H. An, H. Y. Jeong, S. Kang, S. H. Joo, H. Ahn, T. J. Shin and K. T. Kim, Nat. Commun., 2015, 6, 6392 CrossRef PubMed.
- S. D. Aindrilla, M. C. Harsiddhi, R. J. Kisan and J. K. Vilasrao, Curr. Drug Delivery, 2016, 13, 482–493 CrossRef.
- S. Hyde, Z. Blum, T. Landh, S. Lidin, B. Ninham, S. Andersson and K. Larsson, The language of shape: the role of curvature in condensed matter: physics, chemistry and biology, Elsevier, 1996 Search PubMed.
- Y. Deng and M. Mieczkowski, Protoplasma, 1998, 203, 16–25 CrossRef CAS.
- V. Luzzati and P. A. Spegt, Nature, 1967, 215, 701 CrossRef CAS.
- V. Luzzati, A. Tardieu and T. Gulik-Krzywicki, Nature, 1968, 217, 1028 CrossRef CAS PubMed.
- L. Han and S. Che, Adv. Mater., 2018, 30, 1705708 CrossRef PubMed.
- D. J. McClements, Adv. Colloid Interface Sci., 2015, 219, 27–53 CrossRef CAS PubMed.
- E. Elisabetta, D. Markus, N. Claudio and C. Rita, Curr. Pharm. Des., 2016, 22, 5382–5399 CrossRef.
- Z. Karami and M. Hamidi, Drug Discovery Today, 2016, 21, 789–801 CrossRef CAS PubMed.
- J. Mo, G. Milleret and M. Nagaraj, Liq. Cryst. Rev., 2017, 5, 69–85 CrossRef CAS.
- A. Zabara and R. Mezzenga, J. Controlled Release, 2014, 188, 31–43 CrossRef CAS PubMed.
- I. D. M. Azmi, S. M. Moghimi and A. Yaghmur, Ther. Delivery, 2015, 6, 1347–1364 CrossRef CAS PubMed.
- H. Yabu, T. Higuchi and M. Shimomura, Adv. Mater., 2005, 17, 2062–2065 CrossRef CAS.
- T. Higuchi, A. Tajima, H. Yabu and M. Shimomura, Soft Matter, 2008, 4, 1302–1305 RSC.
- H. Yabu, Polym. J., 2012, 45, 261 CrossRef.
- K. H. Ku, J. M. Shin, D. Klinger, S. G. Jang, R. C. Hayward, C. J. Hawker and B. J. Kim, ACS Nano, 2016, 10, 5243–5251 CrossRef CAS PubMed.
- M. H. Shah, S. V. Biradar and A. R. Paradkar, Int. J. Pharm., 2006, 323, 18–26 CrossRef CAS PubMed.
- P. T. Spicer, W. B. Small, M. L. Lynch and J. L. Burns, J. Nanopart. Res., 2002, 4, 297–311 CrossRef CAS.
- A. Chen, Y. Yu, H. Lv, Y. Zhang, T. Xing and Y. Yu, Mater. Lett., 2014, 135, 43–46 CrossRef CAS.
- R. Guillet-Nicolas, J.-L. Bridot, Y. Seo, M.-A. Fortin and F. Kleitz, Adv. Funct. Mater., 2011, 21, 4653–4662 CrossRef CAS.
- R. Longloilert, T. Chaisuwan, A. Luengnaruemitchai and S. Wongkasemjit, J. Sol-Gel Sci. Technol., 2011, 58, 427–435 CrossRef CAS.
- L. Han, D. Xu, Y. Liu, T. Ohsuna, Y. Yao, C. Jiang, Y. Mai, Y. Cao, Y. Duan and S. Che, Chem. Mater., 2014, 26, 7020–7028 CrossRef CAS.
- Y. Cao, K. Kao, C. Mou, L. Han and S. Che, Angew. Chem., Int. Ed., 2016, 55, 2037–2041 CrossRef CAS PubMed.
- C. Lv, C. He and X. Pan, Angew. Chem., Int. Ed., 2018, 57, 9430–9433 CrossRef CAS PubMed.
- H. Li, D. Yang, Y. Gao, H. Li and J. Xu, RSC Adv., 2015, 5, 96377–96386 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09130g |
| This journal is © The Royal Society of Chemistry 2019 |