
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBroadening the scope of sortagging
Xiaolin Dai
ab,
Alexander Böker
 ab and
Ulrich Glebe
*a
ab and
Ulrich Glebe
*a
aFraunhofer Institute for Applied Polymer Research IAP, Geiselbergstr. 69, 14476 Potsdam-Golm, Germany. E-mail: ulrich.glebe@iap.fraunhofer.de
bLehrstuhl für Polymermaterialien und Polymertechnologie, Universität Potsdam, 14476 Potsdam-Golm, Germany
First published on 6th February 2019
Abstract
Sortases are enzymes occurring in the cell wall of Gram-positive bacteria. Sortase A (SrtA), the best studied sortase class, plays a key role in anchoring surface proteins with the recognition sequence LPXTG covalently to oligoglycine units of the bacterial cell wall. This unique transpeptidase activity renders SrtA attractive for various purposes and motivated researchers to study multiple in vivo and in vitro ligations in the last decades. This ligation technique is known as sortase-mediated ligation (SML) or sortagging and developed to a frequently used method in basic research. The advantages are manifold: extremely high substrate specificity, simple access to substrates and enzyme, robust nature and easy handling of sortase A. In addition to the ligation of two proteins or peptides, early studies already included at least one artificial (peptide equipped) substrate into sortagging reactions – which demonstrates the versatility and broad applicability of SML. Thus, SML is not only a biology-related technique, but has found prominence as a major interdisciplinary research tool. In this review, we provide an overview about the use of sortase A in interdisciplinary research, mainly for protein modification, synthesis of protein–polymer conjugates and immobilization of proteins on surfaces.
 Xiaolin Dai | Xiaolin Dai studied Applied Chemistry at Jiangnan University, Wuxi, China and obtained a master of engineering in 2012. After moving to Germany, she worked on her doctoral thesis at RWTH Aachen from 2014–2015 and continued at University of Potsdam from 2015 till her graduation in 2018. She is currently research associate at the Fraunhofer Institute for Applied Polymer Research IAP in Potsdam-Golm, Germany. Her research interests include enzyme catalysis, hybrid materials and applications in cosmetics. |
 Alexander Böker | Alexander Böker is director of the Fraunhofer Institute for Applied Polymer Research (IAP) and holds the Chair for Polymer Materials and Polymer Technology at the University of Potsdam, Germany. He studied chemistry at the Johannes Gutenberg University Mainz and Cornell University, Ithaca, NY, USA and received his PhD from University of Bayreuth in 2002 and his habilitation from the same university in 2007. From 2008–2015, he was deputy scientific director of DWI – Leibniz Institute for Interactive Materials in Aachen. The main research interests of his group include integration of biological functions into polymer materials and guided self-assembly of colloidal and polymer systems. |
 Ulrich Glebe | Ulrich Glebe studied Nanostructure and Molecular Sciences at the University of Kassel, Germany. He accomplished his doctoral thesis from 2008 till 2011 at the University of Kassel. In 2013, he joined the group of Prof. Dr Alexander Böker at the Leibniz Institute for Interactive Materials e.V. in Aachen, Germany as a postdoc. From April 2015 on, he continues his research as a group leader at the Fraunhofer Institute for Applied Polymer Research IAP in Potsdam-Golm, Germany. His research interests include the synthesis of protein–polymer conjugates, enzyme catalysis, self-assembled membranes and protein–polymer materials. |
1. Introduction
Sortases are members of the class of transpeptidases. They are produced by Gram-positive bacteria, and their function is to catalyze the formation of a peptide bond between two specific peptide sequences, a C-terminal recognition sequence and an N-terminal nucleophile. Since the isolation of the first sortase at the end of 1990s,1 the sortase family has been intensively studied. The majority of sortases have been found in conventional Gram-positive bacteria. Based on their recognition sequences and functions, sortases are categorized into A–F different classes.2–4 Main functions of sortases are the attachment of distinct proteins on the cell wall and the assembly of pili. Sortase A (SrtA) is a membrane-bound extracellular enzyme and the best known and studied sortase (enzyme commission number: EC 3.4.22.70). It is responsible for the covalent anchoring of surface proteins with the recognition sequence LPXTG (with X being any amino acid) onto oligoglycine functionalities of the cell wall via a two-step transpeptidation.3,4 First, the cysteine residue in the active site of SrtA nucleophilically attacks and cleaves the peptide bond between threonine and glycine in the LPXTG motif, forming a thioacyl bond within the LPXT–SrtA complex, while the glycine is cleaved off. Second, this thioacyl intermediate is then resolved by the nucleophilic attack of an N-terminal oligoglycine. Thus, the enzyme forms a peptide bond between the carboxylic acid group of threonine in LPXT and the amino group of an oligoglycine. Via this mechanism, SrtA displays virulence factors on the cell wall which mediate the bacterial adhesion to host tissue. In view of the significant health danger by infections through bacteria and their increasing antibiotic resistance, sortase A is considered as a target to develop alternative treatments to antibiotics.5,6 Inhibiting the activity of SrtA leads to a significant reduction of infection as the bacteria can't anchor virulence factors. Therefore, several natural as well as synthetic sortase A inhibitors have been identified yet.5,6 Since the enzyme is not essential in bacterial growth, its inhibition does not place pressure on the bacteria to develop a drug-resistance mechanism.6The structures of many sortase variants including transient enzyme intermediates have been determined by both NMR spectroscopy and X-ray crystallography.7–10 Sortase A from Staphylococcus aureus (S. aureus) consists of an eight-stranded β-barrel core with a hydrophobic pocket for substrate binding. The 3-dimensional solution structure has been determined by NMR spectroscopy (protein data bank, PDB, code 1IJA)7 and crystal structures solved of native SrtAΔ59 (PDB entry 1T2P) and a mutated version in complex with the substrate LPETG (SrtAΔN59Cys184Ala-LPETG, PDB code 1T2W).8 Other sortases like SrtA from Streptococcus pyogenes (SrtAΔ81, PDB 3FN5)9 exhibit similar basic structures, but with small differences.
The expression of sortases can be performed in Escherichia coli (E. coli), and several SrtA variants are commercially available nowadays. While nearly all other sortase classes showed negligible activity in vitro, SrtA proved well suited to catalyze transpeptidation reactions also in non-natural environment. Therefore, this technique became known as sortase-mediated ligation (SML) or sortagging (Scheme 1) and developed to a ubiquitous research tool for protein modification. The high selectivity towards specific peptide motifs, the very robust nature of sortase, synthetic and commercial accessibility of all required compounds (including sortase) and the simplicity to perform sortagging in a lab may have contributed to the extensive in vitro application of sortase A. Furthermore, the recognition sequence of SrtA occurs extremely seldom in natural proteins, which enables specific sortagging also in vivo. The profound progress in the last years demonstrated many advantages of the technique. For instance, the modification at a protein terminus has less impact on folding and functionality of the protein. Likewise, SML allows the immobilization of proteins on surfaces in defined orientation while preserving function or enzymatic activity to a much higher degree than other immobilization techniques.
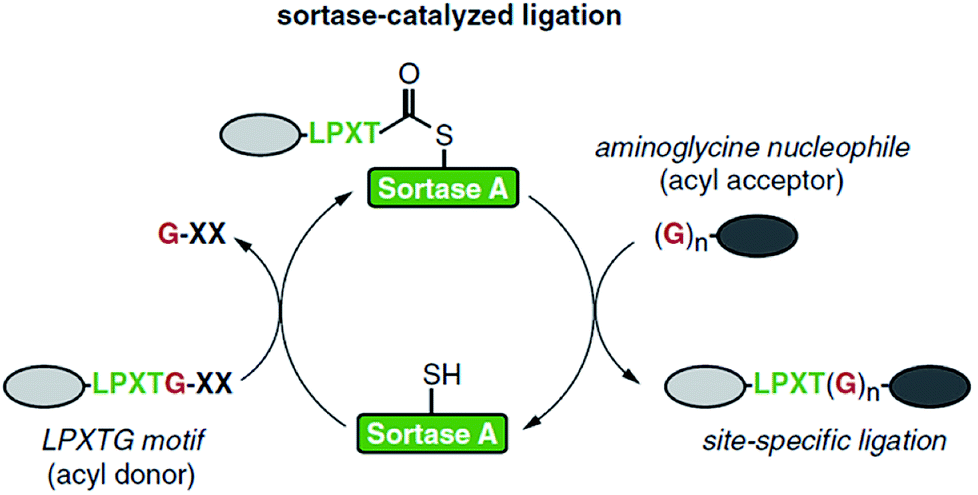 | ||
| Scheme 1 Catalytic cycle of sortase-mediated ligation involving the formation of the thioester intermediate followed by attack of the oligoglycine nucleophile which finally results in the formation of the ligation product. Reprinted from ref. 11, Copyright (2016), with permission from Elsevier. | ||
A major disadvantage of sortagging is the low catalytic efficiency; hence, comparably high enzyme concentrations are required. Furthermore, S. aureus SrtA is Ca2+-dependent and the reaction is reversible: the formed peptide sequence (LPXTGGG) represents again a substrate for the enzyme. These shortcomings were partially solved by biotechnological methods through the development of variants with up to 140-fold higher activity, and Ca2+-independent variants.11 Due to the reversibility of the reaction, a quantitative conversion of a substrate is nearly impossible. Usually, the yield of the reaction product is increased by using an excess of one of the substrates. Furthermore, several approaches were developed to shift the equilibrium of the reaction. The by-product G-XX can be removed from the equilibrium (formation of unreactive fragments, complexation with metal ions, reaction under dialysis conditions).11,12 Moreover, the addition of the amino acids WTWTW at both peptide motifs leads to the formation of a β-hairpin which cannot be recognized by SrtA anymore and is therefore removed from the reverse reaction.13,14
To further improve sortagging, immobilized sortases were utilized which enabled large scale reactions and recyclability of the enzyme.15–20 As a variation of immobilized sortases, the efficiency of the reaction was improved by either performing a flow-based sortagging in a microreactor21 or bringing the enzyme and the target protein in proximity through a SpyTag-SpyCatcher system.22 Diverse mutated sortase A variants with enhanced activity23–26 or altered substrate selectivity27–30 were developed as well as sortase homologues studied.31,32
The controlled ligation between peptides and proteins is an important method in synthetic biology. Normally, two synthetic peptides can be ligated under certain conditions by forming a peptide bond.33,34 However, this can be difficult for large substrates such as proteins from a technical point of view. In 2004, Mao et al. presented for the first time that protein engineering can be realized via SML.35 The authors reported the ligations of peptide–peptide, protein–peptide, protein–protein and even a first example for protein-molecule to demonstrate the utility of SML. After testing various oligoglycine nucleophiles, they concluded that the number of glycines doesn't influence the efficiency of sortagging. This novel approach for protein engineering was extensively expanded in the following years, e.g. for the ligation of fluorescent proteins to a protein of interest,36–38 to develop immunoassays with the aim of monitoring glucose levels,39 to crosslink enzymes,40 to generate fusion proteins that are inaccessible via direct expression,41 to form an enzyme cascade within E. coli42 and to ligate two domains of a protein.43,44 Other studies focused on the ligation of two peptides45–47 and of a peptide to a protein,48–53 among others to demonstrate the purification of recombinant fusion proteins,54 to functionalize amyloid-based polymers,55 to assemble semisynthetic proteins or protein mimics56,57 and to incorporate functional proteins into hydrogels.58 SML also proved helpful in the formation of protein samples for NMR spectroscopic studies. Multi-domain proteins with domain-selective isotope labeling could be formed59–61 as well as a fusion protein of an isotopically labeled protein of interest and a solubility tag in order to facilitate NMR studies of proteins with limited solubility.62
In addition to the ligation of two proteins or peptides, the cyclization of a peptide or protein developed as another application field of sortase catalysis.63 A major aim of such studies is that (therapeutic) proteins have a higher stability in their cyclized than in the native form. Compared with native chemical ligation, cyclic polypeptides can be efficiently formed by SML, which needs only minimal modification on the proteins to be circularized. Antos et al. reported the cyclization of different substrates of the structure Gx-protein-LPXTG (Scheme 2, right).64 It turned out that it depends on the distance of the termini of the protein if cyclization takes place or oligomers are formed. In addition, both cyclization and oligomerization were reversible after addition of an excess of diglycine nucleophile. Hu et al. combined protein cyclization by SML with the conjugation of a synthetic polymer. The formed cyclic green fluorescent protein (GFP)–poly(oligo(ethyleneglycol)methylethermethacrylate) (POEGMA) conjugate showed increased thermal stability and improved tumor retention.65 Furthermore, membrane scaffold proteins (MSPs) were cyclized in order to form covalently circularized nanodiscs (cNDs).66,67 In the last years, the strategy of peptide or protein cyclization by SML was adopted for a myriad of studies.68–74 It was recently demonstrated that a peptide synthesized by SPPS incorporating the SrtA recognition sequence can be cleaved from the resin by the enzyme and in situ cyclized.75
 | ||
| Scheme 2 Labeling (left) as well as circularization (right) of a protein with both peptide motifs for SML. Reprinted with permission from ref. 64. | ||
Apart from the ligation of proteins and peptides, sortases have found many more applications. Excellent articles summarized the different sortase classes with their functions in nature and structures,2,4 mechanistic studies,3,4 sortase inhibitors,5,6 pili construction,76,77 peptide and protein cyclisation,63,78 general application fields and the use for protein engineering,79–85 as well as the improvement of SML technology11,12 and its differentiation with other enzymatic methods.86–90 In this review article, we give an overview about the application of sortases in different interdisciplinary research fields. Using sortase A, a protein can be ligated to another biomolecule, a small synthetic molecule, a polymer or a surface. Therefore, the substrates only need to be equipped with a C-terminal LPXTG and an N-terminal Gx peptide sequence. At present, a wide range of applications have been reported going much beyond standard reactions for protein modification: for instance, the modification of living cells with proteins or labels, the formation of complex protein structures, the immobilization of enzymes on planar surfaces as well as particles while preserving their enzymatic activity. Few studies even demonstrate that two synthetic chemical building blocks can be linked via sortase-mediated ligation. Our overview focuses on important achievements on the border between biology and chemistry with an emphasis on recent examples that show the current developments in the field.
2. Cell surface modification
Since SrtA anchors surface proteins on the cell wall of Gram-positive bacteria in nature, the latter hence can be modified via SML also in lab scale. In addition to proteins, also small molecules were ligated to the cell wall. Nelson et al. reported in 2010 for the first time that the cell surface from wild-type S. aureus can be re-engineered with synthetic small molecules such as fluorescein or proteins like biotin.91 The native SrtA enzymes of the bacteria covalently link non-native molecules bearing the LPETG recognition sequence to the peptidoglycan of the wild-type Gram-positive bacteria (Scheme 3). The anchoring was shown to be covalent and fluorescein linked in an amount comparable to native proteins. | ||
| Scheme 3 Functional small molecules equipped with the recognition sequence LPETG are recognized by native SrtA and covalently linked to the S. aureus cell wall. Reprinted with permission from ref. 91. Copyright (2010) American Chemical Society. | ||
In the following years, much research concerning the engineering of bacterial cells has been done via sortagging. Veerman's group studied the influence of variable recognition sequences on the anchoring efficiency on S. aureus cells.92 The sortagging was more efficient when positively charged amino acids follow after the recognition sequence and also when LPMTG was used instead of LPETG. Park and Jung et al. reported the expression of an optimized SrtA variant and binding of eGFP-LPETG5 model proteins after anchoring of the enzyme in the cell membrane.93 Nguyen et al. displayed recombinant proteins successfully on the Bacillus subtilis cell surface using the sortases of this bacterium.94 Willson et al. similarly demonstrated the binding of recombinant enzymes to the cell wall of Clostridium acetobutylicum by the native sortase system of this organism.95
In addition to modifying bacterial cell walls by using their native sortases, also other living cells can be labeled by sortagging through the addition of a sortase. Appropriate recombinant proteins were expressed and labeled on the surface of living cells at their C-terminus96,97 or N-terminus,98 respectively, by addition of SrtA and the peptide-equiped label. Tomita et al. ligated unnatural lipids in cell membranes with recombinant eGFP-LPETG.99 A GGG-polyethylenglycol (PEG)-lipid was spontaneously incorporated into the cell membrane of HeLa cells and the substrate protein and SrtA were both added to perform the SML (Scheme 4). The successful ligation was proved by confocal microscopy and sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Furthermore, antibodies were displayed on cancer cells using this approach.
 | ||
| Scheme 4 SML of a protein of interest and a GGG-PEG-lipid modified cell membrane. Reprinted with permission from ref. 99. | ||
Swee et al. reported the surface display of engineered proteins or probes on cells which have naturally exposed glycines at the cell surface. The LPETG equipped substrates were added together with sortase A to the cells in buffer solution and reacted for 1 h at RT. The authors demonstrated the conjugation of biotin to mouse hematopoietic cells, yeast cells and Toxoplasma gondii as well as the installation of antibodies onto different cells via the SrtA enzymatic approach.100 Pishesha et al. demonstrated the reverse strategy of linking oligoglycine functionalized peptides to cells with exposed LPETG sequences.101 Membrane proteins with the recognition sequence were installed on red blood cells (RBCs) and antigenic peptides ligated by sortagging. This approach is assessed as promissing for prophylaxis and therapy against autoimmune diseases. Tan et al. used SML to label G-protein coupled receptors (GPCRs) on the surface of living leucocytes with a fluorescent dye.102 With the cells fixed on a substrate, the trafficking of the protein was subsequently observed at different pH values with a microscope.
Sortagging was recently applied to develop an approach to monitor the interaction between cells in vivo.103 One cell type contained a ligand and SrtA while the other cells were functionalized with a receptor and an oligoglycine motif. The acceptor cell is only labeled with a fluorescent substrate if ligand and receptor interact (Scheme 5). A mutated SrtA with lower affinity for the oligoglycine was used to ensure that the driving force for the labeling is the affinity between ligand and receptor instead of sortase and oligoglycine. The approach was demonstrated with immune cells both in vitro and in living mice.
 | ||
| Scheme 5 Overview of the principle to monitor cell–cell interactions: the acceptor cell is labeled by SML when ligand and receptor of two cell types interact. Reprinted by permission from Springer Nature from ref. 103, Copyright (2018). | ||
3. Modification of proteins or peptides with biomolecules such as DNA, lipids, and sugars
In addition to ligations between proteins and peptides, SML became also attractive for protein engineering in respect to the ligation with other biomolecules. Koussa et al. ligated proteins covalently to deoxyribonucleic acid (DNA) via SML.104 Oligoglycine units were first linked to DNA fragments by CuI-catalyzed azide–alkyne cycloaddition (CuAAC; compare Chapter 4.2). Next, protein-LPETG was ligated by sortagging and this enzymatic approach influenced the protein function only slightly. Moreover, different proteins could be sequentially ligated to DNA by using a mixture of GGG and masked GGG. After coupling the first protein, masked GGG sequences were liberated by tobacco etch virus (TEV) protease followed by SML of a second protein. SrtA was also employed for the construction of aptamer–antibody conjugates.105 Therefore, peptide nucleic acids functionalized with oligoglycine unit were ligated to antibody-LPETGG by a mutated eSrtA and the DNA aptamer hybridized subsequently.Antos et al. used SML for the site-specific formation of protein–lipid conjugates.106 Oligoglycine decorated lipids were ligated to the C-terminus of proteins equiped with the LPETG motif. In their work, the purification procedure proposed by Parthasarathy et al.107 was employed to remove the sortase enzyme as well as the excess of the model protein eGFP-LPETG-His6 by the Ni-affinity resin.106 The approach was achieved through a His6-tag after the recognition sequence which is cleaved during sortagging. As SrtA also has a His-tag, only the ligation product does not possess this tag.
Guo et al. ligated a protein to liposomes by SML (Scheme 6a),108 which opened the door for a new method for the modification of liposomes with proteins. Lipids were synthesized with directly attached diglycine or separated by a PEG spacer. The efficiency of the ligation of eGFP-LPATG was improved for higher GG concentrations on the liposomes and for larger distances through the PEG spacer. Tabata et al. focussed on the ligation of biotin to liposomes109 and subsequently showed that liposomes can be functionalized with lung tumor-binding peptide (LTBP) by SML.110 The latter are promissing platforms for cancer-targeting drug delivery systems. Wöll et al. investigated the ligation of single-domain antibody (VHH)-LPETG to different G5-liposome formulations and also demonstrated the pharmaceutical relevance by showing the specific binding of such ligand-functionalized liposomes to cells.111,112 Silvius et al. reported that the binding of proteins to liposomes by sortagging is slow and inefficient but can be enhanced by a “prebinding” strategy.113 Therefore, both protein and sortase A (e.g. GFP-LPETG-His6, SrtA-His6) were reversibly located at the liposome surface by complex formation between their His-tags and Ni(II)-ions from lipids with chelating moieties (Scheme 6b). Ligation of the proteins to GG-(PEG)3-lipid units was shown to be more efficient mainly because “prebinding” enhances the initial reaction between sortase and substrate protein while both ligation to the lipid and reversible detachment of the protein were only slightly influenced.
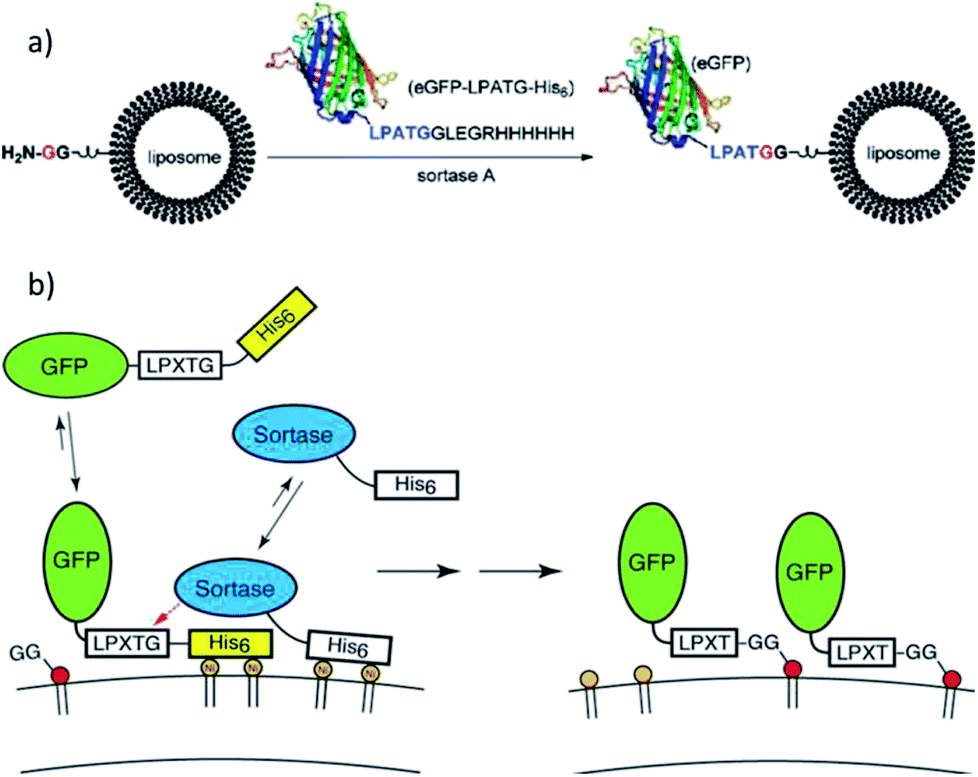 | ||
| Scheme 6 (a) Protein attachment on oligoglycine-functionalized liposomes. Reprinted with permission from ref. 108. Copyright (2012) American Chemical Society. (b) “Prebinding” strategy to enhance the efficiency of sortagging of proteins to liposomes. Reprinted with permission from ref. 113. Copyright (2017) American Chemical Society. | ||
Due to the vital function of glycoconjugates, sugar–peptide and sugar–protein conjugates became popular and well researched in the last years.114 Samantaray et al. proposed that certain sugar amino groups mimic glycine nucleophiles and can be used as substrates for SML.115 The SrtA transpeptidation indeed enabled the ligation of peptides and proteins containing a LPETG sequence to 6-aminohexoses. The linkage was specific to the –CH2–NH2 moieties although other amino groups are present in the sugar units (compare Chapter 4.5 for more details on the nucleophile selectivity).115 The group of Guo presented a new strategy for the chemical synthesis of glycosylphosphatidylinositol (GPI)–peptide/protein conjugates (Scheme 7). GPIs are glycolipids that anchor (glyco)proteins to the cell surface of eukaryotic cells. SML was successful after introducing at least one glycine at the phosphoethanolamine group.116 The approach could be transferred to the synthesis of GPI analogues with either peptides117 or proteins.118 The authors demonstrated that their strategy is successful for the synthesis of human antigen analogues119 as well as the linkage between large proteins and complex GPI anchors.118 Fang et al. also showed the linkage between an antibody and a glycopeptide by means of sortase-mediated ligation.120
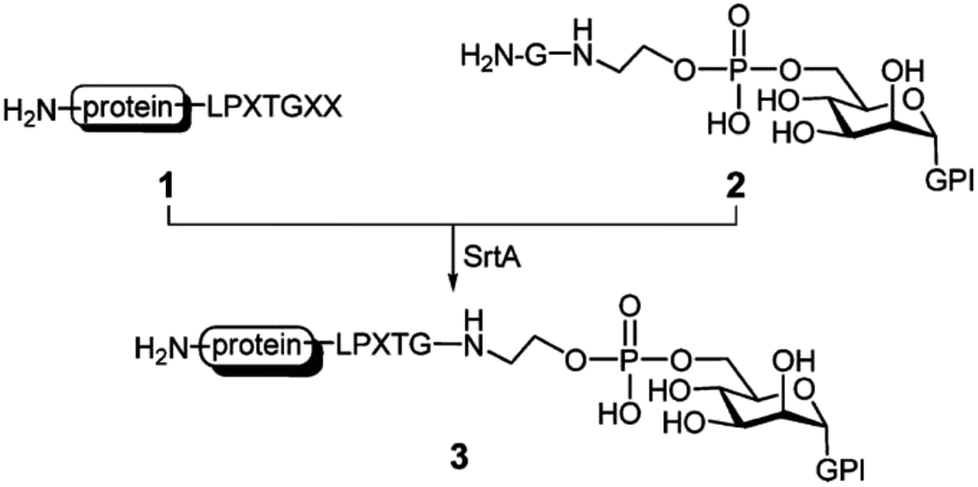 | ||
| Scheme 7 SrtA catalysis applied for the synthesis of GPI–protein conjugates. Reprinted with permission from ref. 117. Copyright (2010) American Chemical Society. | ||
4. Ligation of proteins and small synthetic molecules
In addition to ligating a protein and a biomolecule, the scope of SML has been enlarged for the linkage of small synthetic molecules to proteins. It was shown with multiple examples that the ligation of peptide-functionalized small molecules to both C- and N-terminus of a protein is easily achievable exploiting sortase catalysis. In this respect, SML was most widely used for the attachment of dyes – often called probes in a more general sense – and ligands for metal ions as well as azide or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond functionalities for further click chemical modification of the functionalized proteins. A main advantage of SML is the site specificity of modification which furthermore takes place at the C- or N-terminus of the protein and therefore has less impact on structure and function of the protein. After proof-of-principle, main application fields for SML of proteins with small synthetic compounds have been the formation of antibody conjugates, protein fusions and functional protein assemblies. The next paragraphs summarize the major directions from the last years. A special group of ligated compounds are initiators for a polymerization and will be presented in Chapter 5.1 in the context of forming protein–polymer conjugates aided by SML.
C triple bond functionalities for further click chemical modification of the functionalized proteins. A main advantage of SML is the site specificity of modification which furthermore takes place at the C- or N-terminus of the protein and therefore has less impact on structure and function of the protein. After proof-of-principle, main application fields for SML of proteins with small synthetic compounds have been the formation of antibody conjugates, protein fusions and functional protein assemblies. The next paragraphs summarize the major directions from the last years. A special group of ligated compounds are initiators for a polymerization and will be presented in Chapter 5.1 in the context of forming protein–polymer conjugates aided by SML.
4.1 Labeling of proteins
The site-specific modification of proteins with labels at both the N- and C-termini was demonstrated by the group of Ploegh.121,122 The labeling at one terminus is comparably easy and was shown with the model protein cholera toxin B subunit (CtxB) having N-terminal glycines and fluorescein isothiocyanate (FITC) and biotin labels functionalized with the recognition sequence.121 The installation of two modifications within one protein is much more challenging and required a cumbersome strategy using two different SrtA variants (Scheme 8).121 eGFP and UCHL3 served as model proteins with C-terminal LPXTG-His6 recognition sequence and N-terminal MLVPRG sequence, which functioned as masked glycine. SrtApyogenes (SrtA from Streptococcus pyogenes) accepts alanine nucleophiles (in addition to glycine) and was used at first to ligate tetramethylrhodamine-labeled dialanine (AA-TMR) to the C-terminus of the model proteins. The formed LPXTA sequence can't be cleaved by SrtAaureus (SrtA from Staphylococcus aureus), which was used for the second modification step. Therefore, after removing SrtApyogenes, the N-terminal sequence was cleaved by thrombin setting the glycine nucleophile available. Finally, FITCs could be installed at the N-terminus forming the site-specific dual-labeled model proteins. | ||
| Scheme 8 Strategy to equip both termini of one protein with labels using SrtApyogenes (SrtAstrep) and SrtAaureus (Δ59-SrtAstaph). Reprinted with permission from ref. 121; https://pubs.acs.org/doi/abs/10.1021/ja902681k; further permissions related to the material excerpted should be directed to the ACS. | ||
Warden-Rothman et al. developed a strategy to express a protein followed by modification and purification.123 A chimeric protein, consisting of protein of interest with C-terminal recognition sequence, spacer, and SrtA with His6-tag, was expressed and isolated on beads of a nickel column. Upon addition of calcium ions and GGG-label, the protein of interest (called ligand) is cleaved off and modified with the label (Scheme 9). It was postulated that any kind of cargo can be ligated to the C-terminus of a protein of interest exploiting this strategy. The proof-of-concept was demonstrated with an azide group that was subsequently utilized for click chemical reaction (compare Chapter 4.2). The main advantage compared to SML in solution is omitting an additional step for purification of the desired product from SrtA.
 | ||
| Scheme 9 Strategy from Warden-Rothman et al. for C-terminal labeling of a protein. Reprinted with permission from ref. 123. Copyright (2013) American Chemical Society. | ||
For N-terminal protein labeling, Sarpong et al. developed a procedure that combines TEV protease cleavage, SML and affinity purification.124 The protein of interest with TEV recognition motif is cleaved yielding an N-terminal glycine. A label with sortase recognition sequence and affinity tag for isolation of the modified protein is subsequently linked. This strategy was exemplarily shown with the ligation of fluorescent dyes to epidermal growth factor receptor (EGFR) and membrane scaffold protein (MSP).
Furthermore, several examples exist showing the labeling of proteins with fluorescent dyes utilizing SML. The C-terminal modification with oligoglycine-functionalized dyes was performed53,125–127 as well as the N-terminal modification with LPETG-functionalized dyes.128–130 Wu et al. demonstrated that protein labeling can be also performed in living Caenorhabditis elegans cells.131 Expression of a mutated SrtA variant and a target protein enabled in vivo protein labeling after feeding with oligoglycine-tagged labels. In addition, circularization of G5-GFP-LPETG was also demonstrated.
4.2 Introduction of functional groups for click chemical reactions
In case proteins can't be easily ligated to the compound of interest directly, functional groups need to be introduced that can be addressed by chemical reactions in a subsequent step. The introduction of such functional groups into the proteins can be achieved by SML. The mainly favored chemical reactions for subsequent protein modification are the CuI-catalyzed azide–alkyne cycloaddition or its metal-free analogue strain-promoted azide–alkyne cycloaddition (SPAAC). These cycloadditions are among the prominent examples of a click reaction, which are reactions that stand out with high yield, negligible side products, mild reaction conditions and ease of execution among others.132,133 The 1,3-dipolar azide–alkyne Huisgen cycloaddition is a reaction between substituted azides and alkynes leading to 1,2,3-triazoles which can be executed at RT under CuI catalysis.134 The reaction is bioorthogonal as there are neither azides nor alkynes present in nature, and therefore has been often used for the conjugation of biomolecules.135 A more recent development is SPAAC with exploiting the ring strain of cyclooctynes to lower the energy barrier of the reaction which omits the use of CuI as catalyst – a development especially important for metal-free protein conjugation.136Gupta et al. ligated proteins with LPXTG-His6 C-terminus with N3- or CC-functionalized oligoglycines in order to construct a dendrimer by CuAAC.137 A dendritic scaffold with multivalent display of proteins couldn't be formed directly by ligating proteins to a compound exhibiting several oligoglycine units. The reasons for the latter are most probably steric hindrance and the reversibility of the sortase reaction which does not allow to isolate products with several linked proteins in acceptable yields. However, using SML to link a protein to a compound having more than one azide or alkyne group, enabled to form the dendrimer by CuAAC subsequently. Sortase and unlabeled proteins, exhibiting both a His-tag, could be removed using Ni-nitrilotriacetic acid (NTA) beads.
The same approach was used by Wang et al. who compared four reactions for site-specific introduction of azide functionalities into proteins.138 SML between protein-LPETG-His6 and GG-PEG3-N3 resulted in the highest overall yield, compared to recombinant expression with unnatural amino acid, amidation at the N-terminus, and tyrosine selective three-component Mannich reaction. Furthermore, single site modification preserved the highest activity of the used membrane glycoprotein thrombomodulin (TM456).
A strategy was developed in the group of Ploegh in order to form unnatural N-to-N and C-to-C protein–protein fusions.139 As such fusion proteins are genetically impossible, a chemical strategy was developed. SML was exploited to ligate the click handles azide and strained cyclooctyne to the N-termini of two different proteins. The subsequent SPAAC yielded N-to-N fusion proteins (Scheme 10).139 The click handles can be likewise ligated to proteins' C-termini to form C-to-C fusions.140 As an alternative to the combination of SML and click chemistry, Harmand et al. used the orthogonal enzymes sortase A and butelase-1 to form C-to-C fusion proteins separated by a spacer.141 The PEG or DNA spacers contained the nucleophilic sequences for both enzymes and antibodies bearing the respective recognition sequence were ligated. This latter approach also enabled the modification of an antibody with two different dyes in a one-pot reaction.
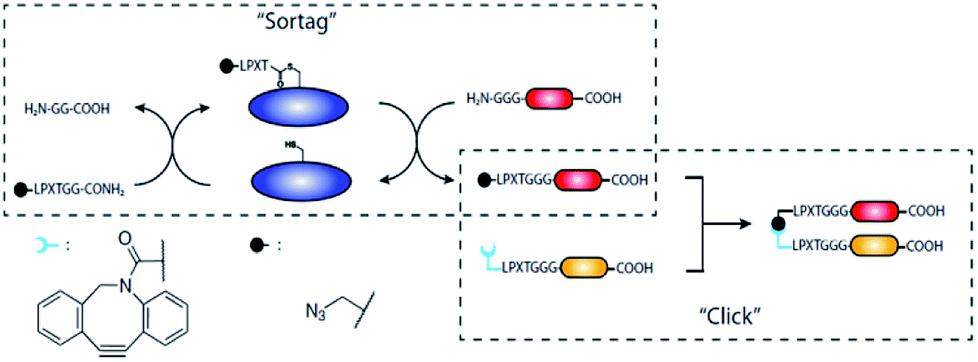 | ||
| Scheme 10 Strategy of preparing unnatural N-to-N and C-to-C fusion proteins. Reprinted from ref. 139. | ||
Matsumoto et al. ligated azide groups to tetrameric streptavidin by SML in order to form a hydrogel with immobilized enzymes.142 The SPAAC reaction with branched PEG exhibiting multiple alkyne groups yielded the hydrogel to which biotin-modified enzymes could be attached. As an application, the coating of the hydrogel on a glassy carbon electrode (GCE) and oxidation of glucose using glucose dehydrogenase (GDH) was demonstrated.
Click chemical approaches became also popular in recent years for the functionalization of antibodies. Alt et al. used the copper-free SPAAC to link an antibody with C-terminal SrtA recognition sequence and a glycine-functionalized strained cyclooctyne by SML.143 The product could be reacted with fluorescent dyes or macrocyclic ligands bearing an azide group. Therefore, this versatile strategy proofed to be well suited for the 2-step ligation of proteins with other biomolecules, labels or any kind of functionalities. Kwon et al. ligated a cyclooctyne to an antibody in order to subsequently form an antibody–cyclotide conjugate by SPAAC.144 Cyclotides are cyclic, crosslinked peptides and the conjugation to an antibody enabled their targeted delivery. Li et al. demonstrated that antibodies can be labeled with multiple (identical) fluorophores to improve fluorescent yields.145 First, a GGG peptide equipped with both a fluorescent dye and an azide was linked to a single-chain antibody fragment (VHH) by means of SML. Next, a DNA Holliday junction with strained cyclooctyne and three fluorescent dyes was reacted with the functionalized antibody fragment in a SPAAC reaction. The semi-rigid DNA structure of the Holliday junction ensures certain spacing between the dyes to avoid self-quenching of fluorophores. Bonnard et al. ligated antibodies with alkyne functionalities by SML to subsequently use SPAAC to incorporate the proteins into particles exclusively based on amino acids.146 These particles showed to be low-fouling, biodegradable and therefore promissing for molecular imaging of vascular diseases. Bartels et al. applied the above presented strategy to form antibody C-to-C fusions.147 In addition to introducing azide and alkyne groups for SPAAC, the authors also ligated click handles for an inverse-electron demand Diels–Alder reaction to the C-termini of antibodies by SML.
4.3 Synthesis of antibody conjugates
In general, the modification of antibodies is an important research field and antibody conjugates can be formed by means of SML. The antibody fragment scFvanti-LIBS was modified at its C-terminus with a chelate ligand using SML. Through complexation of the isotope 64Cu, the antibody fragment was radiolabeled having diagnostic potential.148 In a similar fashion, Massa et al. linked GGGYK-functionalized chelate ligands and a fluorescent dye to camelid single-domain antibody-fragments. The chelate ligands were meant to bind 111In for use in single-photon emission computed tomography (SPECT) and 68Ga for positron emission tomography (PET), respectively, and the fluorescent dye for fluorescence reflectance imaging (FRI).149 Rashidian and Wang et al. showed the introduction of different functionalities into antibodies in order to improve their therapeutic properties.150 GGG-peptides with two functionalities were linked to the C-terminus of single-chain antibody fragments (VHH) via SML. These functionalities were either a fluorophore or served for introduction of 18F isotopes for imaging on the one side and an azide on the other side. The latter was used for SPAAC with cyclooctynes, for instance for PEGylation of the antibody.SLM allows the site-specific formation of antibody–drug conjugates (ADCs) which are potentially anti-tumor drugs. ADCs use a specific antibody for targeting of cancer cells in combination with small-molecule toxic payloads. Beerli et al. linked small molecule toxins to the C-termini of antibodies.151 Compared to classical chemical conjugation to lysine or cysteine side chains, SML forms conjugates in site-specific manner and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The newly formed antibody–drug conjugates showed similar in vitro and in vivo tumor killing activities like traditional conjugates used in clinic. In a following study, the authors could show that ADCs based on anthracycline toxin and anti-HER2 antibody exhibited potencies even exceeding those of marketed products.152
:1 ratio. The newly formed antibody–drug conjugates showed similar in vitro and in vivo tumor killing activities like traditional conjugates used in clinic. In a following study, the authors could show that ADCs based on anthracycline toxin and anti-HER2 antibody exhibited potencies even exceeding those of marketed products.152
Fang et al. used a similar approach to form antibody–dye and antibody–drug conjugates.153 Rapid tumor targeting could be shown using NIR imaging of the fluorescent dye and therapeutic potency demonstrated with the drug conjugate. Van Lith et al. showed the formation of antibody-nanoparticle (NP) structures based on SML and click chemistry.154 Antibody-LPETG proteins were ligated with click handles for SPAAC. The following click reaction formed bispecific C-to-N as well as C-to-C antibody constructs. Click reaction with functionalized PEG2000 enabled to form PEG-based micellar nanoparticles with exposed antibodies. van der Steen et al. developed a new concept for targeting the cancer extracellular matrix with specific antibody-functionalized lyophilisomes.155 Lyophilisomes are albumin-based biocapsules that were loaded with the toxin doxorubicin. The antibody was linked to this drug delivery vehicle in a two-step process consisting of first introducing a clickable group by SML and subsequently linking the protein to functionalized lyophilisomes by SPAAC.
The group of Chen developed approaches for the synthesis of ADCs comparable to strategies explained above. First, ADCs were prepared by SML between antibody-LPETG and GGG-toxin.156 However, a 100-fold excess of toxin is required because of the reversibility of the sortase reaction, but still steric hindrance reduces the efficiency for certain combinations of antibody chains and toxin. Consequently, the group developed a two-step procedure to first ligate small click handles to antibodies followed by SPAAC to link the toxic payload (Scheme 11).157 For the latter, only a two-fold excess of clickable toxin was needed, hence reducing toxic waste.
 | ||
| Scheme 11 Two strategies for generating antibody–drug conjugates: SML and SPAAC (A), only SML (B). Reprinted without change from ref. 157, CC BY 4.0, https://www.mdpi.com/1422-0067/18/11/2284. | ||
4.4 Modification of multiphages
The construction of complex virus-type structures with the help of SML was demonstrated by Hess et al.158,159 In order to attach different dye molecules to capsid proteins of M13 bacteriophage, the sortase variants SrtApyogenes and SrtAaureus were used subsequently like already described in the beginning of this chapter. At first, SrtApyogenes ligated a dye to the body of the phage, followed by linkage of dyes to both ends of the phage by SrtAaureus.158 After labeling, SML was utilized to link LPETGG- or GGGK-functionalized DNA oligonucleotides to the ends of the phage structure. DNA hybridization led to multiphage particles in which phage structures are linked by DNA connections. The labeling with different dyes enabled visualization of the multiphage structures.Compared to small molecules, the ligation of proteins to the virus-like particles (VLPs) has been performed more frequently and should be also shortly introduced in this context. While the group of Chen used recombinant oligoglycine equipped E2 nanocages to ligate different LPETG functionalized proteins,160–162 Tang et al. used the opposed strategy with LPETGG functionalized VLPs to ligate GGG-equipped proteins.163 The latter pointed out that SML is more efficient than chemical coupling and their strategy of ligating antigens to VLPs shows potential as a new approach in the design of vaccines.163 However, it should be noted that the ligation efficiency is limited for SML because of the reversibility of the reaction and other approaches are more promissing to yield nearly quantitative decoration on virus nanocages.164 Further studies in the group of van Hest focused on the encapsulation of cargo and later on the enzyme CalB in virus capsids.165,166 While the activity of the enzyme was unaffected, the enzyme was protected against proteases.166 Patterson et al. demonstrated the ligation of proteins to the exterior of bacteriophage P22 VLPs.167 GG-GFP and GG-HAhead (head domain from the influenza hemagglutinin protein) were ligated to P22 VLPs with coat proteins containing the C-terminal LPETG sequence. Thérien et al. assembled papaya mosaic virus (PapMV) nanoparticles bearing LPETG motifs and coupled GGG-peptide antigens to the NPs.168 The functionalization after NP assembly is preferred as it has no influence on the self-assembly. The approach is promissing as vaccine platform which was demonstrated with mice.
4.5 Non-proteinic amines as nucleophiles
In addition to oligoglycines, it was shown that different non-proteinic amines can function as nucleophiles in SML. Baer et al. studied the sortase-mediated ligation between different proteins with LPETG recognition sequence and dye molecules functionalized with various nucleophilic groups.169 In addition to triglycine and glycine, the ligation was also successful with other amine groups. While substituted ethylenediamine was quite potent, the efficiency for the lysine side chain and functionalized cadaverine was lower. However, the efficiency of the ligation was depending both on the nucleophile and also on the nature of the protein containing the LPETG recognition sequence. In a recent study, diverse commercially available amines were tested for their ligation to model proteins with LPETGG recognition sequence mediated by the engineered sortase variant SrtA7M, which has seven mutations leading to higher activity and Ca2+ independency (Scheme 12a).170 All successful examples – 3-azido-1-propanamine, propargylamine, tetrazine amine, ethylenediamine, aminoethylbenzenesulfonamide, and histamine – were unbranched at the α-carbon, while amines branched at the α-carbon did not show product formation. The latter suggests that SrtA7M prefers unbranched primary amine nucleophiles. Furthermore, the protein modification with diverse amines was not only possible in vitro, but also in living E. coli cell culture.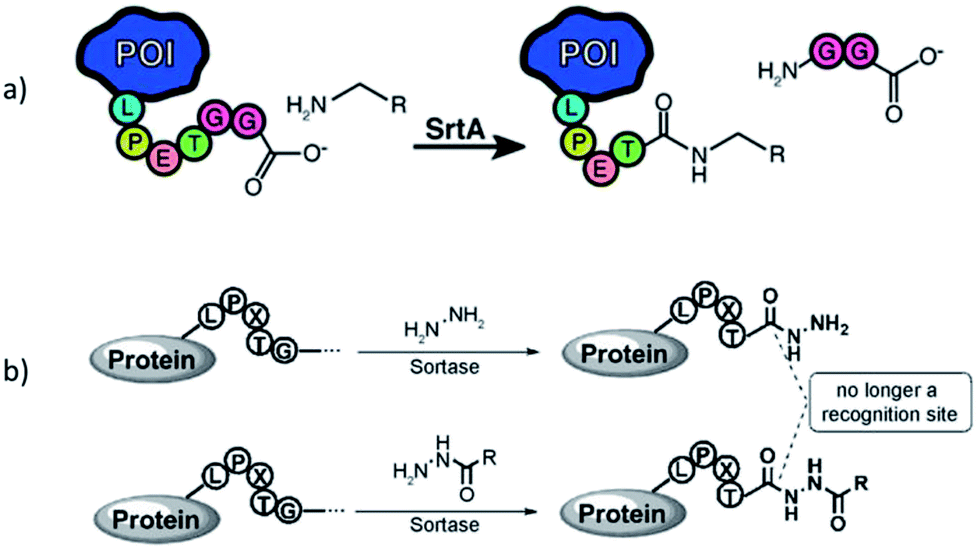 | ||
| Scheme 12 (a) Sortagging with amine nucleophiles being unbranched at the α-carbon atom. Reprinted with permission from ref. 170. Copyright (2016) American Chemical Society. (b) Hydrazinolysis of proteins. Reprinted with permission from ref. 171. | ||
The group of Li ligated hydrazine or derivatives to protein-LPETG C-termini by SML in order to further functionalize the resulting protein hydrazides (Scheme 12b).171,172 Moreover, protein hydrazinolysis is irreversible as the protein hydrazide is no longer a substrate for the enzyme.
Further examples for the sortagging with amine nucleophiles other than oligoglycines represent the use of aminohexoses (see Chapter 3),115 amine-functionalized particles (see Chapter 6.2),107,173,174 H2N-PEG polymer blocks (see Chapter 5.2)107,175,176 and lysine residues in the pilin domain to form isopeptide bonds (see Conclusion).177
5. Formation of protein–polymer conjugates aided by SML
Two different cases have to be distinguished concerning the formation of protein–polymer conjugates using sortase-mediated ligation. First, initiators for atom-transfer radical polymerization (ATRP) were ligated to proteins by SML and the polymerization carried out afterwards. Hence, sortase is only involved in the first step of the conjugate formation and this has to be classified as the ligation of a protein with a small chemical molecule. Second, amine- or peptide-functionalized polymers were also directly ligated to proteins by sortase catalysis.In general, two approaches are commonly used to synthesize protein–polymer conjugates, denoted as grafting-to and grafting-from.178–180 Grafting-to is used since decades to link pre-formed polymers with end-functional groups to proteins. The most prevalent example is PEGylation, the linkage of PEG.181 The attachment of polymer chains usually stabilizes proteins in non-natural environment and improves both solubility and resistance against unfolding.179,182,183 The modification of therapeutic proteins is the main application field of protein–polymer conjugates and they were extensively studied for drug delivery.184 Grafting-to enables the individual synthesis and characterization of polymers without being restricted to reaction conditions that are compatible with proteins. However, the binding of two macromolecules is thermodynamically unfavorable and accompanied by low grafting yields. Furthermore, the purification of the conjugates from unreacted protein and polymer is challenging. In 2005, Maynard and coworkers introduced the grafting-from approach.185,186 Initiators or mediators of a polymerization are linked to a protein and the polymers directly synthesized from the protein. Main advantages are the higher grafting yield and easy purification of the conjugates as only small molecules need to be removed (monomer, eventually catalyst). Controlled radical polymerization (CRP) techniques, mainly ATRP and reversible addition-fragmentation chain transfer (RAFT) polymerization, are the methods of choice for grafting-from. Reaction conditions for ATRP and related techniques were shown to be adapted to biologically relevant conditions.187–191
Although around half of the amino acids can principally be targeted, lysine and cysteine side chains are mainly exploited for protein modification because of their unique reactivity with readily available functional groups.192,193 However, targeting lysines and cysteines often means a loss of control of stoichiometry and position of modification as there are usually more than one of these amino acids available and solvent exposed in a protein.193,194 Alternatively, unnatural amino acids and other tags were on occasion used for a site-specific incorporation of an initiator group for polymerization.184,195 The modification of proteins with polymerization initiators or polymers by sortase-mediated ligation has the advantage over conventional conjugation methods that the functionality is site-specifically and stoichiometrically (1:1) introduced to the protein. Furthermore, modifications at the protein termini have usually negligible influence on activity and folding of a protein.
5.1 Ligation of initiators for a polymerization
Qi et al. ligated an initiator for ATRP to the C-terminus of GFP by SrtA catalysis and conducted the grafting-from polymerization subsequently (Scheme 13a).196 Attachment of the initiator group was performed with 30-fold excess of the GGG-functionalized small molecule and a high yield reached (ca. 95% from quantification of SDS-PAGE band intensities).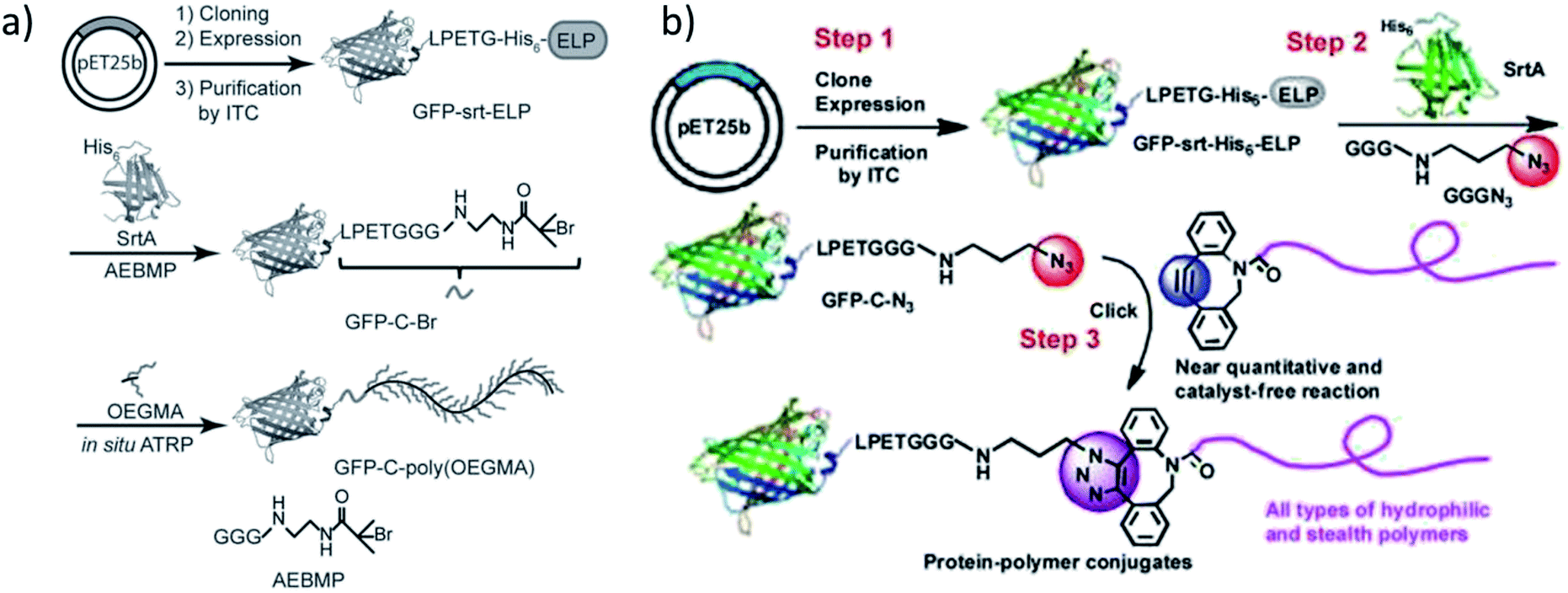 | ||
| Scheme 13 (a) The synthetic route to obtain a GFP-poly(OEGMA) conjugate via SML and grafting-from polymerization; (b) formation of GFP-polymer conjugates by combination of SML and SPAAC. Reprinted with permission from ref. 196 and 197. | ||
For the purification of the product, it was exploited that SrtA and unmodified protein possess a His6-tag which is cleaved off from the protein of interest during SML. Therefore, the reaction product is the only proteinic species without affinity tag – a strategy often exploited for product purification after SML. ATRP of OEGMA was performed in aqueous buffer conditions with a copper-based catalyst system.
In an alternative approach of their strategy, the group of Chilkoti published the SML of azide groups to proteins followed by the attachment of alkyne-functionalized polymers through SPAAC reaction (Scheme 13b).197 Instead of ligating a polymerization initiator to the protein and grafting the polymer directly from the protein, the polymer was individually formed, characterized and subsequently linked to the protein. Diverse polymers were synthesized, end-functionalized with suitable cyclooctynes and finally conjugated to GFP-N3 by SPAAC click reaction. This process was described as a modular approach for the synthesis of defined protein–polymer conjugates and all three steps could be carried out in high yield without (non-enzymatic) catalyst.
Hu et al. used SML to link an ATRP initiator to a therapeutically relevant protein and compared the performance of the subsequently formed protein–polymer conjugate with the clinically approved PEGylated protein.198 The target protein is human interferon alpha (IFN-α), therapeutically used in unmodified and PEGylated form. In order to improve the conjugate formation, SrtA was used to link an ATRP initiator to IFN-LPETGGH6 and subsequently polymerize OEGMA from the C-terminus of IFN-Br. After purification, an overall yield of 66% was reached for IFN-POEGMA. For comparison, the authors also performed post-polymerization linkage of GGG-POEGMA to IFN-LPETGH6 by SML. However, only 1.1% yield was reached when using a 20-fold excess of the polymer. First studies indicated that IFN-POEGMA, possessing a PEG-like polymer, showed a higher efficiency in tumor therapy than unmodified IFN-α and clinically approved forms that are PEGylated in the traditional way. In a further development of their work, the authors grew a block copolymer from the protein macroinitiator which was formed under help of sortase-mediated ligation.199 The second block poly(2-hydroxypropyl methacrylate) (PHPMA) is water insoluble so that the IFN-POEGMA-PHPMA conjugates in situ formed micelles. These conjugate micelles showed a better pharmacological performance than the PEGylated protein therapeutics.
5.2 Ligation of polymer blocks
Although Hu et al. could not reach a satisfactory yield for conjugating a polymer directly to a protein198 – the reason could be the high molecular weight of the polymer (around 66 kDa) – some examples for this strategy exist. Such an approach refers more to the grafting-to strategy. It has the clear advantage over the classical grafting-to technique that the polymer linkage takes place site-specifically and sortase acts as a kind of catalyst for the reaction. However, shift of the equilibrium of the sortase reaction and purification of the reaction product are most probably even more important for the linkage of two macromolecules.Parthasarathy et al. PEGylated eGFP at the C-terminus utilizing SML.107 Commercially available PEG of 10 kDa was used and applied with amine as well as GGG end groups. The formation of eGFP-PEG conjugates could be observed with both polymers and no significant difference noticed for H2N-PEG and GGG-PEG. However, SDS-PAGE indicated low efficiency for the conjugate formation in both cases. This is not surprising when taking into account the low 1.5-fold excess of polymer compared to protein during one of the first reports for SML with one artificial substrate which was published in 2007. Popp et al. used a similar approach and PEGylated proteins at the C-terminus with 10 and 20 kDa PEG.200 The polymers were functionalized with GGGK peptides and ligated to IFN-LPETGGH6 among others using 40-fold excess. Quantitative conversion was not reached, however, after purification by ion exchange chromatography, pure conjugates could be shown by SDS-PAGE.
Qu et al. demonstrated the C-terminal PEGylation of thrombomodulin.175 Therefore, commercially available 5 kDa PEG with amine end group was used. While wildtype SrtA showed unsatisfactorily performance, a SrtA pentamutant (eSrtA) with greatly enhanced activity significantly improved the PEGylation efficiency. Nearly 80% yield could be reached for the reaction between TM-LPETG and H2N-PEG after 2 h when using a 100-fold excess of the nucleophile. It is worth noting that 10-fold excess of H2N-PEG led only to minimal product formation. In a subsequent work, the groups of Liu and Chaikof developed new sortase variants having different peptide recognition sequences (LPXSG, LAXTG).28 Orthogonality of these variants enables the introduction of two substrates into one protein, for instance, fluorophore-protein-PEG conjugates were formed. Hence, some of the shown ligations involve protein PEGylation with 10 kDa GGG-PEG which was synthesized from commercially acquired H2N-PEG. PEGylated conjugates could be isolated after purification with Ni-NTA filtration and concentration using 10 kDa molecular weight cut off (MWCO) membranes in yields of up to 20%.
Hou et al. synthesized different kinds of protein–poly(amino acid) conjugates with unnatural amino acids.201 Among their conjugates are further examples for PEGylation of eGFP and IFNα by SML. Glycine-functionalized commercially available PEG of 2 kDa was reacted with protein-LPETGGLEH6 and about 90% conversion reached (Scheme 14). After purification by Ni-NTA affinity chromatography, conjugates with a yield in the range of approximately 45–55% could be isolated. Optimized conditions for the formation of eGFP-PEG were 30 min reaction time, 0.1 equiv. SrtA and, remarkably, only 5 equiv. G5-PEG. In their subsequent work, it turned out that the cyclization of such an protein–poly(amino acid) conjugate yields particularly interesting protein-based therapeutics.202 A G3-poly(amino acid)-SPh was conjugated to a cystein at the N-terminus of Cys-IFN-LPETG and this conjugate cyclized by SML. The macrocycle combines the advantages of polymer conjugation and cyclization of therapeutic proteins which was evident by greater stability, longer circulation half-life and higher tumor retention compared to the linear counterparts.
 | ||
| Scheme 14 (a) Synthesis of eGFP-PEG conjugate via SML; (b) SDS-PAGE of the protein starting material and the product. Reprinted with permission from ref. 201. Copyright (2016) American Chemical Society. | ||
Another example for protein–polymer conjugates formed by SML was reported by Hui et al.176 The therapeutically used protein human growth hormone (hGH) was PEGylated at its C-terminus with commercially obtained H2N-PEG of 10 kDa by a mutated SrtA enzyme. The conjugates showed clearly increased serum half-life compared to wildtype hGH. However, at least a 5-fold excess of SrtA to the target protein was required for a high ligation efficiency.
The presented studies so far are limited to commercially available synthetic polymers with a suitable end group and only include PEG and derivatives. The ligation of a polymer synthesized by oneself would broaden the range of polymers and enable that any desired polymer could be synthesized by a CRP method and then ligated to a protein in one step.
6. Immobilization of proteins on surfaces by SML
An interesting application of SML is seen in the immobilization of proteins on surfaces. It was shown with many examples that such surfaces can be planar as well as curved particle surfaces. Furthermore, the proteins' immobilization in micro and hydrogels could be achieved as well. All approaches will be presented one after the other in the following paragraphs.The immobilization of proteins on surfaces is always accompanied by the search for a suitable linking chemistry so that the structure and activity of the protein is preserved to a high degree. Nonspecific adsorption or nonspecific covalent binding usually lead to random orientations and reduced activity. Thus, the focus of recent years moved to techniques for site-specific attachment that favor an oriented immobilization of the biomolecule and a homogeneously covered surface.203,204 Sortase-mediated ligation is such a technique which provides the possibility to immobilize recombinant proteins via their termini on surfaces. In contrast to some chemical methods, such an enzymatic approach goes along with reaction conditions that do not disturb protein functionality. Furthermore, design of the peptide sequences allows the incorporation of a short spacer. Hence, SML was studied for immobilization of proteins on various peptide- or amine-functionalized surfaces. The ligation via C-terminus is much more prevalent among the examples for the immobilization of proteins on solid surfaces.
6.1 Immobilization on planar surfaces
The functionalization of biacore sensor chips with proteins was demonstrated by Clow et al.205 Therefore, a peptide with N-terminal GGG was attached to the dextran surface of the chip via a cysteine at the C-terminus. While the SML with LPETG-tagged protein was not successful during flow over the chip surface, incubation overnight at 37 °C yielded in immobilized proteins. Recombinant fibronectin-binding protein (Fba) from the streptococcal cell wall was exemplarily used and ligand–analyte interaction demonstrated with human factor H (FH).Jiang et al. immobilized thrombomodulin on glass surfaces and published two approaches (Scheme 15).206 On the one side, TM-LPETGH6 was directly immobilized on glycine-functionalized glass surfaces via SML. On the other side, the protein was first ligated with biotin via sortase catalysis and the immobilization conducted on streptavidin-functionalized glass slides. Enhanced bioactivity could be observed through the site-specific immobilization of thrombomodulin. The activity measurements indicated that the immobilization via biotin–streptavidin interaction was slightly more efficient. As TM is a membrane protein involved in natural anticoagulant system, the demonstrated approach was postulated as promising for antithrombogenic surfaces for cardiovascular biomaterials to minimize incompatibility and thrombosis.
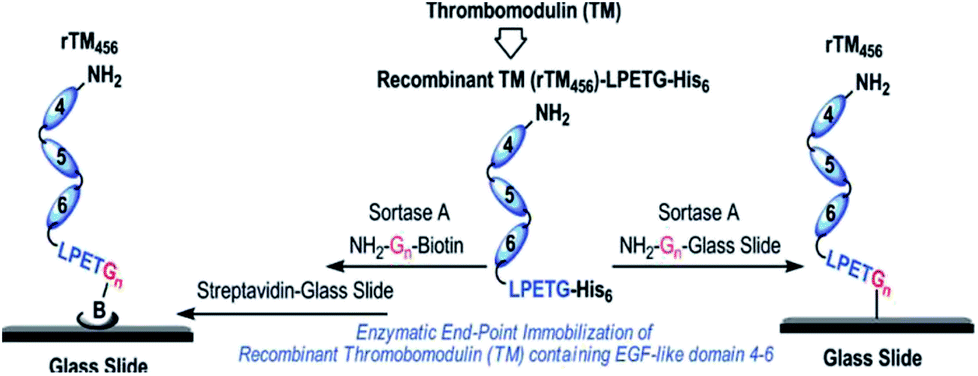 | ||
| Scheme 15 Immobilization of recombinant human thrombomodulin on glass slides via SML. Reprinted with permission from ref. 206. Copyright (2012) American Chemical Society. | ||
Sinisi et al. ligated LPETG-tagged proteins to GGG-functionalized glass surfaces.207 The authors showed with several influenza virus proteins that the ligation is possible directly from a cellular extract without purification of the extracted protein. Antigens from influenza A were used as proof of concept, but such a protein array was postulated to be used for early detection of seasonal flu on the one side and in general for high-throughput screening on the other side.
Dorr et al. demonstrated the simultaneous dual surface modification using two orthogonal SrtA variants.28 GGG-PEG-functionalized 96-well plates were incubated with two newly evolved sortase variants with preference for LPESG and LAETG recognition sequences and two fluorophores equipped with the respective peptide sequences. Hence, two distinct compounds could be simultaneously ligated to a surface.
The immobilization of proteins on crystalline nanocellulose (CNC) was studied by Uth et al.208 CNC has a large surface area, is easily prepared from renewable source cellulose and the introduction of orthogonally addressable aldehyde groups is possible. The authors could show that SML leads to a higher activity of immobilized proteins compared to traditional coupling between protein amino groups and surface carboxylic groups. Peptide sequences bearing a pentaglycine motif for sortase catalysis were introduced by linkage to the mentioned aldehyde groups. Three different recombinant proteins – GFP, the antibody-like domain Lys-vNAR and the enzyme galactose oxidase (GOas) – were expressed with LPETG recognition sequence and ligated to CNC using an evolved SrtA variant.
The attachment of photosystem I (PSI) complex on a conductive gold surface is interesting for biophotovoltaics. In order to overcome limitations from non-uniform and undesired orientations, Le et al. ligated PSI-LPETGH6 to a GGGC-functionalized Au surface (thiol–gold bond utilizing cysteine).209 Through immobilization by SML, 94% PSI could be oriented in the desired manner which is mainly important for the photoinduced electron transfer to the gold surface. Thus, an enhanced electron transfer could be reached compared to other reported immobilization techniques.
The group of Boder continued their work by studying the immobilization of protein layers on gold surfaces using orthogonal SrtA variants.210 SrtAaureus ligating LPETG/GGG and SrtApyogenes ligating LPETA/AAA (and less efficiently also LPETG/GGG) were already introduced in Chapter 4.1. Two fluorescent model proteins, eGFP-LPETG and mCherry-LPETA, were at first independently ligated to gold-coated microscope slides with the mentioned thiol–gold linkage to bind GGGC or AAAC peptides, respectively (Scheme 16a). Next, protein oligomers were formed on gold by using either AAA-mCherry-LPETA or GGG-GFP-LPETG. Atomic force microscopy (AFM) analysis of the surfaces suggested that at least dimers should have been formed. Finally, the controlled formation of two protein layers was performed. Therefore, GGG-mCherry-LPETA was first ligated to AAA-tagged gold by SrtApyogenes. The formed LPETAAA peptide is not a substrate for SrtAaureus, hence the first protein layer will not be cleaved by SrtAaureus. Consequently, eGFP-LPETG could be attached as a second layer catalyzed by SrtAaureus (Scheme 16b). The disadvantage of the used orthogonal sortase variants is the possibility of partially forming immobilized protein oligomers of mCherry in the first step as GGG is a weak nucleophile for SrtApyogenes.
 | ||
| Scheme 16 (a) Immobilization of LPETG-equipped-GFP onto GGG-functionalized gold surface via SML. (b) Strategy of immobilization two protein layers onto a gold surface by using the two different variants SrtApyogenes (Sp-Srt) and SrtAaureus (Sa-Srt). Reprinted from ref. 210, Copyright (2015), with permission from Elsevier. | ||
As an alternative strategy that avoids the use of two different sortase variants, the same group published the protein layer-by-layer immobilization using a protection group for the GGG nucleophilic sequence.211 The peptide sequence DDDDKGGG can be cleaved by enterokinase (EK) after lysine to set the oligoglycine free. At first, eGFP oligomers were formed in solution and up to pentamers reached. On GGG-functionalized polystyrene bead and gold surfaces, protein layers were formed by first ligating DDDDKGGG-eGFP-LPETG, second cleavage of the protecting sequence and third ligation of mCherry-LPETG using the same sortase A variant. However, dimer formation in solution even when the protection group should be present and unequal ratio of the proteins on gold surface suggest an improvement of biotechnological techniques before the strategy can be broadly established.
Single-molecule force spectroscopy (SMFS) is an AFM-related technique that can give detailed insight into biomolecule properties (like unfolding events under tension). The oriented attachment of proteins and polyproteins on surfaces is especially important for such studies. Srinivasan et al. made use of sortagging for the purification and presentation of recombinant proteins on surfaces for single-molecule studies.212 After functionalization with a tetraglycine unit, LPETG-tagged proteins could be immobilized by SML both from purified samples as well as whole cell lysate. Durner et al. developed a method to bind a protein of interest and a receptor for the cantilever tip to the surface by using two orthogonal enzymes, phosphopantetheinyl transferase (Sfp) and sortase A.213 This approach allows the individual expression of the proteins without the need of a fusion protein with eventually low expression yield and risk of improper folding. The groups of Rakshit and Nash independently published strategies for the formation of polyproteins on surfaces.214,215 Liu et al. used a protein with GGG N-terminal and LPETGG C-terminal sequence for the uncontrolled formation of polyproteins on functionalized coverglass surfaces.215 After unmasking of the oligoglycine N-terminus with TEV protease showed very low efficiency in their lab, the authors used immunoglobulin 27 (I27) domains bearing both recognition and nucleophilic sequence at the same time. However, this means that the number of attached protein units is not controllable in this approach and a mixture of polyproteins of different length results. A quantitative analysis revealed that around half of the analyzable proteins consist of only one ligated protein by SML and observed tetramers occurred in less than 1%. Garg and Singaraju et al. used a controlled approach with an alternating use of two enzymes to form defined polyproteins on surfaces.214 The authors used a I272 dimer with LPETGSS Srt recognition sequence and N-terminal TEV protease cleavage site. To optimize SML, the sortase thioester intermediate was formed with the protein substrate at first, then Ca2+ ions removed to reduce SrtA activity and finally the intermediate exposed to the surface and released by oligoglycine nucleophiles on the surface. Hence, the risk that active SrtA can cleave LPETGGGG bonds of linked proteins was reduced. After ligation by SrtA, TEV protease cleaved the sequence at the N-terminus to set free an oligoglycine motif. The formation of polyproteins with up to four-times sequential use of SrtA and TEV protease was demonstrated.
Ott and Durner et al. showed that the enzymatic coupling of proteins to a surface is also advantageous for studying the binding kinetics of receptor–ligand pairs.216 They used three different enzymes, among them SrtA, that allow the covalent and site-specific coupling of proteins to the sensor surface which is a progress compared to the traditionally used immobilization of proteins for binding assays via lysine residues.
In order to form bioactive papers, Hilberg et al. recently ligated proteins onto paper fibers.217 The filter papers were first functionalized by EDC coupling followed by a light-activated Diels–Alder reaction to achieve GG-containing motifs. Three different proteins with C-terminal LPETG sequence were ligated by sortase catalysis under preservation of their functions.
Ham et al. showed that the reversibility of the sortase reaction can be used to repeatedly regenerate immobilized protein films on surfaces.218 GGGGGK-biotin was assembled on streptavidin-functionalized surfaces and thrombomodulin-LPETG ligated. Adding GGG excess together with sortase removed the protein film nearly completely (Scheme 17). The charge/strip cycles could be repeated 10-times in vitro. Evolved sortase A (eSrtA) was significantly more efficient for both immobilization and removal compared to SrtA WT. Moreover, as LPETG and GGG motifs exist rarely in nature, the charge/strip cycles could be likewise performed in 50% v/v whole blood at 37 °C. As the described regeneration of bioactive coatings is interesting for blood-contacting surfaces and TM is known to reduce thrombosis, the process was further conducted on implanted polyurethane catheter in living mice with intravenous administration of eSrtA and either biotin-LPETG or GGG.
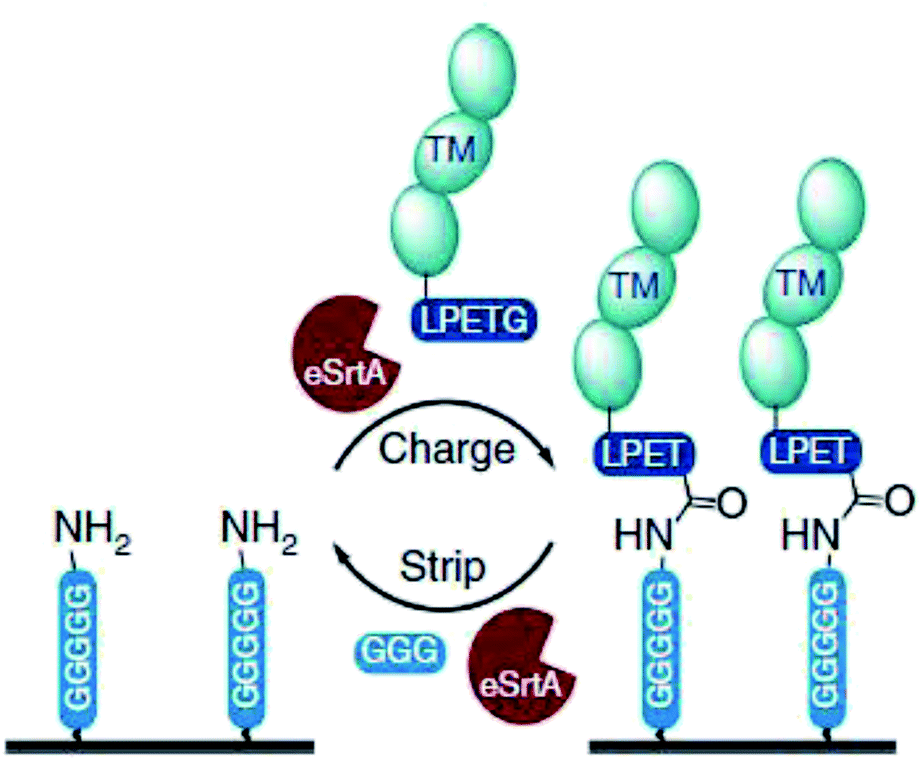 | ||
| Scheme 17 Reaction cycle of immobilizing protein-LPETG on a pentaglycine-equipped surface and its removal by adding eSrtA together with an excess GGG. Reprinted without change from ref. 218, CC BY 4.0, https://www.nature.com/articles/ncomms11140. | ||
6.2 Immobilization on particle surfaces
The ligation of proteins to either planar or curved surfaces from particles showed no obvious difference. Hence, particle surfaces were targeted through SML in parallel and the most important examples will be introduced in the following paragraphs.In addition to eGFP-PEG conjugates (see Chapter 5.2), Parthasarathy et al. also reported the ligation of eGFP-LPETG to polystyrene beads.107 The beads with a diameter of 3 μm were used with either amine groups on the surface or functionalized with GGG peptides. Fluorescence intensity after SML indicated that the efficiency of immobilizing eGFP is higher for GGG-functionalized than NH2-terminated beads.
Chan et al. published one of the first universal studies for the sortase-mediated immobilization of proteins on surfaces and demonstrated the ligation on crosslinked glycidyl methacrylate polymer beads, agarose affinity resins and planar glass surfaces.219 All support materials were functionalized with oligoglycine motifs followed by the immobilization of different fluorescent proteins and the DNA binding protein Tus of which the functionality on polymer beads could be verified.
Ito et al. used SML to find a suitable immobilization technique for the class of extremely unstable glycosyltransferases.220 Two recombinant enzymes, rhGalT and rhFucT, were ligated via their C-termini on alkylamine-functionalized sepharose. This site-specific covalent attachment enabled an immobilization without loss of activity and stability. Furthermore, reuse of the immobilized enzymes with sugar transfer activity was demonstrated.
Adhesive proteins were ligated to microbeads in the group of Proft.221 Adhesins are proteins on the cell wall of bacteria that are responsible for the adhesive properties of bacteria to host cell tissue. While many proteins are possible adhesins, their interaction with cells can be studied after immobilization on microbeads. The in vitro immobilization by sortase has the advantage that the orientation of the proteins on the beads is identical as in nature where sortase enzymes are responsible for the ligation to the bacterial cell wall. Cell binding studies showed that the microspheres with adhesins bound specifically stronger to cells than the negative control samples.
Matsumoto et al. identified a sortase variant from Lactobacillus plantarum with the recognition sequence LPQTSEQ.173 eGFP-LPQTSEQ was ligated to primary amine-modified microbeads by both S. aureus SrtA and SrtLp. Fluorescence microscopy showed that the immobilization was more efficient using SrtLp.
The formation of antibody–drug conjugates by SML was presented in Chapter 4.3. In addition, the ligation of antibodies to particles enables the targeted delivery of imaging agents, drug carrier capsules and therapeutic proteins exposed on micelles. Ta et al. demonstrated the ligation of scFv antibodies to a model protein, particles and cells.222 The single chain antibodies (scFv) expressed with LPETG recognition sequence were ligated to eGFP (Scheme 18a) and iron oxide beads (Scheme 18b), both functionalized with triglycine motifs. The magnetic particles can be used as contrast agents for magnetic resonance imaging (MRI) and the site-specific immobilization of antibodies is crucial for targeted delivery. In case of cells, the antibodies were ligated directly to amino groups on the surface; however, the efficiency was significantly higher for GGG-functionalized cells (Scheme 18c). Bioactivity of antibodies and successful targeting could be demonstrated. The groups of Caruso and Peter expanded their work with the site-specific attachment of antibodies on polymer carrier vehicles that are promising for targeting applications in the medical field.223,224 Low fouling capsules were prepared by layer-by-layer assembly of polymers around silica particles followed by subsequent removal of silica by hydrofluoric acid (HF). Peptides with N-terminal triglycine were linked to the polymer capsules by CuAAC and finally scFv-LPETG antibodies ligated by sortagging. Site-specific immobilization via their C-terminus leaves the antigen-binding sites available for targeting. Furthermore, protein micelles bearing both targeting and therapeutic proteins were formed (Scheme 18d).225 The micelles composed of elastin-like polypeptide (ELP) with a diameter of ca. 50 nm were generated with multiple triglycine units. scFv antibodies for targeting and the catalytically active domain of thrombomodulin for inhibition of thrombus formation were ligated using evolved SrtA. The multifunctional protein micelles could be formed in a one-pot transpeptidation and targeted delivery as well as formation of an inhibitor of the coagulation cascade shown.
 | ||
| Scheme 18 Ligation process of scFv-LPETG antibodies to eGFP (a), magnetic iron oxide particles (b), cells (c) and protein micelles (d). (a–c) Reprinted from ref. 222, https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.111.249375. (d) Reprinted with permission from ref. 225. | ||
The group of Richter used the exemplarily chosen immobilization of eGFP-LPETGGH6 on GGG-polystyrene microbeads for a continuous monitoring of reactions using different sortase variants.226 The real-time flow cytometry assay followed the covalent attachment of the proteins on the surface by an increase of the microbead fluorescence. Interestingly, H6-SrtAΔ59, H6-SrtAΔ25, SrtAΔ59-H6 as well as triple-, tetra- and penta-mutated SrtAΔ59-H6 showed distinct differences in their performance for the investigated reaction.
Hata et al. exploited sortagging for the oriented immobilization of enzymes on particles.227 β-glucosidase (BGL) and α-amylase (AmyA) were ligated to GGG-tagged polystyrene particles of 500 nm in diameter. In parallel, the enzymes were also linked to COOH-functionalized particles through chemical crosslinking using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS). While the same amount of enzymes was attached through both methods, the oriented immobilization by SML provided a higher activity of the enzymes along with better reusability performance.
Qafari et al. published the C-terminal immobilization of protein A on silica and graphene oxide nanoparticles.228 While the amount of immobilized protein was higher for G5-SiO2 NPs than for H2N-SiO2 NPs, the situation was opposite for graphene oxide NPs suggesting that the nature of the nanoparticles influences the immobilization efficiency in addition to the type of amine nucleophile.
6.3 Immobilization in gels and formation of gels
In recent years, it was also demonstrated that sortase is successful in modifying hydrogels and microgels. Moreover, even the formation of a hydrogel network itself could be catalyzed by sortase.The functionalization of PEG-based hydrogels with epidermal growth factor (EGF) was performed by Cambria and Renggli et al.229 The sortase recognition sequence LPRTG was linked with an N-terminal cysteine to acrylate groups in the hydrogel. The amount of ligated GGG-EGF was dependent on the amount of incorporated LPRTG motifs and the concentration of the model protein. Although EGF appeared to be enriched at the surface of the hydrogel, diffusion of the relatively small sortase enzyme into the hydrogel could be concluded. Reversibility of the sortase reaction was exploited to cleave the protein from the hydrogel by adding the used evolved sortase A variant again together with excess GGG nucleophile. Bioactivity of the immobilized proteins could be demonstrated and the shown protein cleavage opens up possibilities for a controlled release. The same group showed that PEG-based hydrogels can be dissolved by SrtA.230 Therefore, the PEG macromers were crosslinked by peptides bearing the sortase recognition sequence. Sequential addition of SrtA and an excess GGG led to dissolution of the gel. Cells incorporated in the hydrogels could be released in nearly all examples without being affected by SrtA dissolution. Similarly, proteins produced by the cells could be recovered to analyze local cell–cell communication networks.
Gau and Mate et al. functionalized stimuli-responsive microgels with the model protein eGFP (Scheme 19).231 Poly(N-vinylcaprolactam) (PVCL) microgels are temperature-responsive and, due to their biocompatibility, interesting candidates for biomedical applications. Microgels were synthesized containing glycidyl methacrylate (GMA) in order to bind peptides with the recognition sequence LPETG via an N-terminal cysteine to the epoxy groups. The reaction kinetics of sortase-mediated ligation of GGG-eGFP could be determined through the fluorescence intensity of ligated protein at different reaction times. After 7 h, no more protein seems to be ligated. The amount of immobilized protein could be further controlled in a linear fashion by the concentration of GGG-eGFP in solution.
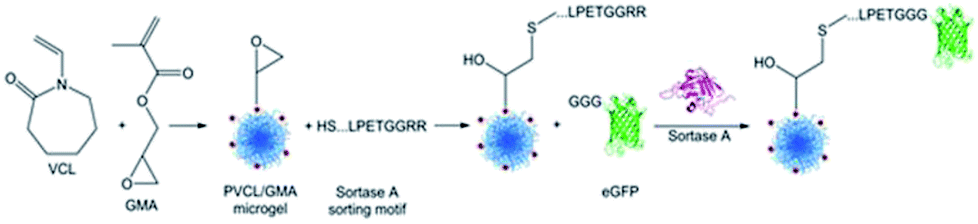 | ||
| Scheme 19 Synthesis of a PNVCL microgel followed by a sortase-mediated immobilization of eGFP. Reprinted with permission from ref. 231. Copyright (2017) American Chemical Society. | ||
Arkenberg et al. demonstrated that sortase is also capable to form a hydrogel network by crosslinking peptide-functionalized precursors.232 Therefore, 8-arm PEG-OH (8 OH-groups in the polymer) was first reacted with 5-norbornene-2-carboxylic acid, followed by reaction of the introduced double bonds with cysteine residues of the used peptides. Crosslinking of LPRTG- or GGGG-functionalized PEGs was performed by addition of SrtA7M for 10 min. The speed of the gelation scaled with the enzyme concentration. An additional mushroom tyrosinase (MT)-triggered secondary crosslinking through dityrosine formation between incorporated tyrosine residues could be performed in order to tune the hydrogel properties. Finally, in situ cell encapsulation was demonstrated under mild conditions. In a subsequent work, it was demonstrated that the reversibility of the sortase reaction can be utilized to soften the hydrogel which can even lead to complete degradation of the gel.233 Furthermore, incorporation of linear peptides with both oligoglycine and LPXTG recognition sequences enabled a reversible softening and stiffening. Applying SrtA led to secondary crosslinks and hence a stiffened gel, while addition of SrtA together with an excess of a glycine substrate softened the hydrogels.
Broguiere et al. studied the crosslinking of peptide-functionalized hyaluronan (HA) to hydrogels using a SrtA pentamutant (Scheme 20) and compared the performance with transglutaminase activated factor XIII (FXIIIa).234 LPETGG and GGGG peptide motifs were linked to vinylsulfone-functionalized hyaluronan via cysteine residues. The authors concluded that SrtA is an excellent crosslinking enzyme for tissue engineering applications because of its unmatched kinetics for hydrogel formation, good enzyme stability, easy access to the enzyme and its cytocompatibility. In a subsequent work, peptide-functionalized poly(2-methyl-2-oxazoline) (PMOXA) and poly(2-ethyl-2-oxazoline) (PEOXA), respectively, were crosslinked by sortase in presence of cells.235 Such biocompatible cell-loaded hydrogels are promissing for tissue engineering.
 | ||
| Scheme 20 Crosslinking of peptide-equiped biopolymers to hydrogels by sortase A. The catalytic cycle is shown over the arrow. Reprinted from ref. 234, Copyright (2018), with permission from Elsevier. | ||
7. Ligation of artificial compounds
In addition to the crosslinking of synthetic polymers to gels, very few other studies demonstrated the linkage of two artificial building blocks by SML. Sijbrandij et al. linked PEG-LPETG (20000 Da) to a functionalized polystyrene surface.236 A surface binding peptide containing a pentaglycine was linked to the polystyrene surface at first and then the polymer ligated by SrtA. Dorr et al. showed the simultaneous dual surface modification with dyes as an application of their orthogonal sortase variants.28 Different dye molecules were equiped with the recognition sequences of these SrtA variants, LAETG and LPESG, respectively, and ligated to GGG-PEG functionalized surfaces of 96-well plates.
We performed a study in our group to demonstrate the universal use of sortase A for the linkage of artificial chemical structures.237 Silica nanoparticles of different size and the polymer blocks poly(ethylene glycol) as well as poly(N-isopropylacrylamide) (PNIPAM) were used as building blocks and functionalized with peptide motifs through thiol–ene reaction between cysteine residues and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds on the building blocks. Subsequently, NP–polymer, NP–NP, and polymer–polymer structures were successfully formed under sortase A catalysis (Scheme 21). After this proof of concept, more detailed investigations on the synthesis of hybrid materials by SML are ongoing.
C double bonds on the building blocks. Subsequently, NP–polymer, NP–NP, and polymer–polymer structures were successfully formed under sortase A catalysis (Scheme 21). After this proof of concept, more detailed investigations on the synthesis of hybrid materials by SML are ongoing.
 | ||
| Scheme 21 Overview of the functionalization of silica nanoparticles, PEG and PNIPAM polymer blocks with peptide sequences and the subsequent formation of NP–NP, NP–polymer and polymer–polymer structures catalyzed by SrtA. Reprinted from ref. 237. | ||
8. Summary and outlook
Sortase-mediated ligations have been extensively exploited in recent years and found a multitude of applications in diverse disciplines. This review gave an overview especially about applications related to chemical techniques like protein modification, synthesis of protein–polymer conjugates and protein immobilization. As sortagging was deeply investigated concerning methods to shift the equilibrium of the reaction, to immobilize sortase for re-use, to engineer variants with enhanced catalytic efficiency as well as orthogonal variants having different recognition sequences, it is obvious that SML is nowadays exploited for more and more advanced studies. For example, apart from recent studies presented in the main text, in the field of biocomputing, peptide logic circuits based on SrtA as logic processor have been developed.238 The ease of functionalization of biomolecules as well as non-biological substrates with peptide motifs facilitates these studies. It can be expected that this trend continues within the next years and the scope of sortagging can be further broadened. For instance, evolved sortase A variants were recently reported with increased resistance and catalytic efficiency in 45% (v/v) dimethylsulfoxide which can enable the ligation of partially hydrophobic substrates in future.239 In addition, a cyclized SrtA was formed with enhanced thermal stability and tolerance towards chemical denaturation.240In contrast to single modification, performing consecutive ligations with a SrtA variant is a difficult task. On the one side, the equilibrium of sortase reactions represents a challenge for obtaining high yields of the desired ligated products. On the other side and significantly important, the formed LPXTGGG bond is again a substrate for the enzyme. Therefore, strategies were developed with the subsequent use of different SrtA variants, mainly SrtApyogenes and SrtAaureus,121,158,159,200,210 as shown with examples in the Chapters 4.1 and 6.1. Furthermore, sortase variants with different substrate selectivities were developed and subsequently used for the ligation of different entities.28 However, using only one sortase variant for multiple ligations is seldom so far. While uncontrolled processes are limited to a low number of repeating units and unavoidable product mixtures,64,107,215 first attempts demonstrate the formation of polyproteins on surfaces in an interplay of SrtA with a protease.211,214
An interesting example for the application of different sortase variants represents the formation of isopeptide bonds. SrtA can form isopeptide bonds between LPXTG-tagged moieties and lysine –NH2 residues. However, the formation of these isopeptide bonds is restricted to lysines in the pilin domain and doesn't occur at other lysine residues in a protein.177 A dual site-specific labeling was shown with isopeptide bond formation in the pilin domain by mutated SrtA from Corynebacterium diphtheriae followed by N-terminal labeling of the target protein by SrtAaureus.241 The two SrtA variants have different recognition sequences which enabled the defined site-specific labeling with different dyes.
These examples illustrate that even more complex and more defined protein structures can be expected by SML in future. Shift of the reaction equilibrium, use of orthogonal variants and interplay with other enzymes provide interesting tools for multiple site-specific modification of proteins as well as formation of polyproteins.
In addition to sortases, other enzymes emerged in the last years that can be used for the modification of proteins.88,90 Among them, ligases like butelase 1, trypsiligase, subtiligase, and peptiligase catalyze peptide ligations, while cysteine residues can be modified using enzymes like formylglycine generating enzyme (FGE) and farnesyltransferase (FTase). For example, butelase 1 has a short C-terminal N/D-HV recognition motif and nearly all amino acids can function as N-terminal ligation partner. Interestingly, butelase 1 has an excellent catalytic activity being superior to sortase A in this respect.242 However, butelase 1 couldn't be recombinantly produced yet and had to be extracted from plants.88,90 Although other enzymes like butelase 1 provide certain advantages compared to sortase, the predominant limitations currently prevent their universal use and it will require intensive work until other enzymes can compete with the broad application fields of sortase-mediated ligations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – project number 414977640. This work was supported as Fraunhofer High Performance Center for Functional Integration in Materials.References
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–764 CrossRef CAS PubMed.
- T. Spirig, E. M. Weiner and R. T. Clubb, Mol. Microbiol., 2011, 82, 1044–1059 CrossRef CAS PubMed.
- K. W. Clancy, J. A. Melvin and D. G. McCafferty, Biopolymers, 2010, 94, 385–396 CrossRef CAS PubMed.
- W. J. Bradshaw, A. H. Davies, C. J. Chambers, A. K. Roberts, C. C. Shone and K. R. Acharya, FEBS J., 2015, 282, 2097–2114 CrossRef CAS PubMed.
- S. Cascioferro, D. Raffa, B. Maggio, M. V. Raimondi, D. Schillaci and G. Daidone, J. Med. Chem., 2015, 58, 9108–9123 CrossRef CAS PubMed.
- Y. Guo, S. Cai, G. Gu, Z. Guo and Z. Long, RSC Adv., 2015, 5, 49880–49889 RSC.
- U. Ilangovan, H. Ton-That, J. Iwahara, O. Schneewind and R. T. Clubb, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6056–6061 CrossRef CAS PubMed.
- Y. Zong, T. W. Bice, H. Ton-That, O. Schneewind and S. V. L. Narayana, J. Biol. Chem., 2004, 279, 31383–31389 CrossRef CAS PubMed.
- P. R. Race, M. L. Bentley, J. A. Melvin, A. Crow, R. K. Hughes, W. D. Smith, R. B. Sessions, M. A. Kehoe, D. G. McCafferty and M. J. Banfield, J. Biol. Chem., 2009, 284, 6924–6933 CrossRef CAS PubMed.
- J. L. Chen, X. Wang, F. Yang, C. Cao, G. Otting and X. C. Su, Angew. Chem., Int. Ed., 2016, 55, 13744–13748 CrossRef CAS PubMed.
- J. M. Antos, M. C. Truttmann and H. L. Ploegh, Curr. Opin. Struct. Biol., 2016, 38, 111–118 CrossRef CAS PubMed.
- L. Schmohl and D. Schwarzer, Curr. Opin. Chem. Biol., 2014, 22, 122–128 CrossRef CAS PubMed.
- Y. Yamamura, H. Hirakawa, S. Yamaguchi and T. Nagamune, Chem. Commun., 2011, 47, 4742–4744 RSC.
- H. Hirakawa, S. Ishikawa and T. Nagamune, Biotechnol. J., 2015, 10, 1487–1492 CrossRef CAS PubMed.
- M. Steinhagen, K. Zunker, K. Nordsieck and A. G. Beck-Sickinger, Bioorg. Med. Chem., 2013, 21, 3504–3510 CrossRef CAS PubMed.
- M. D. Witte, T. Wu, C. P. Guimaraes, C. S. Theile, A. E. M. Blom, J. R. Ingram, Z. Li, L. Kundrat, S. D. Goldberg and H. L. Ploegh, Nat. Protoc., 2015, 10, 508–516 CrossRef CAS PubMed.
- C. B. Rosen, R. L. Kwant, J. I. MacDonald, M. Rao and M. B. Francis, Angew. Chem., Int. Ed., 2016, 55, 8585–8589 CrossRef CAS PubMed.
- X. Zhao, H. Hong, X. Cheng, S. Liu, T. Deng, Z. Guo and Z. Wu, Sci. Rep., 2017, 7, 6561 CrossRef PubMed.
- M. Yang, H. Hong, S. Liu, X. Zhao and Z. Wu, Molecules, 2018, 23, 192 CrossRef PubMed.
- H. Wang, R. Luo, Y. Chen and Q. I. Si, J. Electron. Mater., 2018, 47, 4124–4135 CrossRef CAS.
- R. L. Policarpo, H. Kang, X. Liao, A. E. Rabideau, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2014, 53, 9203–9208 CrossRef CAS PubMed.
- H. H. Wang, B. Altun, K. Nwe and A. Tsourkas, Angew. Chem., Int. Ed., 2017, 56, 5349–5352 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed.
- L. Chen, J. Cohen, X. Song, A. Zhao, Z. Ye, C. J. Feulner, P. Doonan, W. Somers, L. Lin and P. R. Chen, Sci. Rep., 2016, 31899 CrossRef CAS PubMed.
- M. Suliman, V. Santosh, T. C. M. Seegar, A. C. Dalton, M. Schultz, C. S. Klug and W. A. Barton, PLoS One, 2017, 12, e0184271 CrossRef PubMed.
- H. Jeong, G. C. Abhiraman, C. M. Story, J. R. Ingram and K. Dougan, PLoS One, 2017, 12, e0189068 CrossRef PubMed.
- K. Piotukh, B. Geltinger, N. Heinrich, F. Gerth, M. Beyermann, C. Freund and D. Schwarzer, J. Am. Chem. Soc., 2011, 133, 17536–17539 CrossRef CAS PubMed.
- B. M. Dorr, H. O. Ham, C. An, E. L. Chaikof and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13343–13348 CrossRef CAS PubMed.
- N. Li, Z. Yu, Q. Ji, J. Sun, X. Liu and M. Du, Biochem. Biophys. Res. Commun., 2017, 486, 257–263 CrossRef CAS PubMed.
- L. Schmohl, J. Bierlmeier, F. Gerth, C. Freund and D. Schwarzer, J. Pept. Sci., 2017, 23, 631–635 CrossRef CAS PubMed.
- L. Schmohl, J. Bierlmeier, N. Von Kügelgen, L. Kurz, P. Reis, F. Barthels, P. Mach, M. Schutkowski, C. Freund and D. Schwarzer, Bioorg. Med. Chem., 2017, 25, 5002–5007 CrossRef CAS PubMed.
- K. D. Nikghalb, N. M. Horvath, J. L. Prelesnik, O. G. B. Banks, P. A. Filipov, R. D. Row, T. J. Roark and J. M. Antos, ChemBioChem, 2018, 19, 185–195 CrossRef CAS PubMed.
- J. P. Tam, J. Xu and K. D. Eom, Bioorg. Med. Chem., 2001, 60, 194–205 CAS.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- T. Matsumoto, T. Tanaka and A. Kondo, Langmuir, 2012, 28, 3553–3557 CrossRef CAS PubMed.
- W. Ott, T. Nicolaus, H. E. Gaub and M. A. Nash, Biomacromolecules, 2016, 17, 1330–1338 CrossRef CAS PubMed.
- T. Matsumoto, Y. Isogawa, K. Minamihata, T. Tanaka and A. Kondo, J. Biotechnol., 2016, 225, 61–66 CrossRef CAS PubMed.
- N. F. Ismail and T. S. Lim, Sci. Rep., 2016, 6, 19338 CrossRef CAS PubMed.
- K. Li, R. Zhang, Y. Xu, Z. Wu, J. Li and X. Zhou, Sci. Rep., 2017, 7, 3081 CrossRef PubMed.
- V. Ulrich and M. J. Cryle, J. Pept. Sci., 2017, 23, 16–27 CrossRef CAS PubMed.
- T. Matsumoto, K. Furuta, T. Tanaka and A. Kondo, ACS Synth. Biol., 2016, 5, 1284–1289 CrossRef CAS PubMed.
- K. Raltchev, J. Pipercevic and F. Hagn, Chem.–Eur. J., 2018, 24, 5493–5499 CrossRef CAS PubMed.
- K. Omura, Y. Aiba, H. Onoda, J. K. Stanfield, S. Ariyasu, H. Sugimoto, Y. Shiro, O. Shoji and Y. Watanabe, Chem. Commun., 2018, 54, 7892–7895 RSC.
- J. Wang, M. Hu, L. Zhang, X. Shao, C. Lv, Y. Liu, Z. Xu and Z. Guo, Biochimie, 2018, 154, 77–85 CrossRef CAS PubMed.
- A. J. Agwa, L. V Blomster, D. J. Craik, G. F. King and C. I. Schroeder, Bioconjugate Chem., 2018, 29, 3309–3319 CrossRef CAS PubMed.
- X. Cheng, T. Zhu, H. Hong, Z. Zhou and Z. Wu, Org. Chem. Front., 2017, 4, 2058–2062 RSC.
- J. J. Ling, R. L. Policarpo, A. E. Rabideau, X. Liao and B. L. Pentelute, J. Am. Chem. Soc., 2012, 134, 10749–10752 CrossRef CAS PubMed.
- H. Ke, S. Matsumoto, Y. Murashima, H. Taniguchi-tamura, R. Miyamoto, Y. Yoshikawa, C. Tsuda, T. Kumasaka, E. Mizohata and H. Edamatsu, et al., FEBS Lett., 2017, 591, 2470–2481 CrossRef CAS PubMed.
- S. A. M. Van Lith, D. Van Den Brand and R. Wallbrecher, ChemBioChem, 2017, 18, 2390–2394 CrossRef CAS PubMed.
- D. R. White, Z. Khedri, P. Kiptoo, T. J. Siahaan and T. J. Tolbert, Bioconjugate Chem., 2017, 28, 1867–1877 CrossRef CAS PubMed.
- F. Touti, G. Lautrette, K. D. Johnson, J. C. Delaney, A. Wollacott, H. Tissire, K. Viswanathan, Z. Shriver, S. K. Mong and A. J. Mijalis, et al., ChemBioChem, 2018, 19, 2039–2044 CrossRef CAS PubMed.
- D. T. Ta, R. Vanella and M. A. Nash, ACS Appl. Mater. Interfaces, 2018, 10, 30147–30154 CrossRef CAS PubMed.
- J. J. Bellucci, M. Amiram, J. Bhattacharyya, D. McCafferty and A. Chilkoti, Angew. Chem., Int. Ed., 2013, 52, 3703–3708 CrossRef CAS PubMed.
- T. Ohshima and M. Sakono, Bioconjugate Chem., 2017, 28, 2687–2691 CrossRef CAS PubMed.
- S. Pritz, O. Kraetke, A. Klose, J. Klose, S. Rothemund, K. Fechner, M. Bienert and M. Beyermann, Angew. Chem., Int. Ed., 2008, 47, 3642–3645 CrossRef CAS PubMed.
- L. Schmohl and D. Schwarzer, J. Pept. Sci., 2014, 20, 145–151 CrossRef CAS PubMed.
- S. Piluso, H. C. Cassell, J. L. Gibbons, T. E. Waller, N. J. Plant, A. F. Miller and G. Cavalli, Soft Matter, 2013, 9, 6752–6756 RSC.
- L. Freiburger, M. Sonntag, J. Hennig, J. Li, P. Zou and M. Sattler, J. Biomol. NMR, 2015, 63, 1–8 CrossRef CAS PubMed.
- F. P. Williams, A. G. Milbradt, K. J. Embrey and R. Bobby, PLoS One, 2016, 11, e0154607 CrossRef PubMed.
- M. Sonntag, P. Kumar, A. Jagtap, B. Simon, M. Appavou, A. Geerlof, R. Stehle, F. Gabel, J. Hennig and M. Sattler, Angew. Chem., Int. Ed., 2017, 56, 9322–9325 CrossRef CAS PubMed.
- B. R. Amer, R. MacDonald, A. W. Jacobitz, B. Liauw and R. T. Clubb, J. Biomol. NMR, 2016, 64, 197–205 CrossRef CAS PubMed.
- W. Van'T Hof, S. H. Maňásková, E. C. I. Veerman and J. G. M. Bolscher, Biol. Chem., 2015, 396, 283–293 Search PubMed.
- J. M. Antos, M. W. L. Popp, R. Ernst, G. L. Chew, E. Spooner and H. L. Ploegh, J. Biol. Chem., 2009, 284, 16028–16036 CrossRef CAS PubMed.
- J. Hu, W. Zhao, Y. Gao, M. Sun, Y. Wei, H. Deng and W. Gao, Biomaterials, 2015, 47, 13–19 CrossRef CAS PubMed.
- M. L. Nasr, D. Baptista, M. Strauss, Z. J. Sun, S. Grigoriu, S. Huser, A. Plückthun, F. Hagn, T. Walz and J. M. Hogle, et al., Nat. Methods, 2017, 14, 49–52 CrossRef CAS PubMed.
- Y. Yusuf, M. Julien, Y.-T. Chang, P.-H. Wu, V. Yeh, P.-C. Kuo, J. Shiue and T.-Y. Yu, Langmuir, 2018, 34, 3525–3532 CrossRef CAS PubMed.
- J. Zhang, S. Yamaguchi and T. Nagamune, Biotechnol. J., 2015, 10, 1499–1505 CrossRef CAS PubMed.
- N. Rasche, J. Tonillo, M. Rieker, S. Becker, B. Dorr, D. Ter-Ovanesyan, U. A. K. Betz, B. Hock and H. Kolmar, Bioconjugate Chem., 2016, 27, 1341–1347 CrossRef CAS PubMed.
- Z. M. Wu, S. Z. Liu, X. Z. Cheng, X. R. Zhao and H. F. Hong, Chin. Chem. Lett., 2017, 28, 553–557 CrossRef CAS.
- K. Stanger, T. Maurer, H. Kaluarachchi, M. Coons, Y. Franke and R. N. Hannoush, FEBS Lett., 2014, 588, 4487–4496 CrossRef CAS PubMed.
- S. Kwon, F. Bosmans, Q. Kaas, O. Cheneval, A. C. Conibear, K. J. Rosengren, C. K. Wang, C. I. Schroeder and D. J. Craik, Biotechnol. Bioeng., 2016, 113, 2202–2212 CrossRef CAS PubMed.
- N. Voloshchuk, L. Chen, Q. Li and J. F. Liang, Biochem. Biophys Rep., 2017, 10, 1–6 Search PubMed.
- Z. Wu, X. Cheng, H. Hong, X. Zhao and Z. Zhou, Bioorg. Med. Chem. Lett., 2017, 27, 1911–1913 CrossRef CAS PubMed.
- X. Cheng, H. Hong, Z. Zhou and Z. Wu, J. Org. Chem., 2018, 83, 14078–14083 CrossRef CAS PubMed.
- B. Khare and S. V. L. Narayana, Protein Sci., 2017, 26, 1458–1473 CrossRef CAS PubMed.
- I. von Ossowski, Int. J. Mol. Sci., 2017, 18, 1551 CrossRef CAS PubMed.
- M. Schmidt, A. Toplak, P. J. L. M. Quaed, J. H. Van Maarseveen and T. Nuijens, Drug Discovery Today: Technol., 2017, 26, 11–16 CrossRef PubMed.
- S. Tsukiji and T. Nagamune, ChemBioChem, 2009, 10, 787–798 CrossRef CAS PubMed.
- T. Proft, Biotechnol. Lett., 2009, 32, 1–10 CrossRef PubMed.
- M. W.-L. Popp and H. L. Ploegh, Angew. Chem., Int. Ed. Engl., 2011, 50, 5024–5032 CrossRef CAS PubMed.
- V. Haridas, S. Sadanandan and N. U. Dheepthi, ChemBioChem, 2014, 15, 1857–1867 CrossRef CAS PubMed.
- M. Ritzefeld, Chem.–Eur. J., 2014, 20, 8516–8529 CrossRef CAS PubMed.
- N. Pishesha, J. R. Ingram and H. L. Ploegh, Annu. Rev. Cell Dev. Biol., 2018, 34, 163–188 CrossRef CAS PubMed.
- Y. Zhang, K. Park, M. D. Distefano and K. Park, Chem. Soc. Rev., 2018, 47, 9106–9136 RSC.
- T. Matsumoto, T. Tanaka and A. Kondo, Biotechnol. J., 2012, 7, 1137–1146 CrossRef CAS PubMed.
- M. Rashidian, J. K. Dozier and M. D. Distefano, Bioconjugate Chem., 2013, 24, 1277–1294 CrossRef CAS PubMed.
- M. Schmidt, A. Toplak, P. J. Quaedflieg and T. Nuijens, Curr. Opin. Chem. Biol., 2017, 38, 1–7 CrossRef CAS PubMed.
- H. Kim, K. H. Siu, M. Raeeszadeh-Sarmazdeh, Q. Sun, Q. Chen and W. Chen, Biotechnol. Bioeng., 2015, 112, 1495–1505 CrossRef CAS PubMed.
- S. Lin and C. He, Chin. Chem. Lett., 2018, 29, 1017–1021 CrossRef CAS.
- J. W. Nelson, A. G. Chamessian, P. J. McEnaney, R. P. Murelli, B. I. Kazmiercak and D. A. Spiegel, ACS Chem. Biol., 2010, 5, 1147–1155 CrossRef CAS PubMed.
- S. H. Maòásková, K. Nazmi, W. Van'T Hof, A. Van Belkum, N. I. Martin, F. J. Bikker, W. J. B. Van Wamel and E. C. I. Veerman, PLoS One, 2016, 11, 1–14 Search PubMed.
- K. Park, J. Jung, J. Son, S. H. Kim and B. H. Chung, Chem. Commun., 2013, 49, 9585–9587 RSC.
- H. D. Nguyen, T. T. Phan and W. Schumann, AMB Express, 2011, 1, 22 CrossRef PubMed.
- B. J. Willson, K. Kovács, T. W. Steele, R. Markus, K. Winzer and N. P. Minton, Biotechnol. Biofuels, 2016, 9, 109 CrossRef PubMed.
- M. W. Popp, J. M. Antos, G. M. Grotenbreg, E. Spooner and H. L. Ploegh, Nat. Chem. Biol., 2007, 3, 707–708 CrossRef CAS PubMed.
- T. Tanaka, T. Yamamoto, S. Tsukiji and T. Nagamune, ChemBioChem, 2008, 9, 802–807 CrossRef CAS PubMed.
- T. Yamamoto and T. Nagamune, Chem. Commun., 2009, 1022–1024 RSC.
- U. Tomita, S. Yamaguchi, Y. Maeda, K. Chujo, K. Minamihata and T. Nagamune, Biotechnol. Bioeng., 2013, 110, 2785–2789 CrossRef CAS PubMed.
- L. K. Swee, S. Lourido, G. W. Bell, J. R. Ingram and H. L. Ploegh, ACS Chem. Biol., 2015, 10, 460–465 CrossRef CAS PubMed.
- N. Pishesha, A. M. Bilate, M. C. Wibowo, N. Huang, Z. Li, R. Dhesycka, D. Bousbaine, H. Li, H. C. Patterson and S. K. Dougan, et al., Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3157–3162 CrossRef CAS PubMed.
- M. Tan, S. Yamaguchi, M. Nakamura and T. Nagamune, J. Biosci. Bioeng., 2018, 126, 363–370 CrossRef CAS PubMed.
- G. Pasqual, A. Chudnovskiy, J. M. J. Tas, M. Agudelo, L. D. Schweitzer, A. Cui, N. Hacohen and G. D. Victora, Nature, 2018, 553, 496–500 CrossRef CAS PubMed.
- M. A. Koussa, M. Sotomayor and W. P. Wong, Methods, 2014, 67, 134–141 CrossRef CAS PubMed.
- S. Dickgiesser, N. Rasche, D. Nasu, S. Middel, S. Hörner, O. Avrutina, U. Diederichsen and H. Kolmar, ACS Chem. Biol., 2015, 10, 2158–2165 CrossRef CAS PubMed.
- J. M. Antos, G. M. Miller, G. M. Grotenbreg and H. L. Ploegh, J. Am. Chem. Soc., 2008, 130, 16338–16343 CrossRef CAS PubMed.
- R. Parthasarathy, S. Subramanian and E. T. Boder, Bioconjugate Chem., 2007, 18, 469–476 CrossRef CAS PubMed.
- X. Guo, Z. Wu and Z. Guo, Bioconjugate Chem., 2012, 23, 650–655 CrossRef CAS PubMed.
- A. Tabata, N. Anyoji, Y. Ohkubo, T. Tomoyasu and H. Nagamune, Anticancer Res., 2014, 34, 4521–4528 CAS.
- A. Tabata, Y. Ohkubo, N. Anyoji, K. Hojo, T. Tomoyasu, Y. Tatematsu, K. Ohkura and H. Nagamune, Anticancer Res., 2015, 35, 4411–4418 CAS.
- S. Wöll, C. Bachran, S. Schiller, M. Schröder, L. Conrad, L. Kim and R. Scherließ, Eur. J. Pharm. Biopharm., 2018, 133, 138–150 CrossRef PubMed.
- S. Wöll, C. Bachran, S. Schiller, M. Schröder, L. Conrad, R. Scherließ and L. Kim, Eur. J. Pharm. Biopharm., 2019, 134, 190–198 CrossRef PubMed.
- J. R. Silvius and R. Leventis, Bioconjugate Chem., 2017, 28, 1271–1282 CrossRef CAS PubMed.
- M. R. Pratt and C. R. Bertozzi, Chem. Soc. Rev., 2005, 34, 58–68 RSC.
- S. Samantaray, U. Marathe, S. Dasgupta, V. K. Nandicoori and R. P. Roy, J. Am. Chem. Soc., 2008, 130, 2132–2133 CrossRef CAS PubMed.
- X. Guo, Q. Wang, B. M. Swarts and Z. Guo, J. Am. Chem. Soc., 2009, 131, 9878–9879 CrossRef CAS PubMed.
- Z. Wu, X. Guo, Q. Wang, B. M. Swarts and Z. Guo, J. Am. Chem. Soc., 2010, 132, 1567–1571 CrossRef CAS PubMed.
- Z. Wu, X. Guo, J. Gao and Z. Guo, Chem. Commun., 2013, 49, 11689–11691 RSC.
- Z. Wu, X. Guo, G. Gu and Z. Guo, Org. Lett., 2013, 15, 5906–5908 CrossRef CAS PubMed.
- T. Fang, C. H. M. J. Van Elssen, J. N. Duarte, J. S. Guzman, J. S. Chahal and H. L. Ploegh, Chem. Sci., 2017, 8, 5591–5597 RSC.
- J. M. Antos, G. L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W. L. Popp and H. L. Ploegh, J. Am. Chem. Soc., 2009, 131, 10800–10801 CrossRef CAS PubMed.
- C. P. Guimaraes, M. D. Witte, C. S. Theile, G. Bozkurt, L. Kundrat, A. E. M. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1787–1799 CrossRef PubMed.
- R. Warden-Rothman, I. Caturegli, V. Popik and A. Tsourkas, Anal. Chem., 2013, 85, 11090–11097 CrossRef CAS PubMed.
- K. Sarpong and R. Bose, Anal. Biochem., 2017, 521, 55–58 CrossRef CAS PubMed.
- R. Jiang, L. Wang, J. Weingart and X. L. Sun, ChemBioChem, 2014, 15, 42–46 CrossRef CAS PubMed.
- P. N. Atterberry, T. J. Roark, S. Y. Severt, M. L. Schiller, J. M. Antos and A. R. Murphy, Biomacromolecules, 2015, 16, 1582–1589 CrossRef CAS PubMed.
- G. Y. Chen, Z. Li, J. N. Duarte, A. Esteban, R. W. Cheloha, C. S. Theile, G. R. Fink and H. L. Ploegh, Biosens. Bioelectron., 2017, 89, 789–794 CrossRef CAS PubMed.
- P. M. Morrison, M. R. Balmforth, S. W. Ness, D. J. Williamson, M. D. Rugen, W. B. Turnbull and M. E. Webb, ChemBioChem, 2016, 17, 753–758 CrossRef CAS PubMed.
- S. O. Crowe, G. H. Pham, J. C. Ziegler, K. K. Deol, R. G. Guenette, Y. Ge and E. R. Strieter, ChemBioChem, 2016, 1525–1531 CrossRef CAS PubMed.
- A. I. Petrache, D. C. Machin, D. J. Williamson, M. E. Webb and P. A. Beales, Mol. BioSyst., 2016, 12, 1760–1763 RSC.
- Q. Wu, H. L. Ploegh and M. C. Truttmann, ACS Chem. Biol., 2017, 12, 664–673 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- E. Lallana, R. Riguera and E. Fernandez-Megia, Angew. Chem., Int. Ed., 2011, 50, 8794–8804 CrossRef CAS PubMed.
- J. C. M. Van Hest and F. L. Van Delft, ChemBioChem, 2011, 12, 1309–1312 CrossRef CAS PubMed.
- K. Gupta, S. Singh, K. Gupta, N. Khan, D. Sehgal, V. Haridas and R. P. Roy, ChemBioChem, 2012, 13, 2489–2494 CrossRef CAS PubMed.
- L. Wang, R. Jiang, L. Wang, Y. Liu and X. L. Sun, Bioorg. Chem., 2016, 65, 159–166 CrossRef CAS PubMed.
- M. D. Witte, J. J. Cragnolini, S. K. Dougan, N. C. Yoder, M. W. Popp and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11993–11998 CrossRef CAS PubMed.
- M. D. Witte, C. S. Theile, T. Wu, C. P. Guimaraes, A. E. M. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1808–1819 CrossRef PubMed.
- T. J. Harmand, D. Bousbaine, A. Chan, X. Zhang, D. R. Liu, J. P. Tam and H. L. Ploegh, Bioconjugate Chem., 2018, 29, 3245–3249 CrossRef CAS PubMed.
- T. Matsumoto, Y. Isogawa, T. Tanaka and A. Kondo, Biosens. Bioelectron., 2018, 99, 56–61 CrossRef CAS PubMed.
- K. Alt, B. M. Paterson, E. Westein, S. E. Rudd, S. S. Poniger, S. Jagdale, K. Ardipradja, T. U. Connell, G. Y. Krippner and A. K. N. Nair, et al., Angew. Chem., Int. Ed., 2015, 54, 7515–7519 CrossRef CAS PubMed.
- S. Kwon, J. N. Duarte, Z. Li, J. J. Ling, O. Cheneval, T. Durek, C. I. Schroeder, D. J. Craik and H. L. Ploegh, ACS Chem. Biol., 2018, 13, 2973–2980 CrossRef CAS PubMed.
- Z. Li, C. S. Theile, G. Y. Chen, A. M. Bilate, J. N. Duarte, A. M. Avalos, T. Fang, R. Barberena, S. Sato and H. L. Ploegh, Angew. Chem., Int. Ed., 2015, 54, 11706–11710 CrossRef CAS PubMed.
- T. Bonnard, A. Jayapadman, J. A. Putri, J. Cui, Y. Ju, C. Carmichael, T. A. Angelovich, S. H. Cody, S. French and K. Pascaud, et al., ACS Nano, 2018, 12, 6988–6996 CrossRef CAS PubMed.
- L. Bartels, H. L. Ploegh, H. Spits and K. Wagner, Methods, 2019, 154, 93–101 CrossRef CAS PubMed.
- B. M. Paterson, K. Alt, C. M. Jeffery, R. I. Price, S. Jagdale, S. Rigby, C. C. Williams, K. Peter, C. E. Hagemeyer and P. S. Donnelly, Angew. Chem., Int. Ed., 2014, 53, 6115–6119 CrossRef CAS PubMed.
- S. Massa, N. Vikani, C. Betti, S. Ballet, S. Vanderhaegen, J. Steyaert, B. Descamps, C. Vanhove, A. Bunschoten and F. W. B. van Leeuwen, et al., Contrast Media Mol. Imaging, 2016, 11, 328–339 CrossRef CAS PubMed.
- M. Rashidian, L. Wang, J. G. Edens, J. T. Jacobsen, I. Hossain, Q. Wang, G. D. Victora, N. Vasdev, H. Ploegh and S. H. Liang, Angew. Chem., Int. Ed., 2016, 55, 528–533 CrossRef CAS PubMed.
- R. R. Beerli, T. Hell, A. S. Merkel and U. Grawunder, PLoS One, 2015, 10, e0131177 CrossRef PubMed.
- N. Stefan, R. Gébleux, L. Waldmeier, T. Hell, M. Escher, F. I. Wolter, U. Grawunder and R. R. Beerli, Mol. Cancer Ther., 2017, 16, 879–892 CrossRef CAS PubMed.
- T. Fang, J. N. Duarte, J. Ling, Z. Li, J. S. Guzman and H. L. Ploegh, Angew. Chem., Int. Ed., 2016, 55, 2416–2420 CrossRef CAS PubMed.
- S. A. M. Van Lith, S. M. J. Van Duijnhoven, A. C. Navis, W. P. J. Leenders, E. Dolk, J. W. H. Wennink, C. F. Van Nostrum and J. C. M. Van Hest, Bioconjugate Chem., 2017, 28, 539–548 CrossRef CAS PubMed.
- S. C. H. A. van der Steen, R. Raavé, S. Langerak, L. van Houdt, S. M. J. van Duijnhoven, S. A. M. van Lith, L. F. A. G. Massuger, W. F. Daamen, W. P. Leenders and T. H. van Kuppevelt, Eur. J. Pharm. Biopharm., 2017, 113, 229–239 CrossRef CAS PubMed.
- L. Pan, W. Zhao, J. Lai, D. Ding, Q. Zhang, X. Yang, M. Huang, S. Jin, Y. Xu and S. Zeng, et al., Small, 2017, 13, 1602267 CrossRef PubMed.
- Y. Xu, S. Jin, W. Zhao, W. Liu, D. Ding, J. Zhou and S. Chen, Int. J. Mol. Sci., 2017, 18, 2284 CrossRef PubMed.
- G. T. Hess, C. P. Guimaraes, E. Spooner, H. L. Ploegh and A. M. Belcher, ACS Synth. Biol., 2013, 2, 490–496 CrossRef CAS PubMed.
- G. T. Hess, J. J. Cragnolini, M. W. Popp, M. A. Allen, S. K. Dougan, E. Spooner, H. L. Ploegh, A. M. Belcher and C. P. Guimaraes, Bioconjugate Chem., 2012, 23, 1478–1487 CrossRef CAS PubMed.
- Q. Chen, Q. Sun, N. M. Molino, S.-W. Wang, E. T. Boder and W. Chen, Chem. Commun., 2015, 51, 12107–12110 RSC.
- Q. Sun, Q. Chen, D. Blackstock and W. Chen, ACS Nano, 2015, 9, 8554–8561 CrossRef CAS PubMed.
- A. R. Swartz, Q. Sun and W. Chen, Biomacromolecules, 2017, 18, 1654–1659 CrossRef CAS PubMed.
- S. Tang, B. Xuan, X. Ye, Z. Huang and Z. Qian, Sci. Rep., 2016, 6, 25741 CrossRef CAS PubMed.
- A. R. Swartz and W. Chen, Bioconjugate Chem., 2018, 29, 3113–3120 CrossRef CAS PubMed.
- L. Schoonen, J. Pille, A. Borrmann, R. J. M. Nolte and J. C. M. Van Hest, Bioconjugate Chem., 2015, 26, 2429–2434 CrossRef CAS PubMed.
- L. Schoonen, R. J. M. Nolte and J. C. M. van Hest, Nanoscale, 2016, 14467–14472 RSC.
- D. Patterson, B. Schwarz, J. Avera, B. Western, M. Hicks, P. Krugler, M. Terra, M. Uchida, K. Mccoy and T. Douglas, Bioconjugate Chem., 2017, 28, 2114–2124 CrossRef CAS PubMed.
- A. Thérien, M. Bédard, D. Carignan, G. Rioux, L. G. Landry, M. Ève, L. Gagné, M. Bolduc, P. Savard and D. Leclerc, J. Nanobiotechnol., 2017, 15, 54 CrossRef PubMed.
- S. Baer, J. Nigro, M. P. Madej, R. M. Nisbet, R. Suryadinata, G. Coia, L. P. T. Hong, T. E. Adams, C. C. Williams and S. D. Nuttall, Org. Biomol. Chem., 2014, 12, 2675–2685 RSC.
- J. E. Glasgow, M. L. Salit and J. R. Cochran, J. Am. Chem. Soc., 2016, 138, 7496–7499 CrossRef CAS PubMed.
- Y. Li, Y. Li, M. Pan, X. Kong, Y. Huang, Z. Hong and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 2198–2202 CrossRef CAS PubMed.
- X. Tan, M. Pan, Y. Zheng, S. Gao, L. Liang and Y. Li, Chem. Sci., 2017, 8, 6881–6887 RSC.
- T. Matsumoto, R. Takase, T. Tanaka, H. Fukuda and A. Kondo, Biotechnol. J., 2012, 7, 642–648 CrossRef CAS PubMed.
- S. M. Qafari and M. Mohammadi, RSC Adv., 2017, 7, 56006–56015 RSC.
- Z. Qu, V. Krishnamurthy, C. A. Haller, B. M. Dorr, U. M. Marzec, S. Hurst, M. T. Hinds, S. R. Hanson, D. R. Liu and E. L. Chaikof, Adv. Healthcare Mater., 2014, 3, 30–35 CrossRef CAS PubMed.
- H. Shi, Q. Shi, J. T. Oswald, Y. Gao, L. Li and Y. Li, Chem. Res. Chin. Univ., 2018, 34, 428–433 CrossRef CAS.
- J. J. Bellucci, J. Bhattacharyya and A. Chilkoti, Angew. Chem., Int. Ed., 2015, 54, 441–445 CAS.
- R. M. Broyer, G. N. Grover and H. D. Maynard, Chem. Commun., 2011, 47, 2212–2226 RSC.
- L. A. Canalle, D. W. P. M. Löwik and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 329–353 RSC.
- U. Glebe, B. Santos de Miranda, P. van Rijn and A. Böker, RSC Smart Mater., 2015, 1–29 CAS.
- E. M. Pelegri-Oday, E. W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef CAS PubMed.
- Y. Qi and A. Chilkoti, Polym. Chem., 2014, 5, 266–276 RSC.
- P. van Rijn and A. Böker, J. Mater. Chem., 2011, 21, 16735–16747 RSC.
- W. Zhao, F. Liu, Y. Chen, J. Bai and W. Gao, Polymer, 2015, 66, A1–A10 CrossRef CAS.
- D. Bontempo and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 6508–6509 CrossRef CAS PubMed.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS PubMed.
- Q. Zhang, Z. Li, P. Wilson and D. M. Haddleton, Chem. Commun., 2013, 49, 6608–6610 RSC.
- B. S. Sumerlin, ACS Macro Lett., 2012, 1, 141–145 CrossRef CAS.
- B. Zhao, J. Deng and J. Deng, ACS Macro Lett., 2017, 6, 6–10 CrossRef CAS.
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 6371–6379 CrossRef CAS.
- Q. Zhang, M. Li, C. Zhu, G. Nurumbetov, Z. Li, P. Wilson, K. Kempe and D. M. Haddleton, J. Am. Chem. Soc., 2015, 137, 9344–9353 CrossRef CAS PubMed.
- J. D. Wallat, K. A. Rose and J. K. Pokorski, Polym. Chem., 2014, 5, 1545–1558 RSC.
- B. Jung and P. Theato, Adv. Polym. Sci., 2012, 1–34 Search PubMed.
- S. Moelbert, E. Emberly and C. Tang, Protein Sci., 2004, 13, 752–762 CrossRef CAS PubMed.
- R. M. Broyer, G. M. Quaker and H. D. Maynard, J. Am. Chem. Soc., 2008, 130, 1041–1047 CrossRef CAS PubMed.
- Y. Qi, M. Amiram, W. Gao, D. G. McCafferty and A. Chilkoti, Macromol. Rapid Commun., 2013, 34, 1256–1260 CrossRef CAS PubMed.
- Y. Pang, J. Liu, Y. Qi, X. Li and A. Chilkoti, Angew. Chem., Int. Ed., 2016, 55, 10296–10300 CrossRef CAS PubMed.
- J. Hu, G. Wang, W. Zhao, X. Liu, L. Zhang and W. Gao, Biomaterials, 2016, 96, 84–92 CrossRef CAS PubMed.
- X. Liu, M. Sun, J. Sun, J. Hu, Z. Wang, J. Guo and W. Gao, J. Am. Chem. Soc., 2018, 140, 10435–10438 CrossRef CAS PubMed.
- M. W. Popp, S. K. Dougan, T.-Y. Chuang, E. Spooner and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3169–3174 CrossRef CAS PubMed.
- Y. Hou, J. Yuan, Y. Zhou, J. Yu and H. Lu, J. Am. Chem. Soc., 2016, 138, 10995–11000 CrossRef CAS PubMed.
- Y. Hou, Y. Zhou, H. Wang, R. Wang, J. Yuan, Y. Hu and K. Sheng, J. Am. Chem. Soc., 2018, 140, 1170–1178 CrossRef CAS PubMed.
- E. Steen Redeker, D. T. Ta, D. Cortens, B. Billen, W. Guedens and P. Adriaensens, Bioconjugate Chem., 2013, 24, 1761–1777 CrossRef CAS PubMed.
- M. Meldal and S. Schoffelen, F1000Research, 2016, 5, 2303 Search PubMed.
- F. Clow, J. D. Fraser and T. Proft, Biotechnol. Lett., 2008, 30, 1603–1607 CrossRef CAS PubMed.
- R. Jiang, J. Weingart, H. Zhang, Y. Ma and X. L. Sun, Bioconjugate Chem., 2012, 23, 643–649 CrossRef CAS PubMed.
- A. Sinisi, M. W.-L. Popp, J. M. Antos, W. Pansegrau, S. Savino, M. Nissum, R. Rappuoli, H. L. Ploegh and L. Buti, Bioconjugate Chem., 2012, 23, 1119–1126 CrossRef CAS PubMed.
- C. Uth, S. Zielonka, S. Hörner, N. Rasche, A. Plog, H. Orelma, O. Avrutina, K. Zhang and H. Kolmar, Angew. Chem., Int. Ed., 2014, 53, 12618–12623 CAS.
- R. K. Le, M. Raeeszadeh-Sarmazdeh, E. T. Boder and P. D. Frymier, Langmuir, 2015, 31, 1180–1188 CrossRef CAS PubMed.
- M. Raeeszadeh-Sarmazdeh, R. Parthasarathy and E. T. Boder, Colloids Surf., B, 2015, 128, 457–463 CrossRef CAS PubMed.
- M. Raeeszadeh-Sarmazdeh, R. Parthasarathy and E. T. Boder, Biotechnol. Prog., 2017, 33, 824–831 CrossRef CAS PubMed.
- S. Srinivasan, J. P. Hazra, G. S. Singaraju, D. Deb and S. Rakshit, Anal. Biochem., 2017, 535, 35–42 CrossRef CAS PubMed.
- E. Durner, W. Ott, M. A. Nash and H. E. Gaub, ACS Omega, 2017, 2, 3064–3069 CrossRef CAS PubMed.
- S. Garg, G. S. Singaraju, S. Yengkhom and S. Rakshit, Bioconjugate Chem., 2018, 29, 1714–1719 CrossRef CAS PubMed.
- H. Liu, D. T. Ta and M. A. Nash, Small Methods, 2018, 2, 1800039 CrossRef.
- W. Ott, E. Durner and H. E. Gaub, Angew. Chem., Int. Ed., 2018, 57, 12666–12669 CrossRef CAS PubMed.
- V. Hilberg, O. Avrutina, A. Ebenig, D. Yanakieva, T. Meckel, M. Biesalski and H. Kolmar, Chem.–Eur. J., 2018 DOI:10.1002/chem.201804889.
- H. O. Ham, Z. Qu, C. A. Haller, B. M. Dorr, E. Dai, W. Kim, D. R. Liu and E. L. Chaikof, Nat. Commun., 2016, 7, 11140 CrossRef CAS PubMed.
- L. Chan, H. F. Cross, J. K. She, G. Cavalli, H. F. P. Martins and C. Neylon, PLoS One, 2007, 2, e1164 CrossRef PubMed.
- T. Ito, R. Sadamoto, K. Naruchi, H. Togame, H. Takemoto, H. Kondo and S. I. Nishimura, Biochemistry, 2010, 49, 2604–2614 CrossRef CAS PubMed.
- S. Wu and T. Proft, Biotechnol. Lett., 2010, 32, 1713–1718 CrossRef CAS PubMed.
- H. T. Ta, S. Prabhu, E. Leitner, F. Jia, D. Von Elverfeldt, K. E. Jackson, T. Heidt, A. K. N. Nair, H. Pearce and C. Von Zur Muhlen, et al., Circ. Res., 2011, 109, 365–373 CrossRef CAS PubMed.
- M. K. M. Leung, C. E. Hagemeyer, A. P. R. Johnston, C. Gonzales, M. M. J. Kamphuis, K. Ardipradja, G. K. Such, K. Peter and F. Caruso, Angew. Chem., Int. Ed., 2012, 51, 7132–7136 CrossRef CAS PubMed.
- C. E. Hagemeyer, K. Alt, A. P. R. Johnston, G. K. Such, H. T. Ta, M. K. M. Leung, S. Prabhu, X. Wang, F. Caruso and K. Peter, Nat. Protoc., 2014, 10, 90–105 CrossRef PubMed.
- W. Kim, C. Haller, E. Dai, X. Wang, C. E. Hagemeyer, D. R. Liu, K. Peter and E. L. Chaikof, Angew. Chem., Int. Ed., 2015, 54, 1461–1465 CrossRef CAS PubMed.
- T. Heck, P. H. Pham, F. Hammes, L. Thöny-Meyer and M. Richter, Bioconjugate Chem., 2014, 25, 1492–1500 CrossRef CAS PubMed.
- Y. Hata, T. Matsumoto, T. Tanaka and A. Kondo, Macromol. Biosci., 2015, 15, 1375–1380 CrossRef CAS PubMed.
- S. M. Qafari, G. Ahmadian and M. Mohammadi, RSC Adv., 2017, 7, 56006–56015 RSC.
- E. Cambria, K. Renggli, C. C. Ahrens, C. D. Cook, C. Kroll, A. T. Krueger, B. Imperiali and L. G. Griffith, Biomacromolecules, 2015, 16, 2316–2326 CrossRef CAS PubMed.
- J. Valdez, C. D. Cook, C. Chopko, A. J. Wang, A. Brown, M. Kumar, L. Stockdale, D. Rothenberg, K. Renggli and E. Gordon, et al., Biomaterials, 2017, 130, 90–103 CrossRef CAS PubMed.
- E. Gau, D. M. Mate, Z. Zou, A. Oppermann, A. Töpel, F. Jakob, D. Wöll, U. Schwaneberg and A. Pich, Biomacromolecules, 2017, 18, 2789–2798 CrossRef CAS PubMed.
- M. R. Arkenberg and C.-C. Lin, Biomater. Sci., 2017, 5, 2231–2240 RSC.
- M. R. Arkenberg, D. M. Moore and C. Lin, Acta Biomater., 2019, 83, 83–95 CrossRef CAS PubMed.
- N. Broguiere, F. Formica, G. Barreto and M. Zenobi-wong, Acta Biomater., 2018, 77, 182–190 CrossRef CAS PubMed.
- L. Trachsel, N. Broguiere, J. Rosenboom, M. Zenobi-wong and E. M. Benetti, J. Mater. Chem. B, 2018, 6, 7568–7572 RSC.
- T. Sijbrandij, N. Cukkemane, K. Nazmi, E. C. I. Veerman and F. J. Bikker, Bioconjugate Chem., 2013, 24, 828–831 CrossRef CAS PubMed.
- X. Dai, D. Mate, U. Glebe, T. Mirzaei Garakani, A. Körner, U. Schwaneberg and A. Böker, Polymer, 2018, 10, 151 Search PubMed.
- Y. Li, S. Sun, L. Fan, S. Hu, Y. Huang, K. Zhang and Z. Nie, Angew. Chem., Int. Ed., 2017, 56, 14888–14892 CrossRef CAS PubMed.
- Z. Zou, H. Alibiglou, D. M. Mate, M. D. Davari and U. Schwaneberg, Chem. Commun., 2018, 54, 11467–11470 RSC.
- M. Pelay-gimeno, T. Bange, S. Hennig and T. N. Grossmann, Angew. Chem., Int. Ed., 2018, 57, 11164–11170 CrossRef CAS PubMed.
- S. A. Mcconnell, B. R. Amer, J. Muroski, J. Fu, C. Chang, R. R. O. Loo, J. A. Loo, J. Osipiuk, H. Ton-that and R. T. Clubb, J. Am. Chem. Soc., 2018, 140, 8420–8423 CrossRef CAS PubMed.
- G. K. T. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |