DOI:
10.1039/C8RA08588A
(Paper)
RSC Adv., 2019,
9, 206-214
Janus nanofiber array pellicle: facile conjugate electrospinning construction, structure and bifunctionality of enhanced green fluorescence and adjustable magnetism
Received
17th October 2018
, Accepted 10th December 2018
First published on 21st December 2018
Abstract
A [Fe3O4/polyvinyl pyrrolidone (PVP)]//[Tb(BA)3phen/PVP] Janus nanofiber array pellicle (denoted JNAP) was successfully constructed by facile conjugate electrospinning without twisting for the first time. The JNAP offers the dual-functionality of fluorescence and magnetism. This technology entirely solves the dilemma of the magnetic spinning dope and fluorescent spinning dope being easily mixed together during the parallel electrospinning process, as it achieves complete segregation of magnetic nanoparticles and fluorescent molecules. Moreover, conjugate electrospinning without twisting has fewer requirements on the viscosity of the spinning dope compared with parallel electrospinning, in which the two spinning dopes should have the same viscosity. It was satisfactorily found that the JNAP has higher fluorescence intensity than the corresponding non-aligned Janus nanofiber pellicle. The magnetism of the JNAP could be tailored by changing the doping amount of the Fe3O4 NPs. The JNAP has potential applications in nanotechnology and biomedicine, etc., due to its enhanced green fluorescence and adjustable magnetism. In addition, this design concept and manufacturing process provide a facile way for preparing other one-dimensional Janus nanomaterials with multifunctionality.
Introduction
It is becoming harder to meet the needs of many emerging practical technologies using single-functional materials alone. Therefore, research into the preparation and performance of multifunctional materials has become increasingly important in the realm of materials science and technology.1–3 Fluorescent-magnetic bifunctional nanomaterials have attracted much attention of researchers due to their potential uses in cell imaging, cancer studies, sensing and biomolecular detection, etc.4–9 For instance, fluorescent-magnetic bifunctional nanomaterials are ideal candidates for drug delivery because the loaded drug can be transported to a specific location using magnetic navigation, and meanwhile, the real-time position of the drug can be monitored by measuring fluorescence signals emitted from the fluorescent-magnetic bifunctional nanomaterial.10,11 As another example, fluorescent-magnetic bifunctional nanomaterials possess the property of dual-mode imaging, including fluorescent imaging and magnetic resonance imaging, which is beneficial for biological detection.12,13 However, researchers in this field must face the plight that if the magnetic nanoparticles and fluorescent molecules are directly mixed, the fluorescence intensity of the sample will be greatly reduced.14,15 Hence, exceptional structures need to be designed and constructed to separate magnetic nanoparticles from fluorescent molecules in an integrated system. Inspired by Janus, a god in Roman mythology, an exceptive “Janus structure” has been proposed by several research groups.16,17 Researchers have proposed that magnetic nanoparticles and fluorescent molecules should be respectively added to both sides of a Janus material in order to isolate two different substances from each other and reduce the detrimental influence of magnetic nanoparticles on the fluorescence intensity.18,19 Magnetic-fluorescent bifunctional Janus nanofibers are typical Janus materials that realize separation of magnetic nanoparticles from fluorescent molecules. According to the literature, Janus nanofibers can be gained by parallel electrospinning, where two kinds of spinning dopes are respectively loaded into two syringes to generate magnetic-fluorescent bifunctional Janus nanofibers using a specially-made parallel spinneret under electrospinning.20–29 Xi, et al.30,31 fabricated flexible magnetic-photoluminescent bifunctional Janus nanofibers by parallel electrospinning. It has been proven that such Janus nanofibers have stronger fluorescence intensity than the counterpart composite nanofibers. However, parallel electrospinning still has a drawback in that it is difficult to fulfill complete segregation of the magnetic nanoparticles and the fluorescent molecules, since the two spinning dopes are easily mixed at the outlet of spinneret.32 Consequently, it is a pressing subject of study to develop a new technique to overcome this drawback.
Conjugate electrospinning is an excellent technique for constructing one-dimensional nanomaterials. This technique can be divided into two kinds: conjugate electrospinning with twisting33–35 and that without twisting.36,37 Conjugate electrospinning with twisting is used to prepare nanofiber yarns which can be applied to tissue repair, nerve regeneration and electrically conductive material, etc.38–40 Recently, this method has been utilized to construct magnetic-fluorescent bifunctional nanofiber yarns. Fan, et al. used conjugate electrospinning with twisting to fabricate heterogeneous nanofiber yarns to effectively separate magnetic nanoparticles from fluorescent molecules.41,42 To date, there are only a few studies on conjugate electrospinning without twisting, which mainly focus on constructing materials for photocatalysis, and those with mechanical and waterproof properties.43–45 A magnetic-fluorescent bifunctional Janus nanofiber array pellicle built by conjugate electrospinning without twisting has not been reported.
In this work, polyvinyl pyrrolidone (PVP), Fe3O4 nanoparticles (NPs) and Tb(BA)3phen were respectively used as a template, magnetic material and fluorescent compound. Tb(BA)3phen possesses excellent fluorescence properties due to the unique f–f transition of Tb3+. It has become one of the most important fluorescent materials at present. Fe3O4 NPs can be widely used in many fields, such as targeted therapy, magnetically-controlled switches, electronics and biological processes due to their unique superparamagnetism, good biocompatibility and high permeability. A [Fe3O4/PVP]//[Tb(BA)3phen/PVP] Janus nanofiber array pellicle (abbreviated as JNAP) with magnetic-fluorescent bifunctionality was constructed by conjugate electrospinning without twisting (called conjugate electrospinning hereinafter). To highlight the excellent performance of the JNAP, a series of comparative samples were also constructed by conjugate electrospinning and parallel electrospinning. Finally, the as-prepared samples were systematically characterized using the relevant test instruments, and several new findings were obtained.
Experimental
Chemicals
Tb4O7 (99.99%), concentrated nitric acid (HNO3), 1,10-phenanthroline (phen), benzoic acid (BA), FeSO4·7H2O, FeCl3·6H2O, polyethylene glycol (PEG, Mw ≈ 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), NH4NO3, ammonia (NH3·H2O), oleic acid (OA), polyvinyl pyrrolidone (PVP) and ethanol (CH3CH2OH) were used to prepare the samples. All chemicals were of analytical grade.
000), NH4NO3, ammonia (NH3·H2O), oleic acid (OA), polyvinyl pyrrolidone (PVP) and ethanol (CH3CH2OH) were used to prepare the samples. All chemicals were of analytical grade.
Syntheses of OA-modified Fe3O4 NPs and terbium complexes
OA-modified Fe3O4 NPs (denoted as Fe3O4 NPs for short) and Tb(BA)3phen were prepared according to the literature.46,47
Electrospinning process
Two different types of spinning dopes (named spinning dope A and spinning dope B) were used to prepare the Janus nanofibers. Spinning dope A, with fluorescence properties, was prepared as follows: PVP (1 g) was fully dissolved in ethanol (7 g) under magnetic stirring, and then a certain amount of Tb(BA)3phen complex was uniformly dispersed in the solution to form spinning dope A. Spinning dope B, with magnetic properties, was fabricated as follows: Fe3O4 NPs were dispersed in ethanol (7 g) under ultrasonication for 20 min, and then PVP (1 g) was added into the above suspension under mechanical stirring. The actual ingredients of spinning dope A and B are respectively shown in Tables 1 and 2.
Table 1 Ingredients of spinning dope A
| Spinning dope A |
Tb(BA)3phen:PVP (wt%) |
Tb(BA)3phen (g) |
PVP (g) |
CH3CH2OH (g) |
| SA1 |
5% |
0.0500 |
1.0000 |
7.0000 |
| SA2 |
10% |
0.1000 |
1.0000 |
7.0000 |
| SA3 |
15% |
0.1500 |
1.0000 |
7.0000 |
| SA4 |
20% |
0.2000 |
1.0000 |
7.0000 |
| SA5 |
25% |
0.2500 |
1.0000 |
7.0000 |
Table 2 Ingredients of spinning dope B
| Spinning dope B |
Fe3O4 NPs/PVP (mass ratio) |
Fe3O4 NPs (g) |
PVP (g) |
CH3CH2OH (g) |
| SB1 |
0.5:1 |
0.5000 |
1.0000 |
7.0000 |
| SB2 |
1:1 |
1.0000 |
1.0000 |
7.0000 |
| SB3 |
1.5:1 |
1.5000 |
1.0000 |
7.0000 |
| SB4 |
2:1 |
2.0000 |
1.0000 |
7.0000 |
A device diagram for constructing the [Fe3O4/PVP]//[Tb(BA)3phen/PVP] JNAP by conjugate electrospinning is presented in Table 3. The spinning dopes A and B were separately loaded into two syringes with plastic spinnerets. The angle between the syringe and the horizontal line was ca. 45° in the conjugate electrospinning device. To obtain the array pellicle, a rotating drum was used as a collector. The corresponding spinning dopes and detailed spinning conditions are summarized in Table 3.
Table 3 Electrospinning device diagrams, corresponding spinning dopes and electrospinning conditions of samples
| Samples |
Electrospinning device diagrams |
Spinning dopes |
Electrospinning conditions |
| JNAP |
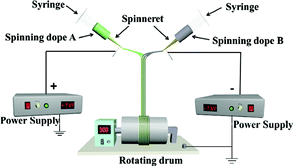 |
Spinning dope A: SA1, SA2, SA3, SA4 and SA 5, Spinning dope B: SB1 |
Collector: rotating drum (10 cm in diameter), rotation speed: 300 rpm, distance between the two spinnerets: 15 cm, curing distance: 25 cm, applied voltages: ± 7 kV, temperature: 15 ± 5 °C, relative humidity: 25 ± 5% |
| JNNP |
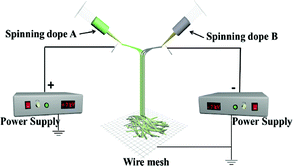 |
Spinning dope A: SA3, Spinning dope B: SB1 |
Collector: wire mesh, distance between the two spinnerets: 15 cm, curing distance: 25 cm, applied voltages: ± 7 kV, temperature: 15 ± 5 °C, relative humidity: 25 ± 5% |
| P-JNAP |
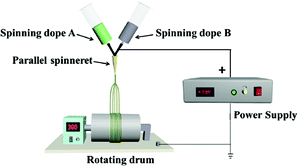 |
Spinning dope A: SA3, Spinning dope B: SB1 |
Collector: rotating drum (10 cm in diameter), rotation speed: 300 rpm, curing distance: 25 cm, applied voltages: + 7 kV, temperature: 15 ± 5 °C, relative humidity: 25 ± 5% |
| HNAP |
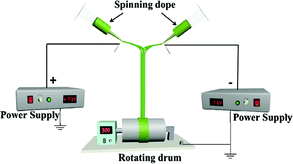 |
Spinning dope: mixing isopyknic SA3 and SB1 |
Collector: rotating drum (10 cm in diameter), rotation speed: 300 rpm, distance between the two spinnerets: 15 cm, curing distance: 25 cm, applied voltages: ± 7 kV, temperature: 15 ± 5 °C, relative humidity: 25 ± 5% |
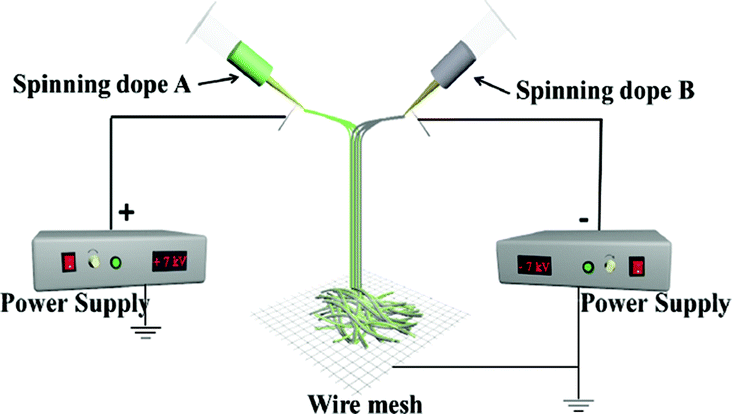
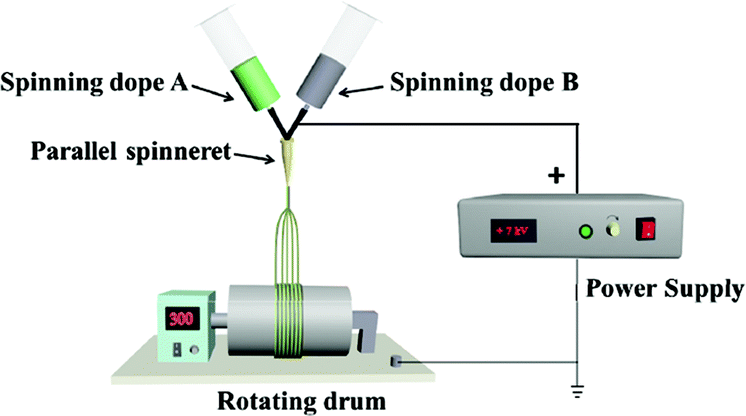
For comparison, a [Fe3O4/PVP]//[Tb(BA)3phen/PVP] Janus nanofiber non-array pellicle (referred to as JNNP) was prepared by conjugate electrospinning with a wire mesh as the collector, and a parallel electrospinning-made [Fe3O4/PVP]//[Tb(BA)3phen/PVP] Janus nanofiber array pellicle (named P-JNAP) was constructed by using spinning dopes SA3 and SB1. Another spinning dope was prepared by evenly mixing equal volumes of spinning dopes SA3 and SB1, and then a [Fe3O4/Tb(BA)3phen/PVP] homogeneous nanofiber array pellicle (called HNAP) was also fabricated by conjugate electrospinning. The device diagrams and specific conditions for the comparative samples are also displayed in Table 3.
Characterization methods
The phase compositions of Fe3O4 NPs, JNAP, JNNP, HNAP and P-JNAP were analyzed using X-ray power diffraction (XRD) with Cu Kα radiation. The operation voltage and current were respectively kept at 40 kV and 20 mA. The morphology of the JNAP was observed using a field-emission scanning electron microscope (FESEM), equipped with energy-dispersive X-ray spectroscopy (EDS). A Hitachi fluorescence spectrophotometer F-7000 was used to investigate the fluorescence of different samples when the excitation and emission slits were 2.5 nm and 2.5 nm. The magnetic properties of the samples were measured using a vibrating sample magnetometer (VSM).
Results and discussion
Phase compositions
Fig. 1 exhibits the XRD patterns of the Fe3O4 NPs, JNAP, JNNP, HNAP and P-JNAP. The XRD patterns of the Fe3O4 NPs conform to the cubic structure of Fe3O4 (PDF#75-0499), and no characteristic peaks of other impurities are found. The corresponding diffraction peaks of Fe3O4 are also found in the four kinds of nanofiber pellicles, which indicates that the nanofiber pellicles contain Fe3O4 NPs. Meanwhile, the diffraction peak of the amorphous PVP (2θ ≈ 22°) can also be observed in the four kinds of nanofiber pellicles.
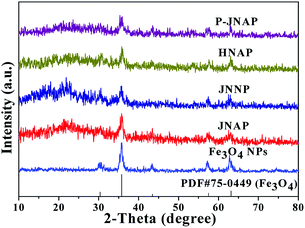 |
| | Fig. 1 XRD patterns of the Fe3O4 NPs, JNAP, JNNP, HNAP and P-JNAP. | |
Morphological and structural analyses
SEM images, EDS spectra and diameter distribution histograms of the Janus nanofibers in JNAP are given in Fig. 2. As seen in Fig. 2a, the nanofibers are in good directional alignment and form an array pellicle. It can be seen in Fig. 2b that exceptive-structured Janus nanofibers are obtained, with every single Janus nanofiber being composed of two nanofibers bound together side-by-side. As shown in Fig. 2b and c, the diameter of each single nanofiber in the Janus nanofibers is almost equal (the average diameter of a single nanofiber is ca. 720 nm). The EDS spectrum of the JNAP (Fig. 2d) indicates that the Janus nanofibers are composed of the elements C, N, O, Fe, Tb and Pt. The Pt peak is attributed to the conductive film sprayed on the surface of the sample for SEM observation. Fig. 2e presents the EDS line-scan analysis results for the Janus nanofibers, where Fe and Tb elements indicate Fe3O4 and Tb(BA)3phen, respectively. The Fe and Tb elements are respectively found in two single nanofibers, further demonstrating that the Janus nanofibers were successfully constructed by conjugate electrospinning and the goal of segregating magnetic nanoparticles and fluorescent molecules has been realized.
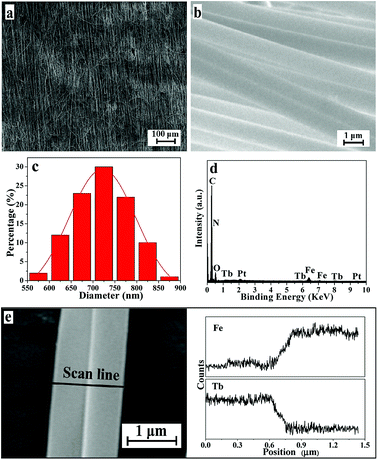 |
| | Fig. 2 SEM images at low magnification (a) and high magnification (b) of the JNAP. Diameter distribution histogram of the nanofibers in Janus nanofibers in the JNAP (c). EDS spectrum of the JNAP (d). EDS line-scan analysis of a single Janus nanofiber in the JNAP (e). | |
Fluorescence performance
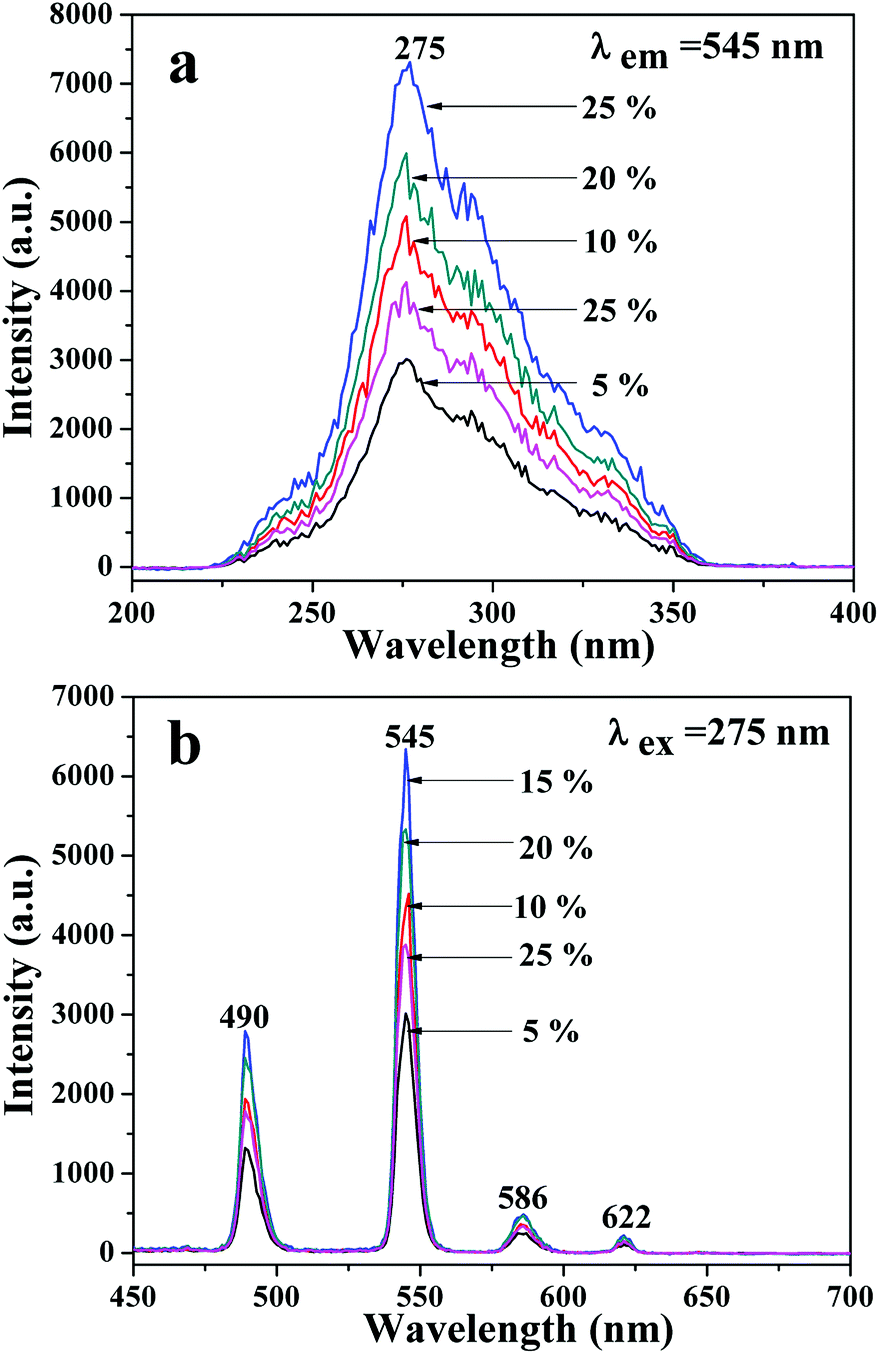
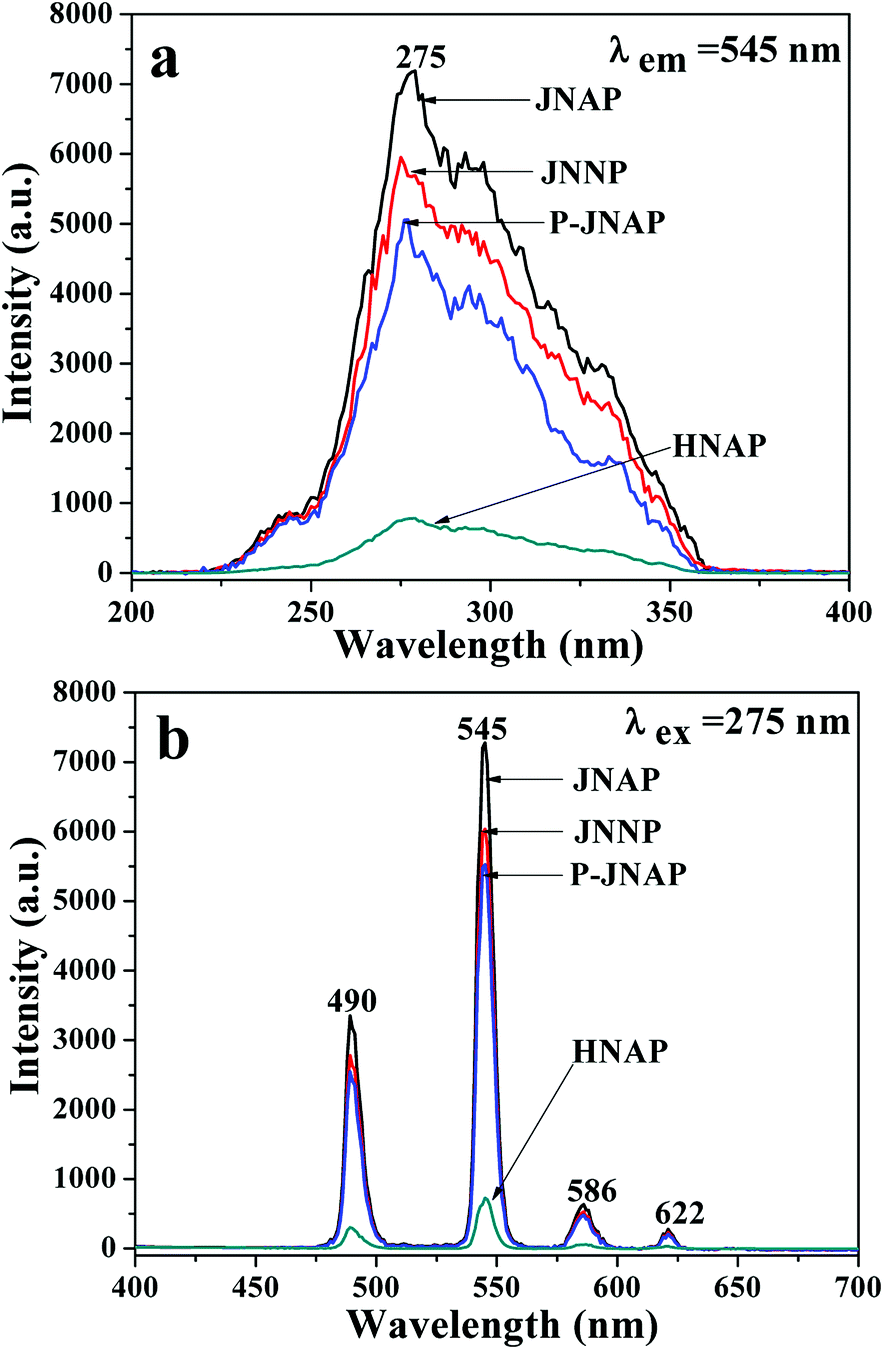
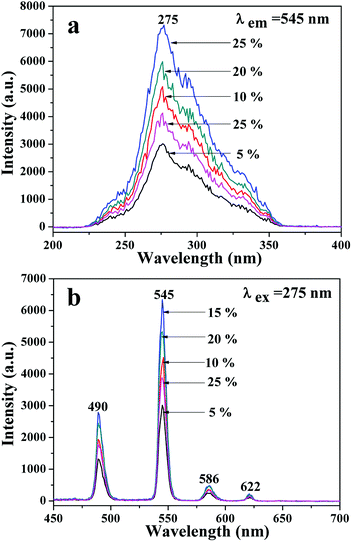
A series of JNAP samples (the mass ratio of Fe3O4 NPs to PVP was 0.5:1, and the mass percentages of Tb(BA)3phen to PVP were 5%, 10%, 15%, 20% and 25%, respectively) were prepared to find the optimal mass percentage of Tb(BA)3phen to PVP. Fig. 3a and b respectively exhibit the excitation and emission spectra of the above-described JNAP samples. A broad excitation band from 200 nm to 400 nm of the JNAP was observed when the monitoring wavelength was 545 nm. The strongest excitation peak at 275 nm can be put down to the π → π* electron transition of the ligands. The strong characteristic emission peaks of Tb3+ were found at 490, 545, 586, 622 nm under 275 nm excitation. These peaks are attributed to the 5D4 → 7F6 (490 nm), 5D4 → 7F5 (545 nm), 5D4 → 7F4 (586 nm), 5D4 → 7F3 (622 nm) energy level transitions of Tb3+, respectively. The 5D4 → 7F5 transition at 545 nm (green light) is the predominant emission peak. The fluorescence intensity of the JNAP first enhances and then weakens with the increase of Tb(BA)3phen content, as revealed in Fig. 3. When the mass percentage of Tb(BA)3phen to PVP is 15%, the JNAP achieves the highest fluorescence intensity. When the mass percentage of Tb(BA)3phen to PVP exceeds 15%, the distribution of the Tb(BA)3phen becomes denser in the polymer matrix, resulting in stronger non-radiative transitions amongst the Tb3+ ions, which causes obvious reductions in the fluorescence intensity. Therefore, the optimum mass percentage of Tb(BA)3phen to PVP is 15%.
 |
| | Fig. 3 Excitation spectra (a) and emission spectra (b) of the JNAP samples doped with different amounts of Tb(BA)3phen when the mass ratio of Fe3O4 to PVP is 0.5:1. | |
Physical photographs of the JNAP are displayed in Fig. 4. From Fig. 4a–c, it can be seen that the JNAP can be easily bent by hand and also possesses the ability of self-recovery, which proves that the JNAP is flexible. Fig. 4d shows a camera photograph of the JNAP under 275 nm UV illumination in darkness, indicating that the JNAP can emit green fluorescence.
 |
| | Fig. 4 Physical digital photos of unbent JNAP (a), bent JNAP (b), recovered JNAP (c), and the JNAP under 275 nm UV illumination in darkness (d). | |
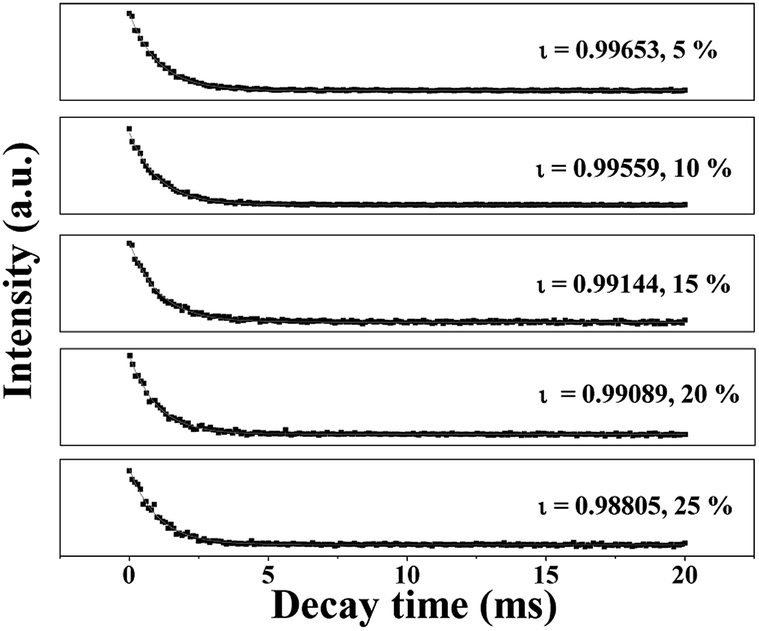
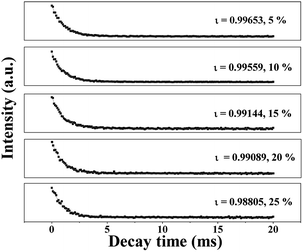
The fluorescence decay curves of Tb3+ ions in the JNAP samples doped with different amounts of Tb(BA)3phen are shown in Fig. 5. The excitation wavelength is set to be 275 nm and the monitoring wavelength is 545 nm. It is generally known that the fluorescence decay curves follow the single-exponential decay:48
| It = I0exp (−t/ι), |
where
It signifies the intensity at time
t,
I0 represents the intensity at
t = 0 and
ι symbolizes the decay lifetime. The average lifetime values (
ι/ms) of the JNAP are revealed in
Fig. 5. As the content of Tb(BA)
3phen increases, the fluorescence lifetime of the
5D
4 →
7F
5 (545 nm) transition is gradually decreased. The introduction of more Tb(BA)
3phen leads to a reduced distance between Tb
3+ ions in Tb(BA)
3phen molecular clusters in the JNAP. Thus, the energy transfer among Tb
3+ ions is increased, and the fluorescence lifetime of Tb
3+ is shortened.
49
 |
| | Fig. 5 Fluorescence decay dynamics of the 5D4 → 7F5 transitions (λem = 545 nm) in the JNAP doped with varying amounts of Tb(BA)3phen. | |
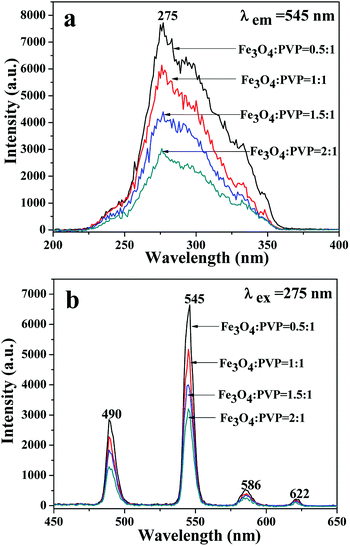
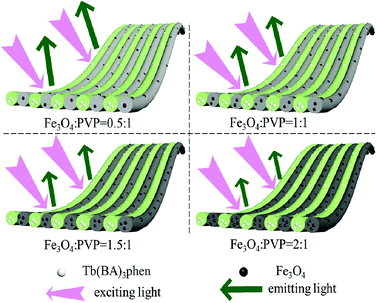
A series of JNAP samples (the mass percentage of Tb(BA)3phen to PVP was 15% and the mass ratios of Fe3O4 NPs to PVP were 0.5:1, 1:1, 1.5:1, and 2:1, respectively) were prepared to explore the effect of different amounts of Fe3O4 NPs on the fluorescence intensity. The intensities of the excitation and emission peaks gradually decrease along with increasing content of Fe3O4 NPs added into the JNAP, as illustrated in Fig. 6. As previously reported, dark-colored Fe3O4 NPs can absorb visible light (400 nm < λ < 700 nm) and ultraviolet light (λ < 400 nm).41 Therefore, Fe3O4 NPs can absorb the excitation light and emission light of the JNAP, and the degree of absorption becomes stronger as the content of Fe3O4 NPs is increased. Moreover, the more Fe3O4 NPs added into the JNAP, the darker the color of the products and the more intense the absorption of excitation emission light by the Fe3O4 NPs, as revealed in Fig. 7, giving rise to lower fluorescence intensity.
 |
| | Fig. 6 Excitation spectra (a) and emission spectra (b) of the JNAP doped with different amounts of Fe3O4 NPs when the mass percentage of Tb(BA)3phen to PVP is 15%. | |
 |
| | Fig. 7 Schematic diagram of the excitation light and emission light in the JNAP doped with different amounts of Fe3O4 NPs when the mass percentage of Tb(BA)3phen to PVP is 15%. | |
Furthermore, the fluorescence intensities of samples with the same ingredients and content prepared by different methods were compared to highlight the superiority of the JNAP. The JNAP has the highest fluorescence intensity compared with three comparative samples. It has stronger fluorescence intensity than the P-JNAP, and HNAP possesses the lowest fluorescence intensity, as shown in Fig. 8. This outcome can be explained by three aspects: the special Janus structure, the arrangement of Janus nanofibers, and the electrospinning method. Fig. 9 presents schematic graphs of the excitation and emission light for the JNAP, JNNP, P-JNAP and HNAP. As seen from Fig. 9a, the JNAP is composed of aligned Janus nanofibers, and the Janus nanofibers are closely arranged. The Janus nanofibers in the upper layer can directly absorb excitation light and the produced emission light can be directly emitted without refraction or reflection, which is the most effective mode for fluorescence. Furthermore, the Fe3O4 NPs and Tb(BA)3phen molecules are absolutely segregated, leading to a reduced effect of the Fe3O4 NPs on the fluorescence performance. As depicted in Fig. 9b, the Fe3O4 NPs and Tb(BA)3phen molecules are also completely segregated in the JNNP. The surface of the JNNP is loose due to the unordered arrangement of Janus nanofibers. In this case, some of the excitation light can pass through the gaps in the upper layer and thereby excite the Janus nanofibers in lower layer. In this process, the Janus nanofibers in the upper layer can absorb some of the excitation light, and thus, the excitation light arriving to the Janus nanofibers in the lower layer is weakened. For the same reason, the emission light emitted from the lower layer is also absorbed by the upper layer in the JNNP. Hence, the fluorescence intensity of the JNNP is lower than that of the JNAP.
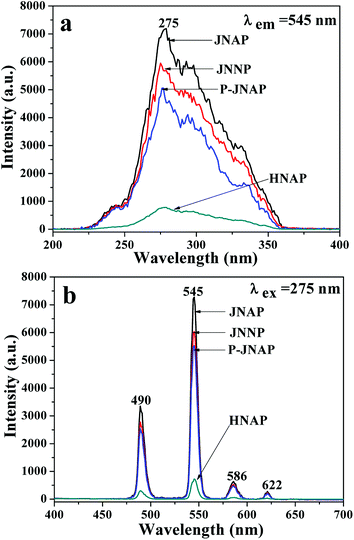 |
| | Fig. 8 Excitation spectra (a) and emission spectra (b) of JNAP, JNNP, P-JNAP and HNAP. | |
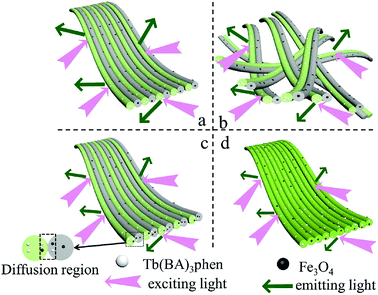 |
| | Fig. 9 Schematic graphs of the excitation and emission light in JNAP (a), JNNP (b), P-JNAP (c) and HNAP (d). | |
P-JNAP was prepared by parallel electrospinning. Fig. 10 describes the actual situation of two spinning dopes in parallel spinneret during the parallel electrospinning process. Inside the parallel spinneret, two spinning dopes mutually diffuse at the contact interfaces, so that diffusion regions exist at the contact interfaces between nanofibers in the Janus nanofibers. This results in an incomplete separation of Fe3O4 NPs from Tb(BA)3phen molecules in the Janus nanofibers. Thus, the fluorescence intensity of P-JNAP is lower than that of JNAP. As seen in Fig. 9d, the HNAP is totally made up of homogeneous nanofibers where Fe3O4 NPs and Tb(BA)3phen molecules are directly mixed, so that the fluorescence intensity of the HNAP is the lowest. The above new findings thoroughly prove that conjugate electrospinning has more advantages than parallel electrospinning in the fabrication of Janus nanofibers.
 |
| | Fig. 10 Digital photograph of parallel spinneret during parallel electrospinning process. | |
Magnetic properties
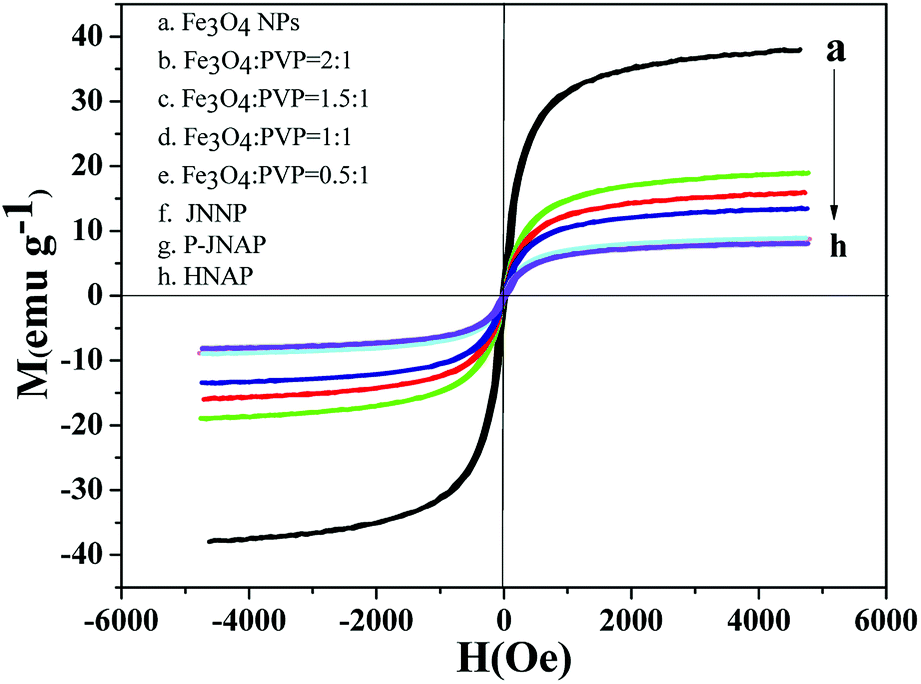
Typical hysteresis loops of the Fe3O4 NPs, the JNAP with different mass ratios of the Fe3O4 NPs and comparative samples are exhibited in Fig. 11, and corresponding saturation magnetization results are summarized in Table 4. It is known that the saturation magnetization of a magnetic compound material depends on the doping mass ratio of the magnetic material. As can be seen in Table 4, the saturation magnetization of the Fe3O4 NPs reaches 37.99 emu g−1. As the content of the Fe3O4 NPs increases in the JNAP, the saturation magnetization of the JNAP also increases from 8.93 to 18.99 emu g−1. Thus, the JNAP possesses adjustable magnetism via changing the doping amount of the Fe3O4 NPs. The magnetism of the comparative samples is similar to that of the JNAP (prepared by SA3/SB1) owing to the same content of Fe3O4 NPs in the samples.
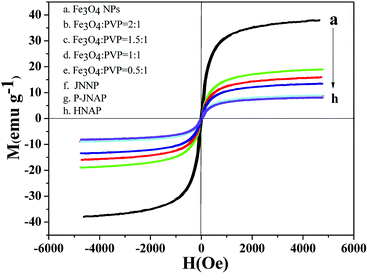 |
| | Fig. 11 Hysteresis loops of the Fe3O4 NPs (a), the JNAP doped with different amounts of Fe3O4 NPs (b–e), JNNP (f), P-JNAP (g) and HNAP (h). | |
Table 4 Saturation magnetization results of the Fe3O4 NPs, the JNAP doped with different amounts of Fe3O4 NPs and comparative samples
| Samples |
Saturation magnetization (emu g−1) |
| Fe3O4 NPs |
37.99 |
| JNAP |
| Fe3O4:PVP = 2:1 |
18.99 |
| Fe3O4:PVP = 1.5:1 |
15.86 |
| Fe3O4:PVP = 1:1 |
13.50 |
| Fe3O4:PVP = 0.5:1 |
8.93 |
| Comparative samples |
| JNNP |
8.77 |
| P-JNAP |
8.07 |
| HNAP |
7.91 |
Formation mechanism for the JNAP
Schematic graphs of the formation mechanism of Janus nanofibers and the JNAP are presented in Fig. 12. Under the effect of an electrostatic field, the spinning dopes in the two spinnerets form two bundles of continuous nanofibers with positive and negative charges after positive and negative power are applied to the two spinnerets, respectively, as seen in Fig. 12a. Here, an electric field is formed between the two spinnerets when positive and negative charges accumulate at the tip of the two spinnerets, and the directions of the electric field lines are indicated by the dotted line (from the positive pole to the negative pole), as shown in Fig. 12b. Magnetic nanofibers and fluorescent nanofibers attract each other along the path of the electric field. After the two kinds of nanofiber bundles meet in the middle of the two spinnerets, the electrical charges in the two kinds of nanofiber bundles are neutralized, and the formed Janus nanofibers move downward by gravity. Due to electrostatic repulsion, the same nanofibers (e.g. magnetic nanofiber and fluorescent nanofiber) cannot get together to form a Janus nanofiber in this process. Therefore, [Fe3O4/PVP]//[Tb(BA)3phen/PVP] Janus nanofibers are obtained. Fig. 12c describes the process of collecting an electrically neutral Janus nanofiber bundle with the rotating drum. At this point, the Janus nanofibers are affected by the vortex generated by the rotating drum in the experimental environment, so that the Janus nanofibers shake slightly and are drawn to and wrap around the rotating drum to form the JNAP.
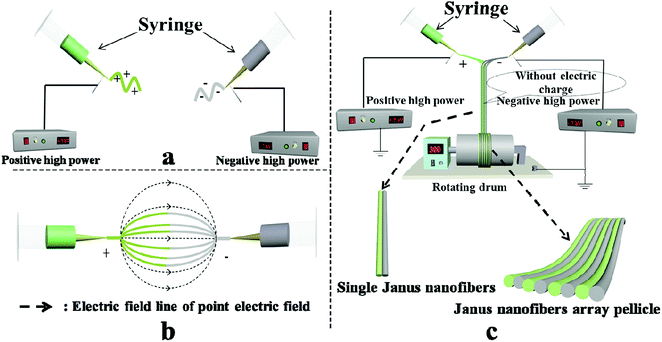 |
| | Fig. 12 Schematic graph of the formation mechanism of Janus nanofibers and the JNAP: (a and c) front view of the device; (b) top view of the device. | |
Conclusions
In summary, a [Fe3O4/PVP]//[Tb(BA)3phen/PVP] magnetic-fluorescent bifunctional Janus nanofiber array pellicle was successfully fabricated via facile conjugate electrospinning with a rotating drum as a collection device. The JNAP is made up of aligned Janus nanofibers. Each Janus nanofiber is composed of a magnetic [Fe3O4/PVP] nanofiber and a fluorescent [Tb(BA)3phen/PVP] nanofiber. The diameter of each single nanofiber in the Janus nanofibers is ca. 720 nm. It is noteworthy that the magnetism of the JNAP can be adjusted by adding diverse amounts of Fe3O4 NPs and the JNAP can emit green light under the excitation of ultraviolet light. Compared with three comparative samples, the JNAP has the highest fluorescence intensity owing to its peculiar Janus structure, which achieves effective segregation of Fe3O4 NPs and Tb(BA)3phen molecules, as well as directional alignment of the Janus nanofibers. More importantly, conjugate electrospinning can be extended to manufacture other Janus nanofibers with multifunctionality.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51573023, 50972020), Natural Science Foundation of Jilin Province (20170101101JC, 20180520011JH), Industrial Technology Research and Development Project of Jilin Province Development and Reform Commission (2017C052-4), Science and Technology Research Planning Project of the Education Department of Jilin Province during the 13th five-year plan period (JJKH20170608KJ, JJKH20181122KJ), and Innovative Foundation (XJJLG-2017-04) and Youth Foundation (XQNJJ-2016-01, XQNJJ-2017-17) of Changchun University of Science and Technology.
References
- N. Lv, J. L. Zhang, G. M. Li, X. Wang and J. Z. Ni, J. Phys. Chem. C, 2017, 121, 11926–11931 CrossRef CAS.
- R. Tao, C. L. Shao, X. H. Li, X. W. Li, S. Liu, S. Yang, C. C. Zhao and Y. C. Liu, J. Colloid Interface Sci., 2018, 529, 404–414 CrossRef CAS.
- C. M. Cobley, J. Y. Chen, E. C. Cho, L. V. Wang and Y. N. Xia, Chem. Soc. Rev., 2011, 42, 44–56 RSC.
- L. Sun, Y. Zang, M. D. Sun, H. G. Wang, X. J. Zhu, S. F. Xu, Q. B. Yang, Y. X. Li and Y. M. Shan, J. Colloid Interface Sci., 2010, 350, 90–98 CrossRef CAS PubMed.
- C. Y. Wen, H. Y. Xie, Z. L. Zhang, L. L. Wu, J. Hu, M. Tang, M. Wu and D. W. Pang, Nanoscale, 2016, 8, 12406–12429 RSC.
- Y. J. Yang, B. Tang, L. Zhang, C. Wang, H. T. Ma, D. W. Pang and Z. L. Zhang, J. Mater. Chem. B, 2018, 6, 961–965 RSC.
- K. Koc, B. Karakus, K. Rajar and E. Alveroglu, Superlattices Microstruct., 2017, 110, 198–204 CrossRef CAS.
- J. W. Lan, J. Y. Chen, N. X. Li, X. H. Ji, M. X. Yu and Z. K. He, Talanta, 2016, 151, 126–131 CrossRef CAS PubMed.
- Y. J. Lu, Y. Zheng, S. S. You, F. Wang, Z. Gao, J. Shen, W. T. Yang and M. Z. Yin, ACS Appl. Mater. Interfaces, 2015, 7, 5226–5232 CrossRef CAS.
- S. L. Gai, P. P. Yang, C. X. Li, W. X. Wang, Y. L. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166–1172 CrossRef CAS.
- P. Sun, H. Y. Zhang, C. Liu, J. Fang, M. Wang, J. Chen, J. P. Zhang, C. B. Mao and S. K. Xu, Langmuir, 2010, 26, 1278–1284 CrossRef CAS PubMed.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372–390 RSC.
- W. H. Di, S. K. P. Velu, A. Lascialfari, C. X. Liu, N. Pinna, P. Arosio, Y. Sakka and W. P. Qin, J. Mater. Chem., 2012, 22, 20641–20648 RSC.
- J. Tian, Q. L. Ma, W. S. Yu, X. T. Dong, Y. Yang, B. Zhao, J. X. Wang and G. X. Liu, New J. Chem., 2017, 41, 13983–13992 RSC.
- P. Jing, Q. Wang, B. C. Liu, G. R. Xu, Y. B. Zhang, J. Zhang and G. De, RSC Adv., 2014, 4, 44575–44582 RSC.
- T. Nisisako, T. Torii, T. Takahashi and Y. Takizawa, Adv. Mater., 2006, 18, 1152–1156 CrossRef CAS.
- N. Lv, Z. G. Wang, W. Z. Bi, G. M. Li, J. L. Zhang and J. Z. Ni, J. Mater. Chem. B, 2016, 4, 4402–4409 RSC.
- Q. L. Ma, W. S. Yu, X. T. Dong, J. X. Wang and G. X. Liu, Nanoscale, 2014, 6, 2945–2952 RSC.
- D. Pan, F. Z. Mou, X. F. Li, Z. Y. Deng, J. Sun, L. L. Xu and J. G. Guan, J. Mater. Chem. A, 2016, 4, 11768–11774 RSC.
- X. J. Zhou, Q. L. Ma, W. S. Yu, T. T. Wang, X. T. Dong, J. X. Wang and G. X. Liu, J. Mater. Sci., 2015, 50, 1–12 CrossRef CAS.
- G. Q. Gai, L. Y. Wang, X. T. Dong, C. M. Zheng, W. S. Yu, J. X. Wang and X. F. Xiao, J. Nanopart. Res., 2013, 15, 1–9 CrossRef.
- Q. T. Wang, Y. T. Geng, J. H. Li, M. Z. Yin, Y. S. Hu, Y. X. Liu and K. Pan, Nanotechnology, 2018, 3(29), 135702 CrossRef PubMed.
- X. Xi, Q. L. Ma, X. T. Dong, D. Li, W. S. Yu, J. X. Wang and G. X. Liu, ChemPlusChem, 2018, 83, 108–116 CrossRef CAS.
- J. Y. Liu, Q. L. Ma, J. Tian, X. Xi, D. Li, X. T. Dong, W. S. Yu, X. L. Wang, J. X. Wang and G. X. Liu, RSC Adv., 2018, 8, 22887–22896 RSC.
- Q. L. Ma, J. X. Wang, X. T. Dong, W. S. Yu and G. X. Liu, Adv. Funct. Mater., 2015, 25, 2436–2443 CrossRef CAS.
- X. B. Li, Q. L. Ma, J. Tian, X. Xi, D. Li, X. T. Dong, W. S. Yu, X. L. Wang, J. X. Wang and G. X. Liu, Nanoscale, 2017, 9, 18918–18930 RSC.
- H. Ji, R. Zhao, N. Zhang, C. X. Jin, X. F. Lu and C. Wang, NPG Asia Mater., 2018, 10, 749–760 CrossRef CAS.
- M. Q. Chi, S. H. Chen, M. X. Zhong, C. Wang and X. F. Lu, Chem. Commun., 2018, 54, 5827–5830 RSC.
- D. Tian, X. F. Lu, G. D. Nie, M. Gao and C. Wang, Inorg. Chem. Front., 2018, 5, 635–642 RSC.
- X. Xi, Q. L. Ma, M. Yang, X. T. Dong, J. X. Wang, W. S. Yu and G. X. Liu, J. Mater. Sci. Eng. A, 2014, 25, 4024–4032 CAS.
- X. Xi, J. X. Wang, X. T Dong, Q. L. Ma, W. S. Yu and G. X. Liu, Chem. Eng. J., 2014, 254, 259–267 CrossRef CAS.
- Y. T. Geng, P. Zhang, Q. T. Wang, Y. X. Liu and K. Pan, J. Mater. Chem. B, 2017, 5, 5390–5396 RSC.
- J. X. He, K. Qi, Y. M. Zhou and S. Z. Cui, J. Appl. Polym. Sci., 2014, 131, 631–644 Search PubMed.
- J. X. He, Y. M. Zhou, K. Qi, L. D. Wang, P. P. Li and S. Z. Cui, Fibers Polym., 2013, 14, 1857–1863 CrossRef CAS.
- S. H. Wu and X. H. Qin, Mater. Lett., 2013, 106, 204–207 CrossRef CAS.
- P. Moradipour, F. Dabirian, L. Rajabi and A. A. Derakhsha, Ceram. Int., 2016, 42, 13449–13458 CrossRef CAS.
- F. Q. Sun, C. Yao, T. Y. Song and X. S. Li, J. Text., 2011, 102, 633–638 CAS.
- D. W. Li, X. Pan, B. B. Sun, T. Wu, W. M. Chen, C. Huang, Q. F. Ke, H. A. EI-Hamshary, S. S. Al-Deyab and X. M. Mo, J. Mater. Chem. B, 2015, 3, 8823–8831 RSC.
- Z. G. Xie, H. T. Niu and T. Lin, RSC Adv., 2015, 5, 15147–15153 RSC.
- Y. M. Zhou, J. X. He, H. B. Wang, K. Qi, B. Ding and S. Z. Cui, Mater. Design, 2016, 95, 591–598 CrossRef CAS.
- L. B. Fan, Q. L. Ma, J. Tian, D. Li, X. Xi, X. T. Dong, W. S. Yu, J. X. Wang and G. X. Liu, Mater. Des., 2018, 53, 2290–2302 CAS.
- L. B. Fan, Q. L. Ma, J. Tian, D. Li, X. Xi, X. T Dong, W. S. Yu, J. X. Wang and G. X. Liu, RSC Adv., 2017, 7, 48702–48711 RSC.
- F. Xu, L. M. Li and X. J. Cui, J. Nanomater., 2012, 2012, 183–190 Search PubMed.
- X. L. Song, W. S. Liu, J. Wang, S. H. Xu, B. Liu, Q. S. Cai and Y. Z. Ma, Mater. Lett., 2017, 212, 20–24 CrossRef.
- Y. L. Chen, Y. M. Cui, Y. S. Jia, K. Zhan, H. Zhang, G. X. Chen, Y. D. Yang, M. Wu and H. M. Ni, J. Appl. Polym. Sci., 2014, 131, 40716–40724 Search PubMed.
- Z. J. Wang, Q. L. Ma, X. T. Dong, D. Li, X. Xi, W. S. Yu, J. X. Wang and G. X. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 26226–26234 CrossRef CAS PubMed.
- Q. L. Ma, J. X. Wang, X. T. Dong, W. S. Yu, G. X. Liu and J. Xu, J. Mater. Chem., 2012, 22, 14438–14442 RSC.
- Y. W. Liu, Q. L. Ma, X. T. Dong, W. S. Yu, J. X. Wang and G. X. Liu, Phys. Chem. Chem. Phys., 2015, 17, 22977–22984 RSC.
- R. Bonzanini, D. T. Dias, E. M. Girotto, E. C. Muniz, M. L. Baesso, J. M. A. Caiut, Y. Messaddeq, S. J. L. Ribeiro, A. C. Bento and A. F. Rubira, J. Lumin., 2006, 117, 61–67 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Wensheng Yu,
Jinxian Wang
*,
Wensheng Yu,
Jinxian Wang