
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew anionic rhodium complexes as catalysts for the reduction of acetophenone and its derivatives
Olga Bartlewicz a,
Magdalena Jankowska-Wajdaa and
Hieronim Maciejewski*ab
a,
Magdalena Jankowska-Wajdaa and
Hieronim Maciejewski*ab
aFaculty of Chemistry, Adam Mickiewicz University in Poznań, Umultowska 89B, 61-614 Poznań, Poland. E-mail: maciejm@amu.edu.pl
bPoznań Science and Technology Park, A. Mickiewicz University Foundation, Rubież 46, 61-612 Poznań, Poland
First published on 4th January 2019
Abstract
New anionic rhodium(III) complexes, obtained by a simple reaction of RhCl3 with organic chlorides (derivatives of imidazole and pyridine), have been employed as catalysts for hydrosilylation (reduction) of acetophenone derivatives. The reactions, in which 1,1,1,3,5,5,5-heptamethyltrisiloxane was a reducing agent, proceeded in a biphasic system because the above complexes are insoluble in the reaction medium. Thereby easy isolation of the complexes from post-reaction mixtures was possible after reaction completion. This is the first example of the application of rhodium complexes of this type as catalysts for ketone reduction. The complexes have shown high activity and enabled obtaining the hydrosilylation product in a very short time and in the range of low concentrations (0.1 mol%). By using FT-IR in situ analysis that enables measuring product concentrations in real time, a comparison has been made of the catalytic activity for hydrosilylation of acetophenone and methoxyacetophenone isomers shown by four rhodium complexes ([C+][RhCl4−]) differing in cations and the most effective catalyst for this process has been distinguished.
Introduction
Catalytic reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is one of the most fundamental transformations in organic chemistry.1 It can be performed using a number of reducing agents with dihydrogen being the most frequently used.2 This transformation can also be achieved by using stoichiometric metal hydrides, although this is concomitant with issues of low product selectivity and poor atom economy. One appealing alternative to these methods is the catalytic hydrosilylation of a carbonyl group. Formally, hydrosilylation of ketones produces (in a single step) silyl ethers, which are protected forms of secondary alcohols, frequently used in organic synthesis. Among them, of particular importance, are chiral secondary alcohols which are of great significance in agrochemicals and pharmaceuticals as well as in fine chemicals.3 Alcohol synthesis requires an additional hydrolysis (deprotection) step, which is realized by one of the standard techniques depending on the specificity of the reaction system. The discovery by Ojima of the high catalytic activity of Wilkinson's catalyst [RhCl(PPh3)3] in hydrosilylation of carbonyl compounds stimulated great development in hydrosilylation of CO bonds, which makes catalytic hydrosilylation one of the most important and convenient reduction methods for obtaining secondary alcohols.4 Up to the present day, a plethora of transition metal catalysts have been exploited for the hydrosilylation of ketones.5 Efficient catalysts include mainly transition metal complexes5 but also main group metal complexes.6 Furthermore, metal-free methods have also been developed using acids,7 bases8 and alkali fluorides9 to effect these transformations, although such methods have mostly required harsh reaction conditions.
O is one of the most fundamental transformations in organic chemistry.1 It can be performed using a number of reducing agents with dihydrogen being the most frequently used.2 This transformation can also be achieved by using stoichiometric metal hydrides, although this is concomitant with issues of low product selectivity and poor atom economy. One appealing alternative to these methods is the catalytic hydrosilylation of a carbonyl group. Formally, hydrosilylation of ketones produces (in a single step) silyl ethers, which are protected forms of secondary alcohols, frequently used in organic synthesis. Among them, of particular importance, are chiral secondary alcohols which are of great significance in agrochemicals and pharmaceuticals as well as in fine chemicals.3 Alcohol synthesis requires an additional hydrolysis (deprotection) step, which is realized by one of the standard techniques depending on the specificity of the reaction system. The discovery by Ojima of the high catalytic activity of Wilkinson's catalyst [RhCl(PPh3)3] in hydrosilylation of carbonyl compounds stimulated great development in hydrosilylation of CO bonds, which makes catalytic hydrosilylation one of the most important and convenient reduction methods for obtaining secondary alcohols.4 Up to the present day, a plethora of transition metal catalysts have been exploited for the hydrosilylation of ketones.5 Efficient catalysts include mainly transition metal complexes5 but also main group metal complexes.6 Furthermore, metal-free methods have also been developed using acids,7 bases8 and alkali fluorides9 to effect these transformations, although such methods have mostly required harsh reaction conditions.
Within transition metal complexes the most efficient catalysts are those based on Zn,10–13 Cu,14–18 Fe,19–23 Re,24–26 Ti27 and Rh,4,28–33 and Mo,34 of which the rhodium ones have been most comprehensively studied. Since the discovery of the activity of Wilkinson's catalyst in hydrosilylation of ketones, rhodium complexes are intensively studied and widely used in hydrosilylation of carbonyl compounds.28 Over the last few years a number of rhodium N-heterocyclic carbene complexes have been developed (including the chiral ones) and proved active catalysts for the reduction of carbonyl compounds.28,29 Examples could be NHC complexes with ferrocenyl substituents which were employed as catalysts for hydrosilylation of acetophenone derivatives and resulted in good selectivity and good yield of the reaction.35 Carbene complexes with alkosilyl groups (that make easier the immobilization on silica surface) have been used in hydrosilylation of acetophenone with 1,1,1,3,5,5,5-heptamethyltrisiloxane and very good catalytic results were obtained.36 Unfortunately, homogeneous N-heterocyclic carbene complexes, although show satisfactory conversion of reactants, not always are characterized by satisfactory selectivity. Moreover, they are very susceptible to process conditions, therefore the process carried out in their presence requires the application of special conditions (inert atmosphere, Schlenk technique).31,37 Yigit et al. have synthesized new carbene complexes in the reaction of rhodium dimer [(Rh(OMe)COD)]2 with 1,3-dialkylimidazolium chloride in tetrahydrofuran and used them in the reaction of acetophenone hydrosilylation with triethylsilane. They reported obtaining good catalytic yield.38 It is worth of mentioning that 1,3-dialkylimidazolium chloride is a representative of a large group of compounds that are called ionic liquids which recently arouse a large interest not only as “green solvents”39,40 but also as immobilizing agents for metal complexes. At present, over 100 types of chemical reactions realized in the medium of ionic liquids and catalyzed by transition metal complexes are known.41,42 We have also developed a number of effective catalytic systems based on diverse platinum and rhodium complexes immobilized in the most important kinds of ionic liquids.43–46 However, in addition to the application of ionic liquids as immobilizing agents, more and more frequently attempts are made at obtaining metal ion-containing ionic liquids with catalytic properties. One of examples are halometallate ionic liquids which can be defined as ionic liquids formed by the reaction of metal halide with an organic halide salt. The first representatives of this group of compounds were chloroaluminate ionic liquids,47 however, at present, examples are known of compounds that contain transition metals such as cobalt, nickel, iridium, gold, palladium as well as platinum.48,49 All the above compounds are basically classified into the group of halometallate ionic liquids, however, their melting points which exceed 150 °C make it difficult to classify them among ionic liquids, whose melting points should be below 100 °C. This is why it would be better to call them anionic metal complexes. Very recently our research group synthesized durable and air-stable anionic rhodium complexes as a result of a direct reaction between rhodium(III) chloride and derivatives of imidazole and pyridine (Scheme 1).50
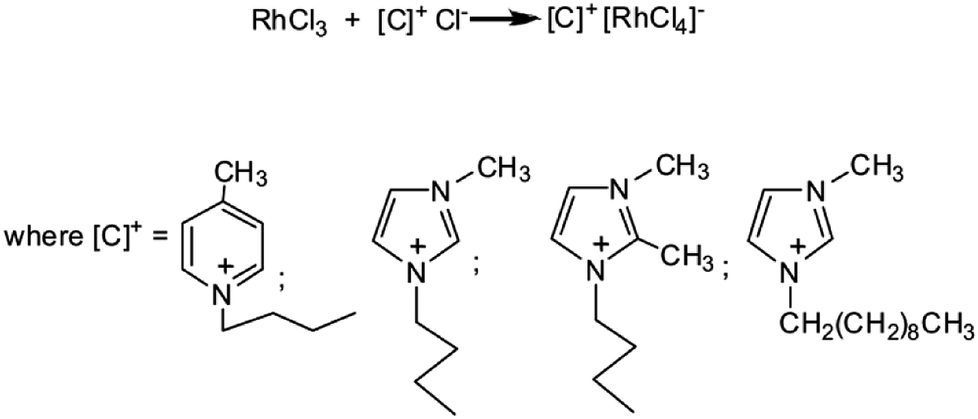 | ||
| Scheme 1 Synthesis of anionic rhodium(III) complexes. | ||
The above complexes were applied as catalysts for hydrosilylation processes. In order to compare the catalytic activity of the obtained new rhodium complexes, acetophenone and its methoxy group-containing derivatives were used. These compounds are present in many fruits and plants and they exhibit a number of interesting chemical and biological properties, due to which they are very attractive for researchers. The other reagent was 1,1,1,3,5,5,5-heptamethyltrisiloxane that is readily available, stable and, first and foremost, cheap compound compared to arylsilanes which were most frequently employed as hydride donors in reduction reactions catalyzed by rhodium complexes. To the best of our knowledge, this study on the above anionic rhodium complexes is the first example of the employment of such complexes in hydrosilylation processes, particularly for reduction of ketones.
Experimental
All reagents used in the catalytic studies, i.e. acetophenone, 2-methoxyacetophenone, 3-methoxyacetophenone, 4-methoxyacetophenone, 4-nitroacetophenone, 4-acetylbenzonitrile, 4-iodoacetophenone, 4-aminoacetophenone, 4-methylacetophenone, benzophenone, 1,1,1,3,5,5,5-heptamethyltrisiloxane, n-decane, toluene, Karstedt's catalyst [Pt2{(CH2CHSi(CH3)2)O}3] and Wilkinson's catalyst [RhCl(PPh3)3], chloro(1,5-cyclooctadiene) rhodium(I) dimer [{Rh(μ-Cl)COD}2] and chlorobis(cyclooctene) rhodium(I) dimer [{Rh(μ-Cl)C8H14}2] were purchased from Sigma Aldrich and used as received. The (1,5-cyclooctadiene)-μ-(trimethylsiloxy) rhodium(I) dimer complex [{Rh(μ-OSiMe3)COD}2] was obtained in accordance to the preparation procedure described by Marciniec et al.,51 whereas anionic platinum complexes [BMPy]2[PtCl4] and [BMIM]2[PtCl4] were prepared following the procedure described by Zhong et al.52
The rhodium(III) chloride, RhCl3 (Pressure Chemicals Co.) was used as the metal precursor for the obtained complexes, while the ligand precursors were ionic liquids: 1-butyl-3-methylimidazolium chloride, 1-butyl-2,3-dimethylimidazolium chloride, 1-butyl-4-methylpyridinium chloride and 1-decyl-3-methylimidazolium chloride, obtained from Iolitec. The yield of a product of a given reaction was determined using a Clarus 680 gas chromatograph (Perkin Elmer) equipped with a 30 m capillary column Agilent VF-5ms and TCD detector, using the temperature program: 60 °C (3 min.), 10 °C min−1, 290 °C (5 min.). FT-IR in situ measurements were performed using a Mettler Toledo ReactIR 15 instrument. For selected samples, spectra were recorded with 256 scans for 1 or 2 hours at 30 second intervals with a resolution of 1 cm−1. For the products obtained, NMR spectra were made with Bruker BioSpin (400 MHz) spectrometer using acetonitrile-d3 as a solvent; chemical shifts are given in ppm.
The GC-MS analysis was conducted using a Varian 3300 chromatograph equipped with a 30 m DB-1 capillary column connected to the Finnigan Mat 700 mass detector. ICP-MS analysis of post-reaction samples was carried out on a PerkinElmer Nexion 300D inductively coupled mass spectrometer.
Synthesis of catalysts
The synthesis of anionic rhodium complexes was performed according to the procedure described in the patent application.50 Anionic rhodium complexes were obtained by placing 1 mmol of ionic liquid and 1 mmol of RhCl3·xH2O in hot CH3CN (2 ml) in a Schlenk tube equipped with a magnetic stirring bar. The mixture was stirred for 3 h under reflux. The obtained solution was cooled to room temperature and the solvent was evaporated. The product was washed with diethyl ether (3 × 5 ml) and dried under vacuum.Spectroscopic characterization of the obtained rhodium complexes:
N–), 7.32 (s, 1H –CHCH–), 7.31 (s, 1H –CHCH), 4.20 (t, 2H N–CH2–), 3.9 (s, 3H –N–CH3), 1.75 (m, 2H, –CH2–), 1.23 (m, 2H, J = 7.5, –CH2–), 1.0 (t, 3H, J = 7.5, –CH3).13C NMR (DMSO-d6): 138 (N–CHN); 123 (–CHCH–), 121 (–CHCH–); 50 (N–CH2), 37 (N–CH3); 32, 31 (–CH2–); 13 (–CH3).
13C NMR: 145 (CAr), 129 (CAr), 62 (–N–CH2), 33 (Ar–CH3), 19, 18 (–CH2–), 14 (–CH3).
N–), 7.32 (s, 1H, –CHCH–), 7.31 (s, 1H, –CHCH), 4.51 (t, 2H,–N–CH2), 4.05 (s, 3H, –CH3), 3.93 (s, 3H, N–CH3), 2.25 (m, 2H, J = 7.5, –CH2–), 1.75 (m, 4H,–CH2–), 1.27 (m, 2H, J = 7.5, –CH2–), 0.9 (t, 3H, J = 7.5, –CH3).13C NMR (DMSO-d6): 138 (C–CH3); 123 (CHCH), 117 (CHCH); 45 (N–CH2), 32 (N–CH3); 29.20 (–CH2–); 25.14 (–CH3).
N), 7.5 (s, 1H, CHCH), 7.5 (s, 1H, CHCH), 5.35 (t, 2H, –N–CH2), 4.1 (s, 3H N–CH3), 2.5–1.2 (m, 14H, –CH2–), 0.8 (t, 3H, J = 7.15, –CH3).13C NMR (DMSO-d6): 138 (N–CHN); 123 (–CHCH–), 121 (CHCH); 47.5(N–CH3), 37 (–N–CH2–); 32–23 (–CH2–), 13 (–CH3).
General procedure for catalytic tests
The catalytic activity of the obtained anionic rhodium complexes was determined in the reactions of hydrosilylation of acetophenone and its derivatives with 1,1,1,3,5,5,5-heptamethyltrisiloxane (HMTS). For this purpose, 0.001 mmol of catalyst, 1 mmol of ketone, 1.25 mmol of HMTS and 1 mmol of n-decane as an internal standard were used. In addition, for the systems with methoxyacetophenone, due to the fact that the substrates were insoluble, it was necessary to add toluene in the amount of 1.88 mmol for 2-methoxyacetophenone, 3.76 mmol for 3-methoxyacetophenone and 5.64 mmol for 4-methoxyacetophenone. The reaction was carried out in a reaction vessel in the presence of air at 110 °C with vigorous stirring for 1 h. The reaction mixture was then cooled down and subjected to GC analysis to determine the reaction yield. The product was isolated and subjected to NMR and GC-MS analyses.C), 4.98 (q; 1H; CH), 1.36 (d; 3H; CH3), 0.07–0.01 (m, 21H, Si–CH3).13C NMR (CD3CN): 146.49 (C–C), 127.93 (C–CC), 126.58, 124.64 (CC–C), 116.67 (C–CN), 69.69 (C–O), 26.29 (C–CH3), 1.16 (Si–CH3), −3.84 (O–Si–CH3).
29Si NMR (CD3CN): 8.58 (OSi(CH3)3), −58.01 (O–Si–O–).
GC-MS: 327.30 [PhCHOSiMe3OSiMeOSiMe3]+, 221.50 [Me3OSiMeOSiMe3]+, 105.20 [PhCHCH3]+.
CH2OSiMeOSiMe3O]+.C–C), 7.24 (m, J = 8.0 Hz, 1H, C–CC), 6.96 (m, 2H, CC–C), 5.40 (q, J = 6.3 Hz, 1H, CH), 3.83 (s, 3H, O–CH3), 1.37 (d, J = 6.3 Hz, 3H, C–CH3), 0.07–0.01 (m, 21H, Si–CH3).13C NMR (CD3CN): 154.91 (C–OCH3), 134.50 (C–CC), 128.26, 120.35 (CC–C), 116.67 (C–CN), 64.26 (C–O), 54.85 (-OCH3), 24.74 (C–CH3), 0.90 (Si–(CH3)3), −3.95 (O–Si–CH3).
29Si NMR (CD3CN): 8.07 (OSi(CH3)3), −58.14 (O–Si–O–).
GC-MS: 356.9 [PhOMeCHOSiMe(OSiMe3)2]+, 221.5 [Me3OSiMeOSiMe3]+, 151.1 [PhOCH3CHOCH3]+, 135.2 [PhOCH3CHCH3]+, 105.1 [PhCHCH3]+, 79.2 [C6H7]+, 78.2 [C6H6]+, 77.2 [C6H5]+, 73.2 [SiMe3]+.
CH2OSiMeOSiMe3]+, 267.0 [PhCCH2OSiMeOSiMe3O]+, 221.5 [Me3OSiMeOSiMe3]+, 149.1 [PhOMeCCH2O]+.C–C), 7.0 (m, 2H, C–CC), 6.83 (m, 1H, CC–C), 5.06 (q, J = 6.4 Hz, 1H, CH), 3.80 (s, 3H, O–CH3), 1.44 (d, J = 6.3 Hz, 3H, C–CH3), 0.19–0.05 (m, 21H, Si–CH3).13C NMR (CD3CN): 159.67 (C–OCH3), 148.06 (C–CHCH3), 129.15, 117.59 (CC–C), 117.24 (C–CN), 112.15, 110.89 (C–CC), 69.85 (C–O), 54.78 (–OCH3), 26.10 (C–CH3), 0.99 (Si–(CH3)3), −3.87 (O–Si–CH3).
29Si NMR (CD3CN): 9.08 (OSi(CH3)3), −57.83 (O–Si–O–).
GC-MS: 356.9 [PhOMeCHOSiMe(OSiMe3)2]+, 221.5 [Me3OSiMeOSiMe3]+, 151.1 [PhOCH3CHOCH3]+, 135.2 [PhOCH3CHCH3]+, 105.1 [PhCHCH3]+, 79.2 [C6H7]+, 78.2 [C6H6]+, 77.2 [C6H5]+, 73.2 [SiMe3]+.
CH2OSiMeOSiMe3]+, 267.0 [PhCCH2OSiMeOSiMe3O]+, 221.5 [Me3OSiMeOSiMe3]+, 149.1 [PhOMeCCH2O]+.C–C), 6.90 (m, 2H, C–CC), 5.02 (q, J = 6.4 Hz, 1H, CH), 3.79 (s, 3H, O–CH3), 1.41 (d, J = 6.45 Hz, 3H, C–CH3), 0.10–0.01 (m, 21H, Si–CH3).13C NMR (CD3CN): 158.89 (C–OCH3), 138.84 (C–CHCH3), 126.56 (CC–C), 117.23 (C–CN), 113.39 (C–CC), 69.58 (C–O), 54.85 (–OCH3), 26.04 (C–CH3), 0.97 (Si–(CH3)3), −3.81 (O–Si–CH3).
29Si NMR (CD3CN): 8.58 (OSi(CH3)3), −58.65 (O–Si–O–).
GC-MS: 356.9 [PhOMeCHOSiMe(OSiMe3)2]+, 221.50 [Me3OSiMeOSiMe3]+, 151.1 [PhOCH3CHOCH3]+, 135.2 [PhOCH3CHCH3]+, 105.1 [PhCHCH3]+, 79.2 [C6H7]+, 78.2 [C6H6]+, 77.2 [C6H5]+, 73.2 [SiMe3]+.
CH2OSiMeOSiMe3]+, 267.0 [PhCCH2OSiMeOSiMe3O]+, 221.5 [Me3OSiMeOSiMe3]+, 149.1 [PhOMeCCH2O]+.Hydrolysis of the hydrosilylation product
The product (1 ml, 2.75 mmol) of hydrosilylation of acetophenone and 1,1,1,3,5,5,5-heptamethyltrisiloxane and 3 ml of 0.4 mol HCl solution in acetone were placed in a round bottom flask equipped with a magnetic stirrer. The reaction of hydrolysis was carried out for 4 h at room temperature. Then acetone was evaporated and the mixture was allowed to stand in the fridge for about 1 hour. After this time, the separation and GC analysis of phases were possible. The obtained chromatograms showed that the upper phase contained mainly by-products of the silyl ether hydrolysis, while the desired product was in the lower phase. After extracting the upper phase, the obtained sample was analyzed by 1H and 13C NMR to uniquely identify the product. 1 ml of hydrosilylation reaction product (product A) gave about 0.25 ml (2 mmol) of 1-phenylethanol (the yield of about 73%).C–), 125.34 (–CC–C–), 117.36 (C–CN), 69.15 (C–OH), 25.05 (C–CH3); GC-MS: 122.1 [PhCHOHCH3]+, 107.1 [PhCHOH]+, 105.1 [PhCHCH3]+, 77.2 [C6H5]+, 78.2 [C6H6]+, 79.2 [C6H7]+, 51.2 [C4H3]+.Results and discussion
The reduction of ketones using rhodium catalysts is a well-known and widely used reaction. This is why the above process has been chosen as a test for determining catalytic activity of four anionic rhodium complexes, the formulae of which are as follows on Fig. 1.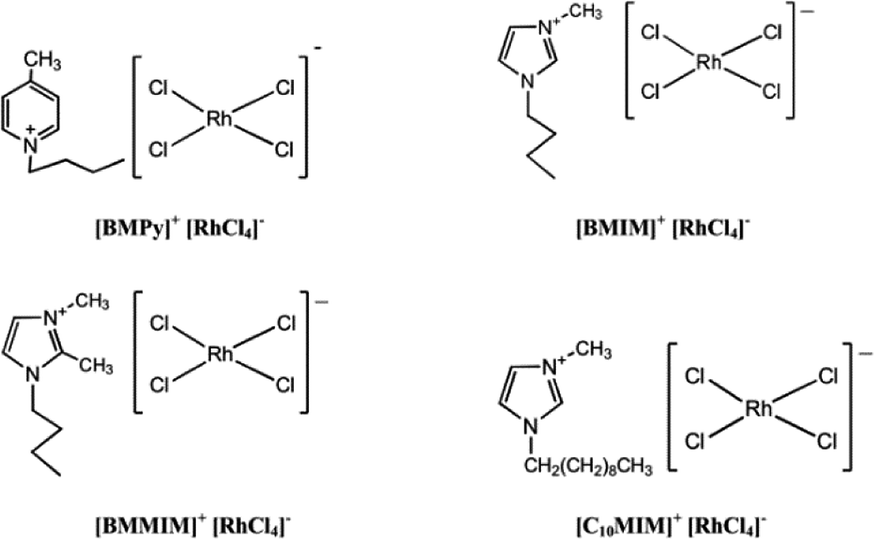 | ||
| Fig. 1 Anionic rhodium complexes studied. | ||
The above complexes were applied as catalysts for the reduction (hydrosilylation) of acetophenone with heptamethyltrisiloxane, which is presented in Scheme 2.
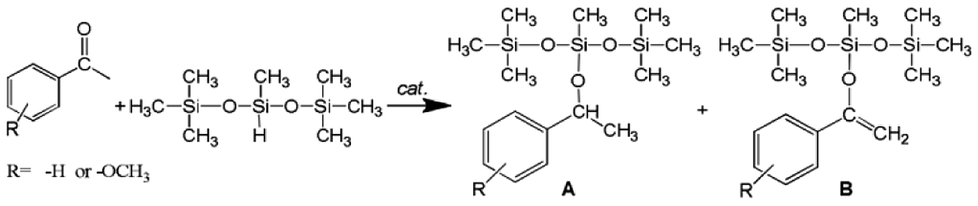 | ||
| Scheme 2 Hydrosilylation of acetophenone with 1,1,1,3,5,5,5-heptamethyltrisiloxane. | ||
According to literature data, hydrosilylation of ketones can be accompanied by dehydrogenative silylation which results in the formation of a respective silyl enol ether (B) in addition to the hydrosilylation product (A).53 Gas chromatographic analysis of post-reaction mixtures of all the systems studied has shown that in some cases the product of dehydrogenative silylation was formed, indeed. However, no other products were found in addition to the main product of hydrosilylation as can be seen in chromatograms of post-reaction mixtures. Exemplary chromatograms of the reaction mixtures before and after the reaction are shown in Fig. 2.
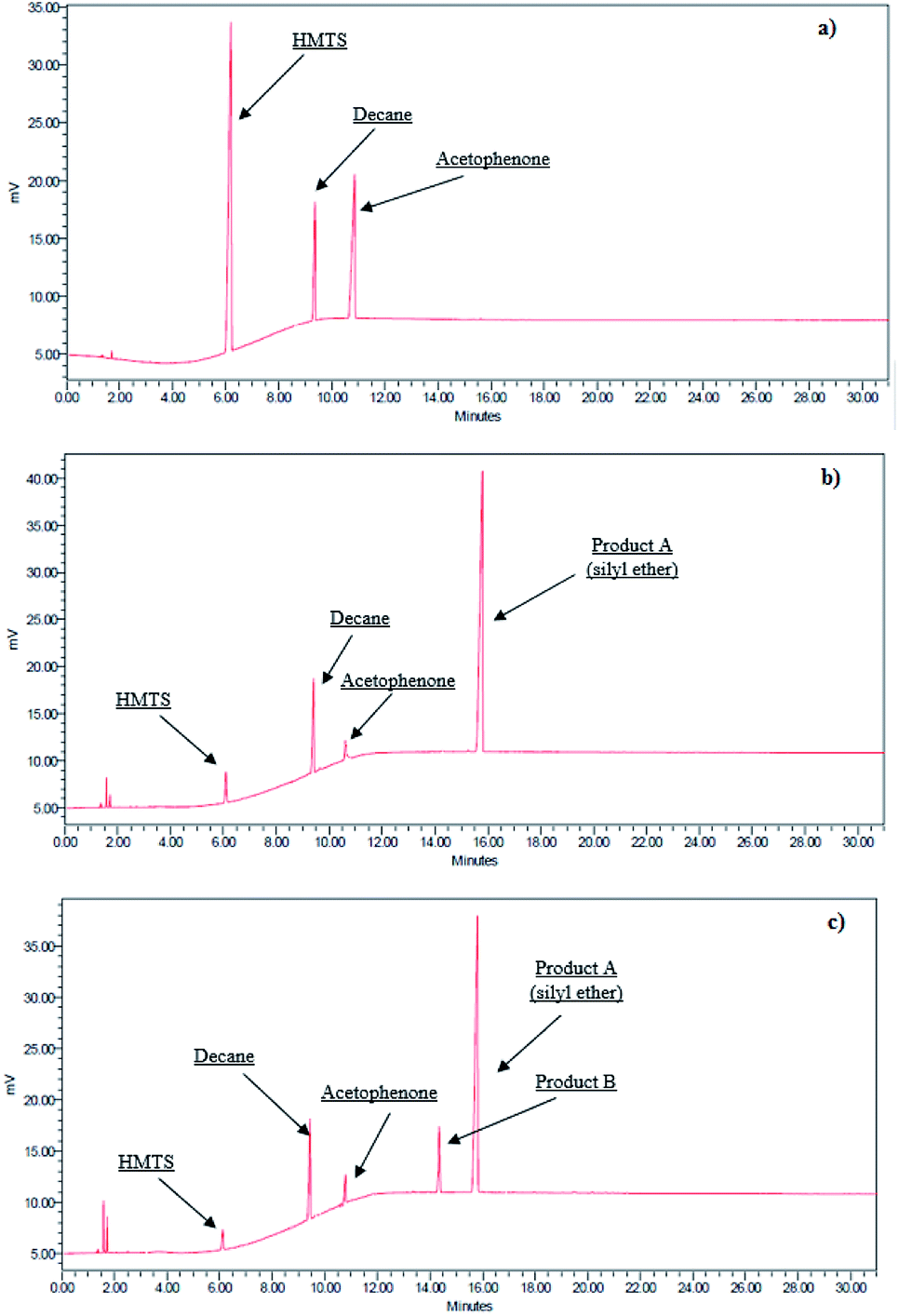 | ||
| Fig. 2 Exemplary chromatograms of hydrosilylation reaction mixtures: (a) before the reaction, (b) after the reaction resulting in one product only, and (c) after the reaction yielding two products (A and B). | ||
The employed anionic rhodium complexes were insoluble in the reaction medium which enabled easy separation of post-reaction mixture from catalyst and analysis of the mixtures composition. The yields of products of the reaction of acetophenone hydrosilylation catalyzed by the studied anionic complexes are shown in Table 1. In all cases one can notice high catalytic activity of the studied complexes in the presence of which the hydrosilylation product (A) was mainly formed in such a short time as 1 hour. Additionally, it should be stressed that the applied concentration of the catalyst was at least 10 times lower than commonly applied concentrations of catalysts for this type of reactions (according to literature data, the concentration is 1–5 mol%). Moreover, frequently a large excess of the reducing agent (Si–H) was used, whereas in our case we have applied the reactant ratio [Si–H]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CO] = 1.25. The highest yield and 100% selectivity was obtained for the reaction catalyzed by the complex [BMPy][RhCl4]. In other cases, yields were somewhat lower (91–83%) and the product of dehydrogenative silylation was also present, albeit its amount was rather small (4–11%). Thereby, one can conclude that complexes containing imidazolium derivatives show somewhat poorer selectivity than pyridinium cation-containing complexes. A comparison of catalytic performances of complexes containing imidazolium derivatives indicate that their activity is quite similar, however, the complex [BMIM][RhCl4] shows poorer selectivity.
:[CO] = 1.25. The highest yield and 100% selectivity was obtained for the reaction catalyzed by the complex [BMPy][RhCl4]. In other cases, yields were somewhat lower (91–83%) and the product of dehydrogenative silylation was also present, albeit its amount was rather small (4–11%). Thereby, one can conclude that complexes containing imidazolium derivatives show somewhat poorer selectivity than pyridinium cation-containing complexes. A comparison of catalytic performances of complexes containing imidazolium derivatives indicate that their activity is quite similar, however, the complex [BMIM][RhCl4] shows poorer selectivity.
| Catalyst | Total yield [%] | Yield of product A [%] | Yield of product B [%] |
|---|---|---|---|
| a Reaction conditions: Rh-catalyst (0.001 mmol), acetophenone (1 mmol), HMTS (1.25 mmol), n-decane (1 mmol), T = 110 °C, t = 1 h. | |||
| [BMPy]+[RhCl4]− | 98 | 98 | 0 |
| [BMIM]+[RhCl4]− | 91 | 80 | 11 |
| [BMMIM]+[RhCl4]− | 89 | 83 | 6 |
| [C10MIM]+[RhCl4]− | 83 | 79 | 4 |
It is known from literature data that rhodium complexes dominate in hydrosilylation of ketones, although in general, the most popular and commonly used catalysts for olefin hydrosilylation are platinum complexes. This is why we have checked catalytic activity of two anionic platinum complexes [BMPy]2[PtCl4] and [BMIM]2[PtCl4] (analogous to the studied rhodium complexes) in the same conditions of the test reaction. To our surprise, the effectiveness of these catalysts was very low and product yields ranged between 3–10%. By comparison, the activity of Karstedt's catalyst (which was also studied in the test reaction) was higher and 30% yield was achieved. Nevertheless, all platinum complexes (anionic ones in particular) showed a lower activity for hydrosilylation of ketones compared to that of anionic rhodium complexes. For this reason we also compared the activity of the studied complexes with that of commercially available rhodium complexes. We have chosen several rhodium compounds which, according to literature data, were characterized by high effectiveness in hydrosilylation processes, namely RhCl3, [RhCl(PPh3)3], [{Rh(μ-OSiMe3)COD}2], [{Rh(μ-Cl)(C8H14)}2] and [{Rh(μ-Cl)COD}2]. To this end, we have carried out the reaction of acetophenone hydrosilylation in conditions analogous to those used for the complexes synthesized by us. The yields of products of the hydrosilylation reaction catalyzed by the above compounds are presented in Table 2.
| Catalyst | Total yield [%] | Yield of product A [%] | Yield of product B [%] |
|---|---|---|---|
| a Reaction conditions: Rh-catalyst (0.001 mmol), acetophenone (1 mmol), HMTS (1.25 mmol), n-decane (1 mmol), T = 110 °C, t = 1 h. | |||
| [RhCl(PPh3)3] | 32 | 29 | 3 |
| [{Rh(μ-Cl)(COD)}2] | 37 | 36 | 1 |
| [{Rh(μ-Cl)(C8H14)}2] | 22 | 21 | 1 |
| RhCl3 | 3 | 3 | 0 |
| [{Rh(μ-OSiMe3)COD}2] | 33 | 31 | 2 |
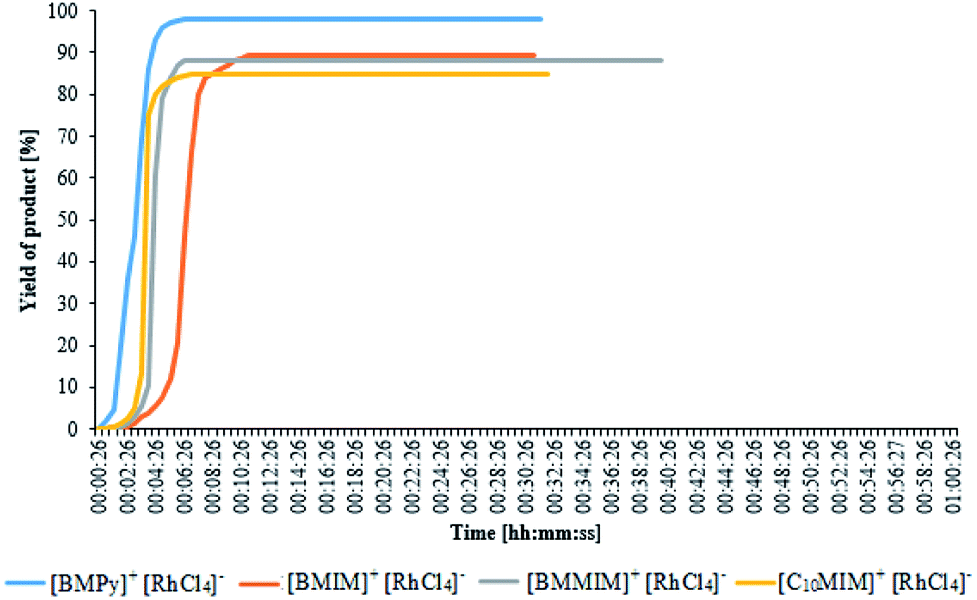
The performed measurements have shown that none of the commercially available catalysts was able to give the product with the yield greater than 37%, although they were highly active in other reactions of hydrosilylation. All the above rhodium complexes, except the simple salt RhCl3, had similar activity, therefore one can be assume that such a low yield was caused by conditions at which the reaction was conducted, namely too low catalyst concentration and too short reaction time. The above assumption was verified by a considerable increase in the yield when the reaction was performed again with tenfold greater concentration of the Wilkinson's catalyst [RhCl(PPh3)3] and twice as long time. Thereby, higher activity of anionic rhodium complexes was confirmed. Real-time monitoring of the reaction catalyzed by the latter complexes using in situ FT-IR probe enabled to determine particular reaction profiles and to show differences in their activity. Fig. 3 presents the change in hydrosilylation product yield as a function of time as observed for particular reactions catalyzed by anionic rhodium complexes (at the concentration of 0.1 mol%), conducted at 110 °C.
 | ||
| Fig. 3 The change in the hydrosilylation product yield as a function of time for the reactions catalyzed by anionic rhodium complexes. | ||
All the complexes make it possible to achieve maximum yield in a very short time. The complex [BMPy][RhCl4] enabled to obtain almost 100% yield already in the third minute of the reaction, whereas the slowest catalyst [BMIM][RhCl4] reached its maximum yield (91%) in the eighth minute. As it has been already mentioned, the complex [BMPy][RhCl4] is characterized not only by the highest yield but also by the fastest reaction rate. Of course, the reaction course is also influenced by the process temperature. The mentioned FT-IR probe was also employed in the studies of reactions catalyzed by [BMPy][RhCl4], but carried out at different temperatures and the obtained results show significant differences in their course, as can be seen in Fig. 4 and Table 3.
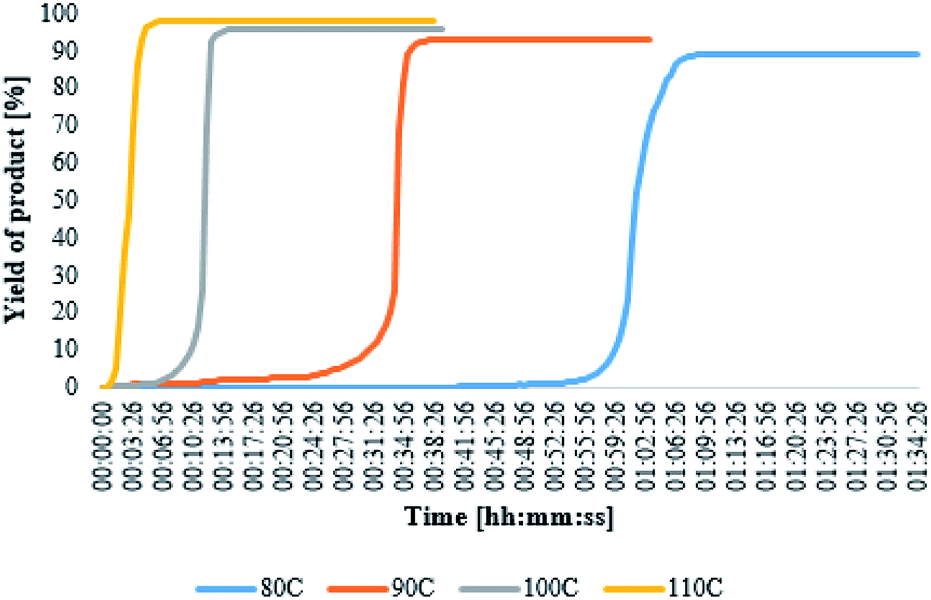 | ||
| Fig. 4 The change in the hydrosilylation product yield as a function of time as observed for the reaction catalyzed by [BMPy][RhCl4], carried out at different temperatures. | ||
| Temperature [°C] | Total yield [%] | Yield of product A [%] | Yield of product B [%] |
|---|---|---|---|
| a Reaction conditions: Rh-catalyst (0.001 mmol), acetophenone (1 mmol), HMTS (1.25 mmol), n-decene (1 mmol). | |||
| 80 | 89 | 86 | 3 |
| 90 | 93 | 90 | 3 |
| 100 | 96 | 96 | 0 |
| 110 | 98 | 98 | 0 |
The results proved that the catalyst was also active at a lower temperature, however, the reaction time has been significantly extended. At the temperature of 80 °C, the catalyst activation time is about one hour. The reaction time is shortened as the temperature rises, as shown by the activation times at 90 °C to 30 min. and 100 °C to 9 min. At 110 °C, the reaction starts almost immediately after the addition of the catalyst and no by-product formation is observed in this case.
In other cases, lower temperatures of the system, except longer reaction times, also affected the formation of the undesirable by-product. Unquestionably, all the studied catalysts are very active as evidenced by almost immediate conversion of substrates (a nearly vertical course of the curves in Fig. 2 and 3 at the moment of the reaction initiation). As concerns the effect of temperature on the reaction initiation time, one can notice that the studied catalysts are insoluble in the reaction medium, i.e. we deal with a heterogeneous catalytic reaction in the presence of a solid catalyst. Such a reaction is a complex process which consists of several steps, the nature of which is physical (diffusion of substrates and products in catalyst pores) or chemical (chemisorption of at least one substrate on the catalyst surface, surface reaction between chemisorbed species and product desorption). With a rise in temperature, both the rate of diffusion and the rate of chemisorption increase. Another factor that seems to affect the activation of the catalysts is their hydrophobicity. As results from Fig. 3, the fastest activation occurs in the case of the complexes [BMPy][RhCl4] and [C10MIM][RhCl4], whose cations are more hydrophobic (due to the presence of a long alkyl substituent or aromatic ring) and the slowest activation proceeds in the case of the complex [BMIM][RhCl4], whose cation is less hydrophobic. This permits to conclude that a greater hydrophobicity and higher temperature favor the reaction studied. Since at 110 °C the reaction starts immediately and no undesirable products are formed, the above temperature can be regarded as optimal for the reaction.
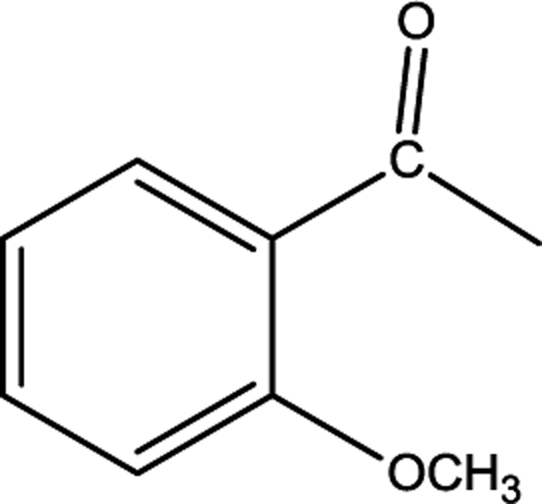
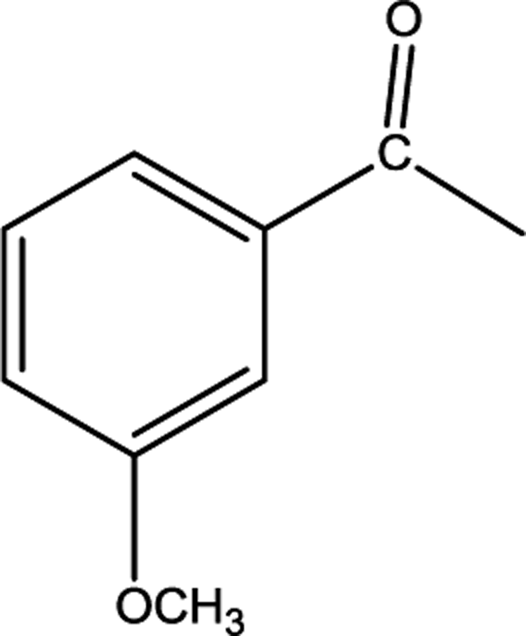
All the above studies concerned hydrosilylation of acetophenone, but at the next stage of our research we determined catalytic performance of anionic rhodium complexes in hydrosilylation of methoxyacetophenone. The effect of the position of the substituent attached to the benzene ring on the catalytic activity of the obtained catalysts was also investigated. For this purpose, acetophenone with the methoxy group in the ortho, meta and para positions was used. The yields achieved in the particular reactions are presented in Table 4.
| Ketone | Catalyst | Total yield [%] | Yields of products A/B [%] |
|---|---|---|---|
| a Reaction conditions: Rh-catalyst (0.001 mmol), acetophenone (1 mmol), HMTS (1.25 mmol), T = 110 °C, t = 1 h. Yields of the reaction carried out for 2 hours were given in parentheses. | |||
 |
[BMPy]+[RhCl4]− | 16 (96) | 16/0 (95/1) |
| [BMIM]+[RhCl4]− | 11 (16) | 11/0 (16/0) | |
| [BMMIM]+[RhCl4]− | 0 (33) | 0/0 (33/0) | |
| [C10MIM]+[RhCl4]− | 89 (100) | 89/0 (96/4) | |
 |
[BMPy]+[RhCl4]− | 100 | 100/0 |
| [BMIM]+[RhCl4]− | 36 | 36/0 | |
| [BMMIM]+[RhCl4]− | 52 | 52/0 | |
| [C10MIM]+[RhCl4]− | 67 | 67/0 | |
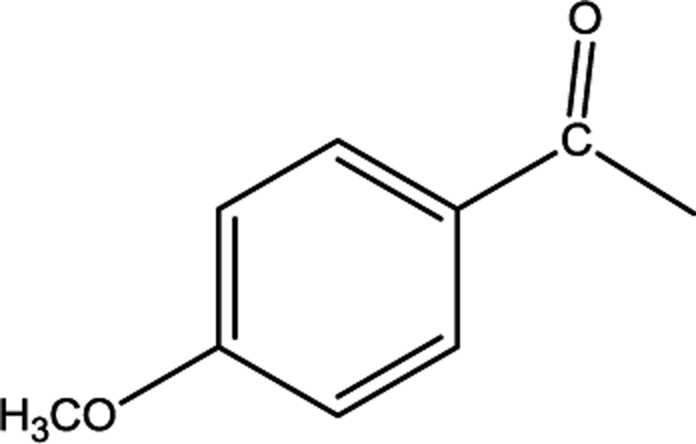 |
[BMPy]+[RhCl4]− | 85 | 81/4 |
| [BMIM]+[RhCl4]− | 24 | 16/8 | |
| [BMMIM]+[RhCl4]− | 72 | 71/1 | |
| [C10MIM]+[RhCl4]− | 82 | 80/2 | |
The obtained results show that catalytic activities of the studied complexes are even more diversified. The complex [BMPy][RhCl4] is one of the most active and selective catalysts, but also the complex [C10MIM][RhCl4] proved to be a very efficient catalyst which, as the only one, made it possible to obtain the product yield of 89% already after 1 hour of the reaction with 2-methoxyacetophenone. Other complexes required longer reaction times, albeit after 2 hours only the complex [BMPy][RhCl4] enabled the achieve the yield of 96% (in Table 4, the values in parentheses present the yields after 2 hours of reaction). Certainly the steric hindrance that makes the reaction progress difficult is the location of the methoxy substituent at the ortho position. In hydrosilylation of the two remaining derivatives having the methoxy substituent in meta and para positions, the greatest effectiveness was shown by [BMPy][RhCl4], which made it possible to obtain after 1 hour of the reaction the yields of 100 and 85%, respectively. Also the complex [C10MIM][RhCl4] enabled to achieve high yields of 67 and 82%, respectively. On the other hand, the complex [BMIM][RhCl4] proved to be definitely the poorest catalyst in all the reactions conducted. The two aforementioned complexes contain imidazole derivatives and differ only in the length of alkyl substituent attached to imidazole ring. Taking into consideration that the alkyl chain length influences hydrophobic properties, one can conclude that the highest hydrophobic properties of the catalytic system the easier the reaction proceeds. Since all the above complexes are insoluble in the reaction medium and catalysis is a surface phenomenon, then surface properties are crucial for the course of catalysis.
In the case of hydrosilylation of other acetophenone derivatives, a significant effect was observed of substituents in acetophenone on the catalytic activity of the studied complexes. Derivatives containing electron-donating and electron-withdrawing substituents were employed in the study and the obtained results showed that in the case of compounds containing electron-withdrawing substituents, namely 4-nitroacetophenone, 4-acetylbenzonitrile, 4-iodoacetophenone and pentafluoroacetophenone, no conversion of the substrates was observed in the presence of the investigated complexes. No reaction also occurred in the case of benzophenone, most likely due to a steric hindrance effect. On the other hand, a better result was obtained in the reaction conducted with the participation of 4-methylacetophenone (Table 5) which contains an electron-donating substituent, just as 4-methoxyacetophenone.
| Ketone | Catalyst | Total yield [%] | Yields of products A/B [%] |
|---|---|---|---|
| a Reaction conditions: Rh-catalyst (0.001 mmol), methylacetophenone (1 mmol), HMTS (1.25 mmol), T = 110 °C, t = 2 h. | |||
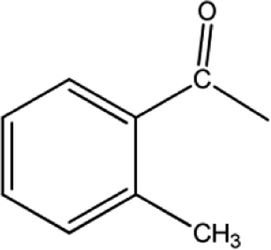 |
[BMPy]+[RhCl4]− | 56 | 54/2 |
| [BMIM]+[RhCl4]− | 14 | 12/2 | |
| [BMMIM]+[RhCl4]− | 25 | 22/3 | |
| [C10MIM]+[RhCl4]− | 51 | 51/0 | |
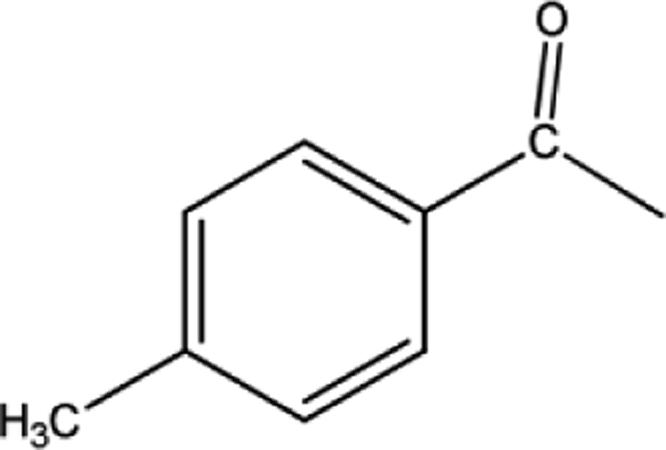 |
[BMPy]+[RhCl4]− | 71 | 71/0 |
| [BMIM]+[RhCl4]− | 23 | 19/4 | |
| [BMMIM]+[RhCl4]− | 45 | 40/5 | |
| [C10MIM]+[RhCl4]− | 67 | 65/2 | |
Also in this case one can notice a similar tendency of activity changes because the best catalysts proved to be the complexes [BMPy]{RhCl4] and [C10MIM][RhCl4]. However, the yields are lower than those obtained with methoxyacetophenone, despite the extension of the reaction time to two hours. A comparison of the two acetophenone derivatives shows that methoxy substituents have stronger electron-donating properties than methyl groups, therefore, one can state that anionic rhodium complexes catalyze reactions of acetophenone derivatives containing electron-donating substituents and the stronger the electron-donating effect, the easier the reaction proceeds. Admittedly, we have also conducted hydrosilylation of 4-aminoacetophenone (amino substituent is classified into strong electron donors), but the achieved yields were low. However, in this case the strong electron-donating effect of amino group probably caused catalyst poisoning, which is frequently observed in hydrosilylation of amino derivatives.5
As was already mentioned, all the studied catalysts are insoluble in the reaction medium, which makes it possible to separate them from post-reaction mixtures. This is why we have made an attempt at their reusing in subsequent catalytic cycles. Table 6 presents the yields acetophenone hydrosilylation products obtained in subsequent reaction runs. After each cycle, the post-reaction mixture was decanted from the solid catalyst and subjected to chromatographic analysis, while the catalyst was reused in a subsequent reaction run with a new portion of substrates.
| Catalyst | Catalytic cycle | Yields of products (A/B) [%] | ||
|---|---|---|---|---|
| 1 h | 2 h | 3 h | ||
| a Reaction conditions: Rh-catalyst (0.001 mmol), acetophenone (1 mmol), HMTS (1.25 mmol), T = 110 °C. | ||||
| [BMPy]+[RhCl4]− | 1 | 98 (98/0) | ||
| 2 | 27 (26/1) | 38 (37/1) | 46 (45/1) | |
| 3 | 6 (6/0) | 12 (10/2) | 15 (13/2) | |
| [BMIM]+[RhCl4]− | 1 | 91 (80/11) | ||
| 2 | 23 (22/1) | 41 (40/1) | 53 (52/1) | |
| 3 | 6 (5/1) | 11 (10/1) | 13 (12/1) | |
| [BMMIM]+[RhCl4]− | 1 | 89 (83/6) | ||
| 2 | 30 (27/3) | 37 (34/3) | 43 (40/3) | |
| 3 | 7 (5/2) | 14 (11/3) | 17 (14/3) | |
| [C10MIM]+[RhCl4]− | 1 | 83 (79/4) | ||
| 2 | 62 (59/3) | 66 (63/3) | 67 (64/3) | |
| 3 | 8 (8/0) | 10 (10/0) | 16 (16/0) | |
The catalytic tests showed that all the studied complexes were catalytically active in the second cycle, albeit their activity was lower than in the first cycle. Subsequent cycles required a longer reaction time, but after the third cycle the yield, even at extended time, was only on the level of a dozen percent. It is worth mentioning that the employed catalysts were in the form of a fine-crystalline powder and, despite being insoluble despite being insoluble in the reagents, they remained dispersed in post-reaction mixtures for quite a long time. Although the post-reaction mixtures were allowed to stand for a long time, placed in a refrigerator and attempts were made at centrifuging, it is possible that a part of the catalyst suspension was removed together with reaction products and this can be the reason for the yield reduction caused by a decrease in the concentration of catalyst. This is why we conducted for the two most active catalysts a subsequent reaction cycle without isolation, i.e. after the reaction completion a new portion of substrates was added. The obtained yields are presented in Table 7.
| Catalyst | Catalytic cycle | Yields of products (A/B) [%] | ||
|---|---|---|---|---|
| 1 h | 2 h | 3 h | ||
| [BMPy]+[RhCl4]− | 1 | 98 (98/0) | ||
| 2 | 82 (80/2) | 84 (82/2) | 87 (83/4) | |
| [C10MIM]+[RhCl4]− | 1 | 84 (80/4) | ||
| 2 | 62 (60/2) | 65 (62/3) | 70 (67/3) | |
The performed experiment proved that the studied complexes do not lose their activity and that there is a possibility of conducting the reaction at even lower catalyst concentration which is an additional economic asset. By using coarse–crystalline complexes or applying efficient sedimentation methods a more effective isolation of catalysts should be achieved, thus creating a possibility of their multiple use.
Further research on a better isolation of catalysts as well as on the mechanism of the reactions catalyzed by anionic rhodium complexes is underway but the studies carried out hitherto permit to designate two complexes [BMPy][RhCl4] and [C10MIM][RhCl4] (particularly the former) as very effective catalysts for hydrosilylation of ketones.
Conclusions
Synthesis of anionic rhodium complexes is very simple and enables obtaining durable air-stable complexes, due to which one can deal with them without any protection measures. The above compounds proved to be catalytically active for the process of ketone reduction. This is the first example of the application of complexes of this type to hydrosilylation of ketones. Additionally, the reactions of hydrosilylation were conducted using heptamethyltrisiloxane which is generally available and cheap compound compared to arylsilanes that were most frequently applied as hydride donors in the reactions catalyzed by rhodium complexes. Anionic rhodium complexes are insoluble in reactions medium, due to which they enable easy separation of catalyst from post-reaction mixtures. The performed tests proved that the catalysts are active in the acetophenone-HMTS system (1 mmol:1.25 mmol) even with a slight excess of siloxane. It has also been shown that the above catalytic complexes are active in the reaction of acetophenone hydrosilylation at much lower concentration (0.1 mol%) than commonly used in numerous studies reported in the literature. Moreover, there is a possibility of catalyst reuse in the next reaction cycle (both with isolation and without isolation of the catalyst). This fact allows to reduce production costs (e.g. secondary alcohols) and also affects the purity of the isolated product. Compared to standard homogeneous rhodium catalysts used in hydrosilylation reactions, this is an important advantage. It was also shown that the hydrosilylation reaction in the presence of anionic rhodium complexes in the acetophenone-HMTS system proceeds very quickly and with very good selectivity. The most effective catalyst for acetophenone reduction proved to be the complex containing the pyridine ligand [BMPy]+[RhCl4]−. In the case of hydrosilylation of methoxyacetophenone isomers visible is the effect of the presence and position of methoxy group on the reaction course and product yields. Hydrosilylation of 2-methoxyacetophenone is more difficult because of a steric hindrance which made necessary to extend the reaction time to obtain yields comparable to those achieved in the reaction of acetophenone. Nevertheless, the obtained yields were similar to those obtained with unsubstituted acetophenone only in the case of two most effective complexes, namely [BMPy][RhCl4] and [C10MIM][RhCl4]. Reactions with other methoxyacetophenone isomers having the CH3O group in the position meta or para proceed easier, however, also in this case the significant superiority of the two above complexes (which are not only the most active but also the most selective) is visible. A similar effect is observed for hydrosilylation of ortho and para isomers of methylacetophenone, however, in this case the activity of the studied complexes is lower because of a weaker electron-donating effect of methyl group, compared to methoxy group. On the other hand, it has been unequivocally established that anionic rhodium complexes effectively catalyze reactions of acetophenone derivatives with electron-donating substituents, whereas are not active for reactions of the derivatives with electron-withdrawing substituents. Summing up, the above complexes can be a very good alternative to hitherto applied ketone reduction catalysts due to their easy synthesis, stability under atmospheric conditions, high effectiveness in the range of low concentrations as well as easy isolation from post-reaction mixtures resulting from their insolubility in reaction medium. Moreover, the complexes catalyze reactions with the use of simple and cheap heptamethyltrisiloxane as a hydride donor.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the National Science Center (Poland), grant OPUS UMO-2014/15/B/ST5/04257, is gratefully acknowledged.References
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986–2012 CrossRef CAS PubMed.
- C. Hedberg in Modern Reduction Methods, ed. P. G.AndCerCson and I. J.Munslow, Wiley-VCH, Weinheim, 2008, pp. 109–134 Search PubMed.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- I. Ojima, M. Nihonyanagi and Y. Nagai, J. Chem. Soc., Chem. Commun., 1972, 938 RSC.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Hydrosilylation. A Comprehensive Review on Recent Advances, Springer, London, 2009, ch. 9 Search PubMed.
- N. J. Lawrence and S. M. Bushell, Tetrahedron Lett., 2000, 41, 4507–4512 CrossRef CAS.
- N. Asao, T. Ohishi, K. Sato and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 6931–6932 CrossRef CAS.
- F. Le Bideau, C. T. D. Gourier, J. Hanique and E. Samuel, Tetrahedron Lett., 2000, 41, 5215–5218 CrossRef CAS.
- Y. Kawanami, H. Yuasa, F. Toriyama, S. Yoshida and T. Baba, Catal. Commun., 2003, 4, 455–459 CrossRef CAS.
- S. Enthaler, K. Schröder, S. Inoue, B. Eckhardt, K. Junge, M. Beller and M. Drieß, Eur. J. Org. Chem., 2010, 25, 4893–4901 CrossRef.
- T. Inagaki, Y. Yamada, L. Thanh Phong, A. Furuta, J. Ito and H. Nishiyama, Synlett, 2009, 2, 253–256 Search PubMed.
- V. Bette, A. Mortreux, C. W. Lehmann and J. F. Carpentier, Chem. Commun., 2003, 332–333 RSC.
- W. Sattler, S. Ruccolo, M. R. Chaijan, T. N. Allah and G. Parkin, Organometallics, 2015, 34, 4717–4731 CrossRef CAS.
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498–504 CrossRef CAS PubMed.
- S. Diez-Gonzáles and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349–358 CrossRef PubMed.
- B. H. Lipshutz in Modern Organocopper Chemistry, ed. N.Krause, Wiley-VCH, Weinheim, 2002, pp. 167–187 Search PubMed.
- F. Lazreg and C. S. J. Cazin in N-Heterocyclic Carbenes, ed. S. P.Nolan, Wiley-VCH, Weinheim, 2014, pp. 199–242 Search PubMed.
- C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed.
- B. Blom, S. Enthaler, S. Inoue, R. Irran and M. Driess, J. Am. Chem. Soc., 2013, 135, 6703–6713 CrossRef CAS PubMed.
- Z. Zuo, L. Zhang, X. Leng and Z. Huang, Chem. Commun., 2015, 51, 5073–5076 RSC.
- F. S. Wekesa, R. Arias-Ugarte, L. Kong, Z. Summer, G. P. McGovern and M. Findlater, Organometallics, 2015, 34, 5051–5056 CrossRef CAS.
- A. Raya-Baron, M. A. Ortuna, P. Ona-Burgos, A. Rodriguez-Dieguez, R. Langer, C. J. Cramer, I. Kuzu and I. Fernandez, Organometallics, 2016, 35, 4083–4089 CrossRef CAS.
- K. Junge, K. Schroeder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC.
- J. J. Kennedy-Smith, K. A. Nolin, H. P. Gunterman and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 4056–4057 CrossRef CAS PubMed.
- K. A. Nolin, J. R. Krumper, M. D. Pluth, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 14684–14696 CrossRef CAS PubMed.
- H. Dong and H. Berke, Adv. Synth. Catal., 2009, 351, 1783–1788 CrossRef CAS.
- A. Heutling, F. Pohlki, I. Bytschkov and S. Doye, Angew. Chem., Int. Ed., 2005, 44, 2951–2954 CrossRef CAS PubMed.
- K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613–621 CrossRef CAS.
- M. Mauduit and H. Clavier in N-Heterocyclic Carbenes in Synthesis, ed. S. P.Nolan, Wiley-VCH, Weinheim, 2006, pp. 207–211 Search PubMed.
- I. Ojima, M. Nihonyanagi, T. Kogure, M. Kumagai, S. Horiuchi, K. Nakatsugawa and Y. Nagai, J. Organomet. Chem., 1975, 94, 449–461 CrossRef CAS.
- K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613–621 CrossRef CAS.
- H. Brunner and P. Brandl, J. Organomet. Chem., 1990, 390, c81–c83 CrossRef CAS.
- M. J. Burk and J. E. Feaster, Tetrahedron Lett., 1992, 33, 2099–2102 CrossRef CAS.
- A. C. Fernandes, R. Fernandes, C. C. Ramao and B. Royo, Chem. Commun., 2005, 213–214 RSC.
- S. Gülcemal, A. Labande, J. C. Daran, B. Çetinkaya and R. Poli, Eur. J. Inorg. Chem., 2009, 1806–1815 CrossRef.
- G. Lázaro, F. J. Fernández-Alvarez, J. Munárriz, V. Polo, M. Iglesias, J. J. Pérez-Torrente and L. A. Oro, Catal. Sci. Technol., 2015, 5, 1878–7887 RSC.
- H. Kaur, F. Kauer Zinn, E. D. Stevens and S. P. Nolan, Organometallics, 2004, 23, 1157–1160 CrossRef CAS.
- B. Yigit, M. Yigit and I. Ozdemir, Inorg. Chim. Acta, 2017, 467, 75–79 CrossRef CAS.
- Ionic Liquids – Industrial Applications to Green Chemistry, ed. R. D.Roger and K. R.Seddon, ACS, Washington, 2002 Search PubMed.
- D. J. Adams, P. J. Dyson and S. I. Travener, Chemistry in Alternative Reaction Media, Wiley, Hoboken, 2004 Search PubMed.
- Catalysis in Ionic Liquids: From Catalyst Synthesis to Application, ed. C.Hardacre and V.Parvulescu, RSC Catalysis Series, 2014 Search PubMed.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44–60 CrossRef CAS.
- H. Maciejewski, K. Szubert, B. Marciniec and J. Pernak, Green Chem., 2009, 11, 1045–1051 RSC.
- H. Maciejewski, K. Szubert, R. Fiedorow, R. Giszter, M. Niemczak and J. Pernak, Appl. Catal., A, 2013, 451, 168–175 CrossRef CAS.
- W. Zieliński, R. Kukawka, H. Maciejewski and M. Śmiglak, Molecules, 2016, 21, 1115–1125 CrossRef PubMed.
- H. Maciejewski, M. Jankowska-Wajda, I. Dąbek and R. Fiedorow, RSC Adv., 2018, 8, 26922–26927 RSC.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- W. Lee, H. B. Jang, J. S. Chun, C. E. Song and S.-G. Lee, Acc. Chem. Res., 2010, 43, 985–994 CrossRef PubMed.
- J. Estager, J. D. Holbrey and M. Swadźba-Kwaśny, Chem. Soc. Rev., 2014, 43, 847–886 RSC.
- H. Maciejewski, M. Jankowska-Wajda, O. Wolna, Pol. Pat. Appl. P. 417232, 2017.
- B. Marciniec, P. Krzyżanowski and M. Kubicki, Polyhedron, 1996, 15, 1–4 CrossRef.
- C. Zhong, T. Sasaki, A. Jimbo-Kobayashi, E. Fujiwara, A. Kobayashi, M. Tada and Y. Iwasawa, Bull. Chem. Soc. Jpn., 2007, 12, 2365–2374 CrossRef.
- K. Garces, R. Lalrempuia, V. Polo, F. J. Fernández-Alvarez, P. Garcia-Orduna, F. J. Lahoz, J. J. Pérez-Torrente and L. A. Oro, Chem.–Eur. J., 2016, 22, 1–14 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |