
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-culture of the fungus Fusarium tricinctum with Streptomyces lividans induces production of cryptic naphthoquinone dimers†
Mariam Moussaa,
Weaam Ebrahimab,
Michele Bonusc,
Holger Gohlke cd,
Attila Mándie,
Tibor Kurtáne,
Rudolf Hartmannf,
Rainer Kalscheuera,
Wenhan Ling,
Zhen Liu*a and
Peter Proksch*a
cd,
Attila Mándie,
Tibor Kurtáne,
Rudolf Hartmannf,
Rainer Kalscheuera,
Wenhan Ling,
Zhen Liu*a and
Peter Proksch*a
aInstitute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine-Universität Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany. E-mail: zhenfeizi0@sina.com; proksch@uni-duesseldorf.de
bDepartment of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
cInstitute of Pharmaceutical and Medicinal Chemistry, Heinrich-Heine-Universität Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany
dJohn von Neumann Institute for Computing (NIC), Jülich Supercomputing Centre (JSC), Institute for Complex Systems – Structural Biochemistry (ICS-6), Forschungszentrum Jülich GmbH, Wilhelm-Johnen-Straße, 52425 Jülich, Germany
eDepartment of Organic Chemistry, University of Debrecen, Egyetem tér 1, Debrecen 4032, Hungary
fInstitute of Complex Systems – Structural Biochemistry, Forschungszentrum Jülich GmbH, Wilhelm-Johnen-Straße, 52428 Jülich, Germany
gState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, China
First published on 11th January 2019
Abstract
Co-cultivation of the endophytic fungus Fusarium tricinctum with Streptomyces lividans on solid rice medium led to the production of four new naphthoquinone dimers, fusatricinones A–D (1–4), and a new lateropyrone derivative, dihydrolateropyrone (5), that were not detected in axenic fungal controls. In addition, four known cryptic compounds, zearalenone (7), (−)-citreoisocoumarin (8), macrocarpon C (9) and 7-hydroxy-2-(2-hydroxypropyl)-5-methylchromone (10), that were likewise undetectable in extracts from fungal controls, were obtained from the co-culture extracts. The known antibiotically active compound lateropyrone (6), the depsipeptides enniatins B (11), B1 (12) and A1 (13), and the lipopeptide fusaristatin A (14), that were present in axenic fungal controls and in co-culture extracts, were upregulated in the latter. The structures of the new compounds were elucidated by 1D and 2D NMR spectra as well as by HRESIMS data. The relative and absolute configuration of dihydrolateropyrone (5) was elucidated by TDDFT-ECD calculations.
Introduction
Cultivating microorganisms under axenic laboratory conditions fails to resemble the natural environment where these microbes are subjected to intense microbial competition. Therefore naturally occurring stimulants and signal molecules that are involved in chemical communication and antagonism between different microbes are absent, which may result in silencing biosynthetic gene clusters and in simplified metabolite patterns.1,2 The endophytic fungus Fusarium tricinctum has already been well investigated with regard to its natural products.3–5 Applying the OSMAC (One Strain MAny Compounds) approach on cultures of this fungus resulted in the accumulation of new bioactive secondary metabolites such as fusarielin congeners3 and in an enhanced production of enniatin derivatives.4 Enniatins are known for their anti-cancer, anti-HIV as well as for their antibacterial activities.5–7 Enniatins A–C (named fusafungin) were even once available on the drug-market as a local antibiotic for the treatment of respiratory infections. However, due to undesired side effects this approval has meanwhile been withdrawn.8Previous co-cultivation experiments with F. tricinctum focused on Bacillus subtilis as a prokaryotic antagonist. Co-culture of both microbes on solid rice medium resulted in a dramatically enhanced (up to 80-fold) accumulation of several fungal metabolites including enniatins, the polyketide lateropyrone and the lipopeptide fusaristatin A.5 Whereas all of the latter compounds were also observed in axenic fungal controls (albeit at much lower concentrations), the co-cultures yielded several additional cryptic metabolites such as macrocarpon C, (−)-citreoisocoumarinol, and (−)-citreoisocoumarin.5
Previous experiments also indicated that fungi when co-cultured with different bacteria respond through accumulation of different cryptic metabolites. For example, treatment of the fungus Chaetomium sp. with autoclaved cultures of Pseudomonas aeruginosa yielded new butenolide derivatives,9 whereas co-culture of the same fungus with live cultures of B. subtilis yielded new shikimic acid derivatives.10
Here we have studied the influence of Streptomyces lividans on accumulation of natural products by F. tricinctum. S. lividans is known both as a plant-endophyte11 or as a soil-bacterium.12,13 Co-cultivation of F. tricinctum and S. lividans resulted in the accumulation of several new compounds, the dimeric naphthoquinones (1–4) and dihydrolateropyrone (5), that were not detected in axenic fungal controls (Fig. 1). In addition, several known metabolites such as enniatin derivatives (11–13) showed an enhanced accumulation in the co-cultures. Structure elucidation of the new compounds by one- and two-dimensional NMR, HRMS, ECD and quantum chemical calculations is discussed.
 | ||
| Fig. 1 Structures of compounds 1–10. | ||
Results and discussion
The EtOAc extracts of both axenic fungal cultures and co-cultures resulting from mixed fermentation of F. tricinctum and S. lividans on solid rice medium were analyzed by HPLC. When fermented axenically on solid rice medium containing liquid YM medium, the fungus accumulates a complex pattern of bioactive metabolites consisting of several enniatines (11–13), polyketides (e.g. lateropyrone, 6) and lipopeptides (e.g. fusaristatin A, 14). By comparison of the chromatograms, a dramatic shift in the metabolic profile of the co-cultures compared to the axenic fungal or bacterial controls was obvious. These changes are presented in Table 1 indicating a pronounced (up to 12-fold) upregulation of constitutively present metabolites and induction of several cryptic compounds. Chromatographic workup of the co-culture extracts yielded five new compounds (1–5) that were not detected in either fungal or bacterial axenic controls.| Compound | Fungal control (mg) | Co-culture (mg) | Increase (fold) |
|---|---|---|---|
| a n.d. = not detected.b n.a. = not available.c Isolated but not detected in all crude extracts. | |||
| 1 | n.d.a | 7.67 ± 0.1 | |
| 2 | n.d. | +c | |
| 3 | n.d. | + | |
| 4 | n.d. | + | |
| 5 | n.d. | + | |
| 6 | 1.3 ± n.a.b | 17.0 ± 0.8 | 12.5 |
| 7 | n.d. | + | |
| 8 | n.d. | 12.5 ± 10.4 | |
| 9 | n.d. | + | |
| 10 | n.d. | + | |
| 11 | 250.7 ± 50.2 | 868.9 ± 38.0 | 3.5 |
| 12 | 203.8 ± 31.8 | 459.5 ± 25.3 | 2.3 |
| 13 | 47.6 ± 32.3 | 95.7 ± 8.9 | 2.0 |
| 14 | 175.2 ± 54.0 | 294.8 ± 11.4 | 1.7 |
Compound 1 exhibited distinct UV absorption maxima at λmax 320, 278 and 223 nm. The HRESIMS data of 1 established the molecular formula C31H24O16, indicating twenty degrees of unsaturation. The 1H NMR spectrum of 1 (Table 2) showed fewer signals in comparison to the molecular formula, suggesting the possible dimeric nature of 1. These signals included two singlet aromatic protons at δH 7.17 (H-8/8′), two singlet methyl groups at δH 1.88 (Me-14) and 1.86 (Me-14′), two singlet methylene groups at δH 3.08 (H2-12′) and 3.03 (H2-12), in addition to three methoxy groups at δH 4.10 (Me-15/15′) and 3.68 (Me-16′). In the HMBC spectrum of 1, 3J-HMBC correlations from H-8 to C-1 (δC 179.5), C-6 (δC 139.8) and C-10 (δC 112.5), and from Me-15 to C-2 (δC 159.0) as well as weak 2J-HMBC correlations from H-8 to C-7 (δC 152.8) and C-9 (δC 127.2) were observed. In addition, H-8 showed long range 4J-HMBC correlations to C-2, C-4 (δC 188.3) and C-5 (δC 145.4), while Me-15 exhibited long range HMBC correlations to C-1, C-3 (δC 121.2), C-4 and C-9. Furthermore, when the spectra were recorded in CDCl3, an additional signal at δH 12.04 assigned for OH-5 was detected which showed HMBC correlations to C-5, C-6 and C-10. These findings confirmed the presence of a dimeric substituted naphthoquinone core structure of 1 through a C-3/3′ linkage (Fig. 1). A methoxy and a hydroxy group were located at C-2/2′ and C-5/5′, respectively. Apart from these, the HMBC correlations from Me-14 to C-11 (δC 120.4) and C-12 (δC 43.5), and from H2-12 to C-11, C-13 (δC 169.9) and C-14 together with the HMBC correlations from Me-14′ to C-11′ (δC 120.1) and C-12′ (δC 43.3), from H2-12′ to C-11′, C-13′ (δC 168.5) and C-14′, and from Me-16′ to C-13′, indicated the presence of two side chains from C-14 to C-13 and from C-14′ to C-16′ as shown. Based on the molecular formula and the chemical shifts of 1, these two side chains are connected to the core structure through two acetonide rings between C-6/6′, C-7/7′ and C-11/11′. Thus, the structure of 1, to which the trivial name fusatricinone A is given, was established as shown.
| Position | 1a | 1b | ||
|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
a Measured in CDCl3–CD3OD (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (1H at 600 MHz and 13C at 150 MHz). :1) (1H at 600 MHz and 13C at 150 MHz). |
||||
| 1/1′ | 179.5, C | 179.5, C | ||
| 2/2′ | 159.0, C | 159.0, C | ||
| 3/3′ | 121.2, C | 121.2, C | ||
| 4/4′ | 188.3, C | 188.3, C | ||
| 5/5′ | 145.4, C | 145.4, C | ||
| 6/6′ | 139.8, C | 139.8, C | ||
| 7/7′ | 152.8, C | 152.8, C | ||
| 8/8′ | 102.5, CH | 7.17, s | 102.5, CH | 7.17, s |
| 9/9′ | 127.2, C | 127.2, C | ||
| 10/10′ | 112.5, C | 112.5, C | ||
| 11 | 120.4, C | 120.4, C | ||
| 12 | 43.5, CH2 | 3.03, s | 43.5, CH2 | 3.03, s |
| 13 | 169.9, C | 169.9, C | ||
| 14 | 24.8, CH3 | 1.88, s | 24.8, CH3 | 1.88, s |
| 15 | 61.6, CH3 | 4.10, s | 61.6, CH3 | 4.09, s |
| 11′ | 120.1, C | 120.1, C | ||
| 12′ | 43.3, CH2 | 3.08, s | 43.3, CH2 | 3.08, s |
| 13′ | 168.5, C | 168.5, C | ||
| 14′ | 25.1, CH3 | 1.86, s | 25.1, CH3 | 1.86, s |
| 15′ | 61.6, CH3 | 4.09, s | 61.6, CH3 | 4.09, s |
| 16′ | 52.5, CH3 | 3.68, s | 52.5, CH3 | 3.68, s |
The UV pattern and NMR data of compound 2 (Table 3) resembled those of 1, suggesting their structural similarity. The molecular formula C30H22O16 of 2 was determined by the HRESIMS data, differing from that of 1 by the loss of 14 amu, which was due to the absence of the carboxymethyl group of 1 as shown by comparison of the 1D and 2D NMR spectra of both compounds. In addition, the 1D NMR of 2 showed the same chemical shifts for related nuclei in both molecule halves, indicating that both molecule halves are constitutionally identical. Thus, 2 was identified as a new natural product and was given the trivial name fusatricinone B.
| Position | 2a | 2b | ||
|---|---|---|---|---|
| δC,b type | δH (J in Hz) | δC,b type | δH (J in Hz) | |
| a Measured in CDCl3 (1H at 600 MHz and 13C at 150 MHz).b Data were extracted from the HSQC and HMBC spectra.c n.d. = not detected. | ||||
| 1/1′ | 179.1, C | 179.1, C | ||
| 2/2′ | 158.6, C | 158.6, C | ||
| 3/3′ | n.d.c | n.d.c | ||
| 4/4′ | 188.3, C | 188.3, C | ||
| 5/5′ | 145.5, C | 145.5, C | ||
| 6/6′ | 139.1, C | 139.1, C | ||
| 7/7′ | 152.1, C | 152.1, C | ||
| 8/8′ | 102.0, CH | 7.21, s | 102.0, CH | 7.20, s |
| 9/9′ | 127.1, C | 127.1, C | ||
| 10/10′ | 112.2, C | 112.2, C | ||
| 11/11′ | 119.2, C | 119.2, C | ||
| 12/12′ | 42.9, CH2 | 3.12, s | 42.9, CH2 | 3.10, s |
| 13/13′ | 171.4, C | 171.4, C | ||
| 14/14′ | 24.6, CH3 | 1.91, s | 24.6, CH3 | 1.88, s |
| 15/15′ | 61.2, CH3 | 4.13, s | 61.2, CH3 | 4.12, s |
| OH-5/OH-5′ | 12.06, s | 12.06, s | ||
The molecular formula of compound 3 was determined as C32H26O16 based on the HRESIMS data, differing from that of 1 by an additional 14 amu and implying the presence of an additional methyl group in 3. The UV and NMR (Table 4) data of 3 were similar to those of 2 and likewise exhibited only one set of NMR signals, indicating methylation of both terminal carboxyl groups to form the respective methyl ester functions. This deduction was further confirmed by the HMBC correlation from Me-16/16′ (δH 3.71) to C-13/13′ (δC 167.6). The trivial name fusatricinone C was assigned to the new compound 3.
| Position | 3a | 3b | ||
|---|---|---|---|---|
| δC,b type | δH (J in Hz) | δC,b type | δH (J in Hz) | |
| a Measured in CDCl3 (1H at 600 MHz and 13C at 150 MHz).b Data were extracted from the HSQC and HMBC spectra.c n.d. = not detected. | ||||
| 1/1′ | 179.3, C | 179.3, C | ||
| 2/2′ | 158.7, C | 158.7, C | ||
| 3/3′ | n.d.c | n.d.c | ||
| 4/4′ | n.d.c | n.d.c | ||
| 5/5′ | 145.4, C | 145.4, C | ||
| 6/6′ | 139.3, C | 139.3, C | ||
| 7/7′ | 152.6, C | 152.6, C | ||
| 8/8′ | 102.0, CH | 7.21, s | 102.0, CH | 7.21, s |
| 9/9′ | 127.6, C | 127.6, C | ||
| 10/10′ | 112.1, C | 112.1, C | ||
| 11/11′ | 119.5, C | 119.5, C | ||
| 12/12′ | 43.3, CH2 | 3.07, s | 43.3, CH2 | 3.06, s |
| 13/13′ | 167.6, C | 167.6, C | ||
| 14/14′ | 24.7, CH3 | 1.90, s | 24.7, CH3 | 1.89, s |
| 15/15′ | 61.2, CH3 | 4.13, s | 61.2, CH3 | 4.12, s |
| 16/16′ | 52.0, CH3 | 3.71, s | 52.0, CH3 | 3.71, s |
| OH-5/OH-5′ | 12.05, s | 12.05, s | ||
Compound 4 had the same molecular formula as that of 3 (C32H26O16) as established by HRESIMS. The UV spectrum was similar to those of compounds 1–3, suggesting the presence of the same naphthoquinone core structure. Unlike 3, however, the 1H and 13C NMR spectra of the monomers of 4 (Table 5) exhibited two sets of signals, one of which (from C-1 to C-16) showed the same pattern as that of the monomer in 3. The second monomer of 4 showed signals accounting for the aromatic proton at δH 7.23 (H-8′) and a hydroxy group at δH 11.10 (OH-7′), which are key signals to determine the substitution pattern of the second monomer. The HMBC spectrum of 4 displayed correlations from H-8′ to C-6′ (δC 140.2), C-7′ (δC 144.3), C-10′ (δC 107.6) and C-1′ (δC 178.6), and from OH-7′ to C-6′, C-7′ and C-8′ (δC 111.4). In addition, a ROESY correlation between H-8′ and OH-7′ was observed. These data indicated that cyclisation of one acetonide moiety of 4 included the oxygens at the C-5′ and C-6′ positions. Thus, the structure of 4 was elucidated as shown and the trivial name fusatricinone D was assigned to this compound.
| Position | 4a | 4b | ||
|---|---|---|---|---|
| δC,b type | δH (J in Hz) | δC,b type | δH (J in Hz) | |
| a Measured in DMSO-d6 (1H at 600 MHz and 13C at 150 MHz).b Data were extracted from the HSQC and HMBC spectra.c n.d. = not detected. | ||||
| 1 | 179.0, C | 179.0, C | ||
| 2 | 158.5, C | 158.5, C | ||
| 3 | n.d.c | n.d.c | ||
| 4 | n.d.c | n.d.c | ||
| 5 | 143.8, C | 143.8, C | ||
| 6 | 139.3, C | 139.3, C | ||
| 7 | 152.4, C | 152.4, C | ||
| 8 | 101.1, CH | 7.16, s | 101.1, CH | 7.16, s |
| 9 | n.d.c | n.d.c | ||
| 10 | 111.8, C | 111.8, C | ||
| 11 | 120.0, C | 120.0, C | ||
| 12 | 42.1, CH2 | 3.29, s | 42.1, CH2 | 3.28, s |
| 13 | 167.9, C | 167.9, C | ||
| 14 | 24.2, CH3 | 1.82, s | 24.2, CH3 | 1.81, s |
| 15 | 60.4, CH3 | 3.96, s | 60.4, CH3 | 3.95, s |
| 16 | 52.0, CH3 | 3.57, s | 52.0, CH3 | 3.56, s |
| OH-5 | 11.80, s | 11.79, s | ||
| 1′ | 178.6, C | 178.6, C | ||
| 2′ | 157.8, C | 157.8, C | ||
| 3′ | n.d.c | n.d.c | ||
| 4′ | n.d.c | n.d.c | ||
| 5′ | n.d.c | n.d.c | ||
| 6′ | 140.2, C | 140.2, C | ||
| 7′ | 144.3, C | 144.3, C | ||
| 8′ | 111.4, CH | 7.23, s | 111.4, CH | 7.23, s |
| 9′ | n.d.c | n.d.c | ||
| 10′ | 107.6, C | 107.6, C | ||
| 11′ | 120.0, C | 120.0, C | ||
| 12′ | 41.8, CH2 | 3.24, s | 41.8, CH2 | 3.24, s |
| 13′ | 167.9, C | 167.9, C | ||
| 14′ | 24.6, CH3 | 1.80, s | 24.6, CH3 | 1.80, s |
| 15′ | 60.7, CH3 | 4.01, s | 60.7, CH3 | 4.00, s |
| 16′ | 52.0, CH3 | 3.59, s | 52.0, CH3 | 3.59, s |
| OH-7′ | 11.10, s | 11.10, s | ||
Compounds 1–4 were detected in the original fungal-bacterial co-culture crude EtOAc extract by LC-MS, indicating that they are true natural products and not formed through methylation during chromatographic workup.
The 1H NMR data of dimeric naphthoquinone core structure of 1–3 were comparable to those of synthetic 3,3′-dimethoxy-8,8′-dihydroxy-2,2′-bi-1,4-naphthoquinone, which exhibited signals of methoxy groups at δH 4.15 and signals of hydroxy groups at δH 12.08.14,15
For the isolated dimeric naphthoqinones 1–4, the 3/3′-biaryl linkage between monomers can lead to axial chirality if the rotational energy barrier is sufficiently high.16 DFT quantum chemical calculations of the rotation energy barrier for the biaryl axis were performed to check the possibility of atropisomers or interconversion of Ra and Sa conformers, which yielded ∼22 kcal mol−1 for the inversion (Fig. 2), indicating that the atropisomers interconvert with a half-life of ∼1.7 × 103 s = ∼28 min at 300 K.17,18
 | ||
| Fig. 2 Calculation of the height of the rotation barrier for compounds 1–4. (A) Conformational energy profile obtained for the gas phase at the B3LYP/6-31G(d) level of theory for the rotation about the central biaryl bond in the model compound 8,8′-dihydroxy-3,3′-dimethoxy-2,2′-binaphthyl-1,1′,4,4′-tetrone. Vertical lines separate regions of values for the torsion angle that correspond to distinct conformations according to the Klyne–Prelog notation. The structures of the Ra and Sa atropisomers are assigned to the corresponding minimum-energy conformers in the anticlinal conformations. (B) Visualization of the distinct conformers according to the Klyne–Prelog notation. The height of the black line indicates the relative energy of the depicted conformer. | ||
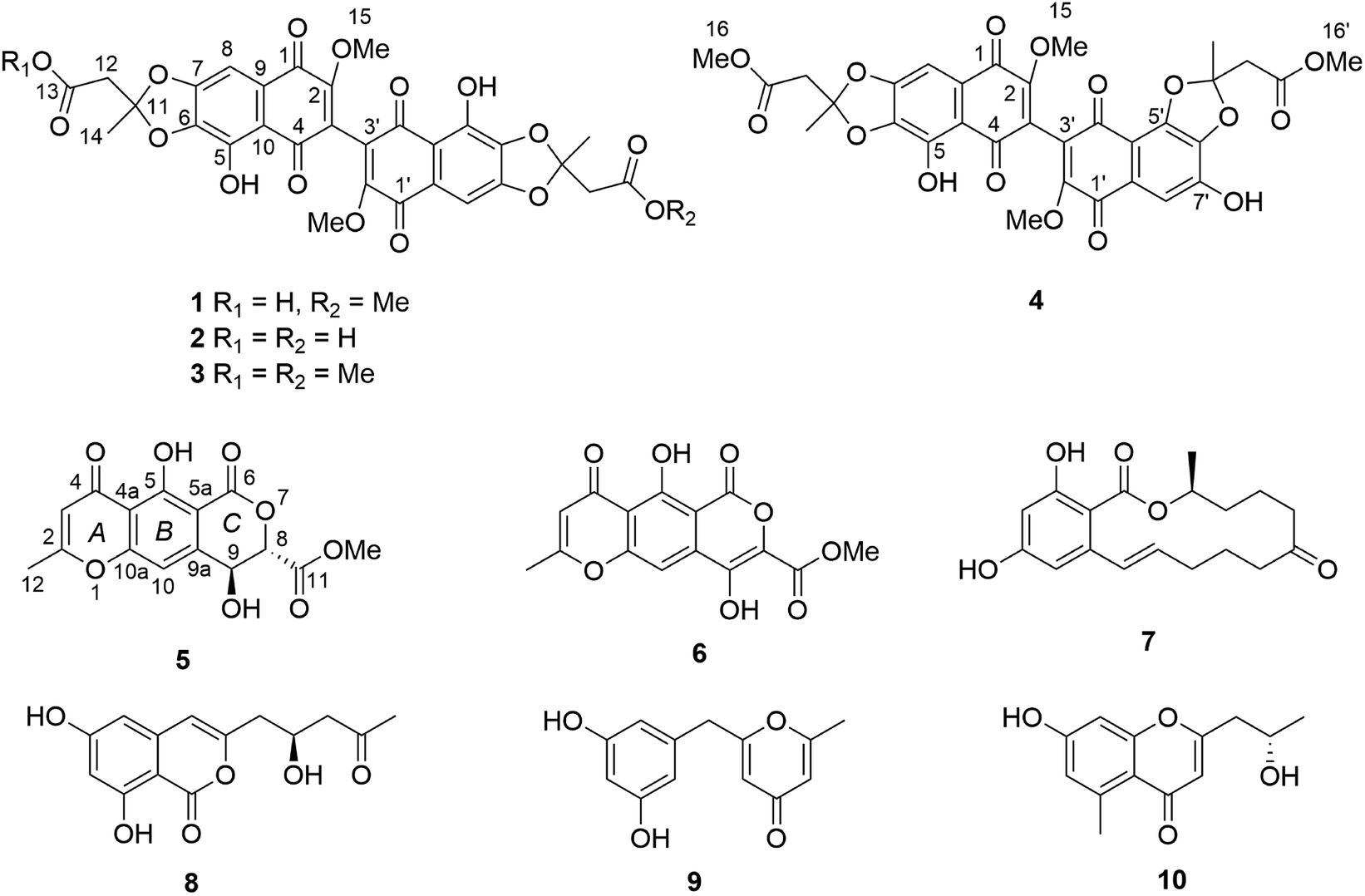
Compounds 1–4 gave baseline ECD spectra and zero specific rotation. Each monomer contains one chiral center (C-11 and C-11′), and there is a configurationally labile biaryl axis, which is expected to allow the interconversion of Ra and Sa conformers in solution. Provided that the monomers have identical absolute configuration for the central chirality elements, namely (11R,11′R) or (11S,11′S) absolute configuration, distinct ECD spectra would be expected for 1–4 as observed for biaryls with central chirality but low rotational energy barrier exemplified by dicerandrol B,19 and versixanthone C.20 Thus the baseline ECD curves suggest stereoisomeric mixtures of 1–4 containing either equimolar amounts of (11R,11′R) and (11S,11′S) stereoisomers or four stereoisomers with (11R,11′R), (11S,11′S), (11S,11′R), (11R,11′S) absolute configuration. If there is a mixture of two quasi-enantiomers [(11R,11′R) and (11S,11′S)], the duplication of NMR signals derives from the slowly interconverting Ra and Sa conformers, which represent rotational diastereomers (Fig. 3A). If there is a mixture of four stereoisomers or two racemates [(11R,11′R), (11S,11′S), (11S,11′R), (11R,11′S)], the duplication can be due to diastereomers that differ in the configuration of the central chirality elements [(11S*,11′S*) versus (11S*,11′R*)], but not due to stereoisomers with enantiomorphic molecule halves [(11S,11′R) versus (11R,11′S)] (Fig. 3B). However, it is more likely that the slowly interconverting Ra and Sa conformers with homomorphic molecule halves are distinguished by the NMR even in this case, and the remote central chirality elements do not result in duplication of NMR signals.
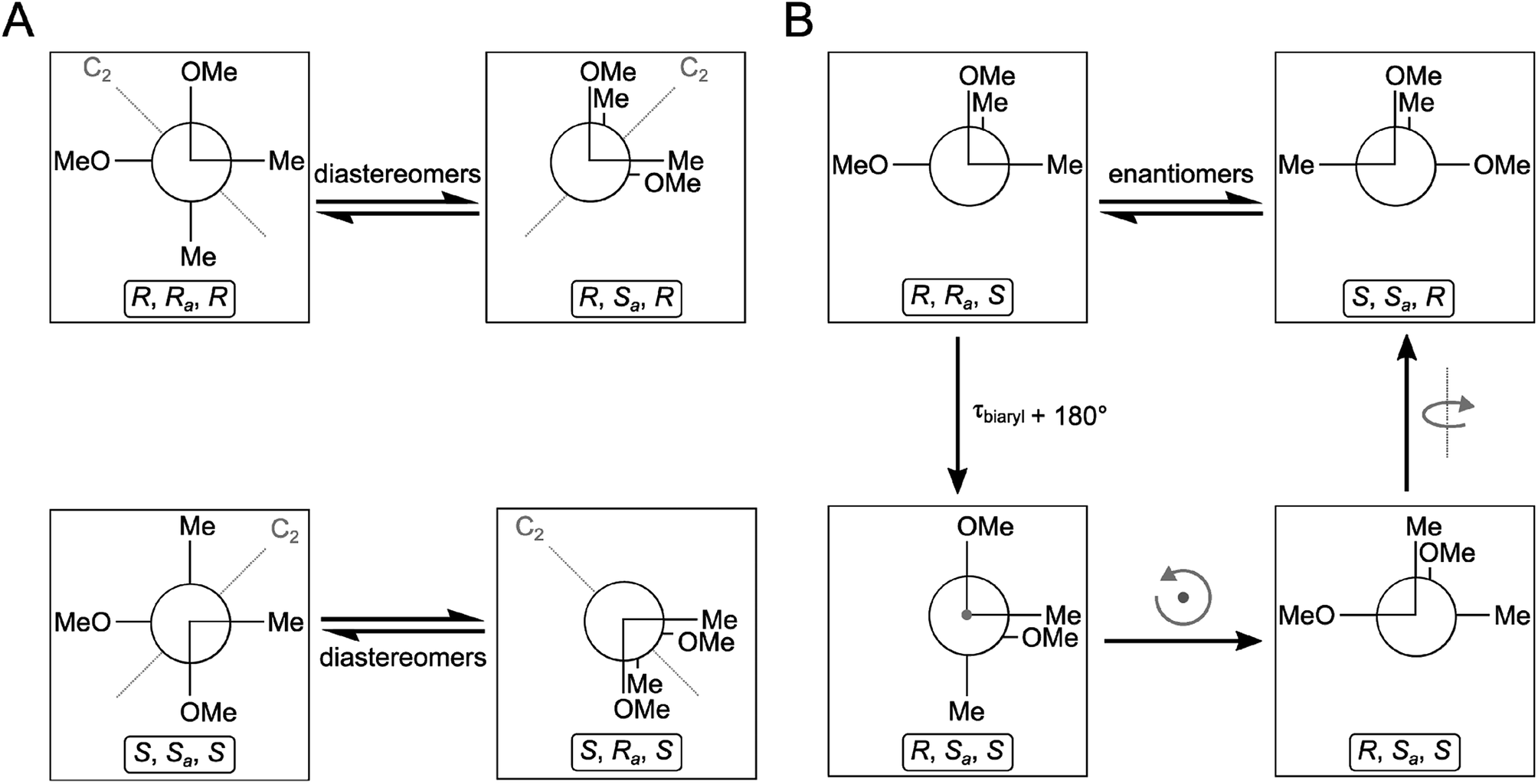 | ||
| Fig. 3 Stereochemical relationship of atropisomers in the case of the symmetric molecules 2 and 3. If both molecule halves are homomorph (A), diastereomeric atropisomers result. If both molecule halves are enantiomorph (B), enantiomeric atropisomers result. “Me” and “OMe” relate to C-14 and 15 as well as C-14′ and 15′ in Fig. 1. The grey arrows in the right panel indicate whole-body rotations; the configuration descriptors relate to the central/axial/central chirality elements in the molecules. | ||
The related biaryl derivative xanthomegnin, which was isolated from several Penicillium and Aspergillus species and occurs naturally as a 1:1 mixture of atropisomers,21 has the same naphthoquinone core structure as compounds 1–4 isolated in this study, but the condensed 1,3-dioxolane ring is replaced by a δ-lactone ring. The biogenetic precursor of xanthomegnin is (R)-semivioxanthin, and xanthomegnin has a distinct ECD spectrum allowing configurational assignment of analogues.22 In the biosynthesis of xanthomegnin, the optically active monomers are formed first and are then dimerized by oxidative coupling.23
Compound 5 showed UV absorption maxima at 350, 263, 243 and 229 nm and had the molecular formula C15H12O8 based on the HRESIMS data. Compound 5 contains two additional protons compared to the co-isolated structurally related known compound lateropyrone (6),24 suggesting hydrogenation of the olefinic double bond of 6 at C-8 and 9, thus yielding 5. This was also evident from the 1H NMR spectrum of 5 (Table 6), which exhibited two aromatic protons at δH 6.36 (s, H-3) and 7.38 (s, H-10), two oxygenated methines at δH 4.92 (br s, H-9) and 6.04 (br s, H-8), an aromatic methyl group at δH 2.51 (s, Me-12) in addition to a methoxy group at δH 3.73 (s, OMe-11). The HMBC correlations from H-3 to C-2 (δC 170.9), C-4 (δC 184.4) and C-4a (δC 113.1), from Me-12 to C-2 and C-3, and from H-10 to C-10a (δC 158.1), C-9a (δC 125.6) and C-4a indicated the presence of the same rings A and B in 5. However, the HMBC correlation from H-8 to C-6 (δC 168.6), C-11 (δC 170.6), C-9a (δC 125.6) and from H-9 to C-11 and C-9a confirmed the disappearance of the double bond of 6 in 5.
| Position | δC,b type | δH (J in Hz) |
|---|---|---|
| a Measured in acetone-d6 (1H at 750 MHz and 13C at 175 MHz).b Data were extracted from the HSQC and HMBC spectra.c n.d. = not detected. | ||
| 2 | 170.9, C | |
| 3 | 109.6, CH | 6.36, s |
| 4 | 184.4, C | |
| 4a | 113.1, C | |
| 5 | n.d.c | |
| 5a | n.d. | |
| 6 | 168.6, CH | |
| 8 | 81.8, CH | 6.04, br s |
| 9 | 70.9, CH | 4.92, br s |
| 9a | 125.6, C | |
| 10 | 103.6, CH | 7.38, s |
| 10a | 158.1, C | |
| 11 | 170.6, C | |
| 12 | 20.3, CH3 | 2.51, s |
| OMe-11 | 52.4, CH3 | 3.73, s |
Based on the small 3JH-8,H-9 coupling constant value, a cis relative configuration of 5 was expected, and hence TDDFT-ECD calculations were performed on the arbitrarily chosen cis-(8R,9S) stereoisomer.25,26 DFT optimization of the 15 initial conformers generated using the Merck Molecular Force Field (MMFF) at various levels of theory (B3LYP/6-31G(d), B97D/TZVP PCM/MeCN and CAM-B3LYP/TZVP PCM/MeCN) resulted in 1, 3 and 5 low-energy DFT conformers (≥1%), respectively. In the lowest-energy CAM-B3LYP/TZVP PCM/MeCN conformer (63.6% population), the C-8 methoxycarbonyl group adopted axial orientation, while the C-9 hydroxy group was axial and formed an intramolecular hydrogen bond to the carbonyl oxygen. Conformers B–D represented geometries with inversed helicity of the α-pyrone ring having equatorial methoxycarbonyl and equatorial hydroxy groups with a total population of 31.4%. Boltzmann-weighted ECD spectra computed for all the sets of conformers of cis-(8R,9S) at various levels (B3LYP/TZVP, BH&HLYP, CAM-B3LYP and PBE0/TZVP) gave a mismatch with the experimental ECD spectrum (Fig. 4). This suggested that the small 3JH-8,H-9 coupling constant may derive from a trans-diequatorial orientation of H-8 and H-9 of the trans relative configuration, which, considering the trans-diaxial orientation of the large substituents, is less common for 1,2-disubstituted benzene-condensed heterocyclic derivatives. However, we have demonstrated recently by ECD and NMR calculation and X-ray analysis that in benzene-condensed chiral heterocycles the large substituents of contiguous chirality centers preferably adopt trans-diaxial orientation both in solution and in the solid-state rendering the protons to trans-diequatorial position.27,28 The same computational ECD protocol was carried out for the arbitrarily chosen trans-(8S,9S) stereoisomer, for which all combinations of the applied levels gave moderate to good agreement with the experimental ECD spectrum. The diequatorial orientation of the methoxycarbonyl and hydroxy groups was found in conformers A and D with a total population of 46%, while the diaxial geometry was found in conformers B–D and G totaling 53% population. Conformers A and B with opposite helicity of the condensed α-pyrone ring had markedly different computed ECD spectra as demonstrated by their rotatory strengths (Fig. 5). Based on the good ECD agreement, (−)-trans-(8S,9S) absolute configuration was determined for dihydrolateropyrone (5).
 | ||
| Fig. 4 Experimental ECD spectrum of 5 in MeCN compared with the Boltzmann-weighted BH&HLYP/TZVP PCM/MeCN//CAM-B3LYP/TZVP PCM/MeCN spectrum of the cis-(8R,9S)-5. | ||
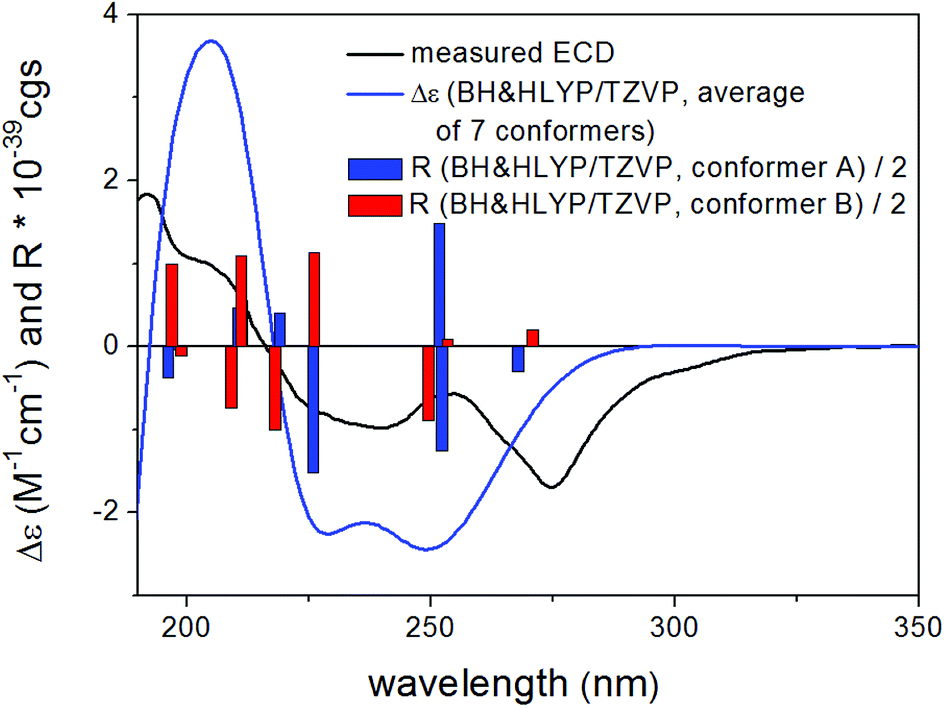 | ||
| Fig. 5 Experimental ECD spectrum of 5 in MeCN compared with the Boltzmann-weighted BH&HLYP/TZVP PCM/MeCN//CAM-B3LYP/TZVP PCM/MeCN spectrum of the trans-(8S,9S)-5. | ||
The five known cryptic compounds that were isolated from the co-culture extracts but were not detected in extracts of the axenic fungal control included lateropyrone (6),24 zearalenone (7),29 (−)-citreoisocoumarin (8),30,31 macrocarpon C (9),5 and 7-hydroxy-2-(2-hydroxypropyl)-5-methylchromone (10).32 In addition, four constitutively present compounds (also detected in axenic fungal controls) including enniatins B, B1 and A1 (11–13)6 and fusaristatin A (14)33 were upregulated in the co-culture extracts.
The new dimeric naphthoquinones 1–4 and the likewise new compound 5 were evaluated for their antibacterial activity against human pathogenic bacterial strains, including Staphylococcus aureus and Pseudomonas aeruginosa but exhibited no antibacterial activity. For compound 5, this is in sharp contrast to the structurally closely related known compound lateropyrone (6), which had previously been noted for its pronounced activity against Gram positive bacteria.5 Apparently, hydrogenation of the double bond at C-8 and 9 leads to a complete loss of antibacterial activity.
The observed upregulation of antibiotically active lateropyrone (6) and enniatins (11–13) during co-cultivation of F. tricinctum and S. lividans (Table 1) may be interpreted as chemical defense of the fungus.5 The most interesting finding from this study is, however, the observation that F. tricinctum responds to the presence of S. lividans in a metabolically different way compared to co-cultures of this fungus with B. subtilis, leading to the accumulation of the new compounds 1–4 that are not detected when the fungus is co-cultured with B. subtilis. This result mirrors data from a similar study with the fungus Chaetomium sp. Co-culture of Chaetomium sp. either with B. subtilis or with P. aeruginosa likewise provoked accumulation of different sets of cryptic fungal compounds.9,10 Future experiments should aim at unravelling the molecular patterns that underlie these specific fungal metabolic responses.
Experimental section
General experimental procedures
For measuring the optical rotations a Perkin-Elmer-241 MC polarimeter was used. ECD spectra were recorded on a J-810 spectropolarimeter. Solvents used for spectroscopic measurements had spectral grade and for other usage solvents were distilled prior to use. For 1D and 2D NMR spectra, Bruker AVANCE DMX 600 or Bruker ARX 700 NMR spectrometers were used. Mass spectra were recorded with a Finnigan LCQ Deca mass spectrometer, while HRMSESI spectra were acquired with a FTHRMS-Orbitrap (Thermo-Finnigan) mass spectrometer. HPLC chromatograms were obtained with a Dionex P580 system connected to a photodiode array detector (UVD340S). Routine UV lengths for detection were set at 235, 254, 280, and 340 nm. The stationary phase of the analytical column (125 × 4 mm) was Eurosphere-10 C18 (Knauer, Germany). The mobile phase was a gradient of acidic nanopure water, set to pH 2 by adding HCOOH and HPLC grade methanol, following the gradient: 0 min, 10% MeOH; 5–35 min, 10–100% MeOH; 35–45 min, 100% MeOH. Semi-preparative HPLC chromatography was performed using the Lachrom–Merck Hitachi semi-preparative HPLC system (UV detector L-7400; pump L-7100; Eurosphere-100 C18, 300 × 8 mm, Knauer, Germany) at a flow rate of 5.0 mL min−1. Column chromatography was carried out using Sephadex LH-20 and Merck MN Silica gel 60 M (0.04–0.063 mm). During the isolation work, precoated silica gel 60 F254 TLC plates (Merck, Darmstadt, Germany) served to control the progress of isolation, under detection at 254 and 365 nm and then sprayed with anisaldehyde reagent.Microbial material
The endophytic fungus F. tricinctum was obtained from healthy fresh rhizomes of A. paucinervis (Aristolochiaceae) adapting standard procedures.34 The fresh plant was collected in January 2006 from the mountains Beni-Mellal, located in Morocco.6 The bacterial strain Streptomyces lividans TK24 (ref. 35) complies to standard laboratory strains.36Identification of fungal material
The fungal microorganism was identified as F. tricinctum using the ITS method according to the protocol of molecular biology, by DNA isolation and amplification of the ITS region as previously described.6 The same strain of the fungus, encoded as F.t., is kept under laboratorial conditions at −80 °C in the Institute of Pharmaceutical Biology and Biotechnology, Heinrich-Heine University, Düsseldorf, Germany.Co-cultivation of F. tricinctum with S. lividans
In order to cultivate the fungus and the bacterium under sterile conditions, ten Erlenmeyer flasks (1 L each) were prefilled with solid rice medium (50 g of milk rice, Oryza) and Yeast Malt (YM) medium (60 mL), and then autoclaved. Four Erlenmeyer flasks were inoculated with only fungus or bacterium, serving as axenic control cultures. S. lividans was precultured using YM medium, which was incubated at 30 °C and shaken at 200 rpm, left overnight to reach exponential mid growth. Since the distribution of the bacterial colonies was not measurable by OD because of agglomeration, the average amount of colonies was estimated visually in order to divide equal amounts of the bacterium to each flask. This has been performed by using a sterilized 5 mL Eppendorf pipette. The bacteria were then added to flasks containing the solid rice medium and left for three days at 30 °C before the fungus was added to the flasks and left at room temperature. This protocol was chosen as the fungus readily killed the bacteria when added simultaneously to the culture flasks. The axenic fungal or bacterial cultures on solid rice media were kept under identical conditions. All flasks were then cultivated at room temperature. Flasks were extracted when the fungus had completely covered the surface of the culture media.Extraction and isolation
After fermentation, 300 mL EtOAc were added to the fully grown cultures which were then extracted for 8 h under continuous shaking. The EtOAc extracts of the co-cultures of F. tricinctum and S. lividans following HPLC chromatography were combined, yielding an amount of 3.8 g. This extract was dissolved in a solvent mixture of MeOH–H2O (9:1) followed by partitioning against n-hexane. The aqueous MeOH extract was subjected to a Sephadex LH-20 column, using acetone as mobile phase, to obtain 12 fractions (Fr.1–14). The second and third fractions (Fr.2 and 3) were combined due to their similar HPLC profiles followed by separation on a silica column. From the first fraction of this chromatographic separation which was eluted with CH2Cl2–MeOH (99:1), compound 3 (2.3 mg) was purified using semi-preparative RP-HPLC with 100% MeOH. Fr.4 was further fractionated using a Sephadex LH-20 column with MeOH as mobile phase which yielded compound 1 (10.8 mg) as the subfraction Fr.4–4. Fraction Fr.4–3 was subjected to semi-preparative RP-HPLC to afford compound 4 (2.1 mg). From Fr.5, compounds 5 (0.8 mg), 6 (3.9 mg) and 7 (1.5 mg) were obtained by semi-preparative RP-HPLC with MeOH/H2O as mobile phase. Compounds 2 and 8 were isolated from Fr.8 by semi-preparative RP-HPLC, where 2 (4.2 mg) was eluted at 100% MeOH and 8 (1.1 mg) at 55% MeOH–H2O. Furthermore, purification of Fr.10 with semi-preparative HPLC yielded compound 9 (1.1 mg) at 40% MeOH–H2O and compound 10 (1.2 mg) at 55% MeOH–H2O elution system.
Calculation of the rotation barrier for compounds 1–4
To obtain an energy profile for the rotation about the central biaryl bond in compounds 1–4 at reasonable computational expense, 8,8′-dihydroxy-3,3′-dimethoxy-2,2′-binaphthyl-1,1′,4,4′-tetrone (Fig. 2B) was used as model compound. First, structures in which the torsion angle defined by atoms C4–C3–C3′-C4′ (Fig. 1) was varied from −180° to 180° in increments of 8° were constructed in the Maestro GUI of the Schrödinger Suite of programs. A single structure with a torsion angle of 0° was added to the resulting series of conformer structures. For each of the resulting 47 structures, a conformational search with a harmonic restraint of 104 kcal mol−1 rad−2 on the corresponding value of the torsion angle was performed using MacroModel and its default settings except for the truncated Newton conjugate gradient method for energy minimization. The minimum energy conformers for each of the 47 searches were then geometry optimized while freezing the respective value of the torsion angle. Geometry optimization was carried out in the gas phase at the B3LYP/6-31G(d) level using the GAUSSIAN 09 software.Conformational analysis and TDDFT-ECD calculations of 5
Mixed torsional/low-frequency mode conformational searches were carried out by means of the MacroModel 10.8.011 software by using the Merck Molecular Force Field (MMFF) with an implicit solvent model for CHCl3.37 Geometry reoptimizations were carried out at the B3LYP/6-31G(d) level in vacuo, the B97D/TZVP38,39 and the CAM-B3LYP/TZVP40 levels with the PCM solvent model for MeCN. TDDFT-ECD calculations were run with various functionals (B3LYP, BH&HLYP, CAM-B3LYP, and PBE0) and the TZVP basis set as implemented in the Gaussian 09 package with the same or no solvent model as in the preceding DFT optimization step.41 Electronic circular dichroism spectra were generated as sums of Gaussians with 2700 and 3300 cm−1 widths at half-height, using dipole-velocity-computed rotational strength values.42 Boltzmann distributions were estimated from the ZPVE-corrected B3LYP/6-31G(d) energies in the gas-phase calculations and from the B97D and CAM-B3LYP energies in the solvated ones. The MOLEKEL software package was used for visualization of the results.43Antibacterial assay
The antibacterial activity was determined by using the microdilution method in alignment with the CLSI guidelines.44 Compounds were prepared using a DMSO stock solution with the highest concentration of 0.64% (64 μg mL−1). This was subjected to a prepared inoculum, in which the direct colony suspension method was applied by using an inoculum of 5 × 105 colony forming units per mL. Simultaneously, antibiotics were serial 2-fold diluted in DMSO with the compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the DFG (GRK 2158) to H. G., R. K. and P. P. is gratefully acknowledged. P. P. also wants to thank the Manchot Foundation for support. H. G. is grateful for computational support and infrastructure provided by the “Zentrum für Informations-und Medientechnologie” (ZIM) at the Heinrich Heine University Düsseldorf and the computing time provided by the John von Neumann Institute for Computing (NIC) on the supercomputer JURECA at Jülich Supercomputing Centre (JSC) (user ID: HKF7). T. K. and A. M. thank the National Research, Development and Innovation Office (NKFI K120181 and PD121020) for financial support and the Governmental Information-Technology Development Agency (KIFÜ) for CPU time.References
- A. Marmann, A. H. Aly, W. Lin, B. Wang and P. Proksch, Mar. Drugs, 2014, 12, 1043–1065 CrossRef PubMed.
- G. Daletos, W. Ebrahim, E. Ancheeva, M. El-Neketi, W. Lin and P. Proksch, in Chemical Biology of Natural Products, ed. D. Newman, G. Cragg and P. Grothaus, CRC Press, Boca Raton, 2017, ch. 8, pp. 233–284 Search PubMed.
- C. F. Pérez Hemphill, P. Sureechatchaiyan, M. U. Kassack, R. S. Orfali, W. Lin, G. Daletos and P. Proksch, J. Antibiot., 2017, 70, 726–732 CrossRef PubMed.
- J. Wang, A. Debbab, C. F. Pérez Hemphill and P. Proksch, Z. Naturforsch., 2013, 68c, 223–230 Search PubMed.
- A. R. B. Ola, D. Thomy, D. Lai, H. Brötz-Oesterhelt and P. Proksch, J. Nat. Prod., 2013, 76, 2094–2099 CrossRef CAS PubMed.
- W. Wätjen, A. Debbab, A. Hohlfeld, Y. Chovolou, A. Kampkötter, R. A. Edrada, R. Ebel, A. Hakiki, M. Mosaddak, F. Totzke, M. H. G. Kubbutat and P. Proksch, Mol. Nutr. Food Res., 2009, 53, 431–440 CrossRef PubMed.
- C. G. Shin, D. G. An, H. H. Song and C. Lee, J. Antibiot., 2009, 62, 687–690 CrossRef CAS PubMed.
- S. Fraeyman, S. Croubels, M. Devreese and G. Antonissen, Toxins, 2017, 9, 228 CrossRef PubMed.
- E. Ancheeva, L. Küppers, S. H. Akone, W. Ebrahim, Z. Liu, A. Mándi, T. Kurtán, W. Lin, R. Orfali, N. Rehberg, R. Kalscheuer, G. Daletos and P. Proksch, Eur. J. Org. Chem., 2017, 3256–3264 CrossRef CAS.
- S. H. Akone, A. Mándi, T. Kurtán, R. Hartmann, W. Lin, G. Daletos and P. Proksch, Tetrahedron, 2016, 72, 6340–6347 CrossRef CAS.
- M. Gulshan, G. Y. Zhao, Z. F. Dong and M. Ghopur, Shengwu Jishu, 2009, 19, 43–46 Search PubMed.
- H. Meschke, S. Walter and H. Schrempf, Environ. Microbiol., 2012, 14, 940–952 CrossRef CAS PubMed.
- R. P. Mellado, World J. Microbiol. Biotechnol., 2011, 27, 2231–2237 CrossRef CAS.
- H. Laatsch, Z. Naturforsch., 1990, 45b, 393–400 Search PubMed.
- H. Laatsch, Liebigs Ann. Chem., 1983, 1886–1900 CrossRef CAS.
- S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed.
- M. Öki, Top. Stereochem., 1983, 14, 1–63 Search PubMed.
- I. Alkorta, J. Elguero, C. Roussel, N. Vanthuyne and P. Piras, Adv. Heterocycl. Chem., 2012, 105, 1–188 CrossRef CAS.
- D. Rönsberg, A. Debbab, A. Mándi, V. Vasylyeva, P. Böhler, B. Stork, L. Engelke, A. Hamacher, R. Sawadogo, M. Diederich, V. Wray, W. Lin, M. U. Kassack, C. Janiak, S. Scheu, S. Wesselborg, T. Kurtán, A. H. Aly and P. Proksch, J. Org. Chem., 2013, 78, 12409–12425 CrossRef PubMed.
- G. Wu, G. Yu, T. Kurtán, A. Mándi, J. Peng, X. Mo, M. Liu, H. Li, X. Sun, J. Li, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2015, 78, 2691–2698 CrossRef CAS PubMed.
- G. Höfle and K. Röser, J. Chem. Soc., Chem. Commun., 1978, 611–612 RSC.
- R. C. Durley, J. MacMillan and T. J. Simpson, J. Chem. Soc., Perkin Trans. 1, 1975, 163–169 RSC.
- I. M. Romaine and G. A. Sulikowski, Tetrahedron Lett., 2015, 56, 3617–3619 CrossRef CAS PubMed.
- G. W. Bushnell, Y. L. Li and G. A. Poulton, Can. J. Chem., 1984, 62, 2101–2106 CrossRef CAS.
- G. Pescitelli and T. Bruhn, Chirality, 2016, 28, 466–474 CrossRef CAS PubMed.
- A. Mándi, I. W. Mudianta, T. Kurtán and M. J. Garson, J. Nat. Prod., 2015, 78, 2051–2056 CrossRef PubMed.
- P. Zhang, L. H. Meng, A. Mándi, T. Kurtán, X. M. Li, Y. Liu, X. Li, C. S. Li and B. G. Wang, Eur. J. Org. Chem., 2014, 4029–4036 CrossRef CAS.
- Y. M. Ren, C. Q. Ke, A. Mándi, T. Kurtán, C. Tang, S. Yao and Y. Ye, Tetrahedron, 2017, 73, 3213–3219 CrossRef CAS.
- W. H. Urry, H. L. Wehrmeister, E. B. Hodge and P. H. Hidy, Tetrahedron Lett., 1966, 27, 3109–3114 CrossRef.
- S. Lai, Y. Shizuri, S. Yamamura, K. Kawai and H. Furukawa, Heterocycles, 1991, 32, 297–305 CrossRef CAS.
- A. Watanabe, Y. Ono, I. Fujii, U. Sankawa, M. E. Mayorga, W. E. Timberlake and Y. Ebizuka, Tetrahedron Lett., 1998, 39, 7733–7736 CrossRef CAS.
- J. Xu, J. Kjer, J. Sendker, V. Wray, H. Guan, R. Edrada, W. Lin, J. Wu and P. Proksch, J. Nat. Prod., 2009, 72, 662–665 CrossRef CAS PubMed.
- Y. Shiono, M. Tsuchinari, K. Shimanuki, T. Miyajima, T. Murayama, T. Koseki, H. Laatsch, T. Funakoshi, K. Takanami and K. Suzuki, J. Antibiot., 2007, 60, 309–316 CrossRef CAS PubMed.
- J. Kjer, A. Debbab, A. H. Aly and P. Proksch, Nat. Protoc., 2010, 5, 479–490 CrossRef CAS PubMed.
- H. Chen, G. Daletos, M. S. Abdel-Aziz, D. Thomy, H. Dai, H. Brötz-Oesterhelt, W. H. Lin and P. Proksch, Phytochem. Lett., 2015, 12, 35–41 CrossRef CAS.
- D. A. Hopwood, T. Kieser, H. M. Wright and M. J. Bibb, J. Gen. Microbiol., 1983, 129, 2257–2269 CAS.
- MacroModel, Schrödinger, LLC, 2015, http://www.schrodinger.com/MacroModel Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- P. Sun, D. X. Xu, A. Mándi, T. Kurtán, T. J. Li, B. Schulz and W. Zhang, J. Org. Chem., 2013, 78, 7030–7047 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
- U. Varetto, MOLEKEL 5.4, Swiss National Supercomputing Centre, Manno, Switzerland, 2009 Search PubMed.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, Approved Standard Ninth ed. CLSI document M07-A9, Clinical and Laboratory Standards Institute, Wayne, PA, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MS, 1D and 2D NMR spectra of compounds 1–5. See DOI: 10.1039/c8ra09067j |
| This journal is © The Royal Society of Chemistry 2019 |