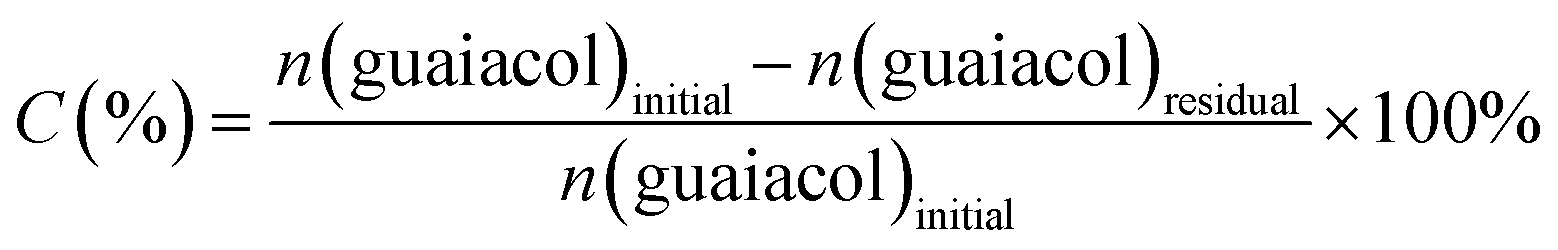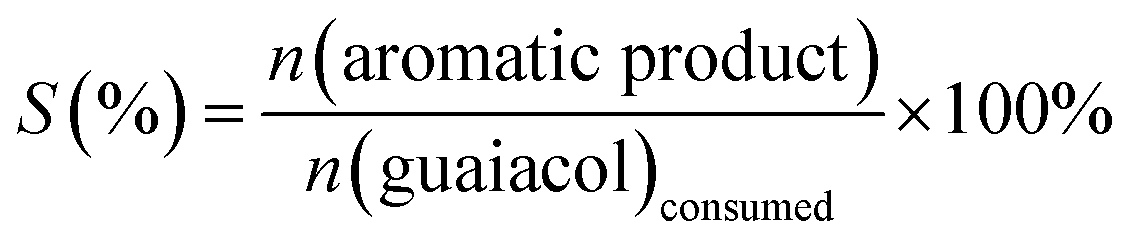DOI:
10.1039/C8RA07962E
(Paper)
RSC Adv., 2019,
9, 2764-2771
Highly selective conversion of guaiacol to tert-butylphenols in supercritical ethanol over a H2WO4 catalyst†
Received
25th September 2018
, Accepted 20th December 2018
First published on 21st January 2019
Abstract
The conversion of guaiacol is examined at 300 °C in supercritical ethanol over a H2WO4 catalyst. Guaiacol is consumed completely, meanwhile, 16.7% aromatic ethers and 80.0% alkylphenols are obtained. Interestingly, tert-butylphenols are produced mainly with a high selectivity of 71.8%, and the overall selectivity of 2,6-di-tert-butylphenol and 2,6-di-tert-butyl-4-ethylphenol is as high as 63.7%. The experimental results indicate that catechol and 2-ethoxyphenol are the intermediates. Meanwhile, the WO3 sites play an important role in the conversion of guaiacol and the Brønsted acid sites on H2WO4 enhance the conversion and favour a high selectivity of the tert-butylphenols. The recycling tests show that the carbon deposition on the catalyst surface, the dehydration and partial reduction of the catalyst itself are responsible for the decay of the H2WO4 catalyst. Finally, the possible reaction pathways proposed involve the transetherification process and the alkylation process during guaiacol conversion.
1 Introduction
The depletion of fossil fuels and the growing demand of energy and chemicals call for practical technologies for biomass, especially lignin, utilization. Lignin is an important component and accounts for 10–35% by weight, and up to 40% by energy in lignocellulosic biomass.1 The available aromatic structure of lignin renders it an attractive material for production of aromatic chemicals. However, due to the strong linkages in its complex structure, only a small amount of lignin is utilized as a low-grade fuel or as a low-value building material additive (lignosulfonate).2 Up to now, a number of techniques have been explored to depolymerize lignin, including pyrolysis,3 hydrogenolysis/hydrogenation,4,5 oxidation,6 hydrolysis,7 reforming8 and ethanolysis.9,10
Guaiacol, which contains both hydroxyl and methoxyl groups, is often chosen as a model compound to verify reaction mechanism during lignin depolymerization.11,12 Early works on guaiacol conversion focused mainly on sulfided CoMo and NiMo catalysts,13,14 and supported noble metal catalysts.15,16 However, the high H2 consumption, high temperature and aromatic ring saturation are not favoured parameters.17 Therefore an alternative catalyst with a relatively strong deoxygenation activity and a weak aromatic ring saturation capacity is called for.5 Transition metals18 and their oxides,19,20 phosphides,21,22 nitrides,23,24 and carbides9,11,25–27 therefore have been examined. Metal phosphide catalysts were found to show excellent activity but tungsten phosphide (WP) showed a relatively disappointing result for gaseous phase hydrodeoxygenation (HDO) of guaiacol.21 Compared with the conventional CoMo/Al2O3 catalyst, a higher conversion and a higher selectivity towards phenolics were obtained in the HDO of guaiacol over W2C/CNF at 55 bar H2 pressure.11 However, metal carbide catalysts are easily oxidized, resulting in deactivation.28
Recently, the conversion of model compounds into value-added chemicals in supercritical alcohol was reported.26,29 Yang et al. reported the transetherification of guaiacol to o-ethoxyphenol over a γ-Al2O3 catalyst in supercritical ethanol.30 Ma et al.9 reported the catalytic conversion of guaiacol in supercritical ethanol over α-MoC1−x/AC without the addition of gaseous hydrogen. Phenols and alkylphenols were obtained as the main products with an overall selectivity of 85%. More recently, Cui et al.28 reported the selective conversion of guaiacol over a MoO3 catalyst to produce various alkylphenols in ethanol. They proposed that the higher alkylphenols formed via a consecutive substitution route. Hansen et al.29 performed the catalytic conversion of 5-hydroxymethylfurfural to 2,5-dimethylfuran and 2,5-dimethyltetrahydrofuran over a Cu-doped porous metal oxide (Cu-PMO) in supercritical methanol. More importantly, supercritical alcohols such as methanol, ethanol and isopropanol were found to be also efficient for lignin depolymerization.30–33
The binary catalyst composed of H2WO4 and Ru/C showed a high activity for the one-pot conversion of cellulose to ethylene glycol.34 Tai et al.35 proposed that H2WO4 was dissolved partially in water under the reaction conditions and transformed into HxWO3, which served as the active site. Jongerius et al.11 found that WOx phase over the W2C/CNF catalyst formed in the reaction process had a beneficial effect on the catalytic activity for gaseous phase HDO (55 bar H2) of guaiacol. In this work, we report the utilization of H2WO4 as a catalyst in the selective conversion of guaiacol in supercritical ethanol. A number of aromatic ethers and alkylphenols are formed with high selectivity, especially for 2,6-di-tert-butylphenol and 2,6-di-tert-butyl-4-ethylphenol.
2 Materials and methods
2.1 Materials
Analytical grade (AR) chemicals and solvents, including guaiacol, phenol, catechol, anisole, ethanol, were purchased from Tianjin Guangfu Technology Development Co., Ltd. and were used without further purification. 2-Ethoxyphenol (AR) was obtained from Heowns (Tianjin) and used as received. Commercial ammonium metatungstate (AMT, (NH4)6H2W12O40·xH2O) and H2WO4 were purchased from Aladdin Industrial Corporation. The water used was provided by an ultrapure water purification system (UPH-1-10).
2.2 Catalyst characterization
The structure of the fresh and spent catalyst samples were identified with an X-ray diffraction (XRD) technique (Bruker AXS, D8-S4), operated at 40 kV and 40 mA, using a Cu-Kα monochromatized radiation source. Diffraction data was recorded between 2θ of 10° and 80° at a rate of 8° min−1. X-ray photoelectron spectroscopy (XPS) was performed with a PHI-1600 ESCA system spectrometer using Mg Kα as the X-ray source under a residual pressure of 5 × 10−6 Pa, with the binding energy calibrated using C1s at 284.6 eV as the standard. Thermogravimetric (TG) curves were measured with a PerkinElmer Diamond analyzer in air from 100 °C to 900 °C with a heating rate of 10 °C min−1. Raman spectra were obtained using a Renishaw inVia reflex spectrometer with a 523 nm argon laser excitation source. The resistivity of the H2WO4 and WO3 catalysts were tested. The catalyst powder was pressed into a thin wafer on a standard abrasive tool and then was tested in a tube furnace at 300 °C. NH3 temperature programmed desorption (NH3-TPD) was performed in a fixed bed reactor using a Thermo Nicolet IS10 FT-IR as the NH3 detector. Nitrogen adsorption measurements at 77 K were performed on an automatic surface analyzer (QuadraSorb Station 3). Pyridine Fourier transform infrared spectroscopy (FT-IR) spectra were recorded in a Nicolet iS50 FT-IR spectrometer. The samples were pressed into thin wafers and treated under vacuum (10−3 torr) at 423 K for 2 h. After cooling to room temperature, the samples were exposed to pyridine steam for 30 min. Then, the spectra (32 scans, 8 cm−1 resolution) were recorded after evacuation for 30 min at room temperature.
2.3 Catalytic activity measurements
H2WO4 was directly applied in guaiacol conversion reactions without any treatment. The WO3 catalyst sample for comparison was prepared with calcining AMT at 500 °C for 4 h in static air.36 All the reactions were carried out in a 250 mL batch reactor (MS 250, Anhui Kemi Machinery Technology Co., LTD.). In a typical test, 1.0 g guaiacol was dissolved in 80 mL ethanol as the reactant. The reactant and 0.5 g catalyst were added into the autoclave. After leakage test, the reactor was purged 5 times with ultrapure N2 to remove the remaining air. The reactor was heated to the reaction temperature (260–310 °C) with a heating rate of 2.8 °C min−1, and kept for the desired time with stirring at 600 rpm. The liquid products and the spent catalyst were separated and recovered after the reaction.
2.4 Analysis of products
| |
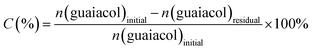 | (1) |
| |
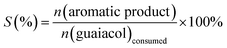 | (2) |
| | |
M (%) = ΣSiquantified aromatics
| (3) |
The organic phase containing liquid products was injected directly with a syringe into a gas chromatograph-mass spectrometer (GC-MS) system for product qualitative analysis. The product was further analysed quantitatively with a GC equipped with a flame ionization detector (FID). The detailed parameters used for the analysis can be found in the previous work.27,28 The identification of the liquid products was achieved through the standard spectrogram database (NIST02). An internal standard method was performed to analyse and quantify the product yield. The subsequent calculation within this context are mainly focused on the aromatic compounds. Hence, the calculation, including guaiacol conversion (C (%)), a specific aromatic product selectivity (S (%)) and mass balance (M (%)), is based on the variations of guaiacol and aromatic compounds before and after the reaction (equation (1)–(3)). In these equations, n presents the amount of the aromatic species in moles. The conversion and yield are determined with averaging the data measured in three times and the error is less than 5%.
3 Results and discussion
3.1 Catalysts characterization
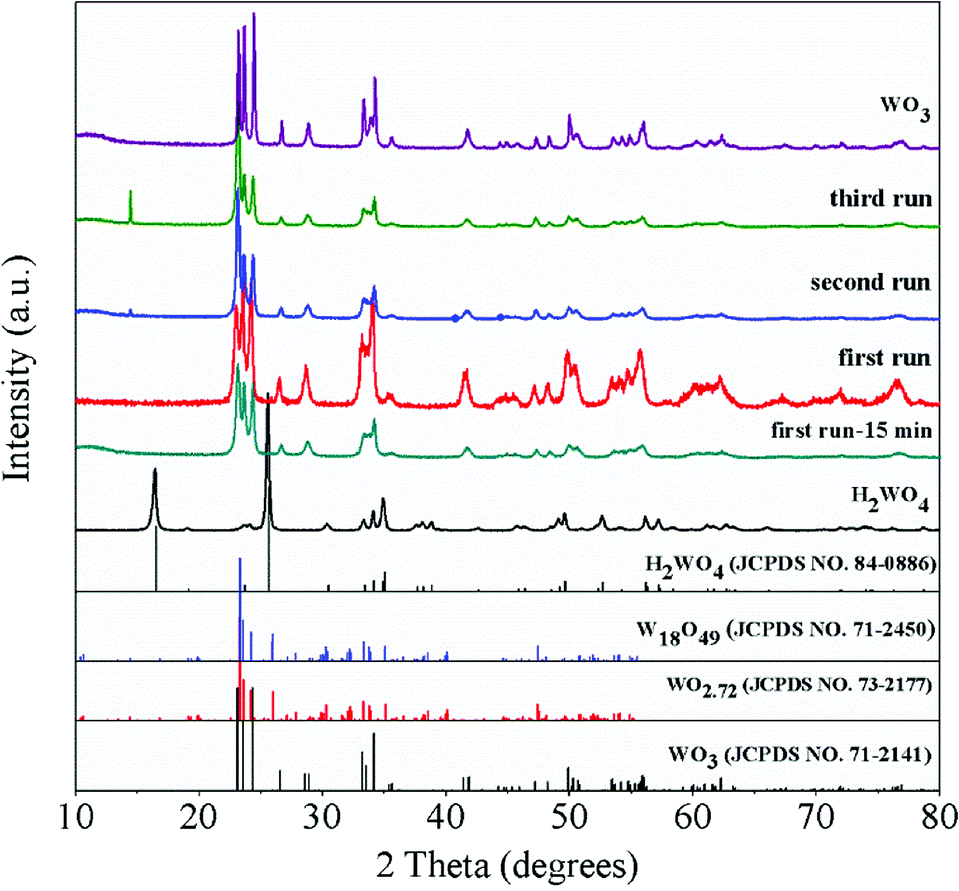
The XRD patterns of the WO3 catalyst, fresh H2WO4 catalyst and spent catalysts are illustrated in Fig. 1. For the mesoporous H2WO4 and WO3 samples (Table S1†), their diffraction peaks corresponded to the standard H2WO4 (WO3·H2O) phase (JCPDS no. 84-0886) and the standard WO3 (JCPDS no. 71-2141) phase, respectively. However, the diffraction peaks of the spent H2WO4 samples (first run-15 min (the H2WO4 catalyst was recovered from the first run after 15 minutes without any treatment) and first run) were basically in accord with those of WO3 catalyst, indicating that H2WO4 was transformed into WO3 under the reaction condition in the first run. The change was again confirmed through Raman spectra (Fig. S1†). For the spent samples, the strength of the X-ray diffraction peaks at 23.6° and 24.4° declined sharply as the cycle time increased, indicating that the spent catalyst may be partly reduced into W18O49 or WO2.72 phase,37,38 and the reduction process was further proved through the XPS profiles (Fig. S2†).
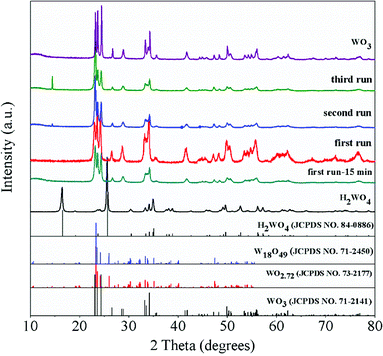 |
| | Fig. 1 XRD patterns of the WO3 catalyst, fresh H2WO4 catalyst and spent catalysts. | |
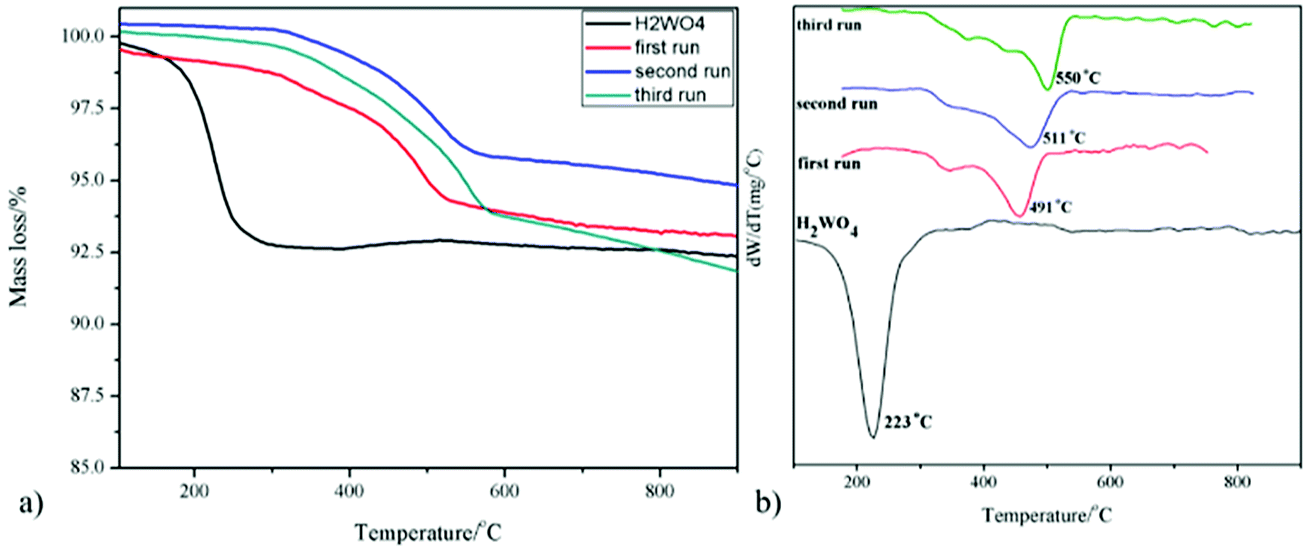
The TG curves of the fresh and spent catalysts in air atmosphere are depicted in Fig. 2. For the fresh H2WO4 catalyst, it can be seen that there is one peak located at below 300 °C due to the lattice water that was the fresh H2WO4 catalyst dehydrated between 170 °C and 260 °C, and the final mass loss reached about 7.5%, which was almost identical to the theoretical value (7.2%).39,40 For the spent samples, there is one main peak respectively located at 491 °C, 511 °C and 550 °C for different runs, corresponding to the accumulation of carbon.20 With the increase of number of cycle time, the mass loss of carbon on the spent catalysts reached 5.9%, 6.5% and 9.2%, respectively. The phenomenon was consistent with the results from Raman spectra. From the Fig. S1,† we can see that as the cycle time increased, the two small bands centered at 1586 cm−1 and 1318 cm−1 in Raman spectra became stronger, showing the accumulation of carbon on the spent catalyst.41
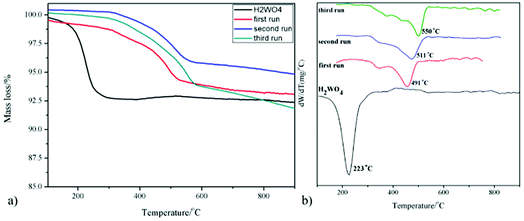 |
| | Fig. 2 (a) TG and (b) DTG profiles of the fresh and spent catalysts. | |
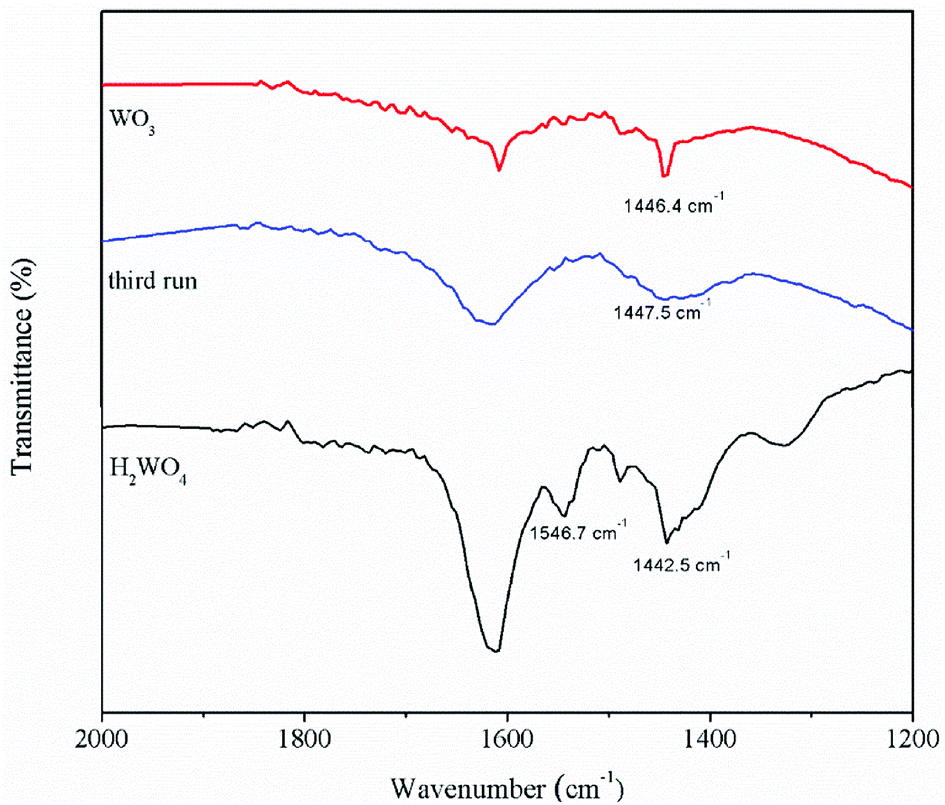
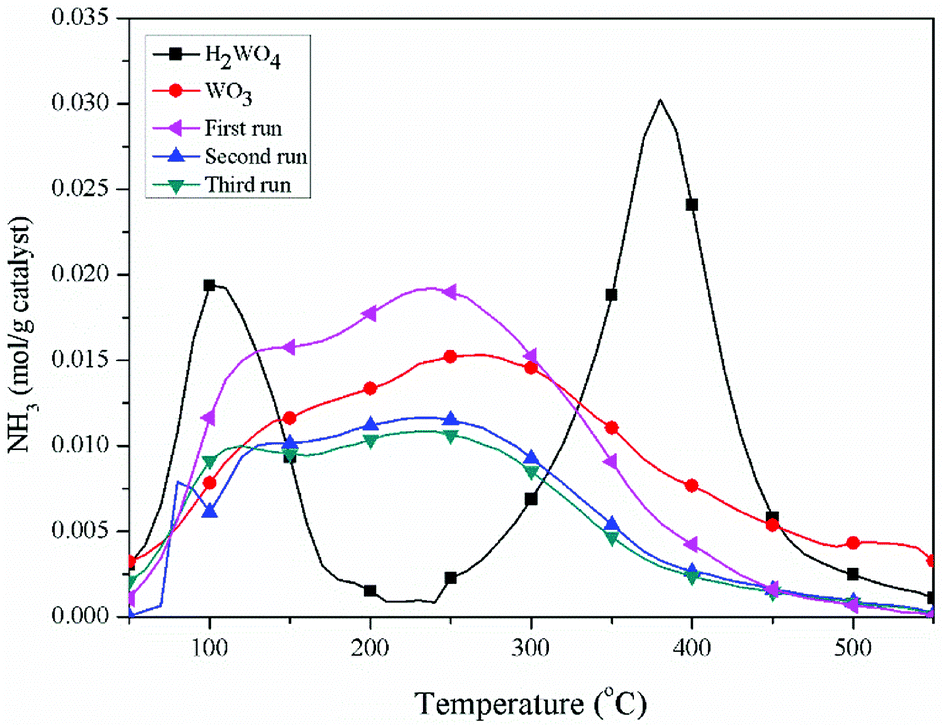
The pyridine FT-IR spectra of the H2WO4 and WO3 samples are shown on Fig. 3. Brønsted acid sites (at 1546.7 cm−1) and Lewis acid sites (between 1442 cm−1 and 1448 cm−1) simultaneously exist on the H2WO4 catalyst, while there are only Lewis acid sites on the WO3 sample. It is well known that WO3 may be a component in the H2WO4 sample. Further, the Brønsted acid sites over the spent H2WO4 catalyst which was recovered from the third run were completely disappeared, while the Lewis acid sites remained. The surface acidity of the fresh and spent catalysts were further characterized with an NH3-TPD method in a temperature rage 100–550 °C, and the results are depicted in Fig. 4. The area and position of the peaks in the TPD curves are correlated to the acid amount and the acid strength, respectively.42,43 For the fresh the H2WO4 sample, two distinct desorption peaks were observed at 110 °C and 380 °C, ascribing to the NH3 desorption from the weak and strong acid sites, respectively.44 For the spent catalysts, the peak ascribed to the strong acid sites disappeared and a peak corresponding to medium strength acid sites between 200 °C and 300 °C formed. Nevertheless, as the increase of the cycle time, the amount of medium acid sites underwent an obvious decrease. Overall, with the increase of number of cycle time, the acid sites over the H2WO4 catalyst gradually decrease, especially for the Brønsted acid sites. Noticeably, the resistivity of the H2WO4 and WO3 catalysts fluctuated between 106 and 107, which belonged to the semiconductor materials. It proved that the results from the pyridine FT-IR spectra and NH3-TPD spectra were reliable to some extent.
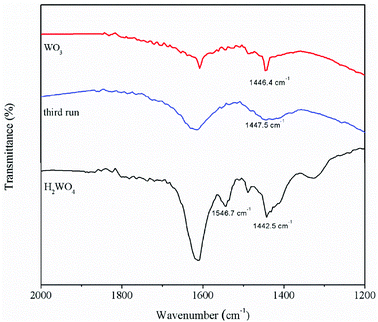 |
| | Fig. 3 The pyridine adsorption infrared spectra of the WO3 catalyst, fresh H2WO4 catalyst and spent catalysts. | |
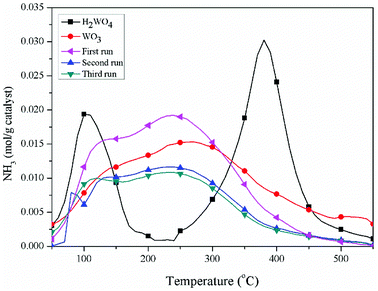 |
| | Fig. 4 NH3-TPD profiles of the WO3 catalyst, fresh H2WO4 catalyst and spent catalysts. | |
3.2 Product distribution
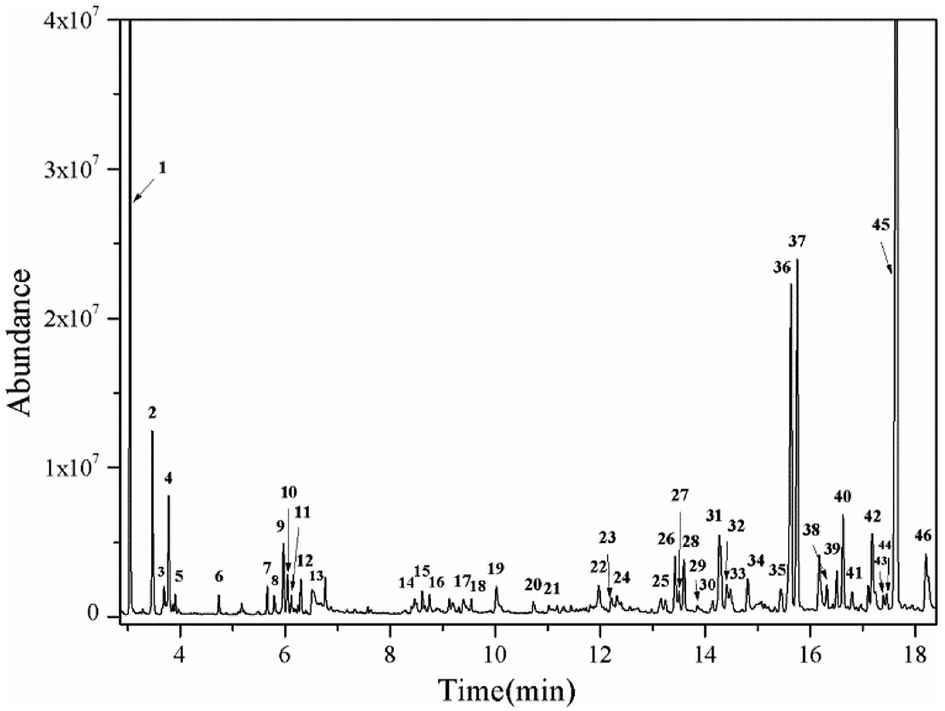
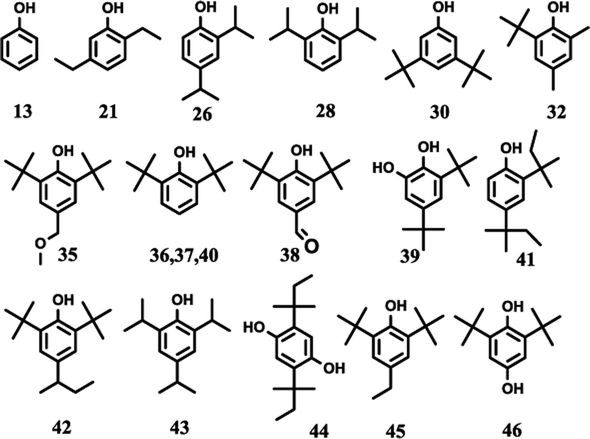
The effects of the reaction parameters such as reaction time, temperature and solvent amount were explored and finally the typical conversion reaction of guaiacol over the H2WO4 catalyst was performed in 80 mL ethanol at 300 °C for 6 h (Fig. S3–S5†). The total-ion chromatogram (TIC) of the products derived from the GC-MS is shown in Fig. 5. 45 compounds (P1–P12, P14–46) were identified after the reaction, and they can be classified into two categories: (1) aliphatic compounds; (2) aromatic compounds. Generally, P1–12, P14–P16 and P18, including aliphatic hydrocarbons and acetal, yielded up to 1535.2 mg and were confirmed to be formed from ethanol with a blank reaction. The detailed molecular structures are listed in Table S2.† In addition to the internal standard (phenol, P13) and the reactant (guaiacol, P17), the other 28 aromatic products (P19–46) were only detected in the guaiacol conversion. The results are listed in Table 1. In terms of aromatic products, there are two sorts: (1) aromatic ethers and (2) alkylphenols. The aromatic ethers were composed of 8 molecules, all containing either ethoxy group or methoxy group, with a total selectivity of 16.7%. Alkylphenols were obtained as the main products with an overall selectivity of 80.0%. The alkylphenols, including ethylphenol, iso-propylphenols, tert-butylphenols and neo-pentylphenols, exhibited selectivities of 0.4%, 6.3%, 71.8%, 1.5%, respectively.
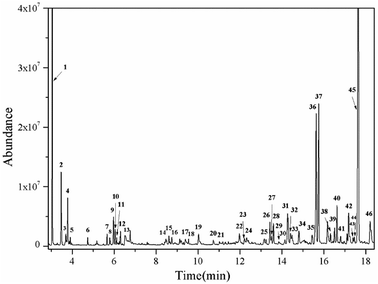 |
| | Fig. 5 TIC of the liquid products obtained from guaiacol conversion over H2WO4 in ethanol at 300 °C for 6 h. The peaks marked with numbers correspond to the products in Tables 1 and S2.† Reaction conditions: guaiacol (1.0 g), catalyst (0.5 g H2WO4), 600 rpm, 0 MPa (gauge) initial N2 pressure at room temperature, 80 ml ethanol. The compound marked with 13 is phenol, which is used as an internal standard to calculate the yield of products. | |
Table 1 Product distribution of the liquid products obtained from guaiacol conversion. a
| Type |
Molecule |
Selectivity |
| The numbers in the table correspond to those product peaks marked in the TIC (Fig. 5). |
| Aromatic ethers |
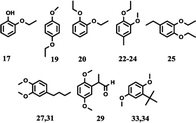 |
16.7% |
| Alkylphenols |
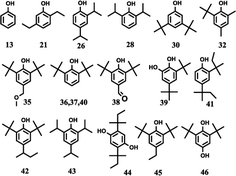 |
80.0% |
The alkylphenols formed through selective deoxygenation and alkylation have been confirmed in supercritical ethanol system.28 Three intensive peaks exist in the TIC of liquid products, which correspond to 2,6-di-tert-butylphenol (P36, P37) and 2,6-di-tert-butyl-4-ethylphenol (P45). The sum selectivity of 2,6-di-tert-butylphenol (28.5%) and 2,6-di-tert-butyl-4-ethylphenol (35.2%) was as high as 63.7%, which accounts almost 79.6% of the yield of the alkylphenols. Notably, no completely deoxygenated product or ring hydrogenation product was detected. Besides, neither phenol nor catechol was found in the liquid products. Compared with the α-MoC1−x/AC and MoO3 catalysts reported in our previous work,9,28 the advantage of high selectivity to tert-butylphenols, especially 2,6-di-tert-butylphenol and 2,6-di-tert-butyl-4-ethylphenol, provides the prospect of lignin depolymerization into high-valued chemicals.
3.3 Reactions with possible intermediates as reactants
Tests with potential intermediates of guaiacol conversion reaction, i.e. catechol, 2-ethoxyphenol, phenol and anisole, as reactants were carried out under the same condition. The results are presented in Table 2. Catechol was completely consumed, and the product distribution was almost identical with that from the reaction with guaiacol, indicating catechol was likely an intermediate. In Fig. S3b,† the selectivity of 2-ethoxyphenol underwent a gradual decrease with time increase, indicating 2-ethoxyphenol may be also an intermediate. The product catagories of using 2-ethoxyphenol as a reactant were indeed identical to those from guaiacol conversion although 12% of 2-ethoxyphenol was not converted and a drop in overall selectivity of alkylphenols were observed. However, the product distribution from phenol conversion was very different from that with guaiacol as the reactant, indicating that phenol is not an intermediate. Similarly, only a small amount of 1-ethoxybenzene, 4-ethylanisole and di-methylanisole were detected when anisole was used as the reactant. Hence, catechol and 2-ethoxyphenol are likely the intermediates of the guaiacol conversion over the H2WO4 catalyst.
Table 2 Conversion of model compounds over H2WO4 in supercritical ethanol. a
| Feedstock |
Cb/% |
Mc/% |
S1d |
S2 |
S3 |
S4 |
S5 |
S6 |
| Reaction conditions: feedstock (1.0 g), catalyst (0.5 g), 300 °C, 6 h, 600 rpm, 80 ml ethanol, 0 MPa (gauge) initial N2 pressure at room temperature. C: conversion. M: carbon balance. S1: aromatic ethers, S2: alkylphenols, S3: tert-butylphenols, S4: 2-ethoxyphenol, S5: 2,6-di-tert-butylphenol, S6: 2,6-di-tert-butyl-4-ethylphenol. Selectivity: 1-ethoxybenzene (53.5%), 4-ethyl-1-ethoxybenzene (2.6%). Selectivity: 2-ethylphenol (2.3%). Selectivity: 1-ethoxybenzene (1.0%), di-methylanisole (7.6%), 4-ethylanisole (4.3%). |
| Guaiacol |
>99 |
96.7 |
16.7 |
80.0 |
71.6 |
2.10 |
28.5 |
35.2 |
| Catechol |
>99 |
88.4 |
19.8 |
68.5 |
63.7 |
3.20 |
26.7 |
28.0 |
| 2-Ethoxyphenol |
88.1 |
92.1 |
28.5 |
62.5 |
55.8 |
— |
22.4 |
24.8 |
| Phenol |
84.8 |
73.5 |
56.1e |
2.3f |
— |
— |
— |
— |
| Anisole |
43.7 |
69.2 |
— |
12.9g |
— |
— |
— |
— |
3.4 Catalytic activity for H2WO4 or WO3
The comparison results for conversion of guaiacol over different catalysts are given in Table 3. When WO3 was utilized as the catalyst, 91.8% guaiacol was consumed and the products were still aromatic ethers and alkylphenols. However, the overall selectivity of aromatic ethers was as high as 67.0%, in which 2-ethoxyphenol was the main product with the selectivity of 46.0%. The overall selectivity of alkylphenols was less than half of that obtained over the H2WO4 catalyst. Obviously, WO3 exhibited a lower yield of alkylphenols than that obtained with H2WO4 as catalyst. No aromatic product was detected when H3PO4 was separately used as the catalyst in the conversion of guaiacol. However, when the WO3 combined with H3PO4 was utilized as the catalyst, guaiacol was completely converted, but the mass balance was 85.3%, which was because Brønsted acid sites facilitated the repolymerization of aromatics.5,45 The combined selectivity of aromatic ethers markedly decreased to 45.8%, in which 2-ethoxyphenol was still a main product but its selectivity decreased to 17.9%. On the contrary, the total selectivity of alkylphenols underwent an increase to 39.5%, and the tert-alkylphenols were still obtained as the dominating alkylphenols with a combined selectivity of 31.8%.
Table 3 Conversion of guaiacol over different catalysts. a
| Catalyst |
Cb/% |
Mc/% |
S1d |
S2 |
S3 |
S4 |
S5 |
S6 |
| Reaction conditions: guaiacol (1.0 g), catalyst (0.5 g), 300 °C, 6 h, 600 rpm, 80 ml ethanol, 0 MPa (gauge) initial N2 pressure at room temperature. C: conversion. M: mass balance. S1: aromatic ethers, S2: alkylphenols, S3: tert-butylphenols, S4: 2-ethoxyphenol, S5: 2,6-di-tert-butylphenol, S6: 2,6-di-tert-butyl-4-ethylphenol. |
| H2WO4 |
>99 |
96.7 |
16.7 |
80.0 |
71.6 |
2.10 |
28.5 |
35.2 |
| WO3 |
91.8 |
98.9 |
67.0 |
31.8 |
28.4 |
46.0 |
4.7 |
21.1 |
| 0.5 g WO3 + 0.1 g H3PO4 |
>99 |
85.3 |
45.8 |
39.5 |
31.8 |
17.9 |
2.3 |
25.2 |
3.5 Recycle tests
The reusability of the H2WO4 catalyst was tested and the results are listed in Table S3.† The spent H2WO4 catalyst was separated from the liquid products and was directly reused in the next run without treatment. The conversion was still as high as 86.0% in the third run despite there was a slight decline. In addition, the product was still aromatic ethers and alkylphenols but the distribution experienced a significant change. As the cycle time increased, the overall selectivity of aromatic ethers increased markedly while that of alkylphenols underwent an obvious decline. In the second run the yield of aromatic ethers was close to that of alkylphenols while the overall selectivity of aromatic ethers increased markedly to 82.1% but that of alkylphenols dropped to 11.8% in the third run. Compared with the first run, the selectivities of 2,6-di-tert-butylphenol and 2,6-di-tert-butyl-4-ethylphenol reduced by 90% and 82% while that of 2-ethoxyphenol increased by more than 26 times in the third run. Further, the decay in activity of the third run decreased more obviously than that of the second run.
3.6 Active sites
Tai et al.34 reported the selective conversion of cellulose to ethylene glycol over H2WO4 and Ru/C catalyst. They found that H2WO4 transformed into HxWO3, which was the active species for the conversion of cellulose. However, H2WO4 was not active for guaiacol conversion when we attempted to utilize H2O as solvent. In addition, the diffraction peaks of the spent H2WO4 sample which was recovered after reaction at once were basically in accord with those of WO3 catalyst. The phenomenon was obviously different from the results reported in the previous work.35 Notably, a higher guaiacol conversion and a higher selectivity of the alkylphenols were obtained over the H2WO4 catalyst than the WO3 catalyst. Besides, Brønsted acid (H3PO4) showed no catalytic activity for guaiacol conversion but the WO3 in combination with the H3PO4 showed a better performance than the WO3 catalyst. Hence, the difference in catalytic activity between H2WO4 catalyst and WO3 catalyst may be related to the presence of Brønsted acid sites. We speculated that WO3 sites played an important role in the conversion of guaiacol, and the Brønsted acid sites on H2WO4 rather than the HxWO3 sites enhanced the conversion and favoured a high selectivity of the alkylphenols, especially for tert-butylphenols. The interaction effect between metal sites and acid sites have been reported.16,46–48
3.7 The mechanism of deoxygenation and alkylation
Two alternative mechanisms have been proposed to describe deoxygenation of phenolics: (1) a direct HDO produces aromatics with water formation.11,13 (2) an indirect hydrogenation of the benzene ring happens, followed by hydroxyl dehydration to form a double bond and hydrogenated hydrocarbons.15,20 No ring saturation product was detected in this work, indicating that the direct HDO reaction occurred. Besides, no phenol or catechol was detected in the product, showing that the deoxygenation and the alkylation reactions performed continuously.28 However, theoretical calculation shows that the bond dissociation energy of Caromatic–OH bond is about 84 kJ mol−1 higher than that of the Caliphatic–OH.16 Thus, the direct deoxygenation path may involve a partial hydrogenation of the benzene ring. A similar reaction pathway was proposed for gaseous phase HDO of phenol on a CoMo catalyst and gaseous phase HDO of anisole over a Pt/H-beta catalyst.16,49
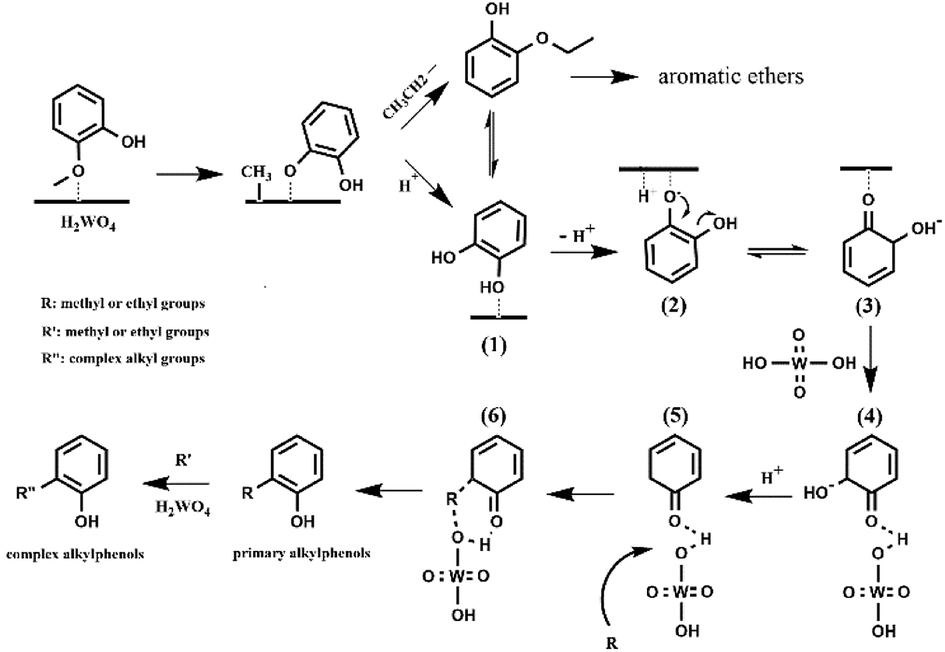
The conversion and product distribution from the different reactant (catechol, 2-ethoxyphenol, phenol and anisole) indicated that catechol and 2-ethoxyphenol were likely the intermediates of the guaiacol conversion over the H2WO4 catalyst. To sum up, the possible deoxygenation and alkylation pathways of guaiacol conversion are proposed as Scheme 1. With the catalysis of H2WO4, the demethylation of guaiacol takes place primarily. The methoxy group (–OMe) in guaiacol forms a hydrogen bond with the acid site over the H2WO4 catalyst, and adsorbs on the H2WO4 catalyst surface.13,14 The guaiacol molecule possesses three types of oxygen–carbon bonds: C(sp3)–OAr, C(sp2)–OMe and C(sp2)–OH with the bond energies about 247, 356 and 414 kJ mol−1 respectively.13 The catalytic cleavage of the C(sp3)–OAr is much easier. Hence, the demethylation step happens subsequently through the catalytic cleavage of the oxygen–carbon bond (C(sp3)–OAr) on the H2WO4 surface.5 The obtained phenoxide ion then reacts with the ethoxy or proton to produce the 2-ethoxyphenol and catechol intermediates. For the 2-ethoxyphenol intermediate, it is transformed into aromatic ethers and catechol soon. For the catechol intermediate, it also reacts with ethanol and produced the 2-ethoxyphenol intermediate. Meanwhile, the deoxygenation and alkylation steps are carried out. The catechol adsorbs on the H2WO4 surface, and one of the hydroxyl groups undergoes deprotonation, producing the electronegative phenolate ion (Scheme 1(2)). Concurrently, the electropositive hydrogen ion is adsorbed on the H2WO4 surface. The unstable phenolate ion (Scheme 1(2)) forms an equivalently charged compound (Scheme 1(3)) containing a ketone group through a resonance hybridization step.50 The formed unstable intermediate (Scheme 1(3)) is adsorbed on the H2WO4 surface by hydrogen bond (Scheme 1(4)). Dai et al.51 investigated direct conversion of cyclohexane to adipic acid over a bifunctional H2WO4/TS-1 catalyst, and also proposed the partial deoxygenation of 1,2-cyclohexanediol involved the activation of cyclohexanone via protonation and tautomerisation. The detail is shown in Scheme S1.† Subsequently, the intermediate (Scheme 1(4)) undergoes a dehydroxylation process. The hydrogen ions on H2WO4 surface, including the adsorbed hydrogen ions and the inherent hydrogen ions over H2WO4 catalyst, can react with the electronegative hydroxyl groups to remove the hydroxyl group in the form of H2O. The intermediate (Scheme 1(4)) can be regarded as the product of partial hydrogenation of the benzene ring, which is beneficial to the subsequent dehydroxylation due to the lower bond dissociation energy.16,49 Therefore, the dehydroxylation reaction becomes relatively easy to proceed. The obtained product (Scheme 1(5)) reacts with the adsorbed methyl or ethyl groups, leading to the formation of primitive alkylphenol. The methyl group can be formed form the demethylation step during guaiacol conversion and the evolved from the ethanol medium.28 Subsequently, the higher alkylphenols, including iso-propylphenols, tert-butylphenols and neo-pentylphenols, are formed through a series of consecutive substitution steps with methyl or ethyl groups supplied by the ethanol medium on the primitive alkylphenol. Finally, the obtained alkylphenol desorbs from the catalyst surface. Notably, no completely deoxygenated compound was detected, because the alkylphenols are more difficult to achieve complete deoxygenation than phenol.13,52 Throughout the reaction, the supercritical ethanol not only severs as a hydrogen-donor for HDO of guaiacol, but plays a role of capping agent to stabilize the highly active phenolic intermediates by the consecutive alkylation steps.32,33
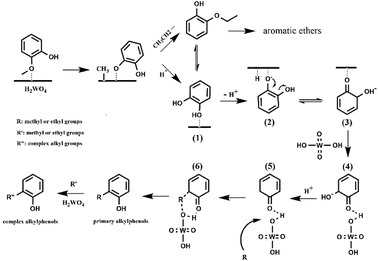 |
| | Scheme 1 The deoxygenation and alkylation pathways of guaiacol conversion. | |
3.8 The reason of deactivation
The XRD profiles (Fig. 1) proved that the H2WO4 catalyst dehydrated into WO3, and further reduced into W18O49 or WO2.72 phase. The TG profiles (Fig. 2) also confirmed the H2WO4 dehydration process. The transformation of H2WO4 to WO3 in a temperature range of 200–310 °C have also been reported.39,40,53 The Raman spectra (Fig. S1†) and TG profiles (Fig. 2) verified that the accumulation of carbon on the spent H2WO4 catalyst was gradually increased with the increase of number of cycle time. Further, we concluded from pyridine FT-IR spectra (Fig. 3) and NH3-TPD profiles (Fig. 4) that with the increase of number of cycle time, the acid sites over the H2WO4 catalyst gradually decrease and the Brønsted acid sites were completely disappeared after third run. Notably, the decrease in the activity of the third run was more obvious than that of the second run. This may be due to the partial reduction of the spent catalyst during the recycle tests and the gradual accumulation of carbon deposited on the spent catalyst. The carbon covered the active sites on the catalyst surface, causing a further decrease in catalytic activity. Overall, the carbon deposition on the catalyst surface, the dehydration and partial reduction of the catalyst itself were responsible for the decay of the catalyst. However, the technique that synthesis H2WO4 through WO3 has already been realized in the recovery of waste tungsten materials.54,55
4 Conclusions
In summary, the H2WO4 catalyst is an effective catalyst for highly selective conversion of guaiacol to tert-butylphenols in supercritical ethanol. The remarkably high selectivity of 2,6-di-tert-butylphenol and 2,6-di-tert-butyl-4-ethylphenol provides an option in the production of high-valued platform chemicals. Catechol and 2-ethoxyphenol are likely the intermediates of the guaiacol conversion in ethanol over the H2WO4 catalyst. The comparative experiment results indicate that the Brønsted acid sites on H2WO4 enhances the conversion and favours a high selectivity of the alkylphenols. The possible reaction pathways of guaiacol conversion are proposed. One route involves a transetherification reaction, and the other route involves a rapid deprotonation step and a rapid resonance hybridization followed by dehydroxylation reaction and alkylation steps. The recycle tests show that the activity for guaiacol conversion over the H2WO4 catalyst decreased with the increase of the cycle time, which is due to the carbon deposition on the catalyst surface, the dehydration and partial reduction of the catalyst itself.
Conflicts of interest
There is no conflicts to declare.
Acknowledgements
The financial support from the Ministry of Science and Technology of China (2011DFA41000) and the National Natural Science Foundation of China (21336008, 21808163 and 21690083) is gratefully acknowledged.
Notes and references
- J. Zakzeski, P. C. Bruijnincx and A. L. Jongerius, Chem. Rev., 2010, 6, 3552–3599 CrossRef PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- M. Saidi, F. Samimi, D. Karimipourfard, T. Nimmanwudipong, B. C. Gates and M. R. Rahimpour, Energy Environ. Sci., 2014, 7, 103–129 RSC.
- K. Stärk, N. Taccardi, A. Bösmann and P. Wasserscheid, ChemSusChem, 2010, 3, 719–723 CrossRef PubMed.
- J. Lavoie, W. Baré and M. Bilodeau, Bioresour. Technol., 2011, 102, 4917–4920 CrossRef CAS PubMed.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- R. Ma, K. Cui, L. Yang, X. Ma and Y. Li, Chem. Commun., 2015, 51, 10299–10301 RSC.
- X. Ma, K. Cui, W. Hao, R. Ma, Y. Tian and Y. Li, Bioresour. Technol., 2015, 192, 17–22 CrossRef CAS PubMed.
- A. L. Jongerius, R. W. Gosselink, J. Dijkstra, J. H. Bitter, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2013, 5, 2964–2972 CrossRef CAS.
- J. Van Haveren, E. L. Scott and J. Sanders, Biofuels, Bioprod. Biorefin., 2008, 2, 41–57 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Delichère and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS.
- A. Gutierrez, R. K. Kaila, M. L. Honkela, R. Slioor and A. O. I. Krause, Catal. Today, 2009, 147, 239–246 CrossRef CAS.
- X. Zhu, L. Lobban, R. Mallinson and R. De, J. Catal., 2011, 281, 21–29 CrossRef CAS.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 RSC.
- M. V. Bykova, D. Y. Ermakov, V. V. Kaichev, O. A. Bulavchenko, A. A. Saraev, M. Y. Lebedev and V. A. Yakovlev, Appl. Catal., B, 2012, 113–114, 296–307 CrossRef CAS.
- T. Prasomsri, T. Nimmanwudipong and Y. R. An-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC.
- X. H. Zhang, Q. Zhang, L. G. Chen, Y. Xu, T. J. Wang and L. L. Ma, Chin. J. Catal., 2014, 35, 302–309 CrossRef CAS.
- H. Y. Zhao, D. Li, P. Bui and S. T. Oyama, Appl. Catal., A, 2011, 391, 305–310 CrossRef CAS.
- X. Ma, Y. Tian, W. Hao, R. Ma and Y. Li, Appl. Catal., A, 2014, 481, 64–70 CrossRef CAS.
- I. T. Ghampson, C. Sepúlveda, R. Garcia, B. G. Frederick, M. C. Wheeler, N. Escalona and W. J. DeSisto, Appl. Catal., A, 2012, 413–414, 78–84 CrossRef CAS.
- I. T. Ghampson, C. Sepúlveda, R. Garcia, L. R. Radovic, J. L. G. Fierro, W. J. DeSisto and N. Escalona, Appl. Catal., A, 2012, 439–440, 111–124 CrossRef CAS.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS PubMed.
- X. Ma, R. Ma, W. Hao, M. Chen, F. Yan, K. Cui, Y. Tian and Y. Li, ACS Catal., 2015, 5, 4803–4813 CrossRef CAS.
- F. Yan, R. Ma, X. Ma, K. Cui, K. Wu, M. Chen and Y. Li, Appl. Catal., B, 2017, 202, 305–313 CrossRef CAS.
- K. Cui, L. Yang, Z. Ma, F. Yan, K. Wu, Y. Sang, H. Chen and Y. Li, Appl. Catal., B, 2017, 219, 592–602 CrossRef CAS.
- T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457–2461 RSC.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2015, 17, 941–4950 Search PubMed.
- X. Huang, C. Atay, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ACS Catal., 2015, 5, 7359–7370 CrossRef CAS.
- Z. Tai, J. Zhang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2012, 48, 7052–7054 RSC.
- Z. Tai, J. Zhang, A. Wang, J. Pang, M. Zheng and T. Zhang, ChemSusChem, 2013, 6, 652–658 CrossRef CAS PubMed.
- Y. Hong, D. Lee, H. Eom and K. Lee, J. Mol. Catal. A: Chem., 2014, 392, 241–246 CrossRef CAS.
- B. Ma, E. Huang, G. Wu, W. Dai, N. Guan and L. Li, RSC Adv., 2017, 7, 2606–2614 RSC.
- J. Park, V. Vo, N. T. V. Hoan, L. H. Hoang and S. J. Kim, J. Mater. Sci.: Mater. Electron., 2016, 27, 2662–2669 CrossRef CAS.
- O. Yamaguchi, D. Tomihisa, H. Kawabata and K. Shimizu, J. Am. Ceram. Soc., 1987, 70, 94–96 Search PubMed.
- J. Cao, B. Luo, H. Lin, B. Xu and S. Chen, Appl. Catal., B, 2012, 111–112, 288–296 CrossRef CAS.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68–72 CrossRef CAS PubMed.
- C. Fang, D. Zhang, L. Shi, R. Gao, H. Li, L. Ye and J. Zhang, Catal. Sci. Technol., 2013, 3, 803–811 RSC.
- S. Echeandia, P. L. Arias, V. L. Barrio, B. Pawelec and J. L. G. Fierro, Appl. Catal., B, 2010, 101, 1–12 CrossRef CAS.
- X. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2010, 271, 88–98 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- G. S. Foo, A. K. Rogers, M. M. Yung and C. Sievers, ACS Catal., 2016, 6, 1292–1307 CrossRef CAS.
- W. Zhang, J. Chen, R. Liu, S. Wang, L. Chen and K. Li, ACS Sustainable Chem. Eng., 2014, 2, 683–691 CrossRef CAS.
- K. L. Luska, P. Migowski, S. El Sayed and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 15750–15755 CrossRef CAS PubMed.
- F. E. Massoth, P. Politzer, M. C. Concha, J. S. Murray, J. Jakowski and J. Simons, J. Phys. Chem. B, 2006, 110, 14283–14291 CrossRef CAS PubMed.
- A. Jha, K. R. Patil and C. V. Rode, ChemPlusChem, 2013, 78, 1384–1392 CrossRef CAS.
- J. Dai, W. Zhong, W. Yi, M. Liu, L. Mao, Q. Xu and D. Yin, Appl. Catal., B, 2016, 192, 325–341 CrossRef CAS.
- B. S. Gevert, J.-E. Otterstedt and F. E. Massotha, Appl. Catal., 1987, 31, 119–131 CrossRef CAS.
- C. Balázsi, M. Farkas-Jahnke, I. Kotsis, L. Petrás and J. Pfeifer, Solid State Ionics, 2001, 141–142, 411–416 CrossRef.
- Y. Qin, China Tungsten Ind., 2002, 17, 40–42 Search PubMed.
- L. Ren, C. Zhao and W. Gao, J. of Petrochem. Univ., 2005, 18, 28–31 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07962e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d and
Yongdan Liabc
*d and
Yongdan Liabc