
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in supported molecular sieve catalysts with wide temperature range for selective catalytic reduction of NOX with C3H6
Junqiang Xu
 a,
Honglin Wang
a,
Fang Guo
*a,
Chuan Zhang
a and
Jiaqing Xie
b
a,
Honglin Wang
a,
Fang Guo
*a,
Chuan Zhang
a and
Jiaqing Xie
b
aSchool of Chemistry and Chemical Engineering, Chongqing University of Technology, Chongqing 400054, China. E-mail: guofang@cqut.edu.cn; Fax: +86-23-62563460; Tel: +86-23-62563460
bSchool of Chemical and Environmental Engineering, Sichuan College of Technology, Zigong 643000, China
First published on 8th January 2019
Abstract
NOX is a major atmospheric pollutant that is emanated by motor vehicles, thermal power plants, and industrial boilers. Therefore, the removal of NOX is a research hotspot in the exhaust gas treatment field. Numerous methods have been used to eliminate NOX: the selective catalytic reduction of NOX using C3H6 as the reducing agent (C3H6-SCR) is an effective method to remove NOX. The key issue in NOX removal in C3H6-SCR is to obtain catalysts with low-temperature activity and wide operating temperatures. Till date, different supported wide-temperature-active molecular sieve catalysts have been prepared and used in C3H6-SCR reactions. Studies have shown that the catalytic performance of supported catalysts is related not only to the active component but also to the structural and textural parameters of the molecular sieve supports. This review summarizes the structural and textural characteristics, catalytic properties, and catalytic mechanism of molecular sieve catalysts with different pore structures for C3H6-SCR reactions. The design strategies of supported molecular sieve catalysts are suggested. The goal of this review is to highlight (1) the structural and textural characteristics and low-temperature catalytic performance of different supported molecular sieve catalysts; (2) the relationship between wide-temperature window and loaded active components, as well as carriers of the supported molecular sieve catalysts; and (3) design strategies and development prospects of supported molecular sieve catalysts with low-temperature activity and wide-temperature operating range for C3H6-SCR reactions.
 Junqiang Xu | Junqiang Xu was born in Sichuan, China, in 1979. He obtained his PhD from Sichuan University, Sichuan, China, in 2007 under the supervision of Prof. W. Chu. Now, he is working at the Chongqing University of Technology as Professor. His research interests include energy, chemical, and environmental chemical industries. |
 Honglin Wang | Honglin Wang was born in Chongqing, China, in 1994. He completed his B.S. degree in the Chongqing University of Technology, Chongqing, China, in 2017 under the supervision of Prof. J. Q. Xu. Now, he is studying for his M.S. degree in the Chongqing University of Technology under the supervision of Prof. J. Q. Xu. His field of study is the syntheses and applications of zeolite catalysts for automobile exhaust degradation. |
 Fang Guo | Fang Guo was born in Sichuan, China, in 1982. She received her B.S. degree in 2005; M.S. degree, in 2008; and PhD, in 2011, from Sichuan University, Sichuan, China, under the supervision of Prof. W. Chu. Now, she is working at the Chongqing University of Technology as Associate Professor. Her research interests include design, development, and applications of new catalytic materials. |
 Chuan Zhang | Chuan Zhang was born in Chongqing, China, in 1992. He received his B.S. degree in 2015 and his M.S. degree in 2018 from the Chongqing University of Technology, Chongqing, China, under the supervision of Prof. J. Q. Xu. Now, he is working at China Coal Science and Engineering Group. His field of study is the syntheses of new catalytic materials and their applications in air pollution control. |
 Jiaqing Xie | Jiaqing Xie was born in Sichuan, China, in 1959. He completed his PhD in Sichuan University, Sichuan, China, in 2004 under the supervision of Prof. X. C. Zeng. Now, he is working at the Sichuan College of Technology as Professor. His research interests include catalytic chemistry, simulated biocatalysts, and nanomaterials. |
1 Introduction
Nitrogen oxides (NOX) in the atmosphere are mainly composed of NO (>90%) and NO2, which mainly come from mobile sources (motor vehicles) and stationary sources (thermal power plants and industrial boilers). Further, they are the main cause for the generation of acid rain and photochemical smog. In order to reduce atmospheric pollution, a large number of researchers have dedicated their focus toward the development of de-NOX technology for high efficiency and low cost. Two methods, NH3-SCR and HC-SCR, have been commonly used to remove NOX. HC-SCR (main component: C3H6) has become an attractive method in the deNOX of mobile sources because hydrocarbon compounds (HC) are nontoxic and coexist with NOX in vehicle exhaust. Among the reported HC-SCR reactions,1–6 reducing agents mainly contain the hydrocarbons of C1, C2, C3, and C4, and universal catalysts mainly include metal oxide catalysts, metal-containing molecular sieve catalysts, and active carbon catalysts.In the 1990s, Iwamoto group and Held group7,8 reported that C3H6 and other olefins could efficiently and selectively reduce NO using the supported molecular sieve catalysts of Cu-ZSM-5 zeolites under an atmosphere of excess oxygen. Currently, numerous supported molecular sieve catalysts have been widely used in HC-SCR reactions due to their large specific surface area, strong adsorptivity, high selectivity, excellent shape selective catalytic performance, relatively wide active temperature range, etc.9,10 The carriers of supported molecular sieve catalysts mainly include ZSM-5, SAPO, SBA, and Al2O3. Further, the loaded metals of catalysts are usually transition metals, precious metals, and so on.
Several reviews regarding the composition and structure of catalysts and the catalytic mechanism of NH3-SCR have been reported in the literature,11–14 but reviews involving the catalytic action of different molecular sieve catalysts have not been reported for HC-SCR so far. This paper reviews the development of supported molecular sieve catalysts for C3H6-SCR. This review mainly (1) summarizes the actions of the carriers (Al2O3 and other carriers) and loaded metals (transition metal, rare earths, or precious metals) and the synthesis methods of the supported molecular sieve catalysts with different pore sizes, (2) evaluates the structural characteristics and catalytic performances of supported molecular sieve catalysts with different compositions and pore sizes, (3) discusses the mechanism of C3H6-SCR over supported molecular sieve catalysts, and (4) attempts to highlight the developing orientations involving supported molecular sieve catalysts with superior low-temperature activity and wider operating temperatures.
To facilitate the understanding of abbreviations in this paper, the nomenclature of the abbreviations is summarized in Table 1.
| Abbreviation | Nomenclature |
|---|---|
| SCR | Selective catalytic reduction |
| C3H6-SCR | SCR of NOX using C3H6 as the reducing agent |
| NTP | Nonthermal plasma |
| C3H6–CO-SCR | SCR with C3H6 and CO as the reducing agents |
| De-NOX | Denitration |
| NH3-SCR | SCR of NOX using NH3 as the reducing agent |
| HC-SCR | SCR of NOX using HC as the reducing agent |
| NH3-TPD | Temperature-programmed desorption with ammonia |
| NO-TPD | TPD with NOX |
| H2-TPR | Temperature-programmed reaction with H2 |
| TPO | Temperature-programmed oxidation |
| CxHyOz | Hydrocarbon oxide |
| CxHyOzNt | Nitrogen-containing hydrocarbon oxide |
| REOs | Rare earth oxides |
2 Microporous molecular sieve catalysts
In the 1970s, Mobil Corporation, United States, first synthesized the ZSM-5 molecular sieve with good thermal stability, short pore length, and uniform distribution, and it was widely used as a catalyst carrier. Since then, different types of microporous molecular sieve catalysts have rapidly become a research hotspot.2.1 ZSM-5-type zeolite catalysts
In order to study the effects of different supported metals on a catalyst, Li et al.9 prepared three catalysts (Cu-ZSM-5, In-ZSM-5, and La-ZSM-5) using H-ZSM-5 as the carrier and Cu, In, and La as the active components. The catalytic reaction results showed that Cu-ZSM-5 exhibited the best catalytic performance in the case of the three catalysts for C3H6-SCR, and the conversion of NO in C3H6-SCR reached above 70% over Cu-ZSM-5 in the temperature range of 375–500 °C. The DRIFTS results (Fig. 1) indicate that the competitive adsorption between NO–O2 and C3H6–O2 occurs on the Brønsted acid site of the Cu-ZSM-5 catalyst, and the amines (–NH2) adsorbed on the Cu-ZSM-5 catalyst surface are the main reaction intermediates that can react with NO or NO2 to produce the final product N2.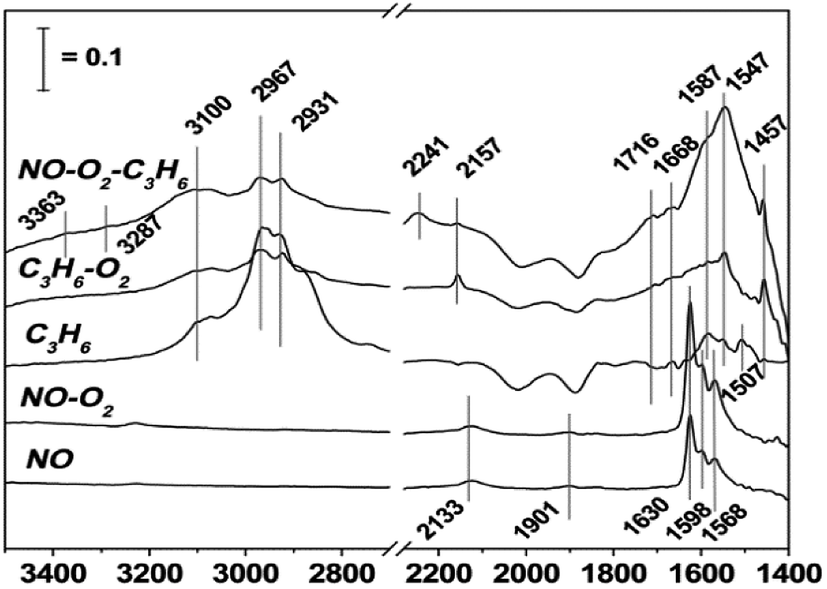 | ||
| Fig. 1 DRIFTS study of the adsorbed species formed on the surface of the Cu-ZSM-5 catalyst for NO reduction by propene under reaction conditions at 400 °C.9 | ||
Schuricht et al.10 prepared the Ag-ZSM-5 catalyst (6 wt%) by the impregnation (IM) method using different Si/Al ratios of H-ZSM-5 zeolite as the carrier, and they studied the catalytic function of the catalyst. The results showed that NO was effectively reduced to N2 over the catalyst in the temperature range of 380–600 °C in the C3H6-SCR reaction, and the NO conversion rate was over 50%. The higher the Si/Al ratio in the ZSM-5 support, the better is the catalyst activity. This can be attributed to the formation of metallic silver clusters and metallic nanosilver particles at higher metal loadings. These clusters and particles promote the quantitative conversion of NO to N2O, and therefore, enhance the activation of the catalyst and NO conversion rate in C3H6-SCR reactions. UV-vis analysis showed that Ag+ is the main species in the Ag–H-ZSM-5 catalyst and is highly dispersed.
Seo et al.15 prepared a supported microporous zeolite catalyst Cu-ZSM-5 by the ion-exchange method and used ZrO2-modified ZSM-5 as the carrier. The results of the C3H6-SCR reaction using this catalyst showed that the NO and NO2 conversion rate was over 50% at 200–500 °C when the ZrO2 content in the catalyst was 2 wt%. These results suggest that the ZrO2 promoter promotes the formation of Cu2+, which favors the storage of NH3 in NH3-TPD, and therefore, improves the acidity of the SCR catalyst, thereby enhancing the catalytic performance of the catalyst. The TEM results (Fig. 2) show that the 2 wt% ZrO2 promoter did not agglomerate on the catalyst, which increased the dispersion of the active component (Cu) on the catalyst.
 | ||
| Fig. 2 TEM images of aged Cu-ZSM-5-ZrO2 catalysts: (a) Cu-ZSM-5-ZrO2 (0 wt%) and (b) Cu-ZSM-5-ZrO2 (2 wt%).15 | ||
Komvokis et al.16 prepared Cu-ZSM-5 by the ion-exchange method and then prepared the CeO2-coated CeO2–Cu-ZSM-5 catalyst by loading CeO2 onto the surface of the Cu-ZSM-5 catalyst. The results of the C3H6-SCR reaction showed that the NO conversion rate was above 50% over the CeO2-coated Cu-ZSM-5 catalyst in the temperature range of 330–390 °C. The highest conversion of NO was increased by 40% as compared to the conversion of uncoated Cu-ZSM-5 at 350 °C. This can be attributed to the synergistic effect of Cu–Ce on the catalyst. The presence of a Ce promoter significantly enhanced the reducibility and electron-giving ability of Cu. XANES-TPR (Fig. 3) also confirmed the synergistic effect between the two metals.
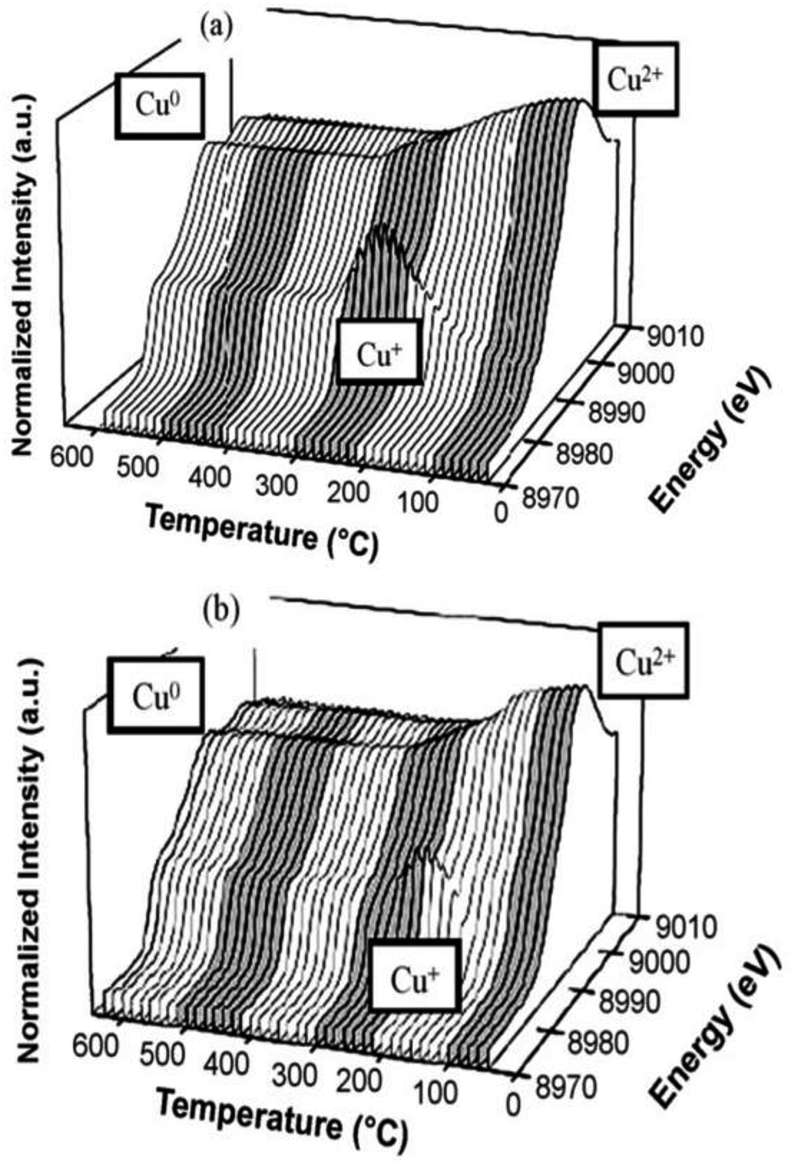 | ||
| Fig. 3 TPR of (a) Cu-ZSM-5 and (b) CeO2–Cu-ZSM-5 as detected by the CuK edge in XANES.16 | ||
Wang et al.17 prepared the La–Cu-ZSM-5 catalyst using a solution ion-exchange method and then obtained the La–Cu-ZSM-5/cordierite catalyst by loading La–Cu-ZSM-5 on cordierite. The results of the selective catalytic reduction with C3H6 and CO as the reducer (C3H6–CO-SCR) indicated that the conversion rates of NO and NO2 reached over 50% in the temperature range of 210–450 °C, and the maximum conversion rate reached 80% at 350 °C under the action of the La–Cu-ZSM-5/cordierite catalyst. In situ FTIR (Fig. 4) spectral results revealed that the 2250 cm−1 band belonged to isocyanate—an important intermediate in the C3H6-SCR reaction. The introduction of CO promoted the formation of reaction intermediates and enhanced the ability of C3H6 activation on La–Cu-ZSM-5/cordierite, and therefore, increased the NO and NO2 conversion rates in C3H6-SCR reactions.
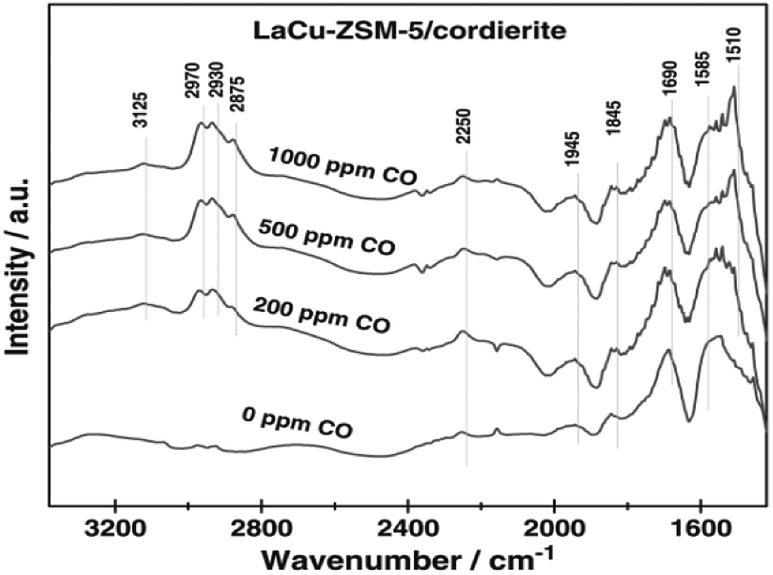 | ||
| Fig. 4 In situ FTIR spectra of the adsorbed species formed on La–Cu-ZSM-5/cordierite at 523 K.17 | ||
The results of the above studies indicate that the microporous molecular sieve catalysts with ZSM-5 as the carrier are mostly prepared by the ion-exchange and IM methods. The carrier active components on the catalysts are mainly Cu, Ag, and Fe. Here, Cu is the best active component to remove NO. The SCR catalytic activity of the molecular sieve catalyst can be improved by loading metal auxiliary agents on the catalyst. For example, the Zr promoter can improve the catalytic activity of the catalyst by adjusting the acidity of the catalyst; additive in the form of Ce enhances the reducibility of Cu through the synergistic effect with Cu, thereby improving the denitration performance of the catalyst. Additive in the form of La can promote the formation of intermediate isocyanates by interacting with Cu, and therefore, enhance the SCR performance of the catalysts. Auxiliaries can also enhance the low-temperature activity of the catalyst through synergy. The ZSM-5 carrier has good ion-exchange ability, high hydrothermal stability, suitable surface acidity, and high shape selectivity. Further, it has a stable structure of a five-membered ring, which favors the interaction with metal active components and auxiliaries. Further, a suitable Si/Al ratio allows the active components and auxiliaries to form highly dispersed isolated ionic active sites, thereby improving the catalytic function of the catalyst for denitrification. However, the microporous structure of the ZSM-5 carrier is not conducive toward the mass transfer of the macromolecules in the reaction, and they can easily lead to the formation of carbon deposits in the pore, thereby reducing the catalytic performance of the catalyst.
2.2 Beta-zeolite catalysts
Pan et al.18 prepared Fe-Beta zeolite catalysts by the IM method with NH4-Beta (Si/Al = 25) as the carrier. The C3H6-SCR reaction results showed that the NO and NO2 conversion rates reached more than 50% over the catalyst in the temperature range of 290–390 °C, and the maximum conversion rate reached 75% at 350 °C. This is attributed to the strong adsorption for C3H6 and the high Fe2+/Fe3+ ratio on the Fe-Beta zeolite catalyst surface. The XPS and H2-TPR results (Fig. 5) confirmed that Fe3+ was almost reduced to Fe2+ on the Beta carrier, which favors the activation of C3H6, and therefore, promoted the denitrification in C3H6-SCR.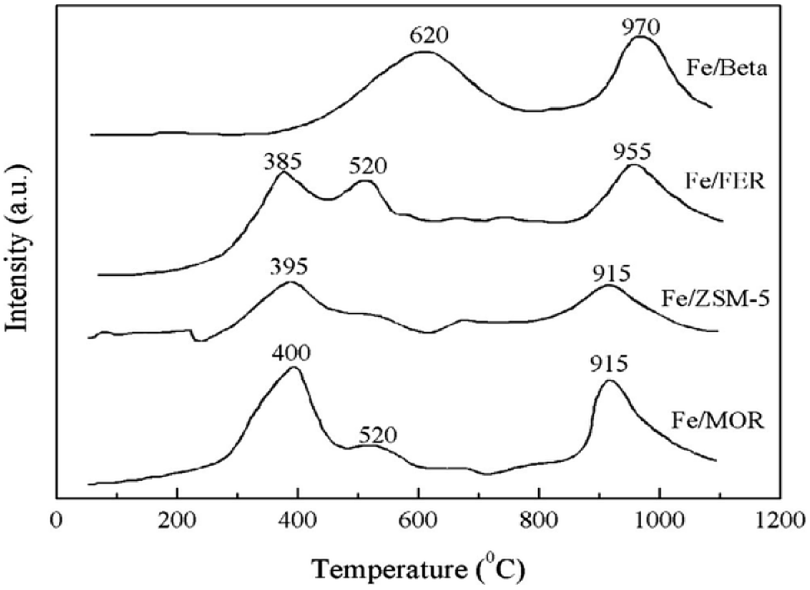 | ||
| Fig. 5 TPR profile of Fe-zeolite catalysts.18 | ||
In order to improve the low-temperature activity of Beta-zeolite catalysts, Cortés et al.19 prepared a Pt-Beta zeolite catalyst by the ion-exchange method with NH4-β (Si/Al = 11.4) as the carrier. The results of the C3H6-SCR reaction using this catalyst showed that the NO and NO2 conversion rates exceeded 50% in the temperature range of 210–260 °C, and the maximum conversion rate was 75% at 220 °C. The XPS and FTIR results reveal that the interaction between Pt and Beta zeolite promoted the adsorption and activation of C3H6 and formed the CxHyOz reaction intermediate, which promoted the denitrification of the catalyst in C3H6-SCR.
In addition, Nejar et al.20 also prepared zeolites K–Cu-Beta and K–Co-Beta with a Beta-zeolite carrier. The C3H6-SCR results revealed that the conversion rates of NO and NO2 on the K–Co-Beta catalyst exceeded 50% in the temperature range of 330–500 °C, and the conversion rates of NO and NO2 on the K–Cu-Beta catalyst exceeded 80% in the temperature range of 300–500 °C. The H2-TPR and XPS results revealed that K enhanced the reduction of CuO. The main peak at 933.6 eV corresponds to CuO; this peak shifts to lower BE due to the interaction of Cu with an electropositive element, such as K. Further, the shoulder at around 936.0 eV can be assigned to ionic-exchanged Cu2+ on the Beta-zeolite or, at least, to CuO with a more intense interaction with the Beta-zeolite support. Therefore the synergistic effect of K–Cu promoted the strong interaction between Beta-zeolite and Cu species (Cu2+, CuO), significantly improving the redox performance of Cu species on K–Cu-Beta catalyst, and therefore, enhances the catalytic performance of the catalyst. For Co-based catalysts, a similar relation between the K content and catalysts activity is observed, but the effect of K is less pronounced than that of Cu catalysts.
From the results of the above literatures, it can be observed that nonnoble metal active components supported by beta molecular sieves are mainly Cu, Fe, and Co in the catalysts, among which the preferred active component is Cu. Beta-zeolite is a unique three-dimensional zeolite with a twelve-membered ring channel and strong acidic characteristic, which can enhance the NOX-adsorbing capability on the surface of the catalyst and the synergy between the carrier and active component, thereby significantly enhancing the redox properties of the active component species, and therefore, improving the catalytic performance of the catalyst. However, its crystal structure is very complex and easily leads to the formation of carbon deposits during the reaction process.
2.3 SAPO-34 molecular sieve catalysts
Li et al.21 fabricated SAPO-34 molecular sieves with different Si/Al ratios by the hydrothermal synthesis method and then prepared the Cu-SAPO-34 catalyst by the ion-exchange method using SAPO-34 as the carrier. The C3H6-SCR results revealed that the greater the Si/Al ratio, the better is the SCR activity. In the presence of the catalyst with a Si/Al ratio of 0.8, the NO conversion increased 5–30% at 300–450 °C (as compared to Si/Al = 0.4) and NO conversion reached 70% at the highest point in the C3H6-SCR results. SEM (Fig. 6) and NH3-TPD results revealed that the molecular sieve (Si/Al = 0.8) had the best morphology regularity and smoothness on the particle surface, and the particles were more evenly dispersed on the catalyst, thereby increasing the number of effective acid sites on the molecular sieve skeleton and acid strength, which favors the increase in the denitrification of the catalyst in C3H6-SCR. | ||
| Fig. 6 SEM images of SAPO-34 zeolites with different Si/Al ratios.21 | ||
In order to compare the catalytic functions of the catalysts prepared by different synthesis methods, Zuo et al.22 adopted hydrothermal synthesis, pore volume IM, and ion-exchange methods to yield Cu-SAPO-34 with SAPO-34 as the carrier. The C3H6-SCR reaction results revealed that the conversion rate of NO on the catalysts prepared by the ion-exchange method increased by 5–22% and 10–30% when compared with the catalysts prepared by the pore volume IM and hydrothermal syntheses, respectively, in the temperature range of 100–300 °C. The SEM results revealed that the SAPO-34 support had a certain degree of regularity and roughness on its crystal structure, and the active constituent (Cu) on the catalyst prepared by the ion-exchange method exhibited high dispersity, thereby providing more active sites, and therefore, improving the catalytic performance of the catalyst.
Furthermore, Lv23 used hydrothermal synthesis to fabricate SAPO-34 molecular sieves with different silicon sources and then prepared the Cu-SAPO-34 catalyst by the ion-exchange method. The C3H6-SCR reaction results revealed that the NO conversion rate on the catalyst prepared using a silica sol as the silicon source reached over 50% in the temperature range of 325–450 °C, and the maximum conversion rate reached 70% at 350 °C. As compared to the catalyst prepared using ethyl orthosilicate as the silicon source, NO conversion increased by 10% over the catalyst in the temperature range of 350–450 °C. The SEM and DRIFTS results revealed that the interaction between the SAPO-34 molecular sieve and the active components of the catalyst prepared with the silica sol as the silicon source promoted the formation of the reaction intermediate –CN, which favored the production of more N2, and therefore, promoted SCR activity.
The above research results show that the main active component supported by SAPO-34 molecular sieve catalyst is mainly Cu; the catalyst preparation method can influence the dispersion degree of the active component supported by SAPO-34, and therefore, change the catalytic performance and catalytic temperature range of the catalyst. The silicon source and Si/Al ratio of the SAPO-34 molecular sieve are also important factors affecting the catalytic performance of the catalyst. The SAPO-34 molecular sieve has intermediate strength with regard to the acid center, a unique pore structure, a large number of micropores, and a high specific surface area; therefore, the catalyst with a SAPO-34 molecular sieve yields additional active sites and promotes interactions between the SAPO-34 molecular sieve and the active component, and therefore, enhances the catalytic performance of the catalyst. However, the synthesis of SAPO-34 molecular sieve is very complicated, such as raw materials, templating agents, and other variable factors; therefore, it is very important to screen suitable synthesis conditions during the synthesis of pure phase and high-crystallinity molecular sieves.
2.4 SSZ-13 molecular sieve catalysts
Raj et al.24 studied the C3H6-SCR reaction on a Cu-SSZ-13 monolithic catalyst using a small-scale flow reactor. The results showed that the NO conversion rate reached over 50% at 380–550 °C under the action of the catalyst, and the highest value obtained was 90%. The DRIFTS results showed that the partially oxidized C3H6 in the hydrocarbon reacted with NO to form the intermediate ANCO, and the intermediate reacted with O2 to form N2, thereby promoting the conversion of NO under catalytic action within a certain temperature range.In addition, Kim et al.25 studied the C3H6-SCR reaction on the Cu-SSZ-13 catalyst using dynamic adsorption and TPD (Fig. 7) methods. The results revealed that there were two different adsorption energies of 4.4 kJ mol−1 and 70.9 kJ mol−1 in the temperature range of 72–544 °C. Therefore, the Cu-SSZ-13 catalyst exhibited a significant adsorption capacity for propylene and was favorable toward the SCR activity of the catalyst.
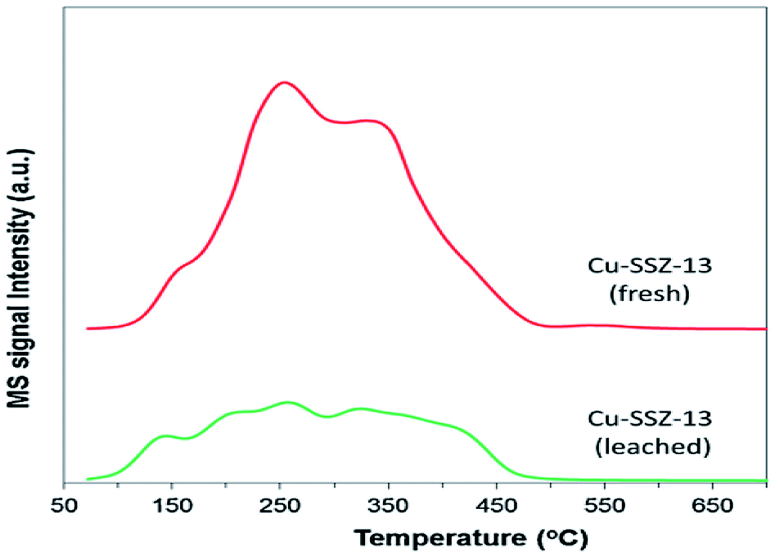 | ||
| Fig. 7 C3H6-TPD profiles of fresh and leached Cu-SSZ-13 catalysts.25 | ||
Raj et al.26 investigated the C3H6-SCR reaction on the Cu-SSZ-13 monolithic catalyst using a benchtop flow reactor. The reaction results revealed that the NO2 conversion rate reached over 80% in the temperature range of 260–550 °C. The DRIFTS results indicated that the interaction of the active component (Cu) with the SSZ-13 support enhanced the selective reaction of NO2 with the oxygen-containing compound, forming a further CxHyOzNt compound. This promoted the formation of the reaction intermediate –NCO and further generation of additional N2.
Studies involving SSZ-13 molecular sieve catalysts are rare so far, and the most common supported active component of the catalyst is Cu. The catalytic performance of the SSZ-13 molecular sieve catalyst is higher than that of the other types of microporous molecular sieve catalysts. The SSZ-13 molecular sieve has an ordered pore structure, with additional surface protonic acidic sites and exchangeable cations. In the C3H6-SCR reaction, the reaction activities of NOX and oxygenated hydrocarbons is enhanced by the strong interaction between the SSZ-13 carriers and the active component (Cu), thereby promoting the formation of the intermediates ANCO and –NCO, which generates additional N2. However, it is difficult to control its acidity and the problem of carbon deposition under high-temperature conditions.
Microporous molecular sieves have large specific surface areas, high hydrothermal stability, and suitable surface acidity; therefore, the support catalyst on the sieves can effectively remove NOX in C3H6-SCR. Nevertheless, the pore structure of the microporous molecular sieves is small and complex, which limits the mass transfer in the SCR reaction, which facilitates the formation of carbon deposits and sulfides in the pores of microporous molecular sieves; this results in the deactivation of the catalyst. Therefore, investigating zeolite catalysts with larger pore size is very important for improving the catalytic performance of the catalyst.
3 Mesoporous molecular sieve catalysts
In 1992, Mobil Petroleum Corporation first synthesized M41s mesoporous molecular sieves, which opened up a new era in the study of mesoporous molecular sieves and supported catalysts.3.1 SBA-type catalysts
SBA-15 is usually used as a mesoporous molecular sieve carrier. Liu et al.27 used the IM method to prepare the Pt-SBA-15 catalyst with SBA-15 as the carrier. The C3H6-SCR results showed that the Pt-SBA-15 catalyst had good catalytic performance in the temperature range of 130–340 °C, and the conversion rates of NO and NO2 in the presence of the catalyst exceeds 50%. This can be attributed to the interaction between the SBA-15 support and the active component (Pt) at lower temperatures. This action promoted the redox properties of the Pt species and enhanced the activation of C3H6 and adsorption rates of NO and NO2. Therefore, the Pt-SBA-15 catalyst exhibited high catalytic function in C3H6-SCR. The XPS results revealed that Pt, PtO, and PtO2 were present in the catalyst, and PtO2 may be the main active center of Pt-SBA-15. NO-TPD (Fig. 8) showed that the lowest desorption amount on Pt-SBA-15 was three times the maximum desorption amount on SBA-15 in the temperature range of 85–260 °C, which suggested that the Pt loaded on SBA-15 favored the adsorption of NO and NO2. The study results also revealed that the introduction of Al assistant suitably enhanced the catalytic function of Pt-SBA-15. The maximum conversion rates of NO and NO2 reached 86%, which, for Pt–Al-SBA-15, is 6% greater than that for pure Pt-SBA-15 at 140 °C. It is believed that the AlO4 tetrahedral coordination in the Pt–Al-SBA-15 catalyst enhanced the surface acidity of the catalyst, thereby improving the catalytic performance and stability of the catalyst.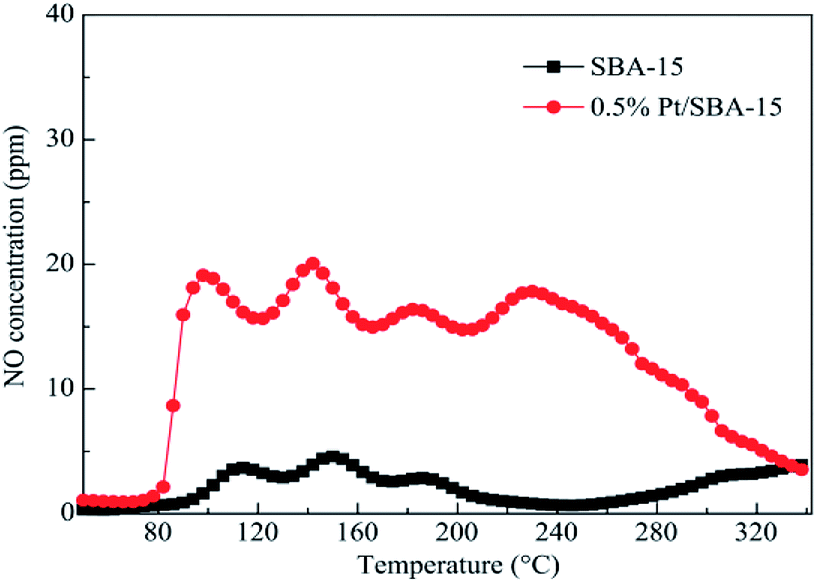 | ||
| Fig. 8 NO-TPD profiles of SBA-15 and 0.5% Pt-SBA-15.27 | ||
In order to study the effect of different supported metals and bimetallic synergistic effect on the catalyst, Zhang et al.28 prepared a series of M-SBA-15 and M2-SBA-15 catalysts using a single metal M (M = Cu, Fe, Cr, or Al) or bimetallic M2 (M2 = Cu–Al, Fe–Al, or Cr–Al) as the active component. The C3H6-SCR results revealed that the Fe-SBA-15 catalyst exhibited the best catalytic performance in the four single-metal catalysts; the NO conversion rate exceeded 50% in the temperature range of 410–600 °C and the maximum reached 90% at 600 °C in the presence of Fe-SBA-15. For the bimetallic catalysts, the Cu–Al-SBA-15 catalyst exhibited the best catalytic function; the NO conversion rate exceeded 50% in the temperature range of 330–600 °C, and the maximum conversion rate was close to 100% at 450 °C in the presence of the Cu–Al-SBA-15 catalyst. The results indicated that the bimetallic synergistic effect enhanced the maximal conversion rate of NO and reduced the optimum catalytic temperature in C3H6-SCR. The H2-TPR results further confirmed that the Cu–Al-SBA-15 catalysts exhibited better redox properties as compared to Fe-SBA-15, and therefore, have superior catalytic performance with regard to SCR.
Sabbaghi et al.29 fabricated SBA-15 molecular sieves by hydrothermal synthesis and the Co–Zr-SBA-15 catalyst by the IM method using SBA-15 as the carrier. The C3H6-SCR results revealed that the NO conversion rate exceeded 50% in the presence of the catalyst within the temperature range of 330–375 °C. The XRD and XPS results reveal that the active components, namely, Zr and Co, were uniformly dispersed on the surface of the support, which enhanced the interaction between the active component and the support, as well as promoted the synergistic redox properties of Zr and Co. This facilitated the improvement in the surface acidity of the catalyst and enhanced the catalytic performance of the catalyst.
The above studies indicate that the commonly supported active components of the SBA-15 catalyst are Fe, Pt, Cu, and Co, and the SCR catalytic activity of the catalyst can be improved by the synergistic interactions between the promoter and the polyactive center in the bimetallic catalyst. Further, the synergy between the multiple active sites can reduce the activation temperature of the catalyst. The SBA-15 molecular sieve has a two-dimensional hexagonal pore structure, and the silica dioxide on the skeleton is generally amorphous. This kind of molecular sieve has a larger surface area, uniform pore diameter distribution, and adjustable pore diameter, which is favorable for the uniform dispersion of the active component on the surface of the carrier, thereby enhancing the interaction between the active component and molecular sieve, as well as promoting the redox performance of the active component. This improves the catalytic performance of the catalyst. However, there are only a few acid active sites in the skeleton, and the problem of poor ion-exchange capacity is more prominent.
3.2 MCM-type catalyst
Kim et al.30 first synthesized a MCM-41 molecular sieve support by hydrothermal synthesis and then prepared the Pt-MCM-41 catalyst by the IM method. The C3H6-SCR reaction results revealed that the NO conversion rate exceeded 50% in the presence of the catalyst within the temperature range of 150–500 °C, and they reached the maximum conversion rate of 79% at 225 °C. The good catalytic performance of the Pt-MCM-41 catalyst in a wide temperature range could be attributed to the good redox performance of the Pt species and the better adsorption of NO on the catalyst surface. In the reduction process, ONO and NCO intermediates are produced, which then react with O2 to generate additional N2. The XPS and DRIFTS results further confirm that Pt had a strong redox performance in the reaction; PtO2 species were the main active sites and promoted the formation of the NO2, ONO, and NCO intermediates in the reaction through the partial oxidation mechanism.Wan et al.31 used the hydrothermal synthesis method to fabricate the Cu–Al-MCM-41 catalyst with MCM-41 as the carrier. The C3H6-SCR reaction results revealed that the NO conversion exceeded 50% in the presence of this catalyst within the temperature range of 300–430 °C, and it reached the maximum of 78% at 360 °C. The H2-TPR (Fig. 9) and NO-TPD results indicate that the strong interaction between Cu–Al and MCM-41 support enhanced the synergistic redox properties between the Cu species (Cu2+, Cu+, CuO, etc.) and Al, consequently promoting the desorption and activity of NO on the catalyst surface.
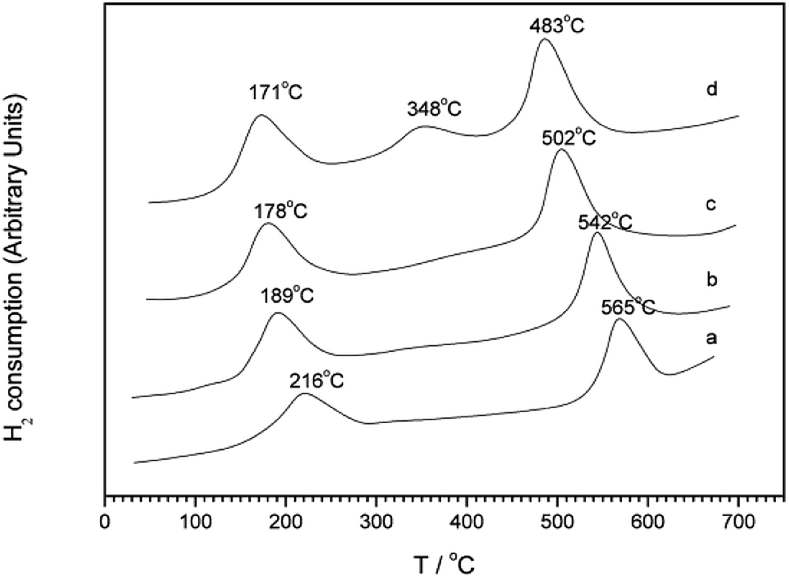 | ||
| Fig. 9 H2-TPR profiles of Cu–Al-MCM-41 samples with Si/Al = 10: (a) Cu–Al-MCM-41-10-31, (b) Cu–Al-MCM-41-10-61, (c) Cu–Al-MCM-41-10-93, and (d) Cu–Al-MCM-41-10-120.31 | ||
Jeon et al.32 prepared a MCM-41 molecular sieve by hydrothermal synthesis and then used the IM method to prepare the Pt–V-MCM-41 catalyst. The C3H6-SCR results revealed that the conversion rate of NO exceeded 50% under the effect of this catalyst in the temperature range of 260–480 °C, particularly achieving the maximum conversion rate of 90% in the temperature range of 270–340 °C. The TPO and FTIR results indicate that the V-doped species on the MCM-41 surface promoted the adsorption of NO on the catalyst surface and improved the redox performance of Pt due to the synergistic effect between Pt, V, and MCM-41 molecular sieve support. Meanwhile, the synergistic effect promoted the adsorption and activity of C3H6; therefore, the Pt–V-MCM-41 catalyst exhibited high catalytic performance.
The above results show that the active components supported by MCM-41 as the carrier are mostly Cu and Pt, where the preferred active component is Pt. The V adjuvant can bind to MCM-41 by bonding with the hydroxyl of the pore wall surface, producing the synergistic effect with the active constituent (Pt). This can significantly improve the catalytic performance of the catalyst for C3H6-SCR in the low-temperature range of 270–340 °C. The MCM-41 molecular sieve has hexagonally arranged pores, high specific surface area, and large adsorption capacity, which is beneficial in facilitating the synergistic effect between the active component and the support, enhancing the redox property of the active component, and promoting the adsorption and oxidation performances of C3H6, thereby promoting the production of intermediates (e.g., ONO and NCO) and enhancing the catalytic performance of the catalyst. The addition of auxiliaries can further enhance the synergistic effect of the active component and the carrier and promote the adsorption and desorption of NO on the catalyst surface. However, the catalyst with MCM-41 as the carrier has fewer acid sites, lower acid strength, and poor hydrothermal stability, resulting in a maximum NO conversion of less than 80%. When compared with microporous molecular sieve catalysts, the pore size distribution of mesoporous molecular sieve catalysts is relatively uniform and can be controlled; when compared with macroporous molecular sieve catalysts, it has high specific surface area and adsorption capacity. However, the disadvantage of mesoporous molecular sieve catalysts is low hydrothermal stability under current. However, the structure and catalytic performance of the catalysts can be significantly improved by adding additives, modifying the preparation process, and other newer methods. Therefore, the current focus is to investigate mesoporous molecular sieve catalysts with high hydrothermal stability and catalytic performance.
4 Macroporous zeolite catalysts
Macroporous molecular sieves have low cost, poor specific surface area, and poor catalytic activity and thermal stability, but a molecular sieve catalyst loaded with noble-metal active components has been widely studied because of its good catalytic performance.Konsolakis et al.33 reported a Pt–Al2O3 catalyst and a REO-modified Pt–Al2O3 catalyst prepared by a coprecipitation method. The study results revealed that as compared to the Pt–Al2O3 catalyst with extremely low catalytic activity, the modified catalyst exhibited good catalytic performance for C3H6-SCR in the wide temperature range of 320–600 °C. The N2O conversion rate exceeded 50%, and the maximum conversion rate was close to 100% in the temperature range of 450–600 °C. The DRIFTS results obtained using CO as the probe molecule revealed that the superior catalytic performance of the promoted samples can be mainly attributed to the increase in the available metal surface area and to the development of new Pt sites with exceptional electron density, located on the metal-support interfacial areas.
Moreover, de Lucas-Consuegra et al.34 prepared the Pt–K-βAl2O3 electrochemical catalyst with K as the promoter by a deposition method. The C3H6-SCR reaction results revealed that the N2O conversion rate exceeded 50% in the temperature range of 440–580 °C, with the highest being 80% achieved under the effect of this catalyst. This can be attributed to the interaction between K and Pt on the catalyst: K enhanced the catalytic activity of Pt, which improved the catalytic properties of the catalyst and further increased the N2O conversion rate. Irani et al.35 also found that the NO2 conversion rate exceeded 50% in the temperature range of 200–350 °C, and it was close to 100% over the catalyst at 240 °C on the Pt–Pd–Al2O3 catalyst in the C3H6-SCR reaction. Here, Pd in the catalyst enhanced the catalytic activity of Pt, and the synergistic action between Pt and Pd favored NO2 reduction within the low-temperature range.
Zhang et al.36 prepared the Ag–Al2O3 catalyst by the IM method using AlOOH as the Al source. The C3H6-SCR results revealed that the Ag–Al2O3 catalyst achieved the best catalytic performance in the temperature range of 300–600 °C, the NO conversion rate exceeded 70%, and the maximum conversion rate was close to 100% in the temperature range of 350–550 °C. Zhang et al. considered that high-density Ag–O–Al species on the Ag–Al2O3 catalyst considerably promoted the selective reduction of NO to N2. The UV-vis and H2-TPR results indicate that Ag+ or Ag2O nanoparticles on the catalyst were highly dispersed on the support.
In addition, Li et al.37 investigated the catalytic function of the Ag–Al2O3 catalyst assisted by NTP for C3H6-SCR. The results revealed that the conversion rate of NO and NO2 exceeded 50% in the temperature range of 310–550 °C, and the maximum conversion rate of NO and NO2 reached 90% in the temperature range of 310–410 °C. The NTP assistance significantly improved the catalytic performance of Ag–Al2O3 in the low-temperature range. Li et al. suggested that NTP assistance promoted the enrichment of intermediates (e.g., NCO and CN), thereby increasing the activity of the catalyst and yield of N2 in the C3H6-SCR reaction. DRIFTS further confirmed that NTP promoted the formation of more nitrogenous organic intermediates, which promoted the formation of more N2 product.
Zhang et al.38 found that the catalytic performance of the Ag–Al2O3 catalyst significantly increased when H2 was introduced into the C3H6-SCR reaction within a wide temperature range of 210–550 °C. The NO and NO2 conversion rates exceeded 70%, and the conversion temperature was broadened by 190 °C using a mixture of H2 and C3H6 rather than pure C3H6 as the reductant. The DRIFTS results revealed that H2 promoted the formation of –NCO intermediates. The presence of the intermediates allowed H2 to act as a coreducing agent for C3H6 in order to increase the NO and NO2 conversion rates.
Moreover, Guo et al.39 found that the catalytic performance of the catalyst was significantly increased in a wide temperature range of 300–550 °C when 1000 ppm H2 was added to the C3H6-SCR reaction under the effect of the Ag–Al2O3 catalyst. The maximum conversion rates of NO and NO2 reached 95% when using a mixture of H2 and C3H6 as the reductant. When the concentration of H2 increased to 5000 ppm, the NO and NO2 conversion rates exceeded 50%, and the conversion temperature was broadened to 75 °C in the low-temperature range. The NO-TPD results revealed that nitrates were the reactive intermediates in the reaction, and most of the nitrate intermediates adsorbed on the catalyst surface were reduced to NO and N2 by reaction with H2. Therefore, the presence of H2 considerably promoted the SCR activity of the catalysts.
Pitukmanorom et al.40 synthesized In2O3–Ga2O3–Al2O3 catalysts with Ga and In as the active components by the controlled use of chemical precipitation technology. The C3H6-SCR reaction results revealed that the NO conversion rate exceeded 50% in the temperature range of 375–550 °C, with the maximum being 80%. This can be attributed to the high surface area of the catalyst and the high dispersibility of the active components (Ga and In) on the catalyst surface, while the synergistic effect between Ga and In promoted the activation of propylene, thereby promoting the formation of the reduction product N2.
Luo et al.41 found that the addition of SO2 in the C3H6-SCR reaction enhanced the catalytic performance of the In2O3–Al2O3 catalyst. In the low-temperature range of 200–300 °C, the NO conversion rate under the effect of the catalyst was increased by 15–25% (as compared to no SO2). The DRIFTS results revealed that SO2 promoted the formation of carboxylate and formate species at lower temperatures, thereby promoting the partial oxidation of C3H6 and promoting the SCR activity.
Liu et al.42 prepared SnO2–Al2O3 catalysts by IM and sol–gel (SG) methods. The C3H6-SCR results revealed that the conversion rates of NO and NO2 on the catalyst prepared by the SG method exceeded 50% in the temperature range of 375–510 °C, the maximum being 76%. Within the same temperature range, NO and NO2 conversion rates increased by 13–21% (when compared with the immersion method). This can be attributed to the more uniform dispersion of SnO2 on the surface of the catalyst prepared by the SG method, which promoted the interaction between the active SnO2 constituent and the Al2O3 support, thereby improving the catalytic performance of the catalyst.
The XRD results (Fig. 10) further confirmed that the SnO2 species were highly dispersed on the Al2O3 support. The XPS results also showed that the Sn on the catalyst prepared by the SG method existed in the form of Sn4+ with a binding energy of 487.2 eV. Simultaneously, Sn4+ could interact with Al, which was beneficial toward the reduction of NO and NO2.
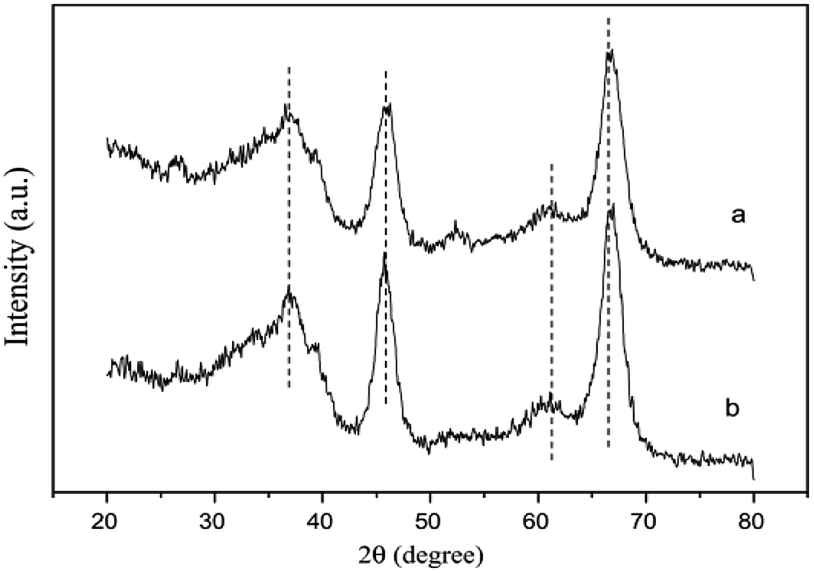 | ||
| Fig. 10 XRD patterns of two different SnO2–Al2O3 catalysts: (a) Sn5 (IM) and (b) Sn5 (SG).42 | ||
The above studies show that the active components of the catalysts supported by the macroporous molecular sieve (Al2O3) are Pt, Ag, In, Ga, and Sn: the preferred active component is Ag. When the noble metal Ag is used as the active component and C3H6 is the reducing agent, the operating temperature range is wider than that of nonnoble metal catalysts, but the low-temperature activity of Ag–Al2O3 for NOX reduction by C3H6 remains to be further explored. Additives (K, Pd, other metals, NTP, other auxiliaries, H2, SO2, etc.) can enhance the synergistic effect between the active components and the carrier and enhance the redox properties of the active components, thereby promoting the redox properties of C3H6 and further promoting the formation of more reactive intermediates (NCO, CN, carboxylates, formates, etc.). This increases the NOX conversion rate. For example, Pd can synergize with Pt to improve the catalytic performance of C3H6-SCR in the low-temperature range of 200–350 °C.
The oxygen ions of the Al2O3 support can be arranged in a hexagonal precision-stacked crystal structure in the preparation process, which does not have a relatively higher specific surface area to provide more active sites, but can enhance the interaction between this carrier and the metal active components dispersed on the surface of the catalyst. This forms the MO–Al species (M = Ag, Pt, etc.), which can promote the adsorption and activation of C3H6 and further promoting the catalytic performance of the catalyst.
In contrast, the macroporous molecular sieve (Al2O3) catalyst has a larger pore size, which is beneficial toward mass transfer and exchange of the reactive molecules. However, its low specific surface area reduces the number of active sites on the catalyst; therefore, the catalytic efficiency of the catalyst is lower than that of mesoporous or microporous molecular sieve catalysts at lower temperatures.
Table 2 summarizes the catalytic temperature ranges and catalytic activities for different molecular sieve de-NOX catalysts synthesized by using different methods.
| Catalysts | Preparation methods, (calcinations temperature) | Reaction conditions | NOX conversion, (temperature range) | Sources |
|---|---|---|---|---|
| Cu-ZSM-5 | Conventional solution ion exchange method, (550 °C) | 0.1%NO, 0.1%C3H6, 5%O2, GHSV = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 000 h−1 |
>70% (NO), (375–500 °C) | 9 |
| Ag-ZSM-5 | Impregnation, (540 °C) | 0.1%NOX, 0.15%C3H6, 5%O2, GHSV = 15000 h−1 |
>50% (NO), (380–600 °C) | 10 |
| Cu-ZSM-5-ZrO2 | Conventional liquid-phase ion exchange method, (500 °C) | 0.05%NO, 0.05%C3H6, 10%O2, GHSV = 28000 h−1 |
>50% (NO + NO2), (200–500 °C) | 15 |
| CeO2–Cu-ZSM-5 | Ion exchange, (500 °C) | 0.1%NO, 0.1%C3H6, 2%O2, GHSV = 59000 h−1 |
>50% (NO), (330–390 °C) | 16 |
| La–Cu-ZSM-5/cordierite | Ion exchange, (500 °C) | 0.05%NO, 0.05%C3H6, 5%O2, GHSV = 15000 h−1 |
>50% (NO + NO2), (210–450 °C) | 17 |
| Fe-Beta | Impregnation, (550 °C) | 0.047%NO, 0.045%C3H6, 3%O2, GHSV = 6529 h−1 | >50% (NO + NO2), (290–390 °C) | 18 |
| Pt-Beta | Ion exchange, (400 °C) | 0.2262%NO, 0.34%C3H6, 5%O2, GHSV = 50000 h−1 |
>50% (NO + NO2), (210–260 °C) | 19 |
| K–Cu-Beta | Impregnation, (400 °C) | 0.06%NO, 0.15%C3H6, 5%O2, GHSV = 12000 h−1 |
>80% (NO + NO2), (300–500 °C) | 20 |
| Cu-SAPO-34 | Ion exchange, (550 °C) | 0.1%NO, 0.1%C3H6, 5%O2, GHSV = 32000 h−1 |
>50% (NO), (350–450 °C) | 21 |
| Cu-SAPO-34 | Ion exchange, (550 °C) | 0.6%NO, 0.7%C3H6, 5%O2, GHSV = 10000 h−1 |
≤50% (NO), (100–300 °C) | 22 |
| Cu-SAPO-34 | Ion exchange, (550 °C) | 0.1%NO, 0.1%C3H6, 5%O2, GHSV = 32000 h−1 |
>50% (NO), (325–450 °C) | 23 |
| Cu-SSZ-13 | — | 0.05%NO, 0.05%C3H6, 5%O2, GHSV = 57000 h−1 |
>50% (NO), (380–500 °C) | 24 |
| Cu-SSZ-13 | — | 0.05%NO2, 0.05%C3H6, 1%O2, GHSV = 57000 h−1 |
>80% (NO2), (260–550 °C) | 26 |
| Pt-SBA-15 | Impregnation, (450 °C) | 0.015%NO, 0.015%C3H6, 18%O2, GHSV = 36000 h−1 |
>50% (NO + NO2), (130–340 °C) | 27 |
| Pt–Al-SBA-15 | Impregnation, (450 °C) | 0.015%NO, 0.015%C3H6, 18%O2, GHSV = 36000 h−1 |
Reached 86%, which increased by 6% (NO + NO2), (140 °C) | 27 |
| Cu–Al-SBA-15 | Impregnation, (550 °C) | 0.3%NO, 0.3%C3H6, 1%O2, GHSV = 60000 h−1 |
>50% (NO), (330–600 °C) | 28 |
| Co–Zr-SBA-15 | Impregnation, (550 °C) | 0.0533%NO2, 0.0533%C3H6, 5.3%O2, GHSV = 18000 h−1 |
>50% (NO), (330–375 °C) | 29 |
| Pt-MCM-41 | Impregnation, (550 °C) | 0.1%NO, 0.4%C3H6,10%O2, GHSV = 30000 h−1 |
>50% (NO), (150–500 °C) | 30 |
| Cu–Al-MCM-41 | A modified hydrothermal method, (540 °C) | 0.1%NO, 0.1%C3H6,2%O2, GHSV = 100000 h−1 |
>50% (NO), (300–430 °C) | 31 |
| Pt–V-MCM-41 | Impregnation, (550 °C) | 0.2%NO, 0.27%C3H6, 3%O2, GHSV = 42000 h−1 |
>50% (NO), (260–480 °C) | 32 |
| Pt–V-MCM-41 | Impregnation, (550 °C) | 0.2%NO, 0.27%C3H6, 3%O2, GHSV = 42000 h−1 |
Reached 90%, (NO), (270–340 °C) | 32 |
| REOS-Pt-Al2O3 | Co-precipitation, (600 °C) | 0.1%N2O, 0.1%C3H6, 0%O2, GHSV = 10000 h−1 |
100% (N2O), (450–600 °C) | 33 |
| Pt–K-βAl2O3 | Deposition, (800 °C) | 0.1%N2O, 0.2%C3H6, 0.2%O2, GHSV = 12000 h−1 |
>50% (N2O), (450–580 °C) | 34 |
| Pt–Pd–Al2O3 | — | 0.02%NO2, 0.08%C3H6, 10%O2, GHSV = 100000 h−1 |
>50% (NO2), (200–350 oC) | 35 |
| Ag–Al2O3(1) | Impregnation, (500 °C) | 0.1%NO, 0.3% C3H6, 10%O2, GHSV = 30000 h−1 |
100% (NO), (350–550 °C) | 36 |
| Ag–Al2O3(2) | A single step sol–gel, (600 °C) | 0.1%NO, 0.1% C3H6, 8%O2, GHSV = 14400 h−1 |
>50% (NO + NO2), (310–550 °C) | 37 |
| Ag–Al2O3(3) | Impregnation, (600 °C) | 0.08%NO, 0.1714%C3H6, 10%O2, GHSV = 50000 h−1 |
>90% (NO + NO2), (200–400 °C) | 38 |
| Ag–Al2O3(4) | Solution-impregnation, (500 °C) | 0.1%NO, 0.15%C3H6, 15%O2, GHSV = 65000 h−1 |
>50% (NO + NO2), (300–550 °C) | 39 |
| In2O3–Ga2O3–Al2O3 | A sequential chemical, precipitation technique, (700 °C) | 0.1%NO, 0.1%C3H6, 15%O2, GHSV = 30000 h−1 |
>50% (NO), (375–550 °C) | 40 |
| In2O3–Al2O3 | A single step sol–gel, (500 °C) | 0.1%NO, 0.1%C3H6, 8%O2, GHSV = 36000 h−1 |
>50% (NO), (380–550 °C) | 41 |
| SnO2–Al2O3 | A single step sol–gel and impregnation, (700 °C) | 0.1%NO, 0.1%C3H6, 8%O2, GHSV = 32000 h−1 |
>50% (NO + NO2), (375–510 °C) | 42 |
In general, the catalytic function of molecular sieve catalysts is related not only to the active component but also to the structure and texture of the support. Molecular sieves with different pore structures have their own advantages and disadvantages. Microporous molecular sieves have a large specific surface area, high hydrothermal stability, and suitable surface acidity. Nevertheless, the pore structure of microporous molecular sieves is small and complex, which leads to limitations in mass transfer. It is well known that carbon deposition, SO2 poisoning, and dealumination are major causes of catalyst deactivation. Mass transfer restrictions in microporous sieves easily lead to carbon deposition on the surface of the active components by the partial oxidation of hydrocarbons, facilitating the production of sulphites (or sulphates) by the reaction of SO2 (or SO2 + O2) and metallic oxides as the active metal component.18 Therefore, carbon deposition and SO2 poisoning may be the major causes of deactivation for microporous molecular sieve catalysts.
The macroporous molecular sieve (Al2O3) catalyst has a large pore size, which is beneficial toward mass transfer and exchange of reactive molecules. However, its low specific surface area and nonuniform pore size distribution lowers the catalytic efficiency of the catalyst as compared to that of mesoporous or microporous molecular sieve catalysts at lower temperatures. Table 2 reveals that the macroporous molecular sieve catalyst usually exhibits high activity in the high-temperature region. High-temperature reactions are bound to facilitate the oxidation of hydrocarbons and reaction of both active metal components and SO2, which may be the main cause of catalyst deactivation. Meanwhile, high-temperature water vapor reacts easily with skeletal Al to form non-skeletal Al species, Al(OH)x, at higher temperatures, yielding structural changes in macroporous molecular sieves and reducing hydrothermal stability and catalytic activity of the catalysts.
When compared with microporous and macroporous molecular sieve catalysts, mesoporous molecular sieves have a higher specific surface area, cation exchange, and hydrocarbon-sorption capacities, and the pore size distribution of mesoporous molecular sieve catalysts is relatively uniform and can be controlled. Table 2 shows that some catalysts such as Pt–V-MCM-41, Pt-MCM-41, and Pt-SBA-15 exhibited good catalytic activity at low temperatures (lower than 300 °C). The low reaction temperature and rapid mass transfer in mesoporous supports can reduce carbon deposition and sulphites (or sulphates), and therefore, reduce the probability of catalyst deactivation. Further, the high specific surface of mesoporous supports is beneficial in increasing the degree of dispersion of the active metal components and the number of active sites on the catalyst surface. This facilitates the increase in the catalytic activity. However, the disadvantage of mesoporous molecular sieve catalysts is low hydrothermal stability under current. Therefore, the thermal and hydrothermal stabilities of these mesoporous materials are crucial factors in their potential applications. As a result of the hydrolysis of bare Si–O–Si bonds in the presence of absorbed water, mesoporous silicate zeolites can lose their structure when exposed to high-temperature steam and boiling water for a long period of time. In order to improve the thermal and hydrothermal stabilities of mesoporous molecular sieve catalyst materials, many approaches have been investigated.
5 C3H6-SCR mechanisms under molecular sieve catalysts
The study of the catalytic reaction mechanism is an important aspect of catalyst research. Since the reaction pathway of the C3H6-SCR reaction over molecular sieve catalysts is very complicated and the type of molecular sieve catalyst is a major factor affecting the reaction pathway, there are different viewpoints on the mechanism of C3H6-SCR under the action of different molecular sieve catalysts. Therefore, numerous researchers have proposed different reaction pathways for C3H6-SCR in the presence of different molecular sieve catalysts.43–54Li et al.43 considered that propylene may serve as a NOX reductant according to the reaction on Fe-ZSN-5 and Cu-SSZ-13 zeolite catalysts:
| C3H6 + NO + 4O2 → 0.5N2 + 3H2O + 3CO2 | (1) |
However, Kotsifa et al.44 proposed that in the presence of oxygen in the feed, Pt–Al2O3 promoted the C3H6-SCR reaction of NO according to the following overall stoichiometric reactions:
| C3H6 + 2NO + (7/2)O2 → N2 + 3H2O + 3CO2 | (2) |
| C3H6 + 2NO + 4O2 → N2O + 3H2O + 3CO2 | (3) |
Similarly, Vaccaro et al.45 reported that the oxidation of propylene (eqn (4)) was one of the two main reactions taking place in the C3H6-SCR process over a Pt–Al2O3 catalyst. The other was the reduction of NO (eqn (5), with n being an integer, 0 < n < 9).
| 2C3H6 + 9O2 → 6H2O + 6CO2 | (4) |
| 2C3H6 + (18 + 2n)NO → 2nN2O + 6H2O + 6CO2 + (9 − n)N2 | (5) |
Rosas et al.46 combined the two main reaction pathways to describe NO reduction over a Pt–SiO2 catalyst in the presence of propylene and oxygen. The first one was the production of molecular nitrogen through NO dissociative adsorption via the redox pathway. The other was the activated propylene reacting with NO to finally decompose into N2. Based on the above results, the following reaction pathways were proposed for the low-temperature SCR:
| * + *NO ↔ *N + *O | (6) |
| * + C3H6 ↔ *C3H6 | (7) |
| *C3H6 + *O → *C3H6O + * | (8) |
| *C3H6 + 4O2 → 3CO2 + 3H2O + * | (9) |
| *N + *N → N2 +2* | (10) |
| *C3H6O + *NO → *C3H6NO2 + * | (11) |
| *C3H6NO2 + *NO + 3O2 → N2 + 3CO2 + 3H2O + 2* | (12) |
C3H6, NO, and O2 are considered to be adsorbed on the same type of Pt adsorption sites (denoted as *).
Furthermore, He et al.47 reported two attractive reaction pathways for the SCR of NO by C3H6 over Ag–Al2O3 and Cu–Al2O3 catalysts. The main reaction pathway over the Ag–Al2O3 catalyst can be summarized as follows:
NO + C3H6 + O2 → CH3COO− + RCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–O− + NO2(NO3−) CH–O− + NO2(NO3−)
| (13) |
|
CH3COO− + RCH CH–O− + NO2(NO3−) → R–NO2 + R–ONO
| (14) |
| R–NO2 + R–ONO → NCO + NO + O2 | (15) |
| NCO + NO + O2 → N2 + CO2 | (16) |
The main reaction pathway over the Cu–Al2O3 catalyst can be summarized as follows:
| NO + C3H6 + O2 → CH3COO− + NO2(NO3−) | (17) |
| CH3COO− + NO2(NO3−) → NO + H2O + CO2 | (18) |
Similarly, Wang et al.48 also summarized and proposed an attractive reaction pathway of the SCR of NO by C3H6 over Pd–Al2O3.
| NO + C3H6 + O2 → NOX(nitrate) + CxHyOz | (19) |
| NOX(nitrate) + CxHyOz → R–NO2 + R–ONO | (20) |
| R–NO2 + R–ONO → –NCO + –CN | (21) |
| –NCO + NO + O2 → N2 | (22) |
NO, O2, and C3H6 were first adsorbed on the catalyst carrier Al2O3. Then, the NO part reacted with oxygen to form nitrogen oxides, such as NOX and nitrate species. Further, C3H6 combined with oxygen to form the corresponding carbon hydroxides, as shown in eqn (19). Then, the active intermediates, R–NO2 and R–ONO, were formed by the catalysis of NOX and carbon hydroxides with Pd, as shown in eqn (20). The active intermediates could be easily transformed into a –NCO species by eqn (21), and –NCO was an active species of NO selective catalytic reduction to N2 and reacting with NO and O2 to generate N2 by eqn (22). In other words, C3H6 adsorption on the catalyst surface is required for the SCR reactions to proceed.
In addition, Capek et al.55 proposed that R-NOX was the main intermediate when the Cu-ZSM-5 zeolite catalyst was used. Sobczak et al.56 and Goscianska et al.57 found that –N2O(g) and –NCO were important organic nitrogenous intermediates when the Nb-MCM-41 molecular sieve catalyst was used. Shimizu et al.58 also found that intermediates such as –NCO and –CN were the major intermediates when Cu–Al2O3 molecular sieve catalysts were used.
At present, a more consistent view is that the mechanism of catalysis and reduction of NOX under the C3H6-SCR system belongs to a redox-type mechanism.59–107 The reaction process (Fig. 11) includes three steps. First, NO is oxidized into NO2 and NOX (ads) or nitrate species on the surface of the catalyst, and the reducing agent is partially oxidized to CxHyOz (ads) or acetate species. The second is the formation of nitrogen-containing organic intermediates. CxHyOz (ads) reacts with NOX (ads) to form nitrogen-containing organic intermediates (R–NO2, R–ONO), and the intermediates are rapidly converted into R–CN, R–NCO, R–NH2, and other species on the surface of the catalyst. Finally, organic intermediates (e.g., R–CN) react with NO + O2 to produce the final product N2.
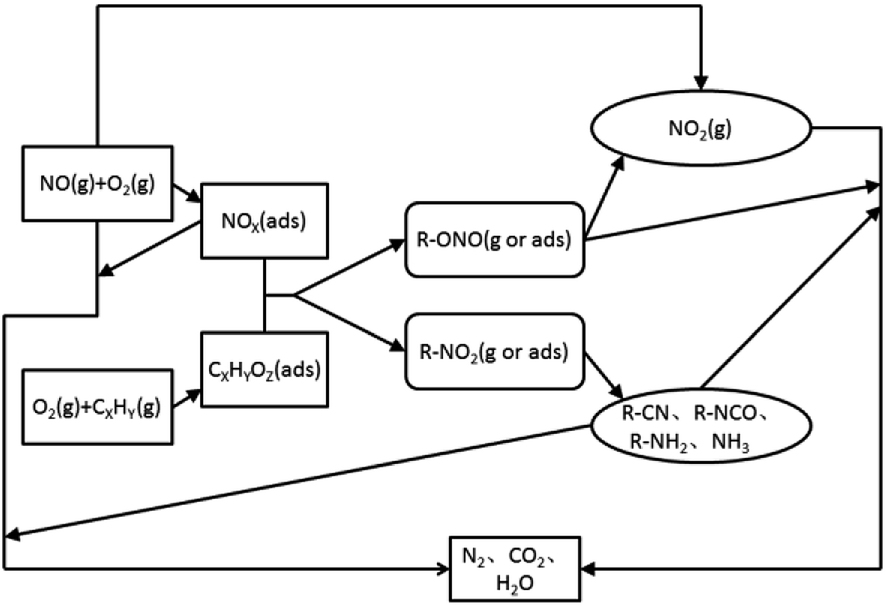 | ||
| Fig. 11 HC-SCR redox mechanism schematic. | ||
Most of the reaction mechanisms reported in the above literature assume that the reaction intermediate is the key to the catalytic reaction. Therefore, on the basis of the above literature and the structure and characteristics of different supported types of molecular sieve catalysts, we suggest that the mechanism of C3H6-SCR under the action of molecular sieve catalysts is as follows: (i) C3H6 is oxidized into CxHyOz (ads) or acetate species on the catalyst surface, and NO is oxidized into NO2 in an oxygen-rich atmosphere; (ii) CxHyOz (ads) reacts with NO2 to produce the nitrogen-containing organic intermediate R–NCO; and (iii) the organic intermediate, R–NCO, reacts with NO2 + O2, yielding the final product N2.
6 Design strategies for supported molecular sieve catalysts
The design of supported molecular sieve catalysts should not only consider the high activity of the catalyst, but also the high stability of the catalyst. From the results of current research, it is evident that the properties of the catalyst are related to the selection of active components, promoter, supports, and so on, and it is necessary to obtain a low-temperature catalyst with high activity to avoid the deactivation of the catalyst at high reaction temperatures.From Table 2, it is evident that Pt and some transition metals are usually selected as the active components of supported molecular sieve catalysts. Pt (Pt-SBA-15), La (La–Cu-ZSM-5/cordierite), Ag (Ag–Al2O3(3)), and Cu (Cu-SAPO-34) catalysts exhibit good low-temperature catalytic activity (Table 2). Therefore, these four metals can be used as the first choice of active components in the design of supported molecular sieve catalysts. However, considering the cost factor, the selection of the transition metal as the active component necessitates a higher performance-to-cost ratio. Further, the reaction of silver with sulfur dioxide is difficult, which can deter SO2 poisoning in the catalyst. The combination of transition metals as the active component and lanthanide elements as the promoter usually exhibits high activity at low temperatures in the catalysts; therefore, this combination deserves further consideration in the design of supported molecular sieve catalysts.
Studies have shown that the addition of a promoter in the catalyst can improve the low-temperature activity and stability of the catalyst due to the synergistic effect between the assistant and active components. For instance, the NOX conversion rate is close to 100% over the Pt–Pd–Al2O3 catalyst at 240 °C in the C3H6-SCR reaction;35 the catalytic efficiency is considerably improved due to the synergistic action of Pd at lower temperatures. Kucherov et al.108 also found that the introduction of lanthanides in Cu-HZSM-5 can improve, to some extent, the catalyst resistance against high-temperature deactivation. Therefore, the selection of proper additives is necessary in the design of catalysts. The auxiliaries of the catalysts in Table 2 (for example, Pd, Al, V, etc.) are worthwhile in the design strategies of supported molecular sieve catalysts.
Based on the previous analysis of the structural characteristics of molecular sieves with different pore sizes, we believe that mesoporous molecules can be used as the preferred support for low-temperature active catalysts. The high specific surface of mesoporous molecular sieves can improve the dispersity of the active component and the number of active sites on the catalyst, thereby increasing the activity of the catalyst. Furthermore, uniformity in the carrier pore size is beneficial toward higher distribution of active components, and high C3H6-sorption capacities facilitate C3H6-SCR. Improvements in the hydrothermal stability of mesoporous molecular sieves is the current focus. Introducing additives in mesoporous molecular sieves is an effective way to achieve stability improvement. Another route for improving the hydrothermal stability is to thicken the pore walls of mesoporous molecular sieves by altering the synthesis conditions (such as changing acid or basic conditions, templating agent, temperature, and reaction time). The recent discovery that inorganic silicates and organic surfactant precursors can self-organize to form ordered materials with nanometer-scale periodicities has created exciting avenues for the synthesis of high hydrothermally stable mesoporous materials.
7 Conclusion
Supported molecular sieve catalysts exhibit wide catalytic temperature windows, and some catalysts such as K–Cu-Beta, Cu-SAPO-34, and Ag–Al2O3(3) (Table 2) exhibit low-temperature catalytic activity. Changes in the synthesis methods can change the structures and textures of the catalysts, such as the number of active sites, pore diameter, and specific area, as well as the dispersion of active components, consequently improving the catalytic function of the catalysts.The choice of the supported metal as the active constituent is very important for the catalytic function of the catalysts. Studies have shown that Pt, Ag, and Cu are usually selected as the active components of different load types of molecular sieve catalysts, and some catalysts exhibit good catalytic performance and wide operating temperatures in the C3H6-SCR reaction; the synergistic effect of multi-metals such as Pt and Pd in the Pt–Pd–Al2O3 catalyst is beneficial toward improving the low-temperature activity of the catalyst. The NOX conversion rate was close to 100% over the catalyst at 240 °C on the catalyst in the C3H6-SCR reaction. NTP assistance can promote the enrichment of intermediates such as NCO and CN in the catalytic reaction. Further, H2 in the mixture of H2 and C3H6 as the reductant can promote the formation of –NCO intermediates, thereby improving the low-temperature activity of the catalyst.
Despite considerable progresses made in the studies involving supported molecular sieve catalysts, some issues need to be investigated further in order to accurately construct supported molecular sieve catalysts for high catalytic activity and wide operating temperatures in the C3H6-SCR reaction. (1) The extraordinary catalytic efficiency of multi-metals generally relies on the cooperative action between the multi-metals and multi-supports; therefore, cooperative catalysis is an important factor to be considered in the design of supported molecular sieve catalysts. (2) The synergistic mechanism between the active components and assistants/supports for supported molecular sieve catalysts is not very clear; therefore, it needs to be exhaustively investigated. (3) The catalytic mechanisms of supported molecular sieve catalysts are associated with many factors. In order to elucidate the catalytic mechanisms of the catalyst, it is necessary to further investigate the relationship between the catalytic efficiency and catalyst structure/component, such as pore diameter and surface area of molecular sieves, dispersion of metal active components of the catalysts, and so on. Perhaps, the most intriguing aspect for studies involving catalytic mechanisms is to determine the structure of intermediate species in the C3H6-SCR reaction by using modern instrumental analysis methods. (4) Enhancements in the low-temperature activity and extending the operating temperatures by introducing other assistive methods and mixing reductants in the C3H6-SCR reaction are worth exploring further.
Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21206202), the Key Industries Common Key Technology Innovation Special of Chongqing City, China (cstc2016zdcy-ztzx0020-02), the Science and Technology Plan Project of Sichuan Province, China (2017GZ0421), Banan Science and Technology Foundation of Chongqing (No. 2017TJ10, 2018TJ03), the Excellent Talent Program for the Fifth Batch of Universities in Chongqing, China (2017-28), Graduate Scientific Research Innovation Project of Chongqing (CYS18314).References
- S. A. Yashnik, A. V. Salnikov, N. T. Vasenin, V. F. Anufrienko and Z. R. Ismagilov, Catal. Today, 2012, 197, 214–227 CrossRef CAS.
- A. V. Boix, S. G. Aspromonte and E. E. Miró, Appl. Catal., A, 2008, 341, 26–34 CrossRef CAS.
- G. Li, X. Wang, C. Jia and Z. Liu, J. Catal., 2008, 257, 291–296 CrossRef CAS.
- F. Lónyi, H. E. Solt, J. Valyon, A. Boix and L. B. Gutierrez, Appl. Catal., B, 2012, 117, 212–223 CrossRef.
- V. Rico-Pérez, J. M. García-Cortés, C. S. M. De Lecea and A. Bueno-López, Chem. Eng. Sci., 2013, 104, 557–564 CrossRef.
- A. N. Mendes, A. Matynia, A. Toullec, S. Capela, M. F. Ribeiro, C. Henriques and P. Da Costa, Appl. Catal., A, 2015, 506, 246–253 CrossRef CAS.
- M. Iwamoto, H. Yahiro, S. Shundo, Y. Yu-u and N. Mizuno, Appl. Catal., 1991, 69, L15–L19 CrossRef CAS.
- W. Held, A. Koenig, T. Richter and L. Puppe, SAE Trans., 1990, 209–216 Search PubMed.
- L. Li and N. Guan, Microporous Mesoporous Mater., 2009, 117, 450–457 CrossRef CAS.
- F. Schuricht and W. Reschetilowski, Microporous Mesoporous Mater., 2012, 164, 135–144 CrossRef CAS.
- W. Shan and H. Song, Catal. Sci. Technol., 2015, 5, 4280–4288 RSC.
- R. M. Heck, Catal. Today, 1999, 53, 519–523 CrossRef CAS.
- L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef.
- Z. Liu and S. Ihl Woo, Catal. Rev., 2006, 48, 43–89 CrossRef CAS.
- C. K. Seo, B. Choi, H. Kim, C. H. Lee and C. B. Lee, Chem. Eng. J., 2012, 191, 331–340 CrossRef CAS.
- V. G. Komvokis, E. F. Iliopoulou, I. A. Vasalos, K. S. Triantafyllidis and C. L. Marshall, Appl. Catal., A, 2007, 325, 345–352 CrossRef CAS.
- T. Wang, L. Li and N. Guan, Fuel Process. Technol., 2013, 108, 41–46 CrossRef CAS.
- H. Pan, Y. Guo and H. T. Bi, Chem. Eng. J., 2015, 280, 66–73 CrossRef CAS.
- J. G. Cortés, M. I. Gómez and C. S. M. de Lecea, Appl. Catal., B, 2007, 74, 313–323 CrossRef.
- N. Nejar and M. J. Illán-Gómez, Catal. Today, 2007, 119, 262–266 CrossRef CAS.
- X. G. Li, H. Z. Yan, J. L. Lv, L. Bo and F. M. Jin, Chem. Ind. Eng., 2017, 34, 8–12 Search PubMed.
- Y. Q. Zuo, L. N. Han, W. R. Bao, L. P. Chang and J. C. Wang, Chin. J. Catal., 2013, 34, 1112–1122 CrossRef CAS.
- J. L. Lv, The preparation of the Cu-SAPO-34 catalyst and its C3H6-SCR performance, Tianjin University, Tianjin City, China, 2014 Search PubMed.
- R. Raj, M. P. Harold and V. Balakotaiah, Chem. Eng. J., 2014, 254, 452–462 CrossRef CAS.
- D. J. Kim, J. Wang and M. Crocker, Catal. Today, 2014, 231, 83–89 CrossRef CAS.
- R. Raj, M. P. Harold and V. Balakotaiah, Ind. Eng. Chem. Res., 2013, 52, 15455–15465 CrossRef CAS.
- X. Liu, Z. Jiang, M. Chen, J. Shi, W. Shangguan and Y. Teraoka, J. Environ. Sci., 2013, 25, 1023–1033 CrossRef CAS.
- R. Zhang, D. Shi, Y. Zhao, B. Chen, J. Xue, X. Liang and Z. Lei, Catal. Today, 2011, 175, 26–33 CrossRef CAS.
- A. Sabbaghi, F. L. Y. Lam and X. Hu, Mol. Catal., 2017, 440, 1–8 CrossRef CAS.
- D. J. Kim, J. W. Kim, S. J. Choung and M. Kang, J. Ind. Eng. Chem., 2008, 14, 308–314 CrossRef CAS.
- Y. Wan, J. Ma, Z. Wang, W. Zhou and S. Kaliaguine, J. Catal., 2004, 227, 242–252 CrossRef CAS.
- J. Y. Jeon, H. Y. Kim and S. I. Woo, Appl. Catal., B, 2003, 44, 301–310 CrossRef CAS.
- M. Konsolakis, C. Drosou and I. V. Yentekakis, Appl. Catal., B, 2012, 123, 405–413 CrossRef.
- A. de Lucas-Consuegra, F. Dorado, C. Jiménez-Borja and J. L. Valverde, Appl. Catal., B, 2008, 78, 222–231 CrossRef CAS.
- K. Irani, W. S. Epling and R. Blint, Appl. Catal., B, 2009, 92, 422–428 CrossRef CAS.
- R. Zhang and S. Kaliaguine, Appl. Catal., B, 2008, 78, 275–287 CrossRef CAS.
- J. Li, R. Ke, W. Li and J. Hao, Catal. Today, 2007, 126, 272–278 CrossRef CAS.
- X. Zhang, Y. Yu and H. He, Appl. Catal., B, 2007, 76, 241–247 CrossRef CAS.
- Y. Guo, J. Chen and H. Kameyama, Appl. Catal., A, 2011, 397, 163–170 CrossRef CAS.
- P. Pitukmanorom and J. Y. Ying, Nano Today, 2009, 4, 220–226 CrossRef CAS.
- C. Luo, J. Li, Y. Zhu and J. Hao, Catal. Today, 2007, 119, 48–51 CrossRef CAS.
- Z. Liu, J. Li and J. Hao, Chem. Eng. J., 2010, 165, 420–425 CrossRef CAS.
- M. Li, V. G. Easterling and M. P. Harold, Appl. Catal., B, 2016, 184, 364–380 CrossRef CAS.
- A. Kotsifa, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2008, 80, 260–270 CrossRef CAS.
- A. R. Vaccaro, G. Mul and J. Pérez-Ramırez, et al.,, Appl. Catal., B, 2003, 46, 687–702 CrossRef CAS.
- J. M. Rosas, R. Ruiz-Rosas and J. Rodríguez-Mirasol, et al.,, Chem. Eng. J., 2017, 327, 351–360 CrossRef CAS.
- H. He, C. B. Zhang and Y. B. Yu, Catal. Today, 2004, 90, 191–197 CrossRef CAS.
- Y. D. Wang, H. J. Zhang and D. N. He, J. Inorg. Mater., 2011, 26, 311–316 CrossRef CAS.
- N. Jagtap, S. B. Umbarkar and P. Miquel, et al.,, Appl. Catal., B, 2009, 90, 416–425 CrossRef CAS.
- J. Wang, Y. Ji and Z. He, et al.,, Appl. Catal., B, 2012, 111, 562–570 CrossRef.
- P. Denton, A. Giroir-Fendler and Y. Schuurman, et al.,, Appl. Catal., A, 2001, 220, 141–152 CrossRef CAS.
- A. Bueno-Lopez, M. J. Illan-Gomez and C. S. M. de Lecea, Appl. Catal., A, 2006, 302, 244–249 CrossRef CAS.
- J. Long, Z. Zhang and Z. Ding, et al.,, J. Phys. Chem. C, 2010, 114, 15713–15727 CrossRef CAS.
- M. Li, V. G. Easterling and M. P. Harold, Catal. Today, 2016, 267, 177–191 CrossRef CAS.
- L. Čapek, K. Novoveska, Z. Sobalik, B. Wichterlová, L. Cider and E. Jobson, Appl. Catal., B, 2005, 60, 201–210 CrossRef.
- I. Sobczak, M. Ziolek and M. Nowacka, Microporous Mesoporous Mater., 2005, 78, 103–116 CrossRef CAS.
- J. Goscianska, P. Bazin, O. Marie, M. Daturi, I. Sobczak and M. Ziolek, Catal. Today, 2007, 119, 78–82 CrossRef CAS.
- K. I. Shimizu, H. Kawabata, H. Maeshima, A. Satsuma and T. Hattori, J. Phys. Chem. B, 2000, 104, 2885–2893 CrossRef CAS.
- S. Chansai, R. Burch, C. Hardacre, J. Breen and F. Meunier, J. Catal., 2011, 281, 98–105 CrossRef CAS.
- Y. P. Chen, D. G. Cheng, F. Q. Chen and Y. L. Zhan, Progress in Chemistry, 2014, 26, 248–258 CAS.
- R. Brosius and J. A. Martens, Top. Catal., 2004, 28, 119–130 CrossRef CAS.
- G. Delahay, A. Guzmán-Vargas and B. Coq, Appl. Catal., B, 2007, 70, 45–52 CrossRef CAS.
- R. Matarrese, H. H. Ingelsten and M. Skoglundh, J. Catal., 2008, 258, 386–392 CrossRef CAS.
- N. W. Cant and I. O. Liu, Catal. Today, 2000, 63, 133–146 CrossRef CAS.
- I. Sobczak, K. Musialska, H. Pawlowski and M. Ziolek, Catal. Today, 2011, 176, 393–398 CrossRef CAS.
- H. H. Ingelsten, A. Palmqvist and M. Skoglundh, J. Phys. Chem. B, 2006, 110, 18392–18400 CrossRef PubMed.
- P. Pietrzyk, C. Dujardin, K. Góra-Marek, P. Granger and Z. Sojka, Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 RSC.
- S. K. Park, Y. K. Park, S. E. Park and L. Kevan, Phys. Chem. Chem. Phys., 2000, 2, 5500–5509 RSC.
- I. Sobczak, A. Kusior and M. Ziolek, Catal. Today, 2008, 137, 203–208 CrossRef CAS.
- M. Misono, H. Niiro and Y. Hirao, Res. Chem. Intermed., 1998, 24, 123–132 CrossRef CAS.
- O. Gorce, F. Baudin, C. Thomas, P. Da Costa and G. Djéga-Mariadassou, Appl. Catal., B, 2004, 54, 69–84 CrossRef CAS.
- R. Burch and J. A. Sullivan, J. Catal., 1999, 182, 489–496 CrossRef CAS.
- R. Burch, A. A. Shestov and J. A. Sullivan, J. Catal., 1999, 182, 497–506 CrossRef CAS.
- G. Xu, J. Ma, G. He, Y. Yu and H. He, Appl. Catal., B, 2017, 207, 60–71 CrossRef CAS.
- T. Gerlach and M. Baerns, Chem. Eng. Sci., 1999, 54, 4379–4384 CrossRef CAS.
- G. Li, S. C. Larsen and V. H. Grassian, J. Mol. Catal. A: Chem., 2005, 227, 25–35 CrossRef CAS.
- J. H. Jang, S. C. Lee, D. J. Kim, M. Kang and S. J. Choung, Appl. Catal., A, 2005, 286, 36–43 CrossRef CAS.
- T. Furusawa, L. Lefferts, K. Seshan and K. I. Aika, Appl. Catal., B, 2003, 42, 25–34 CrossRef CAS.
- A. W. L. Ting, M. P. Harold and V. Balakotaiah, Chem. Eng. Sci., 2018 Search PubMed.
- D. K. Captain and M. D. Amiridis, J. Catal., 1999, 184, 377–389 CrossRef CAS.
- R. Q. Long and R. T. Yang, J. Phys. Chem. B, 1999, 103, 2232–2238 CrossRef CAS.
- V. A. Bell, J. S. Feeley, M. Deeba and R. J. Farrauto, Catal. Lett., 1994, 29, 15–26 CrossRef CAS.
- H. Yasuda, T. Miyamoto and M. Misono, Reduction of Nitrogen Oxide Emissions, 1995, vol. 587, pp. 110–122 Search PubMed.
- K. L. Roberts and M. D. Amiridis, Ind. Eng. Chem. Res., 1997, 36, 3528–3532 CrossRef CAS.
- S. C. Shen and S. Kawi, Catal. Today, 2001, 68, 245–254 CrossRef CAS.
- P. A. Kumar, M. P. Reddy, L. K. Ju, B. Hyun-Sook and H. H. Phil, J. Mol. Catal. A: Chem., 2008, 291, 66–74 CrossRef CAS.
- A. Obuchi, A. Ogata, K. Mizuno, A. Ohi, M. Nakamura and H. Ohuchi, J. Chem. Soc., Chem. Commun., 1992, 247–248 RSC.
- S. C. Shen and S. Kawi, J. Catal., 2003, 213, 241–250 CrossRef CAS.
- G. P. Ansell, A. F. Diwell, S. E. Golunski, J. W. Hayes, R. R. Rajaram, T. J. Truex and A. P. Walker, Appl. Catal., B, 1993, 2, 81–100 CrossRef CAS.
- R. Burch, P. J. Millington and A. P. Walker, Appl. Catal., B, 1994, 4, 65–94 CrossRef CAS.
- A. P. Walker, Catal. Today, 1995, 26, 107–128 CrossRef CAS.
- J. A. Martens, A. Cauvel, A. Francis, C. Hermans, F. Jayat, M. Remy and P. A. Jacobs, Angew. Chem., Int. Ed., 1998, 37, 1901–1903 CrossRef CAS.
- S. Tamm, H. H. Ingelsten and A. E. Palmqvist, J. Catal., 2008, 255, 304–312 CrossRef CAS.
- Y. Wan, J. Ma, Z. Wang, W. Zhou and S. Kaliaguine, Appl. Catal., B, 2005, 59, 235–242 CrossRef CAS.
- Z. Zhang, M. Chen, Z. Jiang and W. Shangguan, J. Environ. Sci., 2010, 22, 1441–1446 CrossRef CAS.
- Y. Zheng, M. Li, D. Wang, M. P. Harold and D. Luss, Catal. Today, 2016, 267, 192–201 CrossRef CAS.
- X. Cheng and X. T. Bi, Chem. Eng. J., 2012, 211, 453–462 CrossRef.
- S. E. Maisuls, L. Lefferts, K. Seshan, T. Furusawa, K. Aika, B. Mosqueda-Jimenez and J. A. Lercher, Catal. Today, 2003, 84, 139–147 CrossRef CAS.
- R. Burch and P. J. Millington, Catal. Today, 1995, 26, 185–206 CrossRef CAS.
- H. Hamada, Catal. Today, 1994, 22, 21–40 CrossRef CAS.
- W. Schießer, H. Vinek and A. Jentys, Appl. Catal., B, 2001, 33, 263–274 CrossRef.
- E. Joubert, X. Courtois, P. Marecot, C. Canaff and D. Duprez, J. Catal., 2006, 243, 252–262 CrossRef CAS.
- Y. Chi and S. S. Chuang, Catal. Today, 2000, 62, 303–318 CrossRef CAS.
- V. A. Sadykov, V. V. Lunin, V. A. Matyshak, E. A. Paukshtis, A. Y. Rozovskii, N. N. Bulgakov and J. R. H. Ross, Kinet. Catal., 2003, 44, 379–400 CrossRef CAS.
- J. T. Miller, E. Glusker, R. Peddi, T. Zheng and J. R. Regalbuto, Catal. Lett., 1998, 51, 15–22 CrossRef CAS.
- D. K. Captain, K. L. Roberts and M. D. Amiridis, Catal. Today, 1998, 42, 93–100 CrossRef CAS.
- M. D. Amiridis, K. L. Roberts and &C. J. Pereira, Appl. Catal., B, 1997, 14, 203–209 CrossRef CAS.
- A. V. Kucherov, C. N. Montreuil, T. N. Kucherova and M. Shelef, Catal. Lett., 1998, 56, 173–181 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |