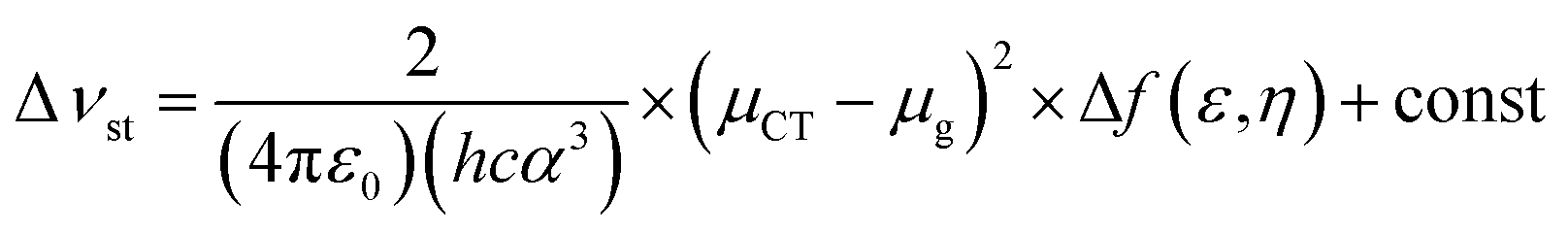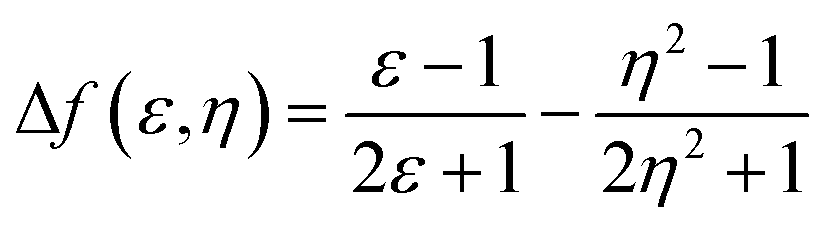DOI:
10.1039/C8RA08439D
(Paper)
RSC Adv., 2019,
9, 1-10
Enhancement of intramolecular charge transfer strength in diphenylamine substituted symmetric 1,3,4-oxadiazole derivatives†
Received
11th October 2018
, Accepted 10th December 2018
First published on 19th December 2018
Abstract
The intramolecular charge transfer characteristic of two diphenylamine substituted symmetric 1,3,4-oxadiazole derivatives (DPAOXD and DPAOXDBEN) was studied through a combination of experimental techniques and theoretical calculations. Significant enhancement of intramolecular charge transfer strength has been found in both these compounds through molecular structure modification. The experimental result found only a small red shift in the absorption spectra (∼15 nm) but a very large red shift in the emission spectra (∼114 nm for DPAOXD and ∼140 nm for DPAOXDBEN) with increasing solvent polarity, indicating a large extent charge transfer occurred in their excited state. The increase of molecular dipole moment from the ground state to the charge transfer excited state is calculated to be 22.10 D in DPAOXD and 26.67 D in DPAOXDBEN, respectively. Theoretical calculations present clear evidence that electrons transfer from the terminal diphenylamine to the bi-1,3,4-oxadiazole rings in DPAOXD, and the two 1,3,4-oxadiazole rings and central benzene ring in DPAOXDBEN. As compared to the methoxy group, the substitution by a diphenylamine group could increase both the transferred charge and distance, which could substantially strengthen the charge transfer character. Further introduction of a central benzene ring in DPAOXDBEN could further increase the transferred distance, and then the charge transfer strength. These findings could provide good guidance for the design of molecules with high intramolecular charge transfer characteristics.
Introduction
Organic π-conjugated materials with intramolecular charge transfer characteristics have drawn considerable attention in recent years for their potential applications in dye-sensitized solar cells (DSSCs),1–3 organic photovoltaic cells (OPVCs),4–7 and organic light-emitting diodes (OLEDs).8–10 As compared to inorganic materials, these organic materials have many advantages, such as flexibility, low cost and easy fabrication. However, deficiencies such as relatively low intramolecular charge transfer strength also limit their widely commercial applications.11 Hence, plenty efforts focus on studying organic materials with intramolecular charge transfer characteristic have been conducted.12–17 Among all these materials investigated, organic small molecules have been considered as an important kind of intramolecular charge transfer materials because of their well-defined structures, easy adjustability and purification.18–20 Commonly, the molecular structures of organic small molecules with intramolecular charge transfer characteristic contain electron donors (D) and electron acceptors (A), which are linked through π-bridge (D–π–A).21,22 It is reported that modifications of the push–pull electronic effect of electron donors and electron acceptors or conjugated linkers are all effective ways to improve the intramolecular charge transfer strength.23–29
The 1,3,4-oxadiazole (OXD) ring is a kind of important structural unit in organic photoelectric functional materials for its good luminance, stable thermal performance and excellent electron-transporting properties.30–33 Significantly, the 1,3,4-oxadiazole ring is also a electron-withdrawing group with excellent electron-withdrawing ability. Our group has been continuously interested in studying molecules with intramolecular charge transfer characteristics looking for approaches which could effectively enhance the intramolecular charge transfer to obtain materials with relatively high strength of intramolecular charge transfer and establish the structure–properties relationship of molecules with intramolecular charge transfer characteristics based on 1,3,4-oxadiazole derivatives. In our previous studies, it has already been proved that alkoxy phenyl substituted 1,3,4-oxadiazole derivatives show obvious intramolecular charge transfer characteristic, in which symmetric molecular structure was demonstrated to be an effective way to enhance their intramolecular charge transfer characteristic.34 Furthermore, it was also found that the substituted positions and quantities of methoxy groups in symmetric bi-1,3,4-oxadiazole derivatives would affect the intramolecular charge transfer characteristic; and the strength of intramolecular charge transfer could be enhanced by the introduction of methoxy group in the meta-position on the benzene ring or increase of the quantities of substituted methoxy groups.35
Herein, in order to further increase the strength of intramolecular charge transfer, two diphenylamine substituted symmetric 1,3,4-oxadiazole derivatives named DPAOXD and DPAOXDBEN were synthesized. As shown in Scheme 1a, both of the two compounds maintain symmetric molecular structures for that could enhance intramolecular charge transfer characteristic through doubling the transferred charge.34 In addition, two key points are also considered in this molecular design: (1) to increase the electron-donating ability of the donors: as compared with BOXD-p-OCH3 in which a methoxy group was introduced in para position on the benzene ring of symmetric bi-1,3-4-oxadiazole derivative (Scheme 1b), the terminal methoxy group was replaced by a stronger donor group, diphenylamine in DPAOXD, which was expected to be able to donate more electrons to the acceptors; (2) to enlarge the charge transfer distance: in DPAOXDBEN, an additional benzene ring was introduced between the two 1,3,4-oxadiazole rings on the basis of the molecular structure of DPAOXD. Experimental studies and theoretical calculations are carried out jointly to illustrate the effect of molecular structural factors on intramolecular charge transfer characteristic. These results would be helpful to establish the structure–property relationships of organic small molecules with high intramolecular charge transfer characteristic.
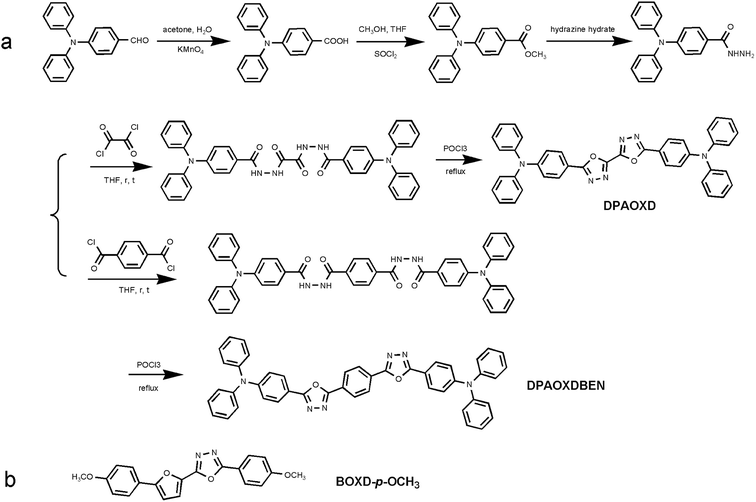 |
| | Scheme 1 (a) Synthetic routes of DPAOXD and DPAOXDBEN; (b) molecular structure of BOXD-p-OCH3. | |
Materials and methods
Synthesis
DPAOXD and DPAOXDBEN are obtained through the synthetic route in Scheme 1a for the first time, while BOXD-p-OCH3 was synthesized at our previous work and present here for comparison. Their purities are verified by FT-IR, 1H NMR, and elemental analysis.
Synthesis of 4-(N,N-diphenylamino)benzoic acid
First, 4-(N,N-diphenylamino)benzaldehyde (5 g) was dissolved into the mixture of acetone and water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); second, slowly added 2.5 times more potassium permanganate powder into the solutions under stirring conditions and heated to reflux for about 8 h. After the reaction complete, cooled the solutions to room temperature and removed the solvent by vacuum-rotary evaporation; third, joined some water and filtered; then, added a few drops of 10% HCl into the filtrate and white floc would be formed; finally, washed the white precipitate several times with water, filtered, and dried to obtain the pure white powder.
:1); second, slowly added 2.5 times more potassium permanganate powder into the solutions under stirring conditions and heated to reflux for about 8 h. After the reaction complete, cooled the solutions to room temperature and removed the solvent by vacuum-rotary evaporation; third, joined some water and filtered; then, added a few drops of 10% HCl into the filtrate and white floc would be formed; finally, washed the white precipitate several times with water, filtered, and dried to obtain the pure white powder.
Synthesis of 4-(N,N-diphenylamino)benzoate
First, the 4-(N,N-diphenylamino)benzoic acid (3 g) was dissolved into the mixture of tetrahydrofuran and methanol; second, slowly added 5 mL thionyl chloride under ice-water bath, stirred for 1 h and heated to reflux for about 10 h. After the reaction complete, cooled the solutions to room temperature and poured into water; third, used dichloromethane to extract the lower liquid and dried with anhydrous magnesium sulfate; then, purification by silica gel column chromatography (dichloromethane:petroleum ether = 4:1); finally, removed the solvent by vacuum-rotary evaporation and obtained the target product.
Synthesis of 4-(N,N-diphenylamino)benzohydrazide
First, the 4-(N,N-diphenylamino)benzoate was dissolved into ethanol; second, added moderate hydrazine hydrate, stirred and heated to reflux for about 30 h; after the reaction complete, cooled the solutions to room temperature and filtration. Then, wash the white precipitate several times with methanol, filtered, and dried to obtain the target product.
Synthesis of 2,2-bis[4-4-(N,N-diphenylamino)]-1,3,4-oxadiazole (DPAOXD)
Firstly, the oxalyl chloride (0.20 g) was slowly dropped into 50 mL tetrahydrofuran (THF) solution of the 4-(N,N-diphenylamino)benzohydrazide (1 g). The mixture was stirred at room temperature for about 10 h and the resulting solution was filtered, yielding oxalyl acid dihydrazide. Secondly, oxalyl acid dihydrazide was dissolved in 50 mL phosphorous oxychloride (POCl3). The mixture was stirred and heated to reflux for about 40 h. After the reaction, the resulting solution was cooled to room temperature and poured into 1000 mL ice water. At last, the product was collected by filtration and recrystallized from ethanol.
1H NMR (300 MHz, DMSO-D6), (ppm, from TMS): 7.97 (m, 4H), 7.43 (t, J = 7.8 Hz, 8H), 7.22 (m, 12H), 7.01 (m, 4H). FT-IR (KBr, pellet, cm−1): 1612, 1590, 1485, 1427, 1335, 1297, 1260, 1185, 1144, 1074, 1029, 1007, 988, 948, 838, 824, 757, 733, 695, 634, 612, 529. Found: C, 76.67; H, 4.35; N, 13.46. Calc. for C40H28N6O2: C, 76.91; H, 4.52; N, 13.45%. Yield > 75%. Melting point: 271 °C.
Synthesis of 1,4-bis{[4-4-(N,N-diphenylamino)]-1,3,4-oxadiazole}benzene (DPAOXDBEN)
Firstly, the terephthaloyl chloride (0.33 g) was slowly dropped into 50 mL tetrahydrofuran (THF) solution of the 4-(N,N-diphenylamino)benzohydrazide (1 g). The mixture was stirred at room temperature for about 10 h and the resulting solution was filtered, yielding oxalyl acid dihydrazide. Secondly, oxalyl acid dihydrazide was dissolved in 50 mL phosphorous oxychloride (POCl3). The mixture was stirred and heated to reflux for about 40 h. After the reaction, the resulting solution was cooled to room temperature and poured into 1000 mL ice water. At last, the product was collected by filtration and recrystallized from ethanol.
1H NMR (300 MHz, DMSO-D6), (ppm, from TMS): 8.32 (s, 4H), 8.00 (d, J = 9.0 Hz, 4H), 7.42 (dd, J = 8.2, 7.4 Hz, 8H), 7.20 (m, 12H), 7.04 (d, J = 8.9 Hz, 4H). FT-IR (KBr, pellet, cm−1): 1611, 1594, 1487, 1449, 1413, 1319, 1275, 1190, 1177, 1155, 1123, 1102, 1072, 1026, 1009, 960, 895, 846, 823, 752, 742, 712, 694, 638, 615, 532, 522. Found: C, 78.69; H, 4.35; N, 11.98. Calc. for C46H32N6O2: C, 78.84; H, 4.60; N, 11.99%. Yield > 70%. Melting point: 253 °C.
Experimental and computational details
All of these solvents for the spectroscopic measurements are of spectroscopic grade and used as received. 1HNMR spectra are recorded with a Mercury-300BB 300 MHz spectrometer, using DMSO-D6 as solvents and tetramethylsilane (TMS) as an internal standard (δ 0.00). FT-IR spectra are recorded with a Perkin-Elmer spectrometer (Spectrum One B), and the sample is in the form of a pressed tablet with KBr. UV-vis absorption spectra are recorded on a Shimazu UV-2550 spectrometer and emission and excitation spectra are measured on a Perkin-Elmer LS 55 spectrometer. The room-temperature luminescence quantum yields in solutions were determined relative to quinine sulfate acid aqueous solution (0.546) and calculated according to the following equation: Φunk = Φstd(Iunk/Aunk)(Astd/Istd)(ηunk/ηstd)2, where Φunk is the radiative quantum yield of the sample; Φstd is the radiative quantum yield of the standard; Iunk and Istd are the integrated emission intensities of the sample and standard, respectively; Aunk and Astd are the absorbance of the sample and standard at the excitation wavelength, respectively; and ηunk and ηstd are the indexes of refraction of the sample and standard solutions (pure solvents were assumed), respectively. Fluorescence lifetimes are measured using FLS920 time-correlated single photon counting (TCSPC) system. In theoretical calculations, all the calculations are carried out with Gaussian 09 software package (version A.02) at the CAM-B3LYP3/6-31G(d,p) level.36 Multiwfn and VMD are employed for visualizing the molecular orbital, electron density variation, and charge transfer.37,38
Results and discussion
Photophysical properties
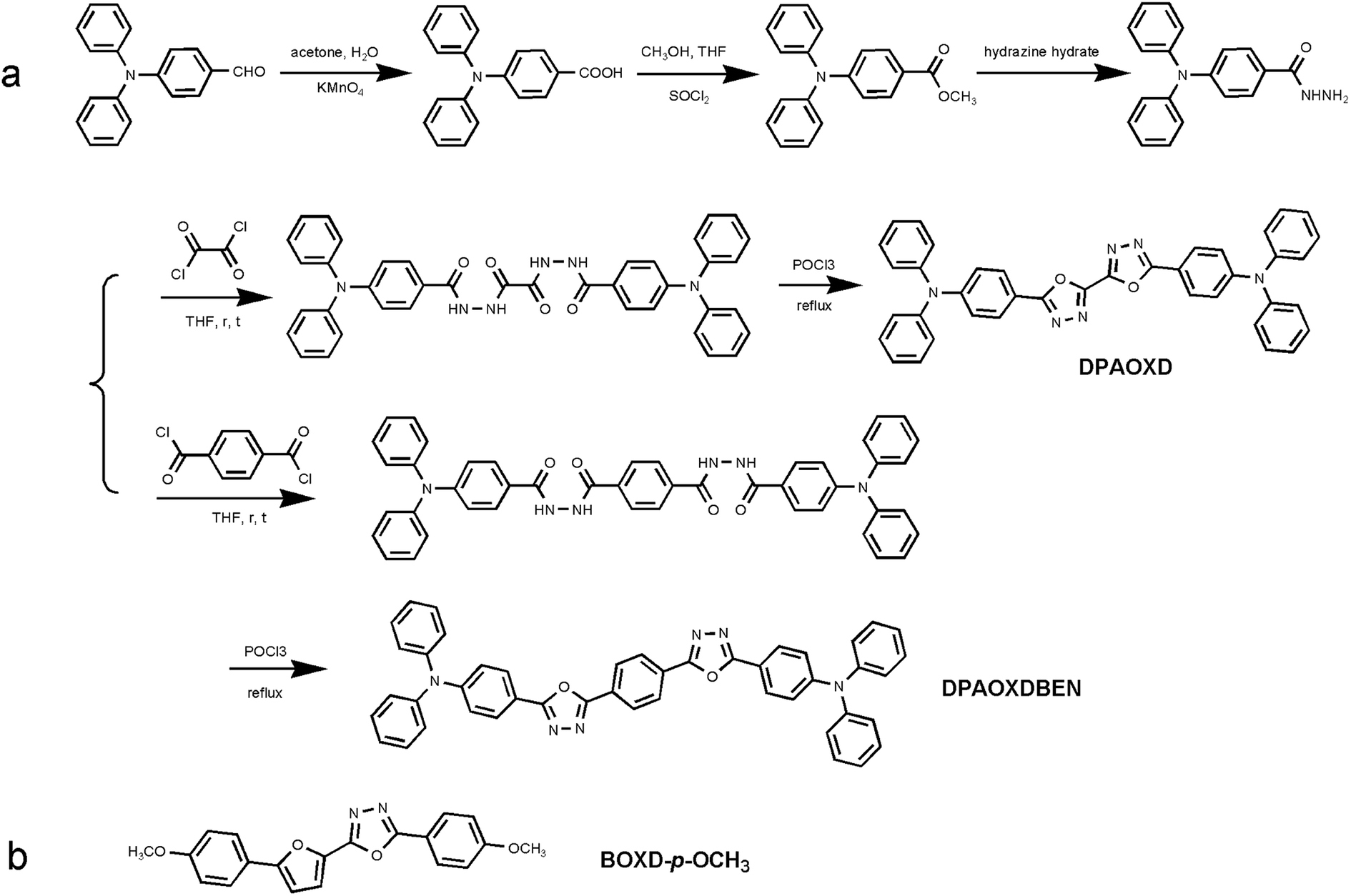
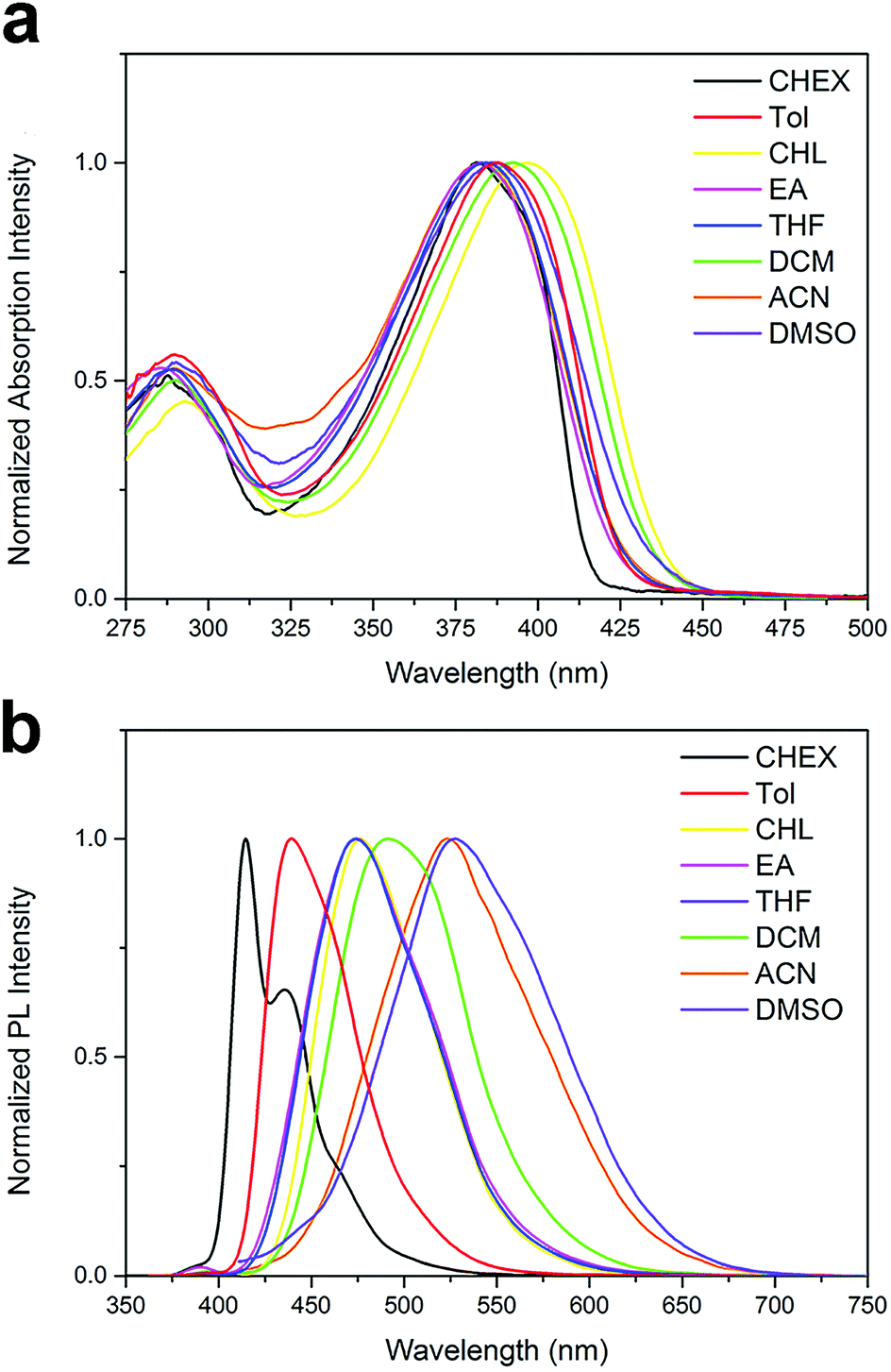
Absorption and fluorescence spectra. The UV-vis absorption and emission spectra of the two compounds are conducted in various solvents with different polarity, including cyclohexane (CHEX), toluene (Tol), chloroform (CHL), ethyl acetate (EA), tetrahydrofuran (THF), dichloromethane (DCM), acetonitrile (ACN) and dimethyl sulfoxide (DMSO). The spectra of the two compounds are shown in Fig. 1 and 2, and their absorption and emission maximum are listed in Table 1. The spectral data of BOXD-p-OCH3 are present in Fig. S1† for comparison. As shown in Fig. 1a and 2a, DPAOXD and DPAOXDBEN display two distinguishable absorption bands concentrated at about 380 nm and 290 nm in CHEX. Theoretical calculations suggest that the two absorption bands could be assigned to S0–S1 transition and S0–S7 transition, respectively, which will be discussed in detail below. From the nonpolar solvent (CHEX) to the strong polar solvent (ACN and DMSO), the maximum absorption peak of both the two compounds only show a slight red shift (∼15 nm), which is similar to BOXD-p-OCH3 (∼5 nm), indicating that the electronic and structural nature of ground state and Franck Condon (FC) excited state do not show obvious change.
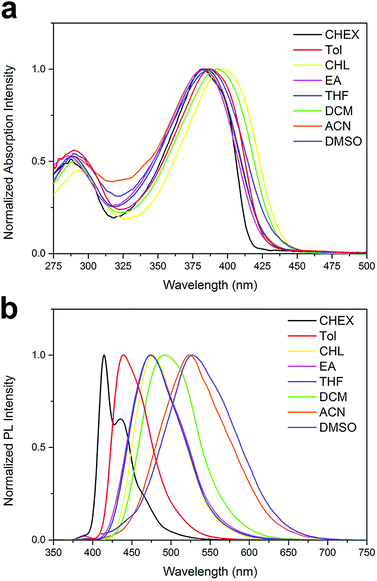 |
| | Fig. 1 Normalized (a) UV-vis absorption spectra and (b) emission spectra of DPAOXD. | |
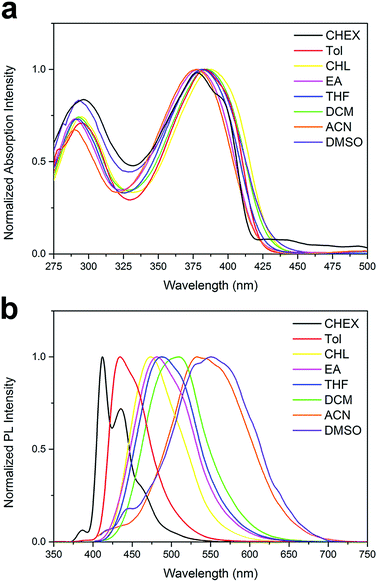 |
| | Fig. 2 Normalized (a) UV-vis absorption spectra and (b) emission spectra of DPAOXDBEN. | |
Table 1 Photophysical characteristics of these compounds investigated in different solvents at 5 × 10−6 M at room temperature
| Compound |
Solvent |
λuv (nm) |
λem (nm) |
Δυst (cm−1) |
Φa |
τf (ns)/χ2 |
kf (×108 s−1) |
knr (×108 s−1) |
| Nonexistent data are due to their poor solubility in DMSO. |
| DPAOXD |
CHEX |
287 |
381 |
414 |
2092 |
0.77 |
1.38/1.42 |
5.58 |
1.67 |
| Tol |
290 |
387 |
439 |
3060 |
0.63 |
1.72/1.33 |
3.66 |
2.15 |
| CHL |
293 |
396 |
476 |
4244 |
0.45 |
2.80/1.04 |
1.61 |
1.96 |
| EA |
286 |
383 |
474 |
5012 |
0.51 |
3.09/1.00 |
1.65 |
1.59 |
| THF |
289 |
384 |
474 |
4944 |
0.74 |
3.18/1.09 |
2.33 |
0.82 |
| DCM |
290 |
392 |
491 |
5143 |
0.39 |
3.81/1.05 |
1.02 |
1.60 |
| ACN |
290 |
383 |
523 |
6989 |
0.05 |
1.80/0.97 |
0.28 |
5.28 |
| DMSO |
290 |
386 |
528 |
6967 |
— |
1.25/1.21 |
— |
— |
| DPAOXDBEN |
CHEX |
296 |
378 |
412 |
2183 |
0.68 |
1.21/1.26 |
5.67 |
2.67 |
| Tol |
296 |
383 |
434 |
3068 |
0.53 |
1.55/1.06 |
3.53 |
3.13 |
| CHL |
295 |
387 |
474 |
4742 |
0.49 |
2.88/1.03 |
1.70 |
1.77 |
| EA |
290 |
378 |
481 |
5665 |
0.43 |
3.44/0.93 |
1.25 |
1.66 |
| THF |
293 |
381 |
487 |
5712 |
0.51 |
3.75/1.07 |
1.36 |
1.31 |
| DCM |
293 |
385 |
510 |
6366 |
0.41 |
4.59/1.15 |
0.89 |
1.29 |
| ACN |
291 |
377 |
533 |
7763 |
0.02 |
1.07/1.49 |
0.19 |
9.25 |
| DMSO |
294 |
383 |
552 |
7993 |
— |
0.68 (89.6), 3.44 (10.4)/1.34 |
— |
— |
However, obvious differences are found in their emission spectra. DPAOXD shows an intense emission at about 414 nm with a fine vibrational structure (∼435 nm) in nonpolar CHEX (Fig. 1b). When the polarity of solvent increase, the maximum emission peak red shift with the increase of solvent polarity and exhibit a large red-shift about 114 nm: 439 nm in Tol, 476 nm in CHL, 474 nm in EA and THF, 491 nm in DCM, 523 nm in ACN and 528 nm in DMSO (Table 1). Such large red shifts indicate that the dipole moment in the excited state is much larger than that in the ground state due to a charge transfer process. While in BOXD-p-OCH3, there are only 29 nm red shift found in the maximum emission (from CHEX to ACN), which is much less than that in DPAOXD, indicating that the replacement of diphenylamine with stronger electron donating ability could effectively enhance the intramolecular charge transfer strength. As present in Fig. 2b, DPAOXDBEN displays a similar solvatochromism behavior as DPAOXD. The emission maximum of DPAOXDBEN in nonpolar CHEX is centered at 412 nm; while in polar DMSO, it red-shifted to 552 nm. Other emission maxima are laid between CHEX and DMSO (Table 1). The red shift of DPAOXDBEN (about 140 nm) is much larger than that of DPAOXD. These results strongly indicate that the introduction of central benzene ring could further enhance the intramolecular charge transfer strength in DPAOXDBEN. In addition, the red shift of the emission spectra with the increasing solvent polarity could also be observed from the emission colors of DPAOXD and DPAOXDBEN in different solvents under 365 nm UV light excitation at 5 × 10−6 M (Fig. 3). It can be seen that solutions of the two compounds exhibit blue light in nonpolar solvents (CHEX and Tol); and then the color of solution gradually become green as the polarity of solvent increases; while in strong polar solvent (ACN and DMSO), their color change to yellow for DPAOXD and orange for DPAOXDBEN. In order to get a better understanding of the origin of the charge transfer character of these derivatives, the excitation spectra of DPAOXD and DPAOXDBEN are measured in solvents with different polarity at their respective maximum emission wavelength (Fig. S2 and S3†). It can be seen that the excitation spectra of both the two compounds show bands at around 290 nm and 390 nm, which are indeed similar to their absorption spectra and independent of the emission wavelengths. These results indicated that the photo-luminescence of DPAOXD and DPAOXDBEN in different solvents follow the same excitation progress. The intramolecular charge transfer state of DPAOXD and DPAOXDBEN should originate from the molecular geometry relaxation from the Franck–Condon excited state and the solvent induced redistribution of the atomic charge.
 |
| | Fig. 3 Images of (a) DPAOXD and (b) DPAOXDBEN solutions at 5 × 10−6 M under 365 nm UV light excitation. | |
Quantum yields and fluorescence decay
The fluorescence quantum yields for all of the two compounds in different solvents have been measured and the data are collected in Table 1. It can be seen that both DPAOXD and DPAOXDBEN exhibit high quantum yields in the nonpolar solvents (∼0.7 in CHEX and Tol). As the increase of the solvent polarity, the fluorescence quantum yields of both the two compounds exhibit a slight decrease. In the moderate polar solvent (CHL, EA, THF and DCM), the fluorescence quantum yields of both DPAOXD and DPAOXDBEN are in a range of 0.4–0.6. However, the fluorescence quantum yields of DPAOXD and DPAOXDBEN in ACN are both less than 0.1. The sharp decrease of the fluorescence quantum yields in strong polar solvents could be induced by the intermolecular interactions between the solute molecular and solvent molecular in polar solvent, which will be detailed discussed below.
In order to get more information about excited state, the fluorescence decay behavior of DPAOXD and DPAOXDBEN was measured in different solutions using the time correlated single photon counting (TCSPC) method. Almost all the fluorescence decay profile of the two compounds fit well with a single-exponential function and lifetimes in the range of 1.07–4.59 ns, except for the fluorescence decay profile of DPAOXDBEN in DMSO, which are fitted by a bi-exponential curve with two lifetimes in 0.6 ns and 3.43 ns, respectively (Table 1). From the nonpolar solvent CHEX to the moderate polar solvent DCM, these lifetimes in both of the two compounds show increase with the solvent polarity, indicating that the excited state could be stabilized in these solvents. While the lifetimes for DPAOXD and DPAOXDBEN decrease a lot in the strong polar solvents (ACN and DMSO). The radiative constants (kr) and non-radiative constants (knr) calculated from the measured fluorescence quantum yield (Φ) and lifetime (τf) are also summarized in Table 1. It can be seen that for the two compounds, the non-radiative constants is obvious large than the radiative constants in strong polar (ACN), thus it could be inferred that the decrease of fluorescence quantum yield and lifetime could be induced by the enhanced nonradiative decay, which is caused by the strong interactions between the compound with intramolecular charge transfer characteristic and the polar solvent.
Solvatochromic measurements and dipole moments
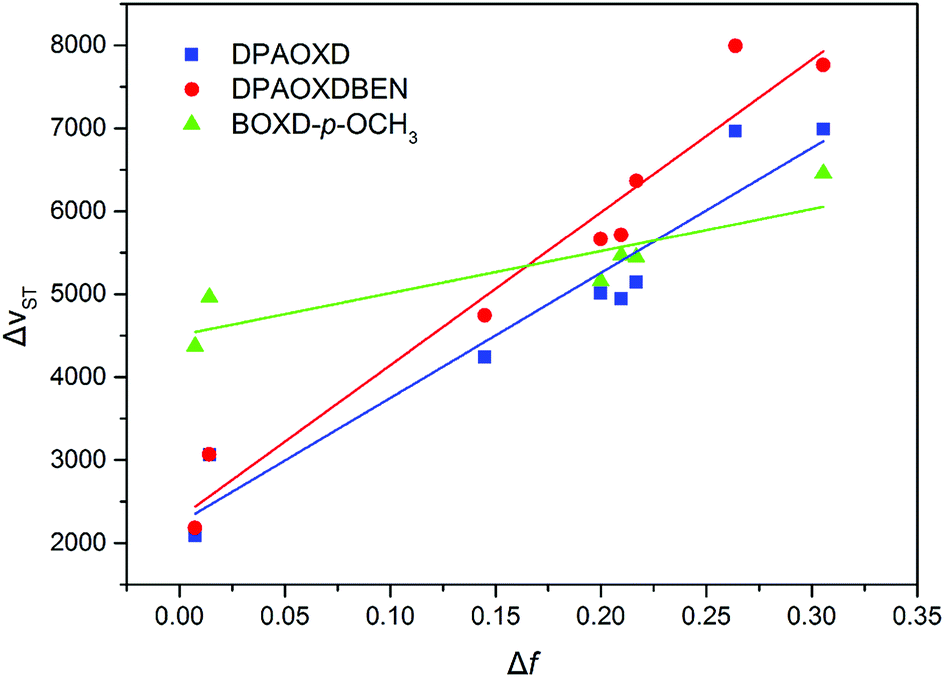
Fig. 4 shows the plot and fit line of stokes shift (Δυst) against solvent polarity parameters (Δf) of DPAOXD and DPAOXDBEN. It can be clearly seen that the stokes shifts of both the two compounds increase along with the increase of solvent polarity, which indicate that the intramolecular charge transfer state could be stable in polar solvents. Lippert–Mataga equation is used to estimated the changes in the dipole moment between the intramolecular charge transfer excited state and the ground state from the slope of the plot of the stokes shift against the solvent parameters.39,40 The equation is:
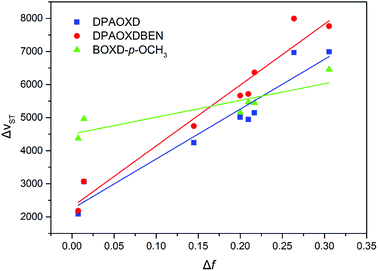 |
| | Fig. 4 Plot and fit line of stokes shift against solvent polarity parameters for these compounds investigated. | |
Δνst is the stokes shifts; h, c and ε0 is the Plank's constant, velocity of light and permittivity of vacuum, respectively; μg is the dipole moment of the ground state, and μCT is the dipole moment of the CT excited state; α is the solvent cavity (Onsager) radius obtained by optimizing the molecular geometry at the CAM-B3LYP/6-31G(d,p) level of theory (DPAOXD: 6.15 Å; DPAOXDBEN: 6.52 Å); ε and η is the dielectric constant and refractive index of the solvent, respectively. As present in Fig. 4, the experimental dots obey the linear relationship predicted by the Lippert–Mataga equation very well in the range of solvents investigated. The slope of DPAOXDBEN is obviously larger than that of DPAOXD, which means that the excited state of DPAOXDBEN is much more sensitive to solvent polarity. The variations of the dipole moment in these compounds are corresponding to the extent of the red shift of their emission maximum. The change in the dipole moment between the charge transfer excited state and the ground state (Δμ) can be calculated as large as 22.10 D and 26.67 D for DPAOXD and DPAOXDBEN, respectively; these values are extremely larger than that of BOXD-p-OCH3 (7.38 D), indicating that increase the electron-donating ability of the terminal substituents and further introduction of the central benzene ring can both enhance excited state intramolecular charge transfer characteristic. Since the molecular structures of the two compounds are centrosymmetric, the calculated ground and excited state dipole moment are almost zero. The two molecules would not exhibit molecule dipole moments, these calculated dipole moment variations should be assigned to the changes in local dipole moments.34 These relatively large changes in the local dipole moment are a direct evidence of the strong intramolecular charge transfer characteristic of the excited state in DPAOXD and DPAOXDBEN.
Theoretical study
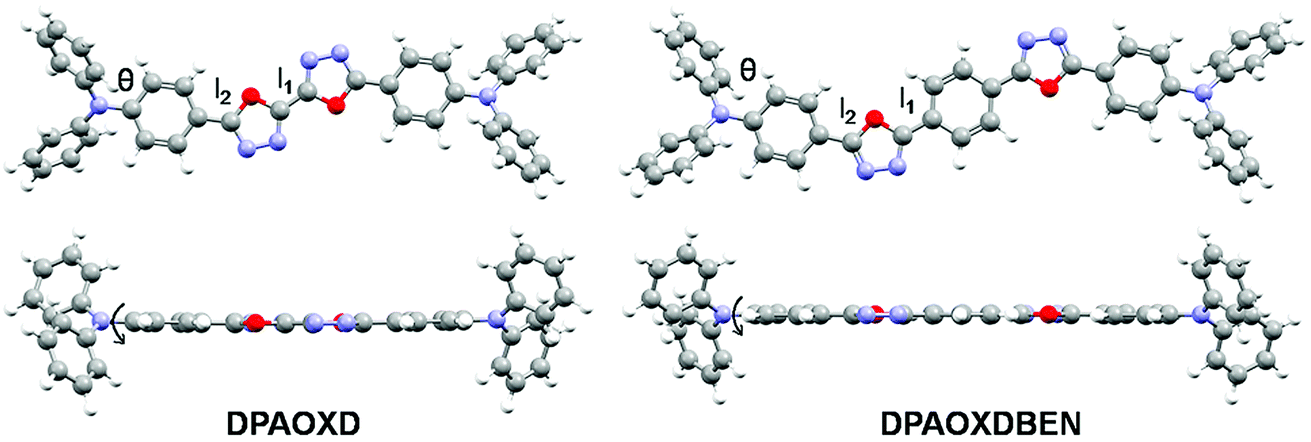
Molecular geometry. The molecular geometries of the two compounds have been fully optimized in both the ground state and the first excited state in gas phase. Fig. 5 present the optimized molecular structures in the ground state of DPAOXD and DPAOXDBEN. The optimized S1 geometries and the detail information about the bonds and dihedral can be obtained in the ESI.† In the ground state of both DPAOXD and DPAOXDBEN, the 1,3,4-oxadiazole rings and their adjacent benzene rings are well co-planar, while the terminal diphenylamine group deviate from the plane with a dihedral angle (θ) of about 30° and 31.7° in DPAOXD and DPAOXDBEN, respectively. In addition, the length of connecting bond (l1 and l2) between the oxadiazole and benzene rings are in a range of 1.44–1.46 Å in both DPAOXD and DPAOXDBEN, suggesting that oxadiazole ring has good conjugating ability with benzene ring. In the optimized S1 geometries, the 1,3,4-oxadiazole rings and their adjacent benzene rings are still well co-planar and conjugated. While the dihedral angles slightly increased as compared with that in the optimized S0 geometries. The values of θ increased up to 35.7° in DPAOXD, and 33.1° in DPAOXDBEN. The bond length of l1 and l2 shorten during the electronic excitation, that l1 decrease about 0.04 Å and l2 decrease about 0.03 Å in DPAOXD and 0.02 Å in DPAOXEBEN, respectively. Other calculated bonds or dihedral angles are found almost no changes from the ground state to the excited state in both of the two compounds, indicating that the two compounds become more quinoid-liked in the excited state. Furthermore, from the viewpoint of molecular geometrical structure, the central introduction of benzene ring does not change the structural feature; only extend the π-conjugation of the molecule to some extent of DPAOXDBEN.
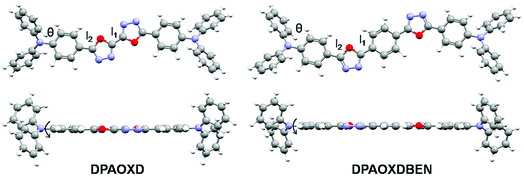 |
| | Fig. 5 Optimized molecular structures in the ground state of DPAOXD and DPAOXDBEN. | |
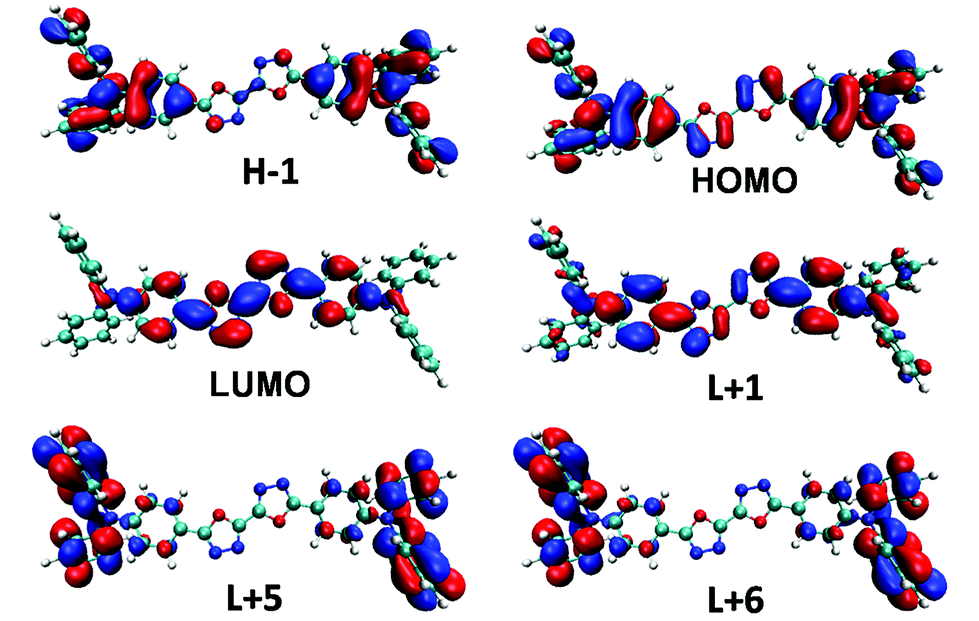
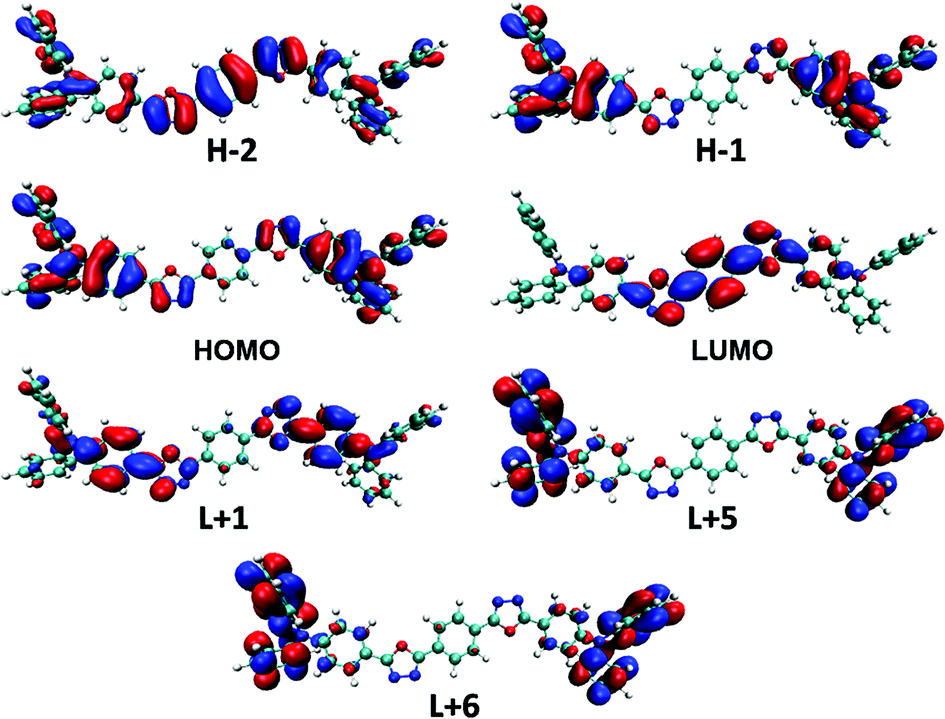
Electronic-state transitions and frontier orbitals. Theoretical calculations at time dependent density functional theory (TD-DFT) level are carried out to reveal the physical picture of the electronic excitation. The excitation energy, oscillator strength and main orbital component of the seven lowest excited states in CHEX calculated by a PCM model of the two compounds are listed in Table 2. The theoretical calculations reveal that the two highest allowed transitions in the two compounds are S0–S1 (3.59 eV, f = 2.33 in DPAOXD, 3.59 eV, f = 2.63 in DPAOXDBEN) and S0–S7 (4.73 eV, f = 0.38 in DPAOXD, 4.68 eV, f = 0.57 in DPAOXDBEN), which are corresponding to the two distinguishable absorption bands in the absorption spectra (Fig. S5 and S6†). In DPAOXD, the S0–S1 transitions are mainly consist of H−1 → L+1 and H → L transitions, and the S0–S7 transitions are mainly consist of H−1 → L+1, H−1 → L+5, and H → L+6 transitions; in DPAOXDBEN, the S0–S1 transitions are mainly consist of H−2 → L, H−1 → L+1 and H → L transitions, and the S0–S7 transitions are mainly consist of H−1 → L+5 and H → L+6 transitions. Fig. 6 and 7 present the frontier molecular orbitals involved in the highest allowed two transitions of the two compounds. As can be seen, these occupied frontier molecular orbitals are all consist of π-bonding orbital, while the unoccupied orbitals are all mainly composed of the π-antibonding orbital. Thus, the two main absorption in these compounds should be assigned as π–π* type and the energy absorption band should show π–π* characteristic. The electron density distributions of all these orbitals involved in the highest allowed two transitions of the two compounds show obvious localized properties except for orbital HOMO of DPAXOD and orbital H−2 of DPAOXDBEN, in which the wave functions are almost delocalized over the whole molecule. In DPAOXD, orbital H−1 has the electron density located on the terminal diphenylamine and its adjacent benzene rings; the electron density in orbital LUMO and L+1 are mainly located on the bi-1,3,4-oxadiazole ring and the benzene ring between the oxadiazole rings and the diphenylamine; orbital L+5 and L+6 have the electron density located on the terminal diphenylamine. Orbitals in DPAOXDBEN have similar electron density distributions to DPAOXD, except for orbital LUMO, in which the distributions of electron density are mainly concentrated on the bi-1,3,4-oxadiazole ring and the central benzene ring between the two oxadiazole rings. The electron density on the benzene ring between the oxadiazole rings and the diphenylamine decrease a lot as compared with DPAOXD. In addition, the electron density distribution of orbitals in DPAOXD and DPAOXDBEN are quite different from BOXD-p-OCH3 in which the wave functions are perfectly delocalized over the whole system for orbital HOMO and LUMO. The well-separated electron densities of these orbitals in these two compounds provide a clear evidence of the obvious intramolecular charge transfer characteristic in the two compounds. Table S7† list the orbitals energy of all these orbitals and the energy gaps involved in the highest allowed two transitions in DPAOXD and DPAOXDBEN. It can be seen that there are almost no changes (0–0.08 eV) in the orbital energy levels in the two compounds except for orbital LUMO and orbital H−2. The orbital energy of LUMO in DPAOXD is larger than that of DPAOXDBEN in a value of 0.15 eV, and the orbital energy of H−2 in DPAOXDBEN is about 0.36 eV larger than that of DPAOXD. The energy gaps between orbitals involved in the highest allowed two transitions of the two compounds also show similar values. The energy gap between HOMO and LUMO (ΔEH–L) is 5.73 and 5.54 eV in DPAOXD and DPAOXDBEN, respectively; the energy gaps between H−1 and L+5 (ΔEH−1–L+5) is 7.45 and 7.37 eV in DPAOXD and DPAOXDBEN, respectively; and the energy gap between HOMO and L+6 (ΔEH–L+6) is 7.32 and 7.29 eV in DPAOXD and DPAOXDBEN, respectively. Considering the H → L transition is the mainly component of the S0–S1 transitions and the H−1 → L+5 and H → L+6 transitions are the mainly component of the S0–S7 transitions in both of the two compounds, such small differences of these energy gaps (<0.2 eV) might be the reason for the close location of the absorption maximum in the two compounds.
Table 2 Computed excitation energy (E, in eV), oscillator strength (f) and main orbital component of the seven lowest allowed excited states in CHEX of DPAOXD and DPAOXDBEN
| State |
DPAOXD |
DPAOXDBEN |
| E (f) |
Orbital component |
E (f) |
Orbital component |
| S1 |
3.59 (2.23) |
H−1 → L+1 (0.19), H → L (0.69) |
3.59 (2.63) |
H−2 → L (0.13), H−1 → L+1 (0.20), H → L (0.55) |
| S2 |
3.93 (0) |
H−1 → L (0.53), H−1 → L+2 (0.06), H → L+1 (0.35) |
3.86 (0) |
H−1 → L (0.42), H−1 → L+2 (0.15), H → L+1 (0.36) |
| S3 |
4.48 (0.03) |
H−1 → L+3 (0.38), H → L+2 (0.13), H → L+4 (0.3) |
4.42 (0.18) |
H−2 → L (0.2), H−1 → L+1 (0.2), H → L+2 (0.25) |
| S4 |
4.48 (0.06) |
H−1 → L+4 (0.30), H → L+3 (0.43) |
4.46 (0) |
H−1 → L+4 (0.39), H → L+3 (0.41) |
| S5 |
4.69 (0.13) |
H−1 → L+1 (0.15), H−1 → L+5 (0.22), H → L+6 (0.19) |
4.49 (0.07) |
H−2 → L (0.10), H−1 → L+3 (0.31), H → L+4 (0.34) |
| S6 |
4.70 (0.07) |
H−1 → L+6 (0.42), H → L+5 (0.45) |
4.67 (0) |
H−1 → L+6 (0.37), H → L+5 (0.39) |
| S7 |
4.73 (0.38) |
H−1 → L+1 (0.16), H−1 → L+5 (0.22), H → L+6 (0.26) |
4.68 (0.57) |
H−1 → L+5 (0.45), H → L+6 (0.44) |
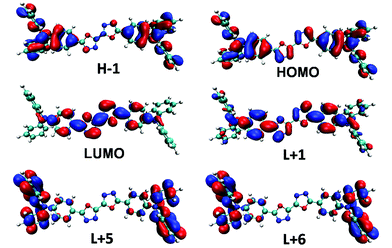 |
| | Fig. 6 Frontier molecular orbitals involved in the highest two allowed transitions of DPAOXD calculated in CHEX. | |
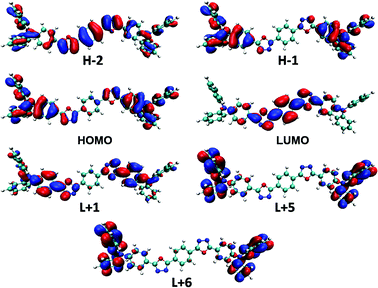 |
| | Fig. 7 Frontier molecular orbitals involved in the highest two allowed transitions of DPAOXDBEN calculated in CHEX. | |
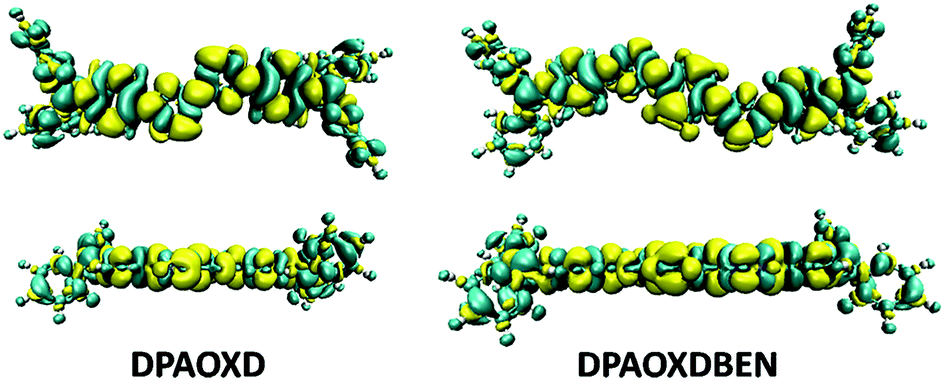
Intramolecular charge transfer. Since DPAOXD and DPAOXDBEN exhibit obvious intramolecular charge transfer characteristic in the first excited states, the electron density differences between the ground state and the first excited state in gas phase (Fig. 8) have been calculated to get a direct view on the intramolecular charge transfer.41 The yellow and cyan regions stand for positive and negative of the electron density differences, which mean the increase and decrease of electron density in the first excited state as compared with the ground state. In DPAOXD, the electron density differences over the terminal diphenylamine are almost negative; in the adjacent benzene ring, the area of the negative parts and the positive parts are nearly the same; and in bi-1,3,4-oxadiazole rings, the electron density differences exhibit obvious positive, indicating that the electrons have been transferred from the terminal diphenylamine (donors) to the bi-1,3,4-oxadiazole rings (acceptors) in DPAOXD. In DPAOXDBEN, the electron density differences over the terminal diphenylamine and its adjacent benzene ring show similar character to DPAOXD; while in the two oxadiazole rings, the proportion of the positive regions is slightly decreased as compared with DPAOXD, but it remain larger than the negative regions, indicating that the accepted electrons of oxadiazole ring in DPAOXDBEN have decreased as compared with DPAOXD; and in the central benzene ring, the positive region takes up the most of the area, which means that the central benzene ring also accept electrons in DPAOXDBEN. Thus, the electrons have transferred from the terminal diphenylamine (donors) to both the 1,3,4-oxadiazole rings and the central benzene ring (acceptors) in DPAOXDBEN. The electron density difference maps clearly show that DPAOXD and DPAOXDBEN exhibit similar intramolecular charge transfer characteristic, in which electrons have transferred from the terminal diphenylamine groups to the central parts. In DPAOXD, electrons are totally accepted by the bi-oxadiazole ring; while there are partial electrons have been accepted by the central benzene ring in DPAOXDBEN.
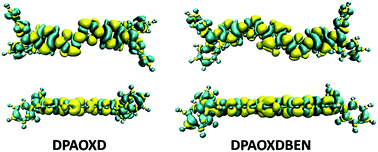 |
| | Fig. 8 Top and side view of electron density difference between the ground state and the first excited state of DPAOXD and DPAOXDBEN. | |
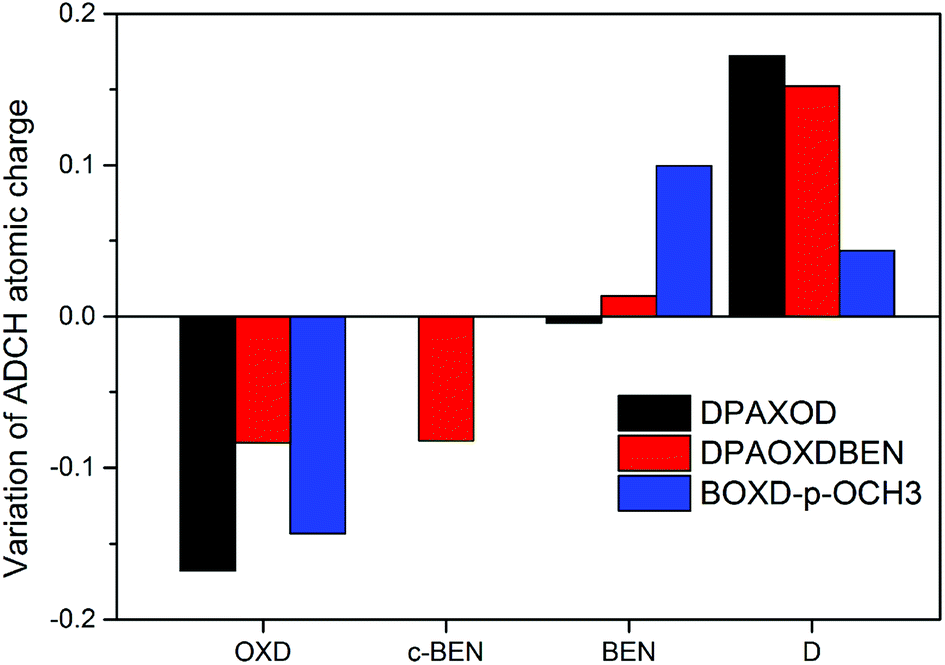
We further calculated the atomic charges of these compounds investigated with atomic dipole moment corrected Hirshfeld charges (ADCH) method to quantify the intramolecular charge transfer in the two compounds.42 Molecular structures of these compounds are divided into several fragment including 1,3,4-oxadiazole rings (OXD), benzene rings (BEN stands for the benzene rings located between the diphenylamine or methoxy group and 1,3,4-oxadiazole ring, c-BEN stands for the central benzene ring located between the two 1,3,4-oxadiazole rings) and substituents (D: diphenylamine or methoxy group). The variations of sum of atomic charges in each fragment from the first excited state compared to the ground state are present in Fig. 9, and the detailed atomic charge of each fragment in both the ground state and the first excited state are summarized in Table S8.† It can be seen that the variations of the sum of atomic charges in each fragment from the first excited state to the ground state are different in the two compounds. On the ground state, each 1,3,4-oxadiazole rings (OXD) in DPAOXD process a negative charge value of about 0.100|e|, each benzene rings (BEN) and diphenylamine (D) have a positive charge value of about 0.028|e| and 0.071|e|, respectively; when going to the S1 state, the charge value of the 1,3,4-oxadiazole rings (OXD) decreased about 0.168|e| accompanied by the charge value of diphenylamine (D) increased about 0.172|e|; as for BEN, there is only about 0.004|e| changes in the sum of atomic charge which is too small to be neglected. Thus, it is clearly that the terminal diphenylamine are donors and the bi-1,3,4-oxadiazole rings are acceptors in DPAOXD, and there is about 16.8% electron transferred from donors to the acceptors. In DPAXODBEN, similar to DPAOXD, in the S1 state compared to the S0 state, the sum atomic charge of diphenylamine (D) and BEN exhibit an increase about 0.152|e| and 0.013|e|, respectively. However, the changes of the atomic charges of the OXD fragment in DPAOXDBEN are much more different from DPAOXD. The atomic charge value of the 1,3,4-oxadiazole rings (OXD) in DPAOXDBEN show a decrease of about 0.083|e|, which is only half of that in DPAOXD; meanwhile, it is found that the atomic charge value of c-BEN decreased about 0.082|e| from the S1 state to the S0 state. Hence, as illustrated by the electron density difference maps, the donors are the terminal diphenylamine, while the acceptors are both the two 1,3,4-oxadiazole rings and the central benzene ring in DPAOXDBEN. Almost equal electrons (16.6%) in DPAOXDBEN are found to transfer from the donors to the acceptors as compared with DPAOXD. Although the introduction of the central benzene ring between the 1,3,4-oxadiazole rings do not change the transferred charges, great part of electron (∼8.2%) was accepted by the central benzene group, which would increase the CT distance (DCT). Since the dipole moment variation (Δμ) between the first excited state and the ground state is the product of both the transferred charge (qCT) and the CT distance (DCT), the strong intramolecular charge transfer character observed in DPAXODBEN might be ascribed to the increased CT distance between donors and the acceptors. In addition, we also calculated the atomic charge of BOXD-p-OCH3 to explore the substituent effect on the charge transfer. The results reveal that about 14.3% electrons have transferred from the methoxy benzene side (donors) to the central bi-1,3,4-oxadiazole ring (acceptors) during the electronic excitation. Thereinto, only about 4.4% electrons are donated by the methoxy groups, while the rest are given by the benzene rings. The transferred charges and transferred distances in BOXD-p-OCH3 are both smaller than that in DPAOXD. Thus, it can be inferred that the enhanced intramolecular charge transfer in DPAOXD would be caused by the increase of the transferred charge and transferred distance simultaneously.
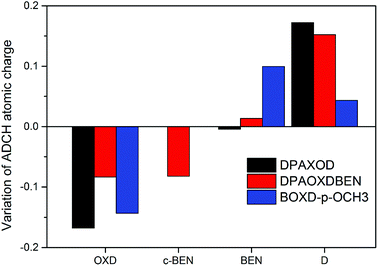 |
| | Fig. 9 Variation of ADCH atomic charges for different fragments of these compounds investigated in the excited state compared to the ground state. | |
Conclusion
Two diphenylamine substituted symmetric 1,3,4-oxadiazole derivatives have been successfully synthesized and the intramolecular charge transfer characteristic has been fully studied though a combination of spectroscopic techniques and theoretical calculations. The experimental results reveal that DPAOXD and DPAOXDBEN exhibit comparable absorption maximum at about 385 nm and 290 nm, and emission maximum at about 412 nm in CHEX. As the solvent polarity increase, there are small red shifts in absorption spectra of the two compounds, indicating that the electronic and structural natures in the ground and FC excited states do not change a lot; while large fluorescence red shifts (∼114 nm for DPAOXD and ∼140 nm for DPAOXDBEN) could be observed, which indicates that a large extent charge transfer has occurred during the excited state relaxation. The increase of molecular dipole moment from the ground state to the charge transfer excited state are calculated to be 22.10 D in DPAOXD and 26.67 D in DPAOXDBEN, respectively, which are especially larger than that in BOXD-p-OCH3 (∼7.38 D), indicating that the substitution by diphenylamine group and further introduction of a central benzene ring could effectively enhance the intramolecular charge transfer strength in 1,3,4-oxadiazole derivatives. Theoretical analysis on electron density difference and atomic charge exhibit a clear map of intramolecular charge transfer in both of the two compounds that electrons have transferred from terminal diphenylamine to the bi-1,3,4-oxadiazole rings in DPAOXD, and both the two 1,3,4-oxadiazole ring and central benzene ring in DPAOXDBEN. It has been proved that the enhanced intramolecular charge transfer strength in DPAOXD are jointly functioned by the increase of the transferred charge and distance through introducing substituent with stronger electron donating ability as compared with BOXD-p-OCH3; and the further enhanced intramolecular charge transfer strength in DPAOXDBEN are caused by the increase of charge transfer distance between donors and the acceptors through the introduction of benzene ring between the 1,3,4-oxadiazole rings as compared with DPAOXD. These findings would be of great value for understanding structure–property relationships and the rational design of functional materials for photoelectric applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Science and Technology Develop Programme of Jilin Province (20170520127JH), Postdoctoral Science Foundation of China (2012T50294), the Fundamental Research Funds for the Central Universities.
References
- Z. S. Wang, Y. Cui, Y. Danoh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2008, 112, 17011–17017 CrossRef CAS.
- Y. Ooyama, Y. Shimada, S. Inoue, T. Nagano, Y. Fujikawa, K. Komaguchi, I. Imae and Y. Harima, New J. Chem., 2011, 35, 111–118 RSC.
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483–5495 RSC.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Mllen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- L. Yao, S. Xue, Q. Wang, W. Dong, W. Yang, H. Wu, M. Zhang, B. Yang and Y. Ma, Chem.–Eur. J., 2012, 18, 2707–2714 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- U. Mitschke and P. BaÈuerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC.
- Y. Cui, P. Li, C. Song and H. Zhang, J. Phys. Chem. C, 2016, 120, 28939–28950 CrossRef CAS.
- A. Singh, A. K. Pati and A. K. Mishra, Phys. Chem. Chem. Phys., 2018, 20, 14889–14898 RSC.
- X. Zhang, X. Sun, C. Wang and Y. Jiang, J. Phys. Chem. A, 2002, 106, 5577–5581 CrossRef CAS.
- X. Zhang and Y. Jiang, Photochem. Photobiol. Sci., 2011, 10, 1791–1796 RSC.
- G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496–4501 CrossRef CAS PubMed.
- G. Haberhauer, Chem.–Eur. J., 2017, 23, 9288–9296 CrossRef CAS PubMed.
- R. K. Konidena, K. R. J. Thomas, D. K. Dubey, S. Sahoob and J.-H. Jou, Chem. Commun., 2017, 53, 11802–11805 RSC.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- H. Gao, Y. Li, L. Wang, C. Ji, Y. Wang, W. Tian, X. Yang and L. Yin, Chem. Commun., 2014, 50, 10251–10254 RSC.
- Y. Chen, X. Wan and G. Long, Acc. Chem. Res., 2013, 46, 2645–2655 CrossRef CAS PubMed.
- H. T. Turan, O. Kucur, B. Kahraman, S. Salman and V. Aviyente, Phys. Chem. Chem. Phys., 2018, 20, 3581–3591 RSC.
- R. Ghosh, A. Nandi and D. K. Palit, Phys. Chem. Chem. Phys., 2016, 18, 7661–7671 RSC.
- H. Zhu, X. Wang, R. Ma, Z. Kuang, Q. Guo and A. Xia, ChemPhysChem, 2016, 17, 3245–3251 CrossRef CAS PubMed.
- H. Staleva-Musto, V. Kuznetsova, R. G. West, G. Kesan, B. Minofar, M. Fuciman, D. Bina, R. Litvin and T. Polivka, J. Phys. Chem. B, 2018, 122, 2922–2930 CrossRef CAS PubMed.
- X. Zhan, Z. Wu, Y. Lin, Y. Xie, Q. Peng, Q. Li, D. Ma and Z. Li, Chem. Sci., 2016, 7, 4355–4363 RSC.
- J. Wu, W. Wang, C. Gong, Q. Li, Z. Li, G. Deng, X. Zhang, K. Chen, Y. Gong and K. s. Chiang, J. Mater. Chem. C, 2017, 5, 7472–7478 RSC.
- R. Ghosh, Phys. Chem. Chem. Phys., 2018, 20, 6347–6353 RSC.
- B. Dereka, D. Svechkarev, A. Rosspeintner, M. Tromayer, R. Liska, A. M. Mohs and E. Vauthey, J. Am. Chem. Soc., 2017, 139, 16885–16893 CrossRef CAS PubMed.
- G. He, L. L. Zhou, H. Song, Z. Kuang, X. Wang, Q. Guo, H. Y. Lu and A. Xia, Phys. Chem. Chem. Phys., 2018, 20, 7514–7522 RSC.
- S. Qu, Q. Lu, S. Wu, L. Wang and X. Liu, J. Mater. Chem., 2012, 22, 24605–24609 RSC.
- M. Thelakkat and H. W. Schmidt, Polym. Adv. Technol., 1998, 9, 429–442 CrossRef CAS.
- B. Schulz, M. Bruma and L. Brehmer, Adv. Mater., 1997, 9, 601–613 CrossRef CAS.
- G. Hughes and M. R. Bryce, J. Mater. Chem., 2004, 15, 94–107 RSC.
- H. Wang, H. Liu, F.-Q. Bai, S. Qu, X. Jia, X. Ran, F. Chen, B. Bai, C. Zhao, Z. Liu, H.-X. Zhang and M. Li, J. Photochem. Photobiol., A, 2015, 312, 20–27 CrossRef CAS.
- F. Chen, W. Zhang, T. Tian, B. Bai, H. Wang and M. Li, J. Phys. Chem. A, 2017, 121, 8399–8407 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 10, 690–691 CrossRef.
- E. Lippert, Z. Naturforsch. A, 1955, 10, 541–545 Search PubMed.
- T. L. Bahers, C. Adamo and I. Ciofin, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef PubMed.
- T. Lu and F. Chen, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and emission spectra of BOXD-p-OCH3, excitation spectra, optimized molecular geometry, calculated orbital energies and atomic charges of DPAOXD and DPAOXDBEN. See DOI: 10.1039/c8ra08439d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Min Li*a
*a and
Min Li*a